

Abstract
Background and Aims
Risk stratification after cure from hepatitis C virus (HCV) infection remains a clinical challenge. We investigated the predictive value of noninvasive surrogates of portal hypertension (liver stiffness measurement [LSM] by vibration‐controlled transient elastography and von Willebrand factor/platelet count ratio [VITRO]) for development of hepatic decompensation and hepatocellular carcinoma in patients with pretreatment advanced chronic liver disease (ACLD) who achieved HCV cure.
Approach and Results
A total of 276 patients with pretreatment ACLD and information on pretreatment and posttreatment follow‐up (FU)‐LSM and FU‐VITRO were followed for a median of 36.6 months after the end of interferon‐free therapy. FU‐LSM (area under the receiver operating characteristic curve [AUROC]: 0.875 [95% confidence interval [CI]: 0.796‐0.954]) and FU‐VITRO (AUROC: 0.925 [95% CI: 0.874‐0.977]) showed an excellent predictive performance for hepatic decompensation. Both parameters provided incremental information and were significantly associated with hepatic decompensation in adjusted models. A previously proposed combined approach (FU‐LSM < 12.4 kPa and/or FU‐VITRO < 0.95) to rule out clinically significant portal hypertension (CSPH, hepatic venous pressure gradient ≥10 mm Hg) at FU assigned most (57.3%) of the patients to the low‐risk group; none of these patients developed hepatic decompensation. In contrast, in patients in whom FU‐CSPH was ruled in (FU‐LSM > 25.3 kPa and/or FU‐VITRO > 3.3; 25.0% of patients), the risk of hepatic decompensation at 3 years following treatment was high (17.4%). Patients within the diagnostic gray‐zone for FU‐CSPH (17.8% of patients) had a very low risk of hepatic decompensation during FU (2.6%). The prognostic value of this algorithm was validated in an internal (n = 86) and external (n = 162) cohort.
Conclusion
FU‐LSM/FU‐VITRO are strongly and independently predictive of posttreatment hepatic decompensation in HCV‐induced ACLD. An algorithm combining these noninvasive markers not only rules in or rules out FU‐CSPH, but also identifies populations at negligible versus high risk for hepatic decompensation. FU‐LSM/FU‐VITRO are readily accessible and enable risk stratification after sustained virological response, and thus facilitate personalized management.
Abbreviations
- ACLD
advanced chronic liver disease
- aHR
adjusted hazard ratio
- AUROC
area under the receiver operating characteristic curve
- BL
baseline
- cACLD
compensated ACLD
- CHC
chronic hepatitis C
- CI
confidence interval
- CLD
chronic liver disease
- CSPH
clinically significant portal hypertension
- CTP
Child‐Turcotte‐Pugh
- dACLD
decompensated ACLD
- FU
follow‐up
- HCC
hepatocellular carcinoma
- HE
hepatic encephalopathy
- HVPG
hepatic venous pressure gradient
- IFN
interferon
- IQR
interquartile range
- LSM
liver stiffness measurement
- MELD
Model for End‐Stage Liver Disease
- NIT
noninvasive test
- NPV
negative predictive value
- NSBB
nonselective betablocker
- OLT
orthotopic liver transplantation
- PH
portal hypertension
- PLT
platelet count
- PPV
positive predictive value
- SVR
sustained virologic response
- VCTE
vibration‐controlled transient elastography
- VITRO
von Willebrand factor antigen/platelet count ratio
- VWF
von Willebrand factor
Interferon (IFN)‐free therapies for chronic hepatitis C are highly effective, achieving sustained virologic response (SVR) (i.e., HCV cure) in almost all patients with advanced chronic liver disease (ACLD).( 1 ) Although SVR ameliorates portal hypertension (PH) in most patients with ACLD treated with IFN‐free regimens,( 2 , 3 , 4 , 5 ) a considerable proportion still remains at risk for hepatic decompensation.( 6 , 7 , 8 ) Thus, risk‐stratification concepts to facilitate personalized follow‐up (FU) in these patients are urgently needed and currently a matter of debate. Recently, we have shown that posttreatment hepatic venous pressure gradient (HVPG) predicts hepatic decompensation in patients who achieved SVR with a higher discriminatory ability, as compared with pretreatment values.( 9 ) Patients without posttreatment clinically significant PH (CSPH) (i.e., HVPG values ≤9 mm Hg) were protected from hepatic decompensation, whereas the risk was highest in patients with HVPG values ≥16 mm Hg. Because HVPG measurement is limited by its invasiveness and restricted availability, simple noninvasive methods for risk stratification after SVR are needed to facilitate personalized management.
Liver stiffness measurement (LSM) by vibration‐controlled transient elastography (VCTE) is a well‐established indicator of the presence of compensated ACLD (cACLD), CSPH, and varices needing treatment, as reflected by the Baveno VI consensus.( 10 ) Moreover, von Willebrand factor (VWF), a marker for endothelial dysfunction,( 11 , 12 ) has been shown to indicate the presence of CSPH in patients with compensated cirrhosis.( 13 ) In a recent study from our group,( 14 ) the VWF antigen/platelet count ratio (VITRO) showed a numerically better diagnostic performance for posttreatment CSPH than plasma VWF levels alone, which may be explained by the high diagnostic accuracy of platelet count (PLT) in the post‐SVR setting.( 15 )
Of note, the predictive value of plasma VWF levels was independent of HVPG in previous studies, indicating that its prognostic significance reaches beyond the mere association with HVPG.( 11 , 16 ) Similarly, changes in LSM have been shown to provide HVPG‐independent prognostic information.( 17 )
However, while the prognostic value of LSM by VCTE and VWF in patients with progressive liver disease is well‐established, data on their use for risk stratification after SVR are scarce. Therefore, we investigated the prognostic values of pretreatment values, changes in, and posttreatment values of LSM, PLT, VWF, and VITRO in patients with ACLD who achieved SVR to IFN‐free therapies.
Patients and Methods
Derivation Cohort
A total of 389 patients receiving IFN‐free treatment at the Medical University of Vienna with pretreatment ACLD (defined as baseline [BL] LSM ≥ 10 kPa, HVPG ≥ 6 mm Hg, or advanced fibrosis/cirrhosis on liver histology), who were registered in a local database, were screened for eligibility for this retrospective study based on prospectively collected data (Fig. 1). Sixteen patients were excluded due to concomitant hepatocellular carcinoma (HCC), 1 patient was diagnosed with porto‐sinusoidal vascular disease, 1 patient underwent orthotopic liver transplantation (OLT) during treatment, and 11 patients were lost to FU. Of note, 84 patients were excluded due to missing information on either LSM or VITRO before or after the end of treatment. Finally, 276 patients were included in the derivation cohort. Of these, 80 patients with paired HVPG measurements were included in a previous study investigating the prognostic value of HVPG after SVR.( 9 ) Importantly, this study did not evaluate the prognostic performance of noninvasive markers.
Fig. 1.
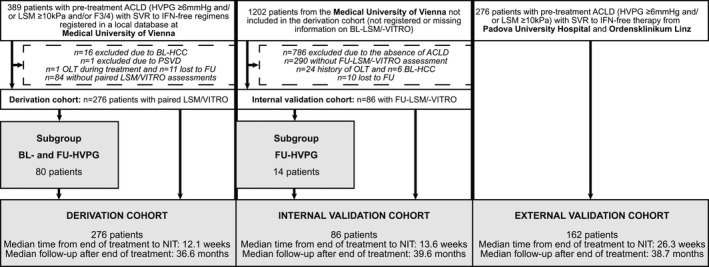
Study flow chart. Abbreviation: PSVD, porto‐sinusoidal vascular disease.
Internal and External Validation Cohorts
By querying the laboratory database of the Division of Clinical Virology of the Medical University of Vienna and applying similar inclusion and exclusion criteria (although only FU‐LSM/FU‐VITRO assessment was required) as in the derivation cohort, 86 additional patients were included in an internal validation cohort.
