

Abstract
Enhancement of the luminescence efficiency of two new diazapentacenium salts (D1 and D2) of more than 55 for D1 and 22 times for D2) in poor solvents, acetonitrile and/or dichloromethane, was observed and rationalized as formation of emissive J‐aggregates. Both compounds displaying 4‐n‐decylphenyl substituents at the 7,14‐carbons and phenyl (D1) or 2,6‐difluorophenyl (D2) substituents at the quaternary nitrogen atoms in 5,12‐positions have been synthetized in a two‐step procedure involving a two‐fold Buchwald‐Hartwig‐type CN cross‐coupling and an electrophilic Friedel‐Crafts‐type cyclization. The optical properties of the dicationic diazapentacenium salts in various solvents and in thin films have been investigated by steady‐state and time‐resolved absorption and photoluminescence spectroscopies. In thin films and in good solvents, isolated molecules coexist with aggregates. Nonetheless, D1 is seven times more emissive than D2, reflecting a higher J‐aggregate contribution in the former.
Keywords: AIE, diazapentacene, fluorescence, J-aggregates, photochemistry
Two new diazapentacenium salts have been obtained from a new synthetic methodology and shown to display a drastic increase of fluorescence quantum yields induced by aggregation (AIE phenomena) in poor solvents and associated to a particular type of aggregates organization: J‐aggregates. A simple structural modification (4 H in D1 vs 4 F in D2) changes the contribution of J‐aggregates which is quantitatively followed by time resolved fluorescence.
Since John and Clar's synthesis of pentacene in the early 1930’s, [1] the synthetic strategies and properties of higher member acenes have challenged chemists. Early on, pentacene and structurally related molecules were of curiosity‐driven research; nowadays, pentacene is considered a reference hole semiconductor in organic field‐effect transistors. [2] Propelled by such curiosity, boron and nitrogen atoms have been introduced into π‐backbones of poly‐aromatic hydrocarbons, structurally similar to their hydrocarbon analogues, resulting in interesting heteroarene molecules showing different properties. [3] Azapentacenes are, thereby, a particularly interesting subgroup of these heteroarenes, [3a] which formally result from inserting nitrogen atoms into the pentacene skeleton; such interest mainly arose from the promise of tuning the electronic structure, molecular packing, stability, and processability of pentacene by replacing C atoms with N atoms of varied number, position and valence state. [4] Synthetic routes to prepare azaacenes often use condensation/alkynylation methods that have been developed for the preparation of all‐carbon‐acenes. [5] Known N‐quaternized azaacenium salts were synthesized by N‐alkylation of azapentacenes or by introducing aryl groups into the precursors of azaacenes. [6] N,N’‐dihydro compounds, which may be obtained by reduction of larger azaacenes, are known for much longer time, and found more stable than longer azaacenes themselves. [7]
Aggregation induced emission (AIE) and AIE‐luminogen systems are, at the moment, topics of high interest and impact due to the increment of the emission properties in aggregate systems, which are usually poorly emissive. The occurrence of AIE with a new fluorescence band originating from intermolecular interactions was first reported by Scheibe et al. [8] and Jelley [9] who independently observed in 1936 an unusual behavior for pseudoisocyanine chloride (also known as 1,1’‐diethyl‐2,2’‐cyanine chloride, PIC chloride) whereas in aqueous solutions the absorption maximum was shifted to lower energies, when compared to the absorption spectrum of the same dye in ethanol, and upon concentration increase (in water), this band became more intense and sharp. [10] Dye aggregates with a narrow absorption band bathochromically shifted, with a very small Stokes’ shift and increased fluorescence intensity relative to the monomer are generally termed Scheibe aggregates or J‐aggregates (J denotes Jelley). [11] In contrast, aggregates with absorption bands shifted to shorter wavelength (hypsochromically shifted) with respect to the monomer band are called H‐aggregates (H denotes hypsochromic), in most cases coupled to the occurrence of low intensity, or absence of fluorescence. [12]
Currently, AIE or aggregation induced enhanced emission (AIEE) caused by the formation of J‐aggregates have been described in several systems. Oelkrug et al. has demonstrated the role of J‐aggregation for enhanced solid‐state emission of a series of oligophenylene vinylenes, with the increase of fluorescence quantum yield from almost zero in solution to 60 % in nanoparticles or in films. [13] They explained this phenomenon as arising from the rigid environments provided by viscous solvents or solid phases that suppress torsion‐induced radiationless deactivation. [14] In this work, we report, to our best knowledge, for the first‐time the formation of J‐aggregates in quaternized 5,12‐diazapentacenium salts, coupled to an increase in fluorescence intensity (AIE properties).