A total of 162 patients from Padua University Hospital and Ordensklinikum Linz Barmherzige Schwestern who met the inclusion and exclusion criteria of the derivation cohort were included as an external validation cohort. Some of these patients have been included in previously published studies focusing on changes in coagulation after HCV cure( 18 ) as well as the predictive value of VITRO for hepatic decompensation.( 19 )
Clinical and Laboratory Parameters
Clinical and laboratory parameters were evaluated by chart review. Plasma VWF antigen levels were measured by a latex agglutination assay (STA LIATEST VWF; Diagnostica Stago, Asnieres, France). VITRO score was calculated by dividing VWF (%) over PLT (g × L−1), as previously described.( 14 )
HCV Therapy
All patients were treated with IFN‐free therapies. The choice of the regimen was at the physicians’ discretion and depended on their availability, reimbursement policies, and national as well as and international clinical practice guidelines at the time of treatment initiation.( 20 , 21 , 22 , 23 , 24 ) Treatment durations ranged from 8 to 24 weeks.
Liver Stiffness and HVPG Measurement
In the derivation cohort, paired measurements of noninvasive markers, and in a subgroup of 80 patients, HVPG, were performed before antiviral therapy as well as at the end of therapy. In the internal validation cohort, noninvasive markers, and ‐ in a subgroup of patients ‐ HVPG, were only perfomed post‐treatment, while in the external validation cohort, only information on noninvasive tests was available. Due to the retrospective design of this study and logistical reasons, the time points were not standardized. VCTE (FibroScan; Echosens, Paris, France) was used for LSM. HVPG measurements were performed by the Vienna Hepatic Hemodynamic Lab at the Medical University of Vienna in accordance with a standardized operating procedure( 25 ) and in the absence of nonselective betablockers (NSBBs) or nitrates. In patients on NSBBs, treatment was interrupted 5 days before HVPG assessment. CSPH and high‐risk PH were defined by HVPG values ≥10 mm Hg and ≥16 mm Hg, respectively.
All LSM/HVPG measurements were performed after a minimum fasting period of 4 hours.
Clinical Events
In patients with cACLD, hepatic decompensation was defined by variceal bleeding, incident ascites, or incident hepatic encephalopathy (HE), whereas in patients with decompensated ACLD (dACLD), further hepatic decompensation was defined by variceal (re)bleeding, requirement of paracentesis, or development of grade 3/4 HE. Moreover, de novo HCC, transjugular intrahepatic portosystemic shunt implantation, and OLT were recorded.
LSM‐Based and VITRO‐Based Risk Stratification Algorithm
Using a previously described step‐wise approach,( 9 ) patients with a LSM and/or VITRO < 12.4 kPa and <0.95, respectively, were assigned to the CSPH ruled‐out/low‐risk group, while patients with a LSM and/or VITRO > 25.3 kPa and >3.3, respectively, were assigned to the CSPH ruled‐in/high‐risk group. Patients not meeting these criteria were analyzed in the gray‐zone group.
Statistical Analyses
See the Supporting Information materials.
Ethics
This study was approved by the ethics committee of the Medical University of Vienna (EK‐Nr 1947/2019), Upper Austria (K‐49‐14), and Padua University Hospital (3103/A0/14). Written informed consent was obtained if the requirement was not waived by the local ethics committee.
Results
Patient Characteristics of the Derivation Cohort
A total of 174 (63.0%) male and 102 (37.0%) female patients with a mean age of 56.0 ± 10.6 years were included. A total of 250 patients (90.6%) had cACLD, while 26 (9.4%) had dACLD. BL Child‐Turcotte‐Pugh (CTP) stage was A in 256 patients (92.8%) and B/C in 20 (7.2%). Overall, 62 patients (22.5%) had varices on gastroscopy. A BL Model for End‐Stage Liver Disease (MELD) score was 8.6 ± 2.6 points, BL‐LSM was 17.1 kPa (interquartile range [IQR]: 15.6), plasma BL‐VWF level was 233% (IQR: 144), and BL‐VITRO was 1.69 (IQR: 2.08). When comparing characteristics of patients with hepatic decompensation during FU (n = 12, 4.3%) to those without (n = 264, 95.7%), varices were significantly more common (n = 10, 83.3% vs. n = 52, 19.7%; P < 0.001), patients had a higher BL‐MELD score (12.1 ± 2.2 vs. 8.5 ± 2.5 points; P < 0.001), and lower serum BL albumin levels (36.2 ± 4.9 vs. 41.4 ± 4.5 g/L; P < 0.001). Moreover, patients developing hepatic decompensation had a higher BL‐LSM (33.4 [IQR: 28.3] vs. 16.6 [IQR: 15.1] kPa; P = 0.001), lower BL‐PLT (74 ± 39 vs. 150 ± 68 g/L; P < 0.001), higher plasma BL‐VWF levels (375% [IQR: 175] vs. 232% [IQR: 133]; P = 0.003), and higher BL‐VITRO (5.40 [IQR: 5.62] vs. 1.60 [IQR: 1.84]; P < 0.001). Importantly, differences in these markers of PH tended to be more pronounced at FU than at BL (FU‐LSM: 12.2 kPa [IQR: 13.5] vs. 44.0 kPa [IQR: 44.9]; P < 0.001; FU‐VITRO: 1.12 [IQR: 1.39] vs. 5.66 [IQR: 6.34]; P < 0.001), suggesting that FU values are more discriminative, and therefore more suitable for risk prediction than BL values (Table 1).
Table 1.
Characteristics and Comparison of Patients in the Derivation Cohort With and Without Hepatic Decompensation During FU
| Patients Characteristics | All Patients, n = 276 | No Hepatic Decompensation During FU, n = 264 | Hepatic Decompensation During FU, n = 12 | P Value |
|---|---|---|---|---|
| Age, years | 56.0 ± 10.6 | 56.1 ± 10.6 | 51.6 ± 9.1 | 0.139 |
| Sex | ||||
| Male | 174 (63.0%) | 166 (62.9%) | 8 (66.7%) | 1.000 |
| Female | 102 (37.0%) | 98 (37.1%) | 4 (33.3%) | |
| History of hepatic decompensation | 26 (9.4%) | 21 (8.0%) | 5 (41.7%) | 0.003 |
| Variceal bleeding | 6 (2.2%) | 5 (1.9%) | 1 (8.3%) | 0.236 |
| Ascites | 18 (6.5%) | 14 (5.3%) | 4 (33.3%) | 0.005 |
| HE | 3 (1.1%) | 3 (1.1%) | 0 (0.0%) | 1.000 |
| Varices | 62 (22.5%) | 52 (19.7%) | 10 (83.3%) | <0.001 |
| Small | 33 (53.2%) | 31 (59.6%) | 2 (20.0%) | <0.001 |
| Large | 29 (46.8%) | 21 (40.4%) | 8 (80.0%) | |
| NSBB users | 70 (25.4%) | 61 (23.1%) | 9 (75.0%) | <0.001 |
| BL‐CTP, points | 5 ± 1 | 5 ± 1 | 6 ± 1 | 0.011 |
| Stage A | 256 (92.8%) | 247 (93.6%) | 9 (75.0%) | 0.047 |
| Stage B/C | 20 (7.2%) | 17 (6.4%) | 3 (25.0%) | |
| Δ CTP, points | 0 (0) | 0 (0) | 0 (1) | 0.722 |
| FU‐CTP, points | 5 ± 1 | 5 ± 1 | 6 ± 1 | <0.001 |
| BL‐MELD, points | 8.6 ± 2.6 | 8.5 ± 2.5 | 12.1 ± 2.2 | <0.001 |
| Δ MELD, points | 0 (2) | 0 (2) | −0.5 (3) | 0.413 |
| FU‐MELD, points | 8.8 ± 3.3 | 8.7 ± 3.3 | 11.6 ± 1.9 | 0.003 |
| BL‐albumin, g × L−1 | 41.1 ± 4.6 | 41.4 ± 4.5 | 36.2 ± 4.9 | <0.001 |
| Absolute Δ albumin, g × L−1 | 1.9 (4.5) | 1.9 (4.5) | −0.1 (5.3) | 0.129 |
| Relative Δ albumin, % | 4.4 (11.9) | 4.6 (11.9) | −0.3 (15.1) | 0.161 |
| FU‐albumin, g × L−1 | 43.1 ± 4.5 | 43.4 ± 4.2 | 36.7 ± 4.9 | <0.001 |
| BL‐LSM, kPa | 17.1 (15.6) | 16.6 (15.1) | 33.4 (28.3) | 0.001 |
| Absolute Δ LSM, kPa | −3.6 (7.4) | −3.6 (7.2) | 8.5 (32.3) | 0.064 |
| Relative Δ LSM, % | −20.7 (39.2) | −21.2 (37.9) | 22.2 (89.5) | 0.010 |
| FU‐LSM, kPa | 12.7 (14.3) | 12.2 (13.5) | 44.0 (44.9) | <0.001 |
| BL‐PLT, g × L−1 | 146 ± 69 | 150 ± 68 | 74 ± 39 | <0.001 |
| Absolute Δ PLT, g × L−1 | 9 (28) | 10 (27) | −6 (38) | 0.021 |
| Relative Δ PLT, % | 6.8 (20.8) | 7.0 (20.2) | −11.5 (43.9) | 0.017 |
| FU‐PLT, g × L−1 | 158 ± 72 | 162 ± 71 | 69 ± 33 | <0.001 |
| BL‐VWF, % | 233 (144) | 232 (133) | 375 (175) | 0.003 |
| Absolute Δ VWF, % | −38 (68) | −41 (71) | 2 (66) | 0.001 |
| Relative Δ VWF, % | −18.4 (25.2) | −19.3 (25.9) | 0.4 (20.1) | <0.001 |
| FU‐VWF, % | 180 (105) | 179 (100) | 379 (174) | <0.001 |
| BL‐VITRO | 1.69 (2.08) | 1.60 (1.84) | 5.40 (5.62) | <0.001 |
| Absolute Δ VITRO | −0.32 (0.80) | −0.33 (0.80) | 0.11 (1.66) | 0.003 |
| Relative Δ VITRO, % | −23.3 (32.4) | −25.1 (30.3) | 3.7 (40.5) | <0.001 |
| FU‐VITRO | 1.15 (1.52) | 1.12 (1.39) | 5.66 (6.34) | <0.001 |
The prevalence of components of the metabolic syndrome, hepatic steatosis, and alcohol consumption is shown in Supporting Table S1. Although the metabolic phenotype was comparable between patients with and without hepatic decompensation during FU, alcohol consumption above the sex‐specific thresholds was more common in patients who developed posttreatment hepatic decompensation (n = 6, 50% vs. n = 24, 9.1%).
Clinical Events During Follow‐up in the Derivation Cohort
Patients were followed for a median of 36.6 (IQR: 22.2) months. The following first decompensating events occurred: variceal bleeding in 2 (0.7%) patients, ascites in 5 (1.8%) patients, and HE in 5 (1.8%) patients. Fourteen (5.1%) patients developed de novo HCC, while 4 patients (1.4%) underwent OLT and 11 (4.0%) patients died. Notably, because six (2.2%) deaths were non–liver‐related (most commonly due to extrahepatic malignancies), only 5 (1.8%) liver‐related deaths occurred. Among these, most patients had been diagnosed with HCC during follow‐up (n = 4, 80.0%).
Overall, 12 (4.3%) patients experienced hepatic decompensation before being censored/devolving a competing event, resulting in an incidence rate of 0.96 per 100 patient‐years for hepatic decompensation and 1.10 for de novo HCC.
Noninvasive Prediction of Hepatic Decompensation in the Derivation Cohort
The median time from the end of treatment to the FU assessment of noninvasive tests (NIT) was 12.1 (IQR: 24) weeks (i.e., the time points clustered around SVR12).
Investigating the area under the receiver operating characteristic curve (AUROC) of noninvasive markers to predict hepatic decompensation during FU, FU‐VITRO (AUROC = 0.925; 95% confidence interval [CI]: 0.874‐0.977), FU‐VWF (AUROC = 0.871; 95% CI: 0.757‐0.986), FU‐LSM (AUROC = 0.875; 95% CI: 0.796‐0.954), FU‐PLT (AUROC = 0.883; 95% CI: 0.815‐0.951), and FU‐albumin (AUROC = 0.858; 95% CI: 0.756‐0.960) yielded the highest accuracies (Table 2). Interestingly, BL values as well as absolute and relative changes of these parameters did not reach the same diagnostic accuracy (Table 2 and Supporting Fig. S1), confirming that noninvasive markers are more accurate if assessed at FU.
Table 2.
AUROC Values, Youden’s Index–Optimized Cutoffs, and Diagnostic Indices of Potential Predictors of Hepatic Decompensation During FU in the Derivation Cohorts
| Parameter | AUROC (95% CI) | Cutoff | Sensitivity | Specificity | PPV | NPV |
|---|---|---|---|---|---|---|
| BL‐MELD | 0.880 (0.804‐0.956) | >9 points* | 91.7% | 77.3% | 15.5% | 99.5% |
| FU‐MELD | 0.879 (0.817‐0.941) | >9 points* | 83.3% | 79.1% | 15.6% | 99.0% |
| BL‐albumin | 0.772 (0.618‐0.927) | <36.9 g × L−1* | 75.0% | 85.2% | 18.8% | 98.7% |
| FU‐albumin | 0.858 (0.756‐0.960) | <42.5 g × L−1* | 91.7% | 66.0% | 11.0% | 99.4% |
| BL‐LSM | 0.812 (0.721‐0.904) | >24.9 kPa* | 83.3% | 72.3% | 12.0% | 99.0% |
| FU‐LSM | 0.875 (0.796‐0.954) | >12.4 kPa | 100% | 50.4% | 8.4% | 100% |
| >25.3 kPa | 66.7% | 80.7% | 13.6% | 98.2% | ||
| BL‐PLT | 0.837 (0.739‐0.935) | <123 g × L−1* | 91.7% | 61.7% | 9.8% | 99.4% |
| FU‐PLT | 0.883 (0.815‐0.951) | <111 g × L−1* | 91.7% | 73.9% | 13.8% | 99.5% |
| BL‐VWF | 0.758 (0.604‐0.911) | >365%* | 66.7% | 87.5% | 19.5% | 98.3% |
| FU‐VWF | 0.871 (0.757‐0.986) | >221%* | 91.7% | 71.6% | 12.8% | 99.5% |
| BL‐VITRO | 0.857 (0.762‐0.952) | >3.58* | 75.0% | 85.2% | 18.8% | 98.7% |
| FU‐VITRO | 0.925 (0.874‐0.977) | >0.95 | 100% | 42.4% | 7.3% | 100% |
| >3.3 | 75.0% | 88.6% | 23.1% | 98.7% |
Youden’s index–optimized cutoff.
Abbreviations: NPV, negative predictive value; PPV, positive predictive value.
In Cox regression investigating factors associated with hepatic decompensation, FU‐VITRO (adjusted hazard ratio [aHR]: 1.044; 95% CI: 1.015‐1.072; P = 0.002) and FU‐LSM (aHR: 1.386; 95% CI: 1.185‐1.622; P < 0.001) were associated with hepatic decompensation after adjusting for history of hepatic decompensation, FU‐MELD score, and serum FU‐albumin levels. In addition, they were independently associated with hepatic decompensation, indicating that both variables provide incremental prognostic information (Table 3), although FU‐LSM and FU‐VITRO showed a positive correlation of moderate strength (Spearman’s ρ = 0.644; P < 0.001).
Table 3.
Cox Regression Analyses Investigating Factors Associated With Hepatic Decompensation During FU in the Derivation Cohort
| Parameter | Model A | Model B | Model C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| aHR | 95% CI | P Value | aHR | 95% CI | P Value | aHR | 95% CI | P Value | |
| History of hepatic decompensation | 1.710 | 0.445‐6.571 | 0.435 | 1.550 | 0.438‐5.491 | 0.497 | — | — | — |
| FU‐MELD, per point | 1.090 | 0.957‐1.242 | 0.193 | 1.141 | 1.006‐1.293 | 0.040 | — | — | — |
| FU‐albumin, per g × L−1 | 0.869 | 0.808‐0.936 | <0.001 | 0.852 | 0.790‐0.919 | <0.001 | — | — | — |
| FU‐LSM, per kPa | 1.044 | 1.015‐1.072 | 0.002 | — | — | — | 1.035 | 1.002‐1.069 | 0.039 |
| FU‐VITRO, per point | — | — | — | 1.386 | 1.185‐1.622 | <0.001 | 1.312 | 1.091‐1.579 | 0.004 |
Model A included FU‐LSM and was adjusted for history of hepatic decompensation and indicators of hepatic dysfunction (FU‐MELD score and serum FU‐albumin levels), whereas model B included FU‐VITRO and was adjusted for the same factors. We computed model C, which included FU‐LSM and FU‐VITRO, to demonstrate that both variables provide independent/incremental prognostic information.