For synthesis of the diazapentacenium salts, N,N’‐diaryl1‐N,N’‐diaryl2‐substituted 2,5‐bis(4‐n‐decylbenzoyl)‐1,4‐phenylene diamine precursors (aryl1: 4‐n‐decylphenyl, aryl2: phenyl (D1) or 2,6‐difluorophenyl (D2)) were generated in Buchwald‐Hartwig‐type aryl‐amine couplings (Scheme 1). The Friedel‐Crafts‐acylation‐type cyclization chemistry was already reported for synthesis of monocationic N‐arylacridinium salts, specifically for reaction of triphenylamines with benzoic acid derivatives or cyclization of 2‐benzoyltriphenylamine into 9,10‐diphenylacridinium salts, both at (nearly) quantitative conversion of the starting compounds into the respective products. [15]
Scheme 1.
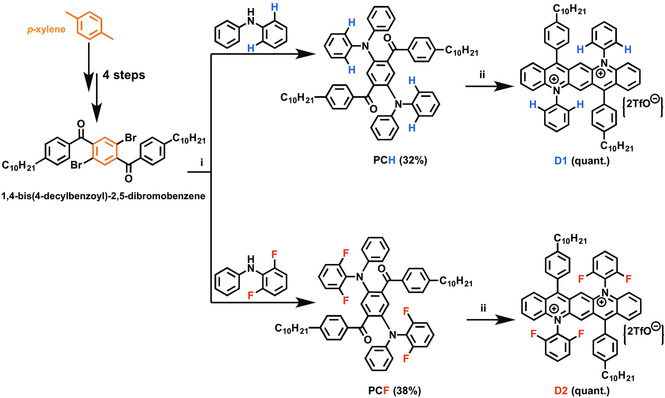
Synthetic route to the diazapentacenium salts D1 and D2. i) NaO‐t‐Bu, Pd(OAC)2, tri‐t‐Bu‐phosphine, toluene, 120 °C, 16 h; ii) trifluoromethanesulfonic acid (TMSA) under argon, toluene, 50 °C, 3 h.
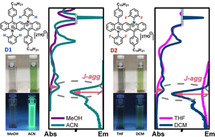
The photophysical properties of the diazapentacenium salts D1 and D2 (Scheme 1) were studied in five solvents of different polarity (here given as solvent's relative permittivity, ϵ): two low polarity aprotic solvents, tetrahydrofuran (THF, ϵ=7.58), and dichloromethane (DCM, ϵ=8.93) and acetonitrile (ACN, ϵ=37.5) as polar aprotic solvents, and methanol (MeOH, ϵ=32.7) as polar protic solvent. Thin films of the diazapentacenium salts prepared by spin‐coating supported in zeonex matrices were also studied. Absorption, fluorescence emission and excitation spectra for the two compounds D1 and D2 are depicted in Figure 1. A first visual observation shows that the solution colour strongly depends from the solvent used (Figure S1 and Table S1). However, in DCM and ACN very similar solution colours for both compounds are observed. In other solvents, also significant differences between D1 and D2 are noticed (see Figure S1).
Figure 1.
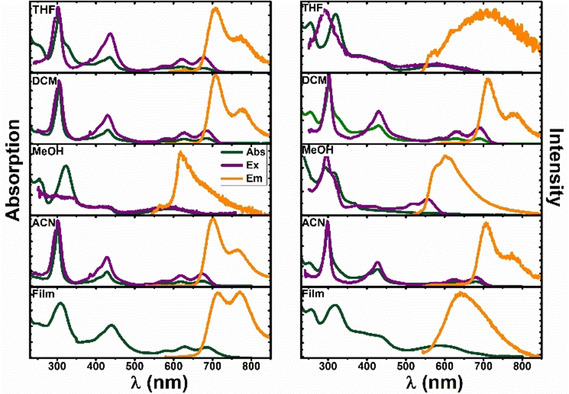
Absorption, fluorescence excitation, and emission spectra of compounds D1 (left) and D2 (right) in THF, DCM, MeOH, ACN and in films. All spectra were recorded with concentration=1.0×10−5 mol L−1. Abs= absorption spectra; Ex= excitation spectra; Em= emission spectra.