Notably, these associations remained unchanged when including alcohol consumption above the threshold in the prognostic model (Supporting Table S2), and when adjusting for ascites instead of history of hepatic decompensation in general (Supporting Table S3).
Risk Stratification for PH and Clinical Events During Follow‐up by an Algorithm Combining LSM and VITRO in the Derivation Cohort
Applying the LSM‐based and VITRO‐based risk stratification algorithm at BL and FU indicated that a considerable proportion of patients who fell into the high‐risk or gray‐zone groups at BL were reclassified into a lower risk group (Supporting Fig. S2), while progression to a higher risk group was comparatively uncommon.
Of note, the patients of the derivation cohort who underwent paired HVPG measurements (n = 80) had more advanced underlying liver disease at BL (LSM: 23.7 kPa [IQR: 18.0] vs. 17.1 kPa [IQR: 15.6] and VITRO: 2.83 [IQR: 2.95] vs. 1.89 [IQR: 2.08]) and at FU (LSM: 17.3 kPa [IQR: 16.4] vs. 12.7 kPa [IQR: 14.3] and VITRO: 1.82 [IQR: 2.08] vs. 1.15 [IQR: 1.52]), as compared with the overall cohort. Among patients with paired HVPG measurements, those meeting the CSPH ruled‐out criteria (n = 29) had a median FU‐HVPG of 5 mm Hg (IQR: 4), and the prevalence rates of CSPH and high‐risk PH were only 6.9% and 3.4%. Accordingly, FU‐CSPH could be ruled out in these patients. In the gray‐zone (n = 21), median FU‐HVPG was 11 mm Hg (IQR: 4), and 76.2% had CSPH; however, high‐risk PH was rare (only 9.5%). Finally, in the CSPH ruled‐in group (n = 30; median FU‐HVPG 17 mm Hg [IQR: 10]), CSPH could be ruled in, as the prevalence attained 90.0%. Furthermore, this group was enriched with patients who had high‐risk PH (60.0%), despite HCV cure. The between‐group differences in FU‐HVPG and the prevalence of CSPH and high‐risk PH at FU attained statistical significance (Fig. 2).
Fig. 2.
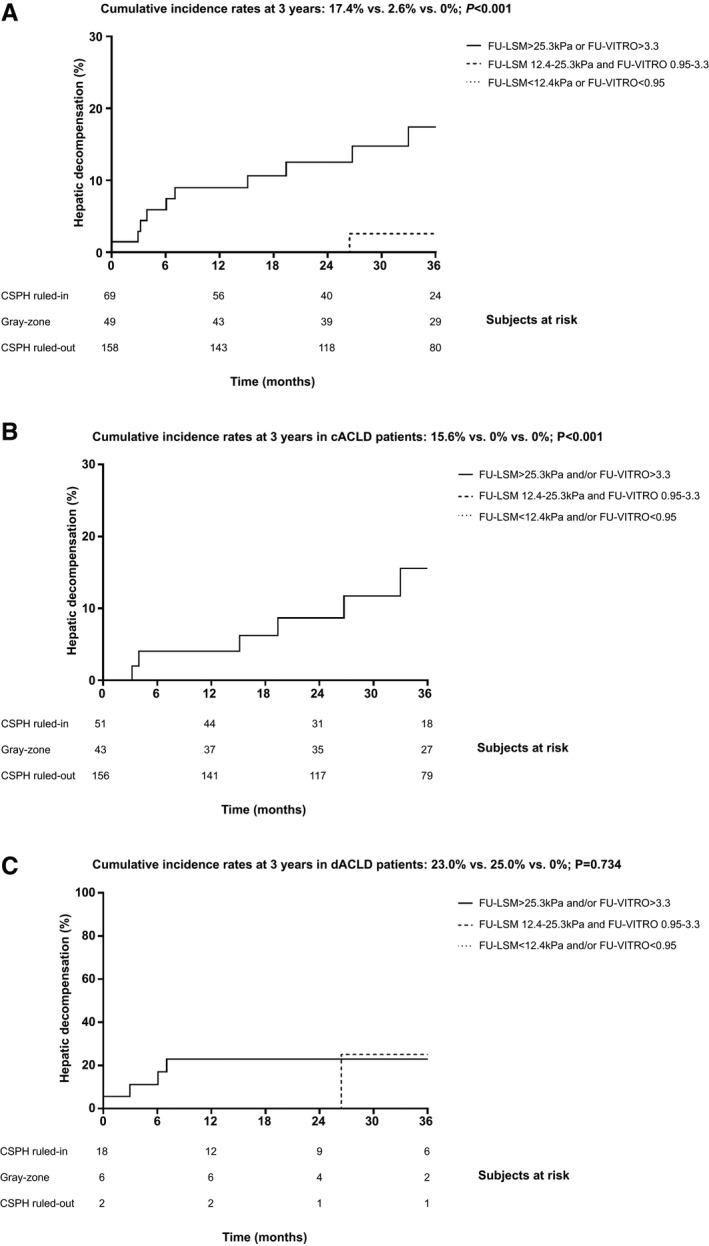
Kapla‐Meier analyses of hepatic decompensation in the derivation cohort. A, All patients, as well as the subgroups of patients with cACLD (i.e., first hepatic decompensation) (B) and dACLD (i.e., further hepatic decompensation) (C), stratified according to their risk of posttreatment clinically significant portal hypertension (CSPH, HVPG ≥ 10 mm Hg).
Because PH is a driver of posttreatment hepatic decompensation, we evaluated whether an LSM‐based and VITRO‐based algorithm, which has previously been developed to stratify patients according to their risk for posttreatment CSPH, is able to predict posttreatment hepatic decompensation.( 9 )
When applying the stepwise FU‐LSM‐based and FU‐VITRO‐based algorithm to the derivation cohort, FU‐LSM < 12.4 kPa and/or FU‐VITRO < 0.95 ruled out CSPH at FU in 158 (57.2%) of 159 (57.6%) patients, depending on whether LSM or VITRO was the first step. In 69 (25.0%) of 68 (24.6%) patients, CSPH was ruled in (FU‐LSM > 25.3 kPa and/or FU‐VITRO > 3.3), while 49 patients (17.8%) remained in the gray‐zone (Table 4). Accordingly, application of LSM or VITRO as a first step yielded very similar results.
Table 4.
FU‐LSM‐Based and FU‐VITRO‐Based Algorithms to Rule In and Rule Out Posttreatment CSPH (HVPG ≥ 10 mm Hg) in the Derivation Cohort
| FU‐LSM First–FU‐VITRO Second | |||||
|---|---|---|---|---|---|
| First Step | FU‐LSM | ||||
| < 12.4kPa (CSPH ruled‐out) | 12.4‐25.3 kPa (gray‐zone) | > 25.3kPa (CSPH ruled‐in) | |||
| 133 (48.2%) | 84 (30.4%) | 59 (21.4%) | |||
| Second Step | FU‐VITRO | ||||
| < 0.95 (CSPH ruled‐out) | 0.95‐3.3 (gray‐zone) | > 3.3 (CSPH ruled‐in) | |||
| 25 (9.1%) | 49 (17.8%) | 10 (3.6%) | |||
| FU‐VITRO First–FU‐LSM Second | |||||
| First Step | FU‐VITRO | ||||
| < 0.95 (CSPH ruled‐in) | 0.95‐3.3 (gray‐zone) | > 3.3 (CSPH ruled‐out) | |||
| 112 (40.6%) | 125 (45.3%) | 39 (14.1%) | |||
| Second Step | FU‐LSM | ||||
| < 12.4 (CSPH ruled‐out) | 12.4‐25.3 (gray‐zone) | > 25.3 (CSPH ruled‐in) | |||
| 47 (17.0%) | 49 (17.8%) | 29 (10.5%) | |||
If patients within the gray‐zone were excluded, both approaches (“FU‐LSM first–FU‐VITRO second” and “FU‐VITRO first–FU‐LSM second”) had a negative predictive value of 100% and a positive predictive value of 14.5%/14.7% for hepatic decompensation during FU. Patients in whom posttreatment CSPH was ruled out (n = 158, 57.2%) and those in the gray‐zone (n = 49, 17.8%) had a significantly lower cumulative incidence of hepatic decompensation at 3 years (0% and 2.6%, respectively) than patients in whom posttreatment CSPH was ruled in (n = 69, 25.0%; cumulative incidence: 17.4%; P < 0.001; Fig. 3A). This finding was confirmed by competing risks analysis, with hepatic decompensation as the event of interest, and HCC development, OLT, as well as death as competing risks, which yielded nearly identical results, as compared with the Kaplan‐Meier method/log‐rank test (hepatic decompensation at 3 years: 0% vs. 2.4% vs. 16.8%; P < 0.001) (Supporting Fig. S3). Of note, stratification by LSM or VITRO as a first step yielded similar results (data not shown), indicating that both approaches can be used interchangeably.
Fig. 3.
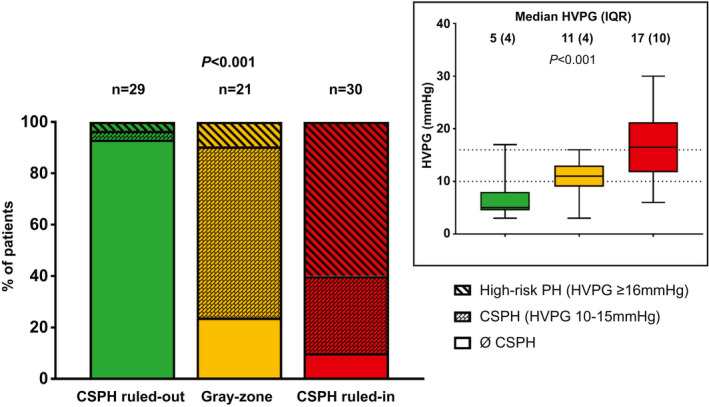
Prevalence of clinically significant portal hypertension (HVPG ≥ 10 mm Hg) and high‐risk portal hypertension (HVPG ≥ 16 mm Hg) and comparison of HVPG levels throughout the risk groups in the derivation cohort, as defined by noninvasive markers.
A similar pattern was observed in patients with cACLD (n = 250, 90.6%), with a cumulative incidence of hepatic decompensation at 3 years of 15.6% in patients in whom posttreatment CSPH was ruled in versus 0% in patients in the gray‐zone or in whom CSPH was ruled out (P < 0.001; Fig. 3B). However, there was a comparable incidence of hepatic decompensation at 3 years in patients with dACLD (n = 26, 9.4%) within the gray‐zone and in patients in whom CSPH was ruled in (25.0% and 23.0%, respectively), while no events occurred in the CSPH ruled‐out group (P = 0.734; Fig. 3C).
A sensitivity analysis restricted to patients with cirrhosis (n = 168), as diagnosed by BL‐LSM ≥ 15 kPa or histology, confirmed the discriminatory ability of the FU‐LSM‐based and FU‐VITRO‐based risk‐stratification algorithm (hepatic decompensation at 3 years: 0% vs. 2.9% vs. 18.4%; P = 0.001) (Supporting Fig. S4). Finally, applying other previously proposed highly sensitive/specific FU‐LSM cutoffs (13.6( 26 ))/21 kPa( 27 )) had no relevant effect on the results (Supporting Fig. S5).
Internal Validation
See the Supporting Information materials for information on BL characteristics and FU.
The prognostic performance of FU‐LSM (AUROC: 0.896 [95% CI 0.807‐0.985]) and FU‐VITRO (AUROC: 0.803 [95% CI 0.604‐1.000]) for posttreatment hepatic decompensation in the internal validation cohort was very similar to that observed in the derivation cohort. In line with these findings, the rates of hepatic decompensation at 3 years in the different groups of our FU‐LSM‐based and FU‐VITRO‐based risk stratification algorithm were as follows: high risk (38.2%) versus gray‐zone (0%) versus low risk (0%); P < 0.001 (Supporting Fig. S6).
Although the number of patients in the validation cohort who underwent paired HVPG measurements was small (n = 14), we also attempted to replicate our previous findings regarding prevalence of CSPH and high‐risk PH at FU. Among patients meeting the noninvasive CSPH ruled‐out (i.e., low‐risk) criteria (n = 3), median FU‐HVPG was 4 mm Hg (IQR: 5), and no patient had CSPH. Accordingly, FU‐CSPH could be ruled out in these patients. In the gray‐zone (n = 4), median FU‐HVPG was 15 mm Hg (IQR: 17), and the prevalence rates of CPSH and high‐risk PH were 75% and 50%. Finally, in the CSPH ruled‐in (i.e., high‐risk) group (n = 7; median FU‐HVPG = 21 mm Hg [IQR: 9]), CSPH could be ruled in, as the prevalence attained 100%. Furthermore, this group was enriched with patients who had high‐risk PH (57.1%) after HCV cure. Despite the small number of patients, the between‐group differences in HVPG, the prevalence of CSPH, and high‐risk PH at FU attained statistical significance (Supporting Fig. S7).
External Validation
See the Supporting Information materials for information on BL characteristics and FU.
In line with the findings in the derivation and the internal validation cohort, FU‐LSM of (AUROC: 0.943 [95% CI: 0.886‐1.000]) and FU‐VITRO of (AUROC: 0.844 [95% CI: 0.758‐0.930]) were predictive of hepatic decompensation. The FU‐LSM‐based and FU‐VITRO‐based algorithm stratified patients into groups that differed in their risk for hepatic decompensation during FU (high risk [13.4%] vs. gray‐zone [4.5%] vs. low risk [0%] [P < 0.001] when comparing only high‐risk versus low‐risk group due to crossing curves) (Supporting Fig. S8).
Recompensation in Patients with dACLD in the Derivation Cohort
See the Supporting Information materials.
Noninvasive Prediction of de Novo HCC and Liver‐Related Transplant‐Free Mortality in the Derivation Cohort
See the Supporting Information materials.
Discussion
Our study demonstrates that a combination of noninvasive markers of PH (i.e., LSM and VITRO), determined after HCV cure, predicts posttreatment hepatic decompensation in patients with ACLD. Importantly, our noninvasive algorithm is based on readily accessible parameters that are easily applicable in clinical practice, and thus facilitate risk stratification and personalized therapy.
A number of imaging, laboratory, and functional tests have been shown to correlate with HVPG.( 28 ) However, LSM and VWF are particularly promising, as their diagnostic value for CSPH has been confirmed in patients with cACLD (i.e., the main target population),( 28 ) and they have been shown to predict outcomes in patients with ACLD, even independently of HVPG.( 11 , 16 , 17 ) Importantly, the diagnostic performance as well as the prognostic ability of these and other noninvasive markers have primarily been studied in progressive liver disease. However, with the advent of etiological therapies, regressive liver disease after the removal of the primary etiological factor (i.e., HCV cure) is becoming increasingly common. Regardless of prevention strategies, the number of individuals who are projected to be treated for and cured from HCV infection worldwide will be around or even considerably higher than 1 million per year for the next decade.( 29 ) Because a relevant proportion of patients have pretreatment ACLD and the risk for complications is decreased but not completely abolished( 6 , 7 , 8 ) by HCV cure, risk stratification is a key to decrease resource use by individualizing posttreatment management. We have recently established the role of HVPG as a prognostic marker after HCV cure, and although HVPG measurement is unsuitable to guide patient management after SVR on a large scale, our findings highlight the central role of PH.( 9 ) In contrast to HVPG measurement, LSM and VWF/VITRO are broadly available and easily applicable in clinical routine.