MeOH for D1 and THF for D2 represent the best solvents for each compound. The difference between D1 and D2 is expected to be caused by the introduction of the fluorine atoms into the N‐phenyl substituents. Here, absorption and emission, associated to a π‐π* transition, lack vibronic resolution and the compounds are practically non‐emissive, with φF≤1 % (Table 2). In contrast, ACN and DCM, behave as a poor solvent for the two salts. Now, mirror‐symmetry between absorption and fluorescence excitation spectra is observed. Especially D1 is much more emissive in ACN and DCM if compared to MeOH as good solvent (see the φF values in Table 2). The long‐wavelength absorption bands are vibronically resolved and red shifted, when compared to the spectra in the good solvents MeOH and THF at the same concentrations. For similar cationic compounds, e. g., quaternary N‐phenyl azaacenium salts in dichloromethane, also small Stokes’ shifts and vibronically well‐resolved long wavelength absorption bands have been reported. [7] The formation of the vibronically structured, red shifted absorption bands, together with a small Stokes’ shift (ΔSS ∼600 cm−1, Table 1) and an increase of the fluorescence quantum yield (φF), indicates the formation of J‐aggregates in the poor solvents ACN and DCM (Figure 1). In thin films, although the longest absorption and emission bands are vibronically resolved for D1, the φF value is lower than in poor solvents but higher than in good solvents (Table 2). For thin films of D2, a broad absorption and emission bands are observed, with a large Stokes shift and low φF value. The data indicate that the C10H21 chain (present in D1 and D2) and the fluorine atoms (only in D2) contribute differently to the degree of organization of the salts in the solid state and in solutions of poor solvents. To summarize at this stage, a comparison of all spectra for D1 and D2 shows significant differences for the solutions in different solvents, but also some differences between D1 and D2: THF is a good solvent for D2, but not for D1, while MeOH is a good solvent for D1 (Figure 2).
Table 2.
Room temperature fluorescence quantum yields (ϕF) and emission lifetimesa (τi) for compounds D1 and D2 in pure solvents. Also presented are the associated pre‐exponential factors (ai), the fractional contributions for each decay time (%Ci), and the chi‐square values (χ2) for the judgment of the quality of the fits. Radiative (kF) and radiationless rate (kNR) constants associated with the third decay component (τ3) are also listed.
|
Dye |
Solvent |
Concentration [μM] |
ϕF |
τ1 [ns] |
a1 [C1%] |
τ2 [ns] |
a2 [C2%] |
τ3 [ns] |
a3 [C3%] |
χ2 |
b)kF ×10−6 [s−1] |
c) kNR ×10−8 [s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
D1 |
THF |
50 |
0.013 |
|
|
|
|
4.07 |
1 (100) |
0.69 |
3.19 |
2.43 |
|
DCM |
50 |
0.102 |
|
|
|
|
5.41 |
1 (100) |
0.81 |
18.9 |
1.66 |
|
|
MeOH |
50 |
0.002 |
0.16 |
0.889 (44) |
1.18 |
0.096 (35) |
4.40 |
0.015 (21) |
1.04 |
0.46 |
2.27 |
|
|
ACN |
50 |
0.11 |
|
|
|
|
4.36 |
1 (100) |
0.70 |
25.2 |
2.04 |
|
|
Film |
– |
0.007 |
0.14 |
0.968 (50) |
|
|
4.27 |
0.032 (50) |
1.09 |
1.64 |
2.36 |
|
|
D2 |
THF |
50 |
0.001 |
0.34 |
0.902 (62) |
1.51 |
0.089 (27) |
5.65 |
0.010 (11) |
0.92 |
0.18 |
1.77 |
|
DCM |
50 |
0.022 |
1.21 |
0.146 (5.5) |
|
|
3.55 |
0.854 (94.5) |
0.68 |
6.2 |
2.75 |
|
|
MeOH |
5 |
0.067 |
|
|
13.31 |
0.026 (7.0) |
4.56 |
0.974 (93) |
1.16 |
14.7 |
2.05 |
|
|
10 |
0.018 |
|
|
13.30 |
0.028 (8.0) |
4.5 |
0.972 (92) |
1.04 |
4.0 |
2.18 |
||
|
25 |
0.003 |
0.42 |
0.894(30) |
18.65 |
0.025 (37) |
5.03 |
0.081 (33) |
1.01 |
0.60 |
1.98 |
||
|
50 |
0.001 |
0.48 |
0.797 (22) |
21.21 |
0.019 (24) |
5.02 |
0.184 (54) |
1.11 |
0.20 |
1.99 |
||
|
ACN |
50 |
0.008 |
|
|
|
|
2.36 |
1 (100) |
0.6 |
3.4 |
4.20 |
|
|
Film |
– |
0.001 |
0.19 |
0.996 (92) |
|
|
4.17 |
0.004 (8) |
0.82 |
0.24 |
2.40 |
[a] Experimental conditions: λex=460 nm [b] . [c] .