LSM, which has already found its way into the recommendations for the management of PH in 2015,( 10 ) has been used to predict liver‐related events following SVR in two recent studies. Corma‐Gomez et al.( 30 ) reported an independent association of LSM with a composite endpoint of hepatic decompensation, HCC, OLT, and mortality in patients co‐infected with HIV/HCV who achieved HCV cure. However, although such a composite endpoint increases statistical power, it negatively affects the granularity of results, as the dynamics of PH do not necessarily relate to HCC risk after HCV cure.( 9 ) Even more recently, Pons et al.( 31 ) investigated the risk factors for hepatic decompensation and de novo HCC in patients with pretreatment cACLD. They reported an incidence rate for hepatic decompensation of 0.31/100 patient‐years, with hepatic decompensation occurring only in patients with BL‐LSM > 20 kPa. Despite the higher incidence of hepatic decompensation (0.96 per 100 patient‐years) in our study, de novo HCC development was less common (1.10 vs. 1.50/100 patient‐years) than in the study by Pons et al. Importantly, differences in incidence rates are likely explained by differences in patient characteristics, as we also included a small proportion of patients with a history of hepatic decompensation, resulting in a higher rate of PH‐induced complications, and our patients were considerably younger (56.0 vs. 63.7 years), potentially contributing to the lower incidence of HCC.
In line with a recent study that included a small subgroup of patients following SVR,( 19 ) FU‐VITRO showed a consistently good performance for predicting posttreatment hepatic decompensation throughout the study cohorts (AUROC values ranged from 0.803 to 0.925) and an AUROC value that was numerically higher than that of LSM as well as PLT or plasma VWF levels alone in the derivation cohort. Interestingly, VITRO added prognostic information to LSM. This may be explained by the contribution of systemic inflammation (and possibly also endothelial dysfunction and procoagulant imbalance) (i.e., additional factors promoting hepatic decompensation, to plasma VWF levels).( 11 , 12 , 16 ) Based on a previously developed two‐step algorithm for ruling in and ruling out posttreatment CSPH,( 9 ) we stratified patients into three risk groups, which may have important implications for patient management:
Patients with FU‐LSM < 12.4 kPa and/or FU‐VITRO < 0.95 had a CSPH prevalence of <10% (i.e., CSPH ruled‐out). Importantly, most patients fulfilled these criteria, and these patients did not develop first or further hepatic decompensation (i.e., low risk of hepatic decompensation). Due to the absence of CSPH, there might be no need for endoscopic surveillance, even in centers that use NSBB in patients with low‐risk varices. Moreover, there is no potential for strategies aiming to prevent hepatic decompensation (e.g., NSBB therapy( 32 , 33 )), as these patients are not at a relevant risk.
Patients within the diagnostic gray‐zone (i.e., patients with FU‐LSM and FU‐VITRO of 12.4‐25.3 kPa and 0.95‐3.3, respectively) had a low probability of hepatic decompensation (2.6% at 3 years), despite three quarters of patients undergoing paired HVPG measurements having CSPH. This may be explained by the very low (<10%) prevalence of high‐risk PH (i.e., HVPG ≥ 16 mm Hg) in this subgroup and the previously mentioned HVPG‐independent prognostic value of LSM and VWF. Surveillance for varices in these patients is clearly warranted, possibly at longer time intervals, than currently recommended.( 10 )
In patients with FU‐LSM > 25.3 kPa and/or FU‐VITRO > 3.3, CSPH was ruled in (prevalence of 90%), and the prevalence of high‐risk PH was as high as 60%. This translated into an exceedingly high risk of hepatic decompensation (17.4% at 3 years). Besides variceal surveillance, compensated patients classified as being at high risk based on noninvasive markers may benefit from NSBB treatment to prevent hepatic decompensation.( 32 , 33 )
Moreover, considering that LSM is the most well‐established and broadly available elastography technique and that VWF is an inexpensive routine laboratory test,( 28 ) our suggested algorithm appears easy to implement. Because the “FU‐LSM first” and the “FU‐VITRO first” approaches yielded similar results, the sequence of testing can be adopted according to local availability. For instance, using the “FU‐VITRO first” approach, most of the patients can be assigned to the low‐risk or high‐risk group, without the need of VCTE, which may be helpful in settings where VCTE is not readily available.
Of note, the time point of the FU assessment of NIT was not standardized in our study but clustered around SVR12. Accordingly, the predictive value of our risk‐stratification algorithm was not critically dependent on the time point of assessment, thereby facilitating its use in clinical routine. Studies performing repeated HVPG measurements within the posttreatment period revealed that the regression of PH may continue beyond the first months after the end of treatment,( 9 , 34 ) although CSPH commonly persists 48 weeks after the end of treatment.( 5 ) An important strength of noninvasive methods for the long‐term management of patients is the possibility of repeated assessments, which provides updated information on PH and may even increase their prognostic value. However, despite the relatively short period of time, a considerable proportion of patients had stage migration from a higher to a lower risk group; this proportion may have further increased, if assessed at a later time point. Accordingly, it should be investigated in future studies whether sequential reassessments of FU‐LSM and FU‐VITRO further increase the proportion of patients assigned to lower risk groups and improve prognostication.
Although we did not observe an association between features of the metabolic syndrome or hepatic steatosis and posttreatment hepatic decompensation in our study, alcohol consumption was linked to adverse outcomes, highlighting the importance of harm reduction measures.
Apart from PH‐related complications, HCC is a main determinant of morbidity and mortality in patients with ACLD who achieved SVR.( 35 ) Interestingly, Pons et al.( 31 ) found FU‐albumin and FU‐LSM to be independently associated with de novo HCC, and therefore developed a model combining these two parameters to predict HCC development during FU. Although our study confirms the ability of serum albumin levels at FU to predict de novo HCC (AUROC = 0.786), both BL‐LSM and FU‐LSM were not predictive, suggesting that the model by Pons et al. requires further validation. Of note, FU examinations were performed around SVR12 (i.e., at a median of 12.1 weeks after end of treatment) in our study, compared with 12 months in the Spanish study, possibly contributing to the partially conflicting results. Interestingly, of all the evaluated variables, VITRO showed the highest AUROC (around 0.8) for de novo HCC development in our derivation cohort, and BL‐VITRO and FU‐VITRO cutoffs of >2.66 and >1.82, respectively, stratified patients into groups at low risk and high risk of HCC. These findings are in line with a previous study, reporting that plasma VWF levels are predictive of HCC development, independently of liver fibrosis stage.( 36 ) Moreover, VITRO is a noninvasive marker of CSPH, and the latter condition has previously been linked to de novo HCC in patients with cirrhosis.( 37 ) These promising results indicate that VITRO should be further evaluated as a predictor of HCC development in future studies.
Our study has limitations. First, we combined patients with cACLD and dACLD. However, the concept of cACLD/dACLD has been developed in the setting of progressive disease and will likely be updated with Baveno VII, also considering liver disease regression/recompensation, which is increasingly common. Patients may recompensate after etiological cure, which is not always clinically evident, as drugs for ascites and/or hepatic encephalopathy are commonly not withdrawn. Moreover, a patient who has already bled may not be at risk for variceal bleeding anymore, due to a decrease of HVPG below the bleeding threshold or even resolution of CSPH. Accordingly, similar to our previous study, we chose to combine patients with cACLD and dACLD for the main analyses and subsequently confirmed the prognostic value of our noninvasive risk stratification algorithm in the substantially larger subgroup of patients with cACLD.( 9 ) Unfortunately, the number of patients with dACLD with complications other than variceal bleeding was small, which limited the ability of our study to investigate recompensation. Second, not all patients with pretreatment ACLD who underwent antiviral therapy at our center had paired information on LSM/VITRO at BL and FU, and therefore were excluded from the derivation cohort. This could have led to some selection bias, which we tried to reduce by including patients with information on FU‐LSM/FU‐VITRO in the internal validation cohort, in which we observed very similar results. Third, there were no predefined selection criteria for paired HVPG measurements, and therefore the main decisive factors were the physician’s preference/recommendation as well as the patient’s willingness. As a result, patients in the derivation cohort who underwent paired HVPG measurements had higher LSM and VITRO at BL and FU, which is indicative of a higher prevalence of CSPH. This may implicate that the application of our algorithm to an unselected ACLD population (i.e., a population comparable to our overall study population) with a lower prevalence of CSPH could also lead to a lower prevalence of CSPH throughout all CSPH risk groups. Although this is unproblematic for the CSPH ruled‐out group, it could be of concern for the patients in whom FU‐CSPH is ruled in, as it could lead to a prevalence of CSPH lower than 90%. However, this potential limitation is very hard to overcome (or possibly unavoidable), as even a prospectively designed study performing HVPG measurements would be unlikely to recruit an unselected/representative population of patients with ACLD. Fourth, we applied LSM/VITRO cutoffs that were not identified on the basis of their association with the direct clinical endpoint of hepatic decompensation. However, these cutoffs provide additional clinically relevant information on the presence or absence of CSPH. Moreover, choosing cutoffs based on their diagnostic indices for hepatic decompensation could have led to overfitting. Finally, the algorithm applying our previously established cutoffs showed an excellent discriminatory ability throughout all assessed cohorts.
In conclusion, FU‐LSM and FU‐VITRO are strongly and independently predictive of posttreatment hepatic decompensation in patients with ACLD who have achieved SVR using IFN‐free therapies. Collectively, our results across the derivation of internal and external validation cohorts indicate that the proposed risk stratification algorithm provides robust information regarding the risk of hepatic decompensation after SVR as well as the extent of PH. In particular, it reliably identified a considerable proportion (overall, 57.2%) of patients with ACLD who are at negligible risk for CSPH and hepatic decompensation and who may be easily managed in a primary/secondary‐care setting, given that adequate HCC surveillance is ascertained. This approach could help to take the load off tertiary care centers. At the same time, the FU‐LSM‐based and FU‐VITRO‐based algorithm also identified patients who are at high risk of hepatic decompensation, despite etiological cure, and who should be the focus of our clinical and scientific attention. Accordingly, this algorithm enables risk stratification after SVR, and therefore facilitates personalized therapy in this steadily increasing patient population.
Author Contributions
M.M. was responsible for the study concept and design. All authors were responsible for the data acquisition. G.S., T.R., and M.M. were responsible for the analysis, data interpretation, and drafting of the manuscript. All authors were responsible for critical revision of the manuscript for important intellectual content.
Supporting information
Supporting Information
Supported by a grant from the Medical Scientific Fund of the Major of the City of Vienna (No. 17035) as well as the Andrew K. Burroughs short‐term training fellowship of the European Association for the Study of the Liver.
Potential conflict of interest: Dr. Ferenci consults, advises, is on the speakers’ bureau, and received grants from Gilead. He consults, advises, and is on the speakers’ bureau for AbbVie, Bristol‐Myers Squibb and MSD. Dr. Kozbial received grants from AbbVie, Bristol‐Myers Squibb, and Gilead. Dr. Schwabl is on the speakers’ bureau for Bristol‐Myers Squibb and Boehringer Ingelheim. He received grants from Falk and Gilead. Dr. Hametner‐Schreil consults, advises, is on the speakers’ bureau, and received grants from AbbVie and Gilead. She consults, advises, and is on the speakers’ bureau for Eisai, Bristol‐Myers Squibb, and Intercept. Dr. Gavasso received grants from Stago. Dr. Chromy consults, advises, is on the speakers’ bureau, and received grants from AbbVie, MSD, and Gilead. He received grants from Viiv. Dr. Bauer received grants from AbbVie and Gilead. Dr. Simbrunner received grants from AbbVie and Gilead. Dr. Scheiner received grants from AbbVie, Gilead, and Ipsen. Dr. Bucsics is on the speakers’ bureau and received grants from Bristol‐Myers Squibb. She received grants from AbbVie and Medis. Dr. Stättermayer consults, advises, and is on the speakers’ bureau for MSD, Gilead, and Boehringer Ingelheim. Dr. Pinter consults, advises, is on the speakers’ bureau, and received grants from Bayer and Bristol‐Myers Squibb. He consults, advises, and is on the speakers’ bureau for Ipsen, Eisai, Lilly, MSD, and Roche. Dr. Steindl‐Munda consults, advises, is on the speakers’ bureau, and received grants from AbbVie and Gilead. She consults, advises, and is on the speakers’ bureau for Intercept and MSD. She received grants from Falk. Dr. Russo consults, advises, is on the speakers’ bureau, and received grants from AbbVie, Biotest, and Gilead. He consults, advises, and is on the speakers’ bureau for MSD. Dr. Simioni consults, advises, is on the speakers’ bureau, and received grants from CSL Behring. He consults, advises, and is on the speakers’ bureau for Stago, uniQure, and Werfen. Dr. Trauner consults, is on the speakers’ bureau, and received grants from Falk, Gilead, Intercept, and MSD. He consults and received grants from Albireo. He is on the speakers’ bureau and received grants from Roche. He consults for BiomX, Boehringer Ingelheim, Genfit, Novartis, Phenex, and Regulus. He received grants from CymaBay, Takeda, and AbbVie. Dr. Reiberger consults, advises, is on the speakers’ bureau, and received grants from AbbVie, Boehringer Ingelheim, MSD, Gore, and Gilead. He consults, advises, and is on the speakers’ bureau for Bayer, Intercept, and Siemens. He received grants from Phillips. Dr. Mandorfer consults, advises, is on the speakers’ bureau, and received grants from AbbVie, Bristol‐Myers Squibb, and Gilead. He consults, advises, and is on the speakers’ bureau for Collective Acumen and Gore.
References
Author names in bold designate shared co‐first authorship.
- 1. Mandorfer M, Kozbial K, Freissmuth C, Schwabl P, Stattermayer AF, Reiberger T, et al. Interferon‐free regimens for chronic hepatitis C overcome the effects of portal hypertension on virological responses. Aliment Pharmacol Ther 2015;42:707‐718. [DOI] [PubMed] [Google Scholar]
- 2. Mandorfer M, Kozbial K, Schwabl P, Freissmuth C, Schwarzer R, Stern R, et al. Sustained virologic response to interferon‐free therapies ameliorates HCV‐induced portal hypertension. J Hepatol 2016;65:692‐699. [DOI] [PubMed] [Google Scholar]
- 3. Schwabl P, Mandorfer M, Steiner S, Scheiner B, Chromy D, Herac M, et al. Interferon‐free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment Pharmacol Ther 2017;45:139‐149. [DOI] [PubMed] [Google Scholar]
- 4. Lens S, Alvarado‐Tapias E, Marino Z, Londono MC, Elba LL, Martinez J, et al. Effects of all‐oral anti‐viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus‐associated cirrhosis. Gastroenterology 2017;153:1273‐1283. [DOI] [PubMed] [Google Scholar]
- 5. Diez C, Berenguer J, Ibanez‐Samaniego L, Llop E, Perez‐Latorre L, Catalina MV, et al. Persistence of clinically significant portal hypertension after eradication of HCV in patients with advanced cirrhosis. Clin Infect Dis 2020. May 9. 10.1093/cid/ciaa502. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6. Thabut D, Bureau C, Layese R, Bourcier V, Hammouche M, Cagnot C, et al. Validation of Baveno VI criteria for screening and surveillance of esophageal varices in patients with compensated cirrhosis and a sustained response to antiviral therapy. Gastroenterology 2019;156:997‐1009. [DOI] [PubMed] [Google Scholar]
- 7. McDonald SA, Pollock KG, Barclay ST, Goldberg DJ, Bathgate A, Bramley P, et al. Real‐world impact following initiation of interferon‐free hepatitis C regimens on liver‐related outcomes and all‐cause mortality among patients with compensated cirrhosis. J Viral Hepat 2020;27:270‐280. [DOI] [PubMed] [Google Scholar]
- 8. Moon AM, Green PK, Rockey DC, Berry K, Ioannou GN. Hepatitis C eradication with direct‐acting anti‐virals reduces the risk of variceal bleeding. Aliment Pharmacol Ther 2020;51:364‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandorfer M, Kozbial K, Schwabl P, Chromy D, Semmler G, Stattermayer AF, et al. Changes in HVPG predict hepatic decompensation in patients who achieved SVR to IFN‐free therapy. Hepatology 2020;71:1023‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Franchis R, Faculty BVI. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J Hepatol 2015;63:743‐752. [DOI] [PubMed] [Google Scholar]