Table 1.
Spectroscopic data (Absorption, λabs, emission maxima, λem, and Stokes shift, ΔSS) of diazapentacenium salts D1 and D2 in different solvents.[a]
|
Dye |
Solvent |
λabs [nm] |
λem [nm][b] |
ΔSS [cm−1] |
|---|---|---|---|---|
|
D1 |
THF |
304/435/575/620/678 |
708/773(s) |
625 |
|
DCM |
305/431/581/627/685 |
709/773(s) |
494 |
|
|
MeOH |
323/411/589 |
683 |
2337 |
|
|
ACN |
302/428/569/618/674 |
701/765(s) |
571 |
|
|
Film |
309/440/580/630/685 |
713/770 |
573 |
|
|
D2 |
THF |
319/386/583 |
716 |
3186 |
|
DCM |
302/429/581/628/688 |
710/778(s) |
450 |
|
|
MeOH |
317/384/555 |
572/618(s) |
536 |
|
|
ACN |
300/427/576/620/680 |
709/773(s) |
602 |
|
|
Film |
318/379/430/586 |
642 |
1489 |
[a] All data were collected with a concentration=1.0×10−5 mol L−1. [b] Emission maxima were collected with with excitation into the S0→S1 band (λex=530 nm). s=shoulder.
Figure 2.
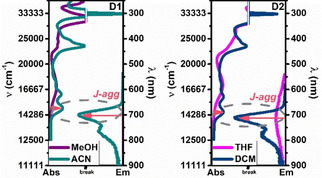
Absorption and emission spectra of compounds D1 and D2 in good and poor solvents pairs. [D1, D2]: 1.0×10−5 mol L−1. Highlighted bands were assigned to J aggregates (J‐agg) of D1 and D2. Line breaks were added to magnify visualization of the new absorption bands (at lower wavenumbers) present in poor solvents.
The observed spectral characteristics for D1 or D2 in ACN and DCM (especially for the absorption and fluorescence excitation spectra) together with the red shifted fluorescence and increased quantum yields, φF, indicates J‐aggregate formation in these poor solvents. However, the most attractive feature comes from the distinct increase, by 55 times (for D1) and 22 times (for D2) in φF when going from the good solvents to ACN as poor solvent (D1: MeOH→ACN; D2: THF→DCM).
Next, we varied the concentrations of D1 and D2; the results (concentration range from 2.0 to 50×10−6 mol L−1) show that D1 (Figure S2) and D2 (Figure S3), both in THF or ACN, display similar spectral profiles during the variation of the concentration, with the intensities of absorption, fluorescence excitation and emission linearly increasing with increasing concentration (Figures S2 and S3).
However, for D2 in MeOH (Fig. S3), the spectral profile changes, and the intensity of the observed bands does not linearly respond to an increasing concentration, as indication for the presence of mixtures of different absorbing/emitting species (isolated molecules, aggregates). The φF also decreases with the concentration (Table 2). At the lowest concentration (2 μM), the emission spectrum is poorly resolved (presence of isolated, non‐aggregated molecules), whereas at 5 μM the spectrum displays the characteristic (vibronically structured) emission of either D1 or D2 J‐aggregates observed in poor solvents such as DCM and ACN. Further increase in concentration leads, best seen in the fluorescence excitation spectra, to a progressive loss of the vibronic resolution and the appearance of an additional broad emission band at emission wavelengths >700 nm, that increases in intensity with increasing concentration. We attribute this behaviour to an initial coexistence of monomers and, possibly, a few J‐aggregates at low concentrations, with the relative concentration of J‐aggregates increasing with increasing concentration and reaching its maximum for a concentration of ca. 5 μM. Further increase of the concentration leads to the progressive formation of disordered and/or ill‐defined, non‐emissive aggregates, reflected in a gradual decrease of φF with the rise in concentration for D2 in MeOH.
To further rationalize these observations, time‐resolved studies were performed (Figure S4 and S5). The complete photophysical data (quantum yields and decay times) of the two compounds in different organic solvents and in films, together with the concentration dependence for D2 also in methanol, are presented in Table 2.
In DCM and ACN, the emission of both D1 and D2 decays mono‐exponentially with lifetimes τ=5.41 ns (DCM) and 4.36 ns (ACN) for D1, and τ=3.55 ns (DCM) and 2.36 ns (ACN) for D2. This emission behaviour is assigned to J‐aggregates. Also in THF, as poor solvent for D1, a single‐exponential decay (τ=4.07 ns) is observed, indicative of J‐aggregation. In MeOH (for D1) and THF (for D2), as good solvents, it seems that, a few amount of aggregated species coexist with the monomeric component (showing the shortest lifetime of τ1<1 ns). As expected, D1 and D2 show the lowest φF in these solvents. In MeOH, D2 displays a much higher lifetime decay component (τ2>13 ns). The contribution associated to this lifetime increases with increasing concentration, accompanied by the occurrence of an additional emission band at longer wavelengths, at >700 nm, and by an ongoing fluorescence quenching (Figure S3). In thin films, the fluorescence decays for D1 and D2 are bi‐exponential, showing that both monomers and J‐aggregated coexist, yet with different contributions. Indeed, for D1 J‐aggregates and monomers are found with equal contribution to fluorescence (as seen by the %C1 and %C3, in Table 2), whereas for D2, J‐aggregates contribution is now of 8 %. These findings corroborate the steady‐state data with D1 presenting higher φF values (higher J‐aggregate contribution), while D2 is poorly emissive in the solid state (lower J‐aggregates contribution). The radiative (kF) and non‐radiative rate (kNR) constants were determined by considering the lifetime τ3 as indicator of J‐aggregate formation. The kNR are at least 1 order of magnitude higher than kF in solvents where the compounds show a higher φF. The photophysical data (Table 2) also display an increase in the radiative rate constant (when going from good to poor solvents) of more than one order of magnitude with a nearly constant rate constant of radiation‐less deactivation. For D1, as example, kF goes from 0.5×106 s−1 to 25×106 s−1 (MeOH→ACN), whereas kNR only varies between 2.3×108 s−1 and 2.0×108 s−1 in these solvents. This also reflects the enhancement of the photoluminescence emission intensity caused by J‐aggregate formation.
We hereby describe the synthesis and optical characterization of two cationic diazapentacenium dyes, with particular emphasis to the photoluminescence properties in different solvents. Our data indicate formation of emissive J‐aggregates in poor solvents, while in good solvents and in thin films, isolated molecules coexist with aggregates. This is evidenced by a strong increase of the fluorescence quantum yields along with the appearance of a narrow absorption band bathochromically shifted in poor solvents. Therefore, the nature of the fluorescence decay changes from single exponential (when J‐aggregates dominate, in poor solvents) to triexponential decays, reflecting the coexistence of isolated molecules, J‐aggregates, and, probably, other disordered aggregate species; the last presented especially at higher concentrations. The simple structural change of the N‐aryl substituents from phenyl (D1) to 2,6‐difluorophenyl (D2) also influences the optical properties. As an example, THF is found to be the best solvent for fluorinated derivative D2, while it is a poor solvent for D1. This illustrates that simple structural modifications in these compounds can dramatically change their optical properties, thus paving the way for a rational control of the supramolecular ordering, and consequently, of the luminescence efficiency.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by Project “Hylight” (no. 031625) 02/SAICT/2017, PTDC/QUI‐QFI/31625/2017, which is funded by the Portuguese Science Foundation (Fundação para Ciência e Tecnologia, FCT) and Compete Centro 2020. We acknowledge funding by Fundo Europeu de Desenvolvimento Regional (FEDER) through COMPETE and project ROTEIRO/0152/2013. CQC acknowledges FCT for financial support (Projects UIDB/00313/2020, UIDP/00313/2020). We also acknowledge funding from Laserlab‐Europe (no. 284464, EC's 7thFramework Programme). Open access funding enabled and organized by Projekt DEAL.