- 11. La Mura V, Reverter JC, Flores‐Arroyo A, Raffa S, Reverter E, Seijo S, et al. Von Willebrand factor levels predict clinical outcome in patients with cirrhosis and portal hypertension. Gut 2011;60:1133‐1138. [DOI] [PubMed] [Google Scholar]
- 12. Ferro D, Quintarelli C, Lattuada A, Leo R, Alessandroni M, Mannucci PM, et al. High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: relationship to endotoxemia. Hepatology 1996;23:1377‐1383. [DOI] [PubMed] [Google Scholar]
- 13. Ferlitsch M, Reiberger T, Hoke M, Salzl P, Schwengerer B, Ulbrich G, et al. von Willebrand factor as new noninvasive predictor of portal hypertension, decompensation and mortality in patients with liver cirrhosis. Hepatology 2012;56:1439‐1447. [DOI] [PubMed] [Google Scholar]
- 14. Maieron A, Salzl P, Peck‐Radosavljevic M, Trauner M, Hametner S, Schofl R, et al. Von Willebrand factor as a new marker for non‐invasive assessment of liver fibrosis and cirrhosis in patients with chronic hepatitis C. Aliment Pharmacol Ther 2014;39:331‐338. [DOI] [PubMed] [Google Scholar]
- 15. Mandorfer M, Peck‐Radosavljevic M, Ferenci P, Reiberger T. Dynamics of platelet count after sustained virologic response do not mirror those of hepatic venous pressure gradient. Liver Int 2020;40:988‐989. [DOI] [PubMed] [Google Scholar]
- 16. Mandorfer M, Schwabl P, Paternostro R, Pomej K, Bauer D, Thaler J, et al. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment Pharmacol Ther 2018;47:980‐988. [DOI] [PubMed] [Google Scholar]
- 17. Piecha F, Mandorfer M, Peccerella T, Ozga AK, Poth T, Vonbank A, et al. Pharmacological decrease of liver stiffness is pressure‐related and predicts long‐term clinical outcome. Am J Physiol Gastrointest Liver Physiol 2018;315:G484‐G494. [DOI] [PubMed] [Google Scholar]
- 18. Russo FP, Zanetto A, Campello E, Bulato C, Shalaby S, Spiezia L, et al. Reversal of hypercoagulability in patients with HCV‐related cirrhosis after treatment with direct‐acting antivirals. Liver Int 2018;38:2210‐2218. [DOI] [PubMed] [Google Scholar]
- 19. Schwarzer R, Reiberger T, Mandorfer M, Kivaranovic D, Hametner S, Hametner S, et al. The von Willebrand factor antigen to platelet ratio (VITRO) score predicts hepatic decompensation and mortality in cirrhosis. J Gastroenterol 2020;55:533‐542. [DOI] [PubMed] [Google Scholar]
- 20. Sarrazin C, Berg T, Buggisch P, Dollinger MM, Hinrichsen H, Hofer H, et al. S3 guideline hepatitis C addendum. Z Gastroenterol 2015;53:320‐334. [DOI] [PubMed] [Google Scholar]
- 21. Sarrazin C, Zimmermann T, Berg T, Neumann UP, Schirmacher P, Schmidt H, et al. Prophylaxis, diagnosis and therapy of hepatitis‐C‐virus (HCV) infection: the German guidelines on the management of HCV infection—AWMF‐Register‐No.: 021/012. Z Gastroenterol 2018;56:756‐838. [DOI] [PubMed] [Google Scholar]
- 22. European Association for the Study of the Liver . EASL Recommendations on Treatment of Hepatitis C 2015. J Hepatol 2015;63:199‐236. [DOI] [PubMed] [Google Scholar]
- 23. European Association for the Study of the Liver . EASL Recommendations on Treatment of Hepatitis C 2016. J Hepatol 2017;66:153‐194. [DOI] [PubMed] [Google Scholar]
- 24. European Association for the Study of the Liver . EASL Recommendations on Treatment of Hepatitis C 2018. J Hepatol 2018;69:461‐511 . [DOI] [PubMed] [Google Scholar]
- 25. Reiberger T, Schwabl P, Trauner M, Peck‐Radosavljevic M, Mandorfer M. Measurement of the hepatic venous pressure gradient and transjugular liver biopsy. J Vis Exp 2020. Jun 18. 10.3791/58819. [DOI] [PubMed] [Google Scholar]
- 26. Vizzutti F, Arena U, Romanelli RG, Rega L, Foschi M, Colagrande S, et al. Liver stiffness measurement predicts severe portal hypertension in patients with HCV‐related cirrhosis. Hepatology 2007;45:1290‐1297. [DOI] [PubMed] [Google Scholar]
- 27. Bureau C, Metivier S, Peron JM, Selves J, Robic MA, Gourraud PA, et al. Transient elastography accurately predicts presence of significant portal hypertension in patients with chronic liver disease. Aliment Pharmacol Ther 2008;27:1261‐1268. [DOI] [PubMed] [Google Scholar]
- 28. Mandorfer M, Hernandez‐Gea V, Garcia‐Pagan JC, Reiberger T. Non‐invasive diagnostics for portal hypertension: a comprehensive review. Semin Liver Dis 2020. Jun 18. 10.1055/s-0040-1708806. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29. Heffernan A, Cooke GS, Nayagam S, Thursz M, Hallett TB. Scaling up prevention and treatment towards the elimination of hepatitis C: a global mathematical model. Lancet 2019;393:1319‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corma‐Gomez A, Macias J, Tellez F, Freyre‐Carrillo C, Morano L, Rivero‐Juarez A, et al. Liver stiffness at the time of sustained virological response predicts the clinical outcome in HIV/HCV‐coinfected patients with advanced fibrosis treated with direct‐acting antivirals. Clin Infect Dis 2019. Nov 22. 10.1093/cid/ciz1140. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31. Pons M, Rodriguez‐Tajes S, Esteban JI, Marino Z, Vargas V, Lens S, et al. Non‐invasive prediction of liver‐related events in patients with HCV‐associated compensated advanced chronic liver disease after oral antivirals. J Hepatol 2020;72:472‐480. [DOI] [PubMed] [Google Scholar]
- 32. Mandorfer M, Reiberger T. Beta blockers and cirrhosis, 2016. Dig Liver Dis 2017;49:3‐10. [DOI] [PubMed] [Google Scholar]
- 33. Villanueva C, Albillos A, Genesca J, Garcia‐Pagan JC, Calleja JL, Aracil C, et al. β blockers to prevent decompensation of cirrhosis in patients with clinically significant portal hypertension (PREDESCI): a randomised, double‐blind, placebo‐controlled, multicentre trial. Lancet 2019;393:1597‐1608. [DOI] [PubMed] [Google Scholar]
- 34. Afdhal N, Everson GT, Calleja JL, McCaughan GW, Bosch J, Brainard DM, et al. Effect of viral suppression on hepatic venous pressure gradient in hepatitis C with cirrhosis and portal hypertension. J Viral Hepat 2017;24:823‐831. [DOI] [PubMed] [Google Scholar]
- 35. Ji F, Yeo YH, Wei MT, Ogawa E, Enomoto M, Lee DH, et al. Sustained virologic response to direct‐acting antiviral therapy in patients with chronic hepatitis C and hepatocellular carcinoma: a systematic review and meta‐analysis. J Hepatol 2019;71:473‐485. [DOI] [PubMed] [Google Scholar]
- 36. Takaya H, Kawaratani H, Tsuji Y, Nakanishi K, Saikawa S, Sato S, et al. von Willebrand factor is a useful biomarker for liver fibrosis and prediction of hepatocellular carcinoma development in patients with hepatitis B and C. United European Gastroenterol J 2018;6:1401‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ripoll C, Groszmann RJ, Garcia‐Tsao G, Bosch J, Grace N, Burroughs A, et al. Hepatic venous pressure gradient predicts development of hepatocellular carcinoma independently of severity of cirrhosis. J Hepatol 2009;50:923‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information