A. C. B. Rodrigues, D. Wetterling, U. Scherf, J. S. Seixas de Melo, Chem. Eur. J. 2021, 27, 7826.
Contributor Information
Prof. Ullrich Scherf, Email: scherf@uni-wuppertal.de.
Prof. J. Sérgio Seixas de Melo, Email: sseixas@ci.uc.pt.
References
- 1. Clar E., John F., Chem. Ber. 1929, 62, 3021–3029 [Google Scholar]
- 2. Müller M., Ahrens L., Brosius V., Freudenberg J., Bunz U. H. F., J. Mater. Chem. C 2019, 7, 14011–14034. [Google Scholar]
- 3.
- 3a. Bunz U. H., Engelhart J. U., Lindner B. D., Schaffroth M., Angew. Chem. Int. Ed. 2013, 52, 3810–3821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3898–3910; [Google Scholar]
- 3b. Bunz U. H., Acc. Chem. Res. 2015, 48, 1676–1686. [DOI] [PubMed] [Google Scholar]
- 4. Miao Q., Adv. Mater. 2014, 26, 5541–5549. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. He Z., Liu D., Mao R., Tang Q., Miao Q., Org. Lett. 2012, 14, 1050–1053; [DOI] [PubMed] [Google Scholar]
- 5b. Wu Y., Jin Y., Xu J., Lv Y., Yu J., Curr. Org. Chem. 2020, 24, 885–899. [Google Scholar]
- 6. Gu X., Shan B., He Z., Miao Q., ChemPlusChem 2017, 82, 1034–1038. [DOI] [PubMed] [Google Scholar]
- 7. Xie G., Brosius V., Han J., Rominger F., Dreuw A., Freudenberg J., Bunz U. H. F., Chem. Eur. J. 2020, 26, 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scheibe G., Angew. Chem. 1937, 49, 212–219. [Google Scholar]
- 9.
- 9a. Jelley E. E., Nature 1936, 138, 1009–1010; [Google Scholar]
- 9b. Jelley E. E., Nature 1937, 139, 631–631. [Google Scholar]
- 10. Wurthner F., Kaiser T. E., Saha-Moller C. R., Angew. Chem. Int. Ed. 2011, 50, 3376; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3436–3410; [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 3376–3410. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Kasha M., Rawls H. R., Ashraf El-Bayoumi M., Pure Appl. Chem. 1965, 11, 371–392; [Google Scholar]
- 11b. Möbius D., Adv. Mater. 1995, 7, 437–444. [Google Scholar]
- 12. Brixner T., Hildner R., Köhler J., Lambert C., Würthner F., Adv. Energy Mater. 2017, 7, 1700236. [Google Scholar]
- 13. Oelkrug D., Tompert A., Egelhaaf H.-J., Hanack M., Steinhuber E., Hohloch M., Meier H., Stalmach U., Synth. Met. 1996, 83, 231–237. [Google Scholar]
- 14. Oelkrug D., Tompert A., Gierschner J., Egelhaaf H.-J., Hanack M., Hohloch M., Steinhuber E., J. Phys. Chem. B 1998, 102, 1902–1907. [Google Scholar]
- 15.
- 15a. Vaid T. P., Lytton-Jean A. K., Barnes B. C., Chem. Mater. 2003, 15, 4292–4299; [Google Scholar]
- 15b. Staskun B., J. Org. Chem. 1964, 29, 2856–2860; [Google Scholar]
- 15c. Staskun B., J. Org. Chem. 1968, 33, 3031–3036. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary