

Abstract
Background and Aims
Adeno‐associated viral (AAV) gene therapy has shown great promise as an alternative treatment for metabolic disorders managed using liver transplantation, but remains limited by transgene loss and genotoxicity. Our study aims to test an AAV vector with a promoterless integrating cassette, designed to provide sustained hepatic transgene expression and reduced toxicity in comparison to canonical AAV therapy.
Approach and Results
Our AAV vector was designed to insert a methylmalonyl‐CoA mutase (MMUT) transgene into the 3′ end of the albumin locus and tested in mouse models of methylmalonic acidemia (MMA). After neonatal delivery, we longitudinally evaluated hepatic transgene expression, plasma levels of methylmalonate, and the MMA biomarker, fibroblast growth factor 21 (Fgf21), as well as integration of MMUT in the albumin locus. At necropsy, we surveyed for AAV‐related hepatocellular carcinoma (HCC) in all treated MMA mice and control littermates. AAV‐mediated genome editing of MMUT into the albumin locus resulted in permanent hepatic correction in MMA mouse models, which was accompanied by decreased levels of methylmalonate and Fgf21, and improved survival without HCC. With time, levels of transgene expression increased and methylmalonate progressively decreased, whereas the number of albumin‐MMUT integrations and corrected hepatocytes in MMA mice increased, but not in similarly treated wild‐type animals. Additionally, expression of MMUT in the setting of MMA conferred a selective growth advantage upon edited cells, which potentiates the therapeutic response.
Conclusions
In conclusion, our findings demonstrate that AAV‐mediated, promoterless, nuclease‐free genome editing at the albumin locus provides safe and durable therapeutic benefit in neonatally treated MMA mice.
Abbreviations
- AAV
adeno‐associated virus
- AAV8
AAV serotype 8
- Afp
alpha‐fetoprotein
- Alb
albumin
- ANOVA
analysis of variance
- cDNA
complementary DNA
- Fgf21
fibroblast growth factor 21
- Gapdh
glyceraldehyde 3‐phosphate dehydrogenase
- gDNA
genomic DNA
- HCC
hepatocellular carcinoma
- IMDs
inherited metabolic diseases
- LT
liver transplantation
- MMA
methylmalonic acidemia
- MMUT
methylmalonyl‐CoA mutase
- RISH
RNA in situ hybridization
- VG
vector genomes
- WT
wild type
A diverse group of patients with inherited metabolic diseases (IMDs), such as urea cycle disorders and organic acidemias, can benefit from elective liver transplantation (LT).( 1 ) Despite the clinical and biochemical improvement commonly experienced after successful transplantation, the procedural risks, requirement for lifelong immunosuppression after surgery, and finite organ donor pool have led to the exploration of alternative approaches to treat these patients, including systemic adeno‐associated viral (AAV) gene therapy. In fact, a number of clinical trials using AAV for liver‐directed gene therapy have begun, with promising results emerging.( 2 , 3 , 4 ) Yet questions concerning the therapeutic persistence of the transgene( 5 , 6 , 7 ) and especially genotoxicity remain as major obstacles to the widespread application of systemic gene therapy to infants and children with IMDs, where vector redosing may be required because of the hepatic growth and concomitant loss of AAV episomes.( 8 , 9 , 10 , 11 , 12 , 13 )
A representative IMD is hereditary or classical methylmalonic acidemia (MMA), a severe organic acidemia, most often caused by mutations in methylmalonyl‐CoA mutase (MMUT).( 14 ) Patients with classical MMA resulting from MMUT deficiency have a clinical phenotype characterized by recurrent episodes of life‐threatening metabolic crises and multiorgan disease.( 15 ) Despite diligent dietary and medical management,( 16 , 17 , 18 , 19 ) patients remain at risk for disease progression, disability, and death. The high mortality and morbidity of classical MMA has led to the use of LT as an elective surgical intervention, even in young children.( 20 , 21 , 22 ) LT can stabilize MMA patients, greatly reduces metabolite burden, and eliminates metabolic crises.( 22 , 23 , 24 , 25 , 26 ) However, given that transplantation procedures bear grave risks and complications,( 27 ) an alternative to early LT that is less invasive, durable, and therapeutic would represent an important treatment for classical MMA.
Several mouse models that accurately recapitulate the severe clinical symptoms displayed by MMA patients have been developed and used to explore therapeutic approaches.( 28 , 29 , 30 ) Although conventional AAV‐mediated gene delivery effectively rescued neonatal mice with the most severe form of MMA,( 31 , 32 , 33 , 34 ) a rapid loss of hepatic transgene expression was observed and a majority of treated animals developed hepatocellular carcinoma (HCC), which was subsequently determined to be caused by AAV‐mediated insertional mutagenesis of the Rian locus.( 8 , 9 ) Unfortunately, the correlation between AAV exposure and HCC formation predominantly occurred following vector administration in the neonatal period, a time when treatment of patients with MMA, as well as those with other IMDs, would have the greatest potential benefit.( 1 , 20 , 21 ) Whereas genotoxicity from recombinant AAV vector clinical trials has not been observed, recent studies have demonstrated that wild‐type (WT) AAV can be associated with oncogenic events in humans.( 12 , 13 ) This mandates the consideration of possible side effects of canonical AAV integration in human gene therapy, especially in younger patients.
To increase the stability of transgene expression and reduce the potential risk of AAV‐mediated genotoxicity, we created a vector coding for a promoterless integrating cassette,( 35 , 36 , 37 , 38 ) designed to express MMUT in hepatocytes. Our approach relies upon endogenous homologous recombination to direct site‐specific genome insertion of a human codon‐optimized MMUT into the mouse albumin (Alb) locus, immediately upstream of the native Alb stop codon (Fig. 1A). In the edited hepatocytes, fused mRNA transcripts contain Alb, a P2A sequence, and the MMUT transgene expressed through the endogenous Alb promoter. The fused mRNA produces two separate proteins attributable to ribosomal skipping induced by the P2A sequence (Fig. 1A).( 39 , 40 , 41 , 42 ) Here, we present results from efficacy, durability, and safety studies in severe and hypomorphic mouse models of MMA to demonstrate that AAV‐mediated genome editing into Alb can achieve durable and therapeutic expression of MMUT, without genotoxicity. In the course of our investigations, we also found that edited hepatocytes expanded in vivo because of selective growth advantage, which enhanced corrective effects and mitigated the low efficiency of nuclease‐free editing. Our studies establish AAV‐mediated integration of MMUT into Alb as an immediately translatable, genomic therapy for patients with isolated MMA, which should be broadly extendable to related IMDs that are currently managed using elective LT.
FIG. 1.
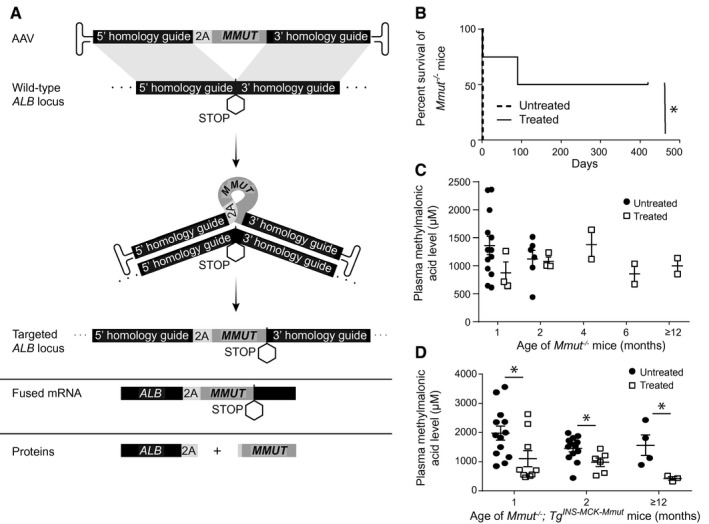
AAV‐Alb‐2A‐MMUT therapy improves the phenotype of MMA mice in a pilot study. (A) Schematic of AAV‐Alb‐2A‐MMUT vector integration and expression. Homologous recombination allows for site‐specific gene addition of human codon‐optimized MMUT into the mouse Alb locus upstream of the stop codon. This generates a fused allele under the control of the native Alb promoter producing a fused mRNA. The 2A peptide mediates ribosomal skipping, which generates two separate, functional proteins; Alb‐2A and MMUT. (B) Survival curve of Mmut−/− mice treated (n = 4) with 8.6e11 VG of AAVDJ‐Alb‐2A‐MMUT versus untreated Mmut−/− mice (n = 7; *P < 0.02; log‐rank [Mantel‐Cox] test). (C) Plasma methylmalonic acid levels of neonatal lethal Mmut−/− mice (n = 3, 3, 2, 2, and 2) were not significantly reduced compared to untreated historical Mmut−/− animals (n = 13, 6, 0, 0, and 0). (D) Plasma methylmalonic acid levels in treated Mmut–/–;TgINS‐MCK‐Mmut mice (n = 9, 6, and 3) treated with 8.6e11‐2.5e12 VG of AAV8‐Alb‐2A‐MMUT were significantly lower than untreated Mmut–/–;TgINS‐MCK‐Mmut mice (n = 13, 13, and 4; *P < 0.05, ±). Error bars are ± the SEM.
Materials and Methods
Animal Studies
Animal work was performed in accordance with the guidelines for animal care at the National Institutes of Health and the Guide for the Care and Use of Laboratory Animals. Both mouse models were on a mixed C57BL/6 × 129SV/Ev × FvBN genetic background and were described; Mmut−/− ( 28 , 30 ) and Mmut–/–;TgINS‐MCK‐Mmut.( 29 ) Experimental groups (n = 81) contained roughly equal numbers of both sexes ranging from 1 to 26 months old. Treated mice received AAV by intrahepatic injection 12‐24 hours postpartum. Mice were bled by retro‐orbital sinus plexus sampling using a sterile glass capillary tube and weighed monthly. Controls were cohoused mice from different litters with the same breeding pairs. All mice are housed in microisolator cages on positive‐pressure ventilated racks (Lab Products, Inc., Seaford, DE), and cages are bedded with steam‐sterilized hardwood bedding (NEPCO Beta‐Chips). Standard environmental enrichment for all mouse cages containing ≥2 animals is a pulped virgin cotton Nestlet (Ancare, Bellmore, NY). Environmental enrichment for cages containing singly housed animals consists of a Nestlet and a paper nesting product (Bed‐r’Nest; The Andersons Lab Bedding, Maumee, OH). Mice are fed autoclaved or irradiated Purina Lab Diet (Product #5R31; LabDiet, St. Louis, MO).
Plasmid Vector and Recombinant AAV Production
AAV‐Alb‐2A‐MMUT was generated by replacing the FIX complementary DNA (cDNA) with a human codon‐optimized MMUT cDNA from the pAB288 plasmid.( 35 ) AAV serotype 8 (AAV8) and AAVDJ vectors were produced by triple transfection with Ca3(PO4)2 followed by CsCl gradient purification( 43 ) and titered as described.( 44 )
Methylmalonic Acid Measurement
Methylmalonic acid was measured in plasma using gas chromatography/mass spectrometry with stable isotopic internal calibration, as described.( 45 )
Western Blotting Analysis
Samples were prepared and run according to standard protocols. The following antibodies were used for the detection of MMUT (ab134956; Abcam, Cambridge, MA), ß‐Actin (ab8227; Abcam), Cytochrome C (4280; Cell Signaling Technology, Danvers, MA; or 66009‐1‐Ig; Proteintech, Rosemont, IL), and alpha‐fetoprotein (Afp; ab169552; Abcam) in conjunction with the following secondary antibodies: (ab6721; Abcam), (925‐32211; LI‐COR, Lincoln, NE), and (926‐68072; LI‐COR). Quantification of western blotting bands was performed using ImageJ (http://rsb.info.nih.gov/ij/).
Hepatic MMUT RNAscope and Immunohistochemistry
Livers were fixed in 4% paraformaldehyde and processed into paraffin blocks. Five‐micron sections were cut and stained with either the RNAScope 2.5 HD Assay‐Brown (322300; ACDBio, Newark, CA), following the manufacturer’s instructions, or with rabbit polyclonal anti‐MMUT antibody (17034‐1‐AP; Proteintech), according to the manufacturer’s recommendations. Slide images were captured using the Zeiss AxioScan Z1 slide scanner and analyzed using Image Pro premier 3D version 9.3 by Media Cybernetics.
Histology and HCC Screening
Livers were visually inspected for abnormalities at the time of necropsy, then fixed in formalin, embedded, serially sectioned, stained with hematoxylin and eosin (H&E), and reviewed by a veterinary pathologist.
Vector Genomes Quantitation
Genomic DNA (gDNA) from liver samples was extracted using the DNeasy Blood & Tissue Kit (69506; Qiagen, Germantown, MD). ddPCR was performed, according to the manufacturer’s recommendations, for the Bio‐Rad QX200 AutoDG ddPCR system using 10 or 20 ng of DNA as input and the following probes: BIO‐RAD (Hercules, CA) ddPCR CNV assay (glyceraldehyde 3‐phosphate dehydrogenase [Gapdh]; catalogue no.: 10042961, dMumCNS300520369) and MMUT (catalogue no.: 10042958, dCNS513322846).
Albumin Integration Assay
gDNA from liver samples was extracted using the DNeasy Blood & Tissue Kit (69504; Qiagen). A synthesized DNA fragment representing the genomic integration between the Alb locus and 5′ end of MMUT was used to generate a standard curve for integration efficiency from 100% to 0.01%. gDNA (200 ng) from various mouse liver samples was used as a template for the DNA integration assay.
Both standard curve and sample gDNA were run in parallel by LR‐PCR with primers (F: 5′‐ATGTTCCACGAAGAAGCCA‐3′, R: 5′‐TCAGCAGGCTGAAATTGGT‐3). PCR product from LR‐PCR was purified with SPRI beads (G950; ABM, Richmond, BC, Canada) and then used as a template for qPCR (F: 5′‐ATGTTCCACGAAGAAGCCA‐3′, R: 5′‐AGCTGTTTCTTACTCCATTCTCA‐3′, Probe: 5′‐AGGCAACGTCATGGGTGTGACTTT‐3′).
Sanger Sequencing of Alb‐2A‐MMUT cDNA
RNA from mouse liver samples was extracted using the conventional TRIzol protocol and cleaned using an RNeasy kit and treated with DNAse I. cDNA was generated by the High‐Capacity cDNA Reverse Transcription Kit (4368814; ThermoFisherScientific, Waltham, MA). The integration event was captured with PCR primers surrounding the 5′ (F: 5′‐CTGTCTGCAATCCTGAACCGTG‐3′ and R: 5′‐GGCTTGATTGAAATCCCCTCTG‐3′) and 3′ (F: 5′‐GAGCTGAACTCTCTGGGCAGA‐3′ and R: 5′‐GCAGCACAGAGACAAGAAGTC‐3′) integration junctions and submitted for Sanger sequencing.
Enzyme‐Linked Immunosorbent Assays
ALB‐2A in plasma was measured by chemiluminescence sandwich enzyme‐linked immunosorbent assay (ELISA), using a proprietary rabbit polyclonal anti‐2A antibody for capture, and horseradish peroxidase–labeled antimouse Alb for detection. Fibroblast growth factor 21 (Fgf21) protein was measured using an ELISA kit (MF2100; R&D Systems) as per the manufacturer’s instructions.
1‐13C‐Propionate Oxidation
In vivo 1‐13C‐propionate oxidation was measured using the described protocol.( 32 )
Statistical Analyses
Prism 8 by GraphPad was used to analyze all data. Results are expressed as mean ± SEM. Values of P < 0.05 were considered statistically significant. Depending on the experimental design, an unpaired t test, log‐rank (Mantel‐Cox) test, or one‐way analysis of variance (ANOVA) was used as indicated in the legends to the figures.
Results
AAV‐Alb‐2A‐MMUT Therapy: Pilot Study in Murine Models of MMA
To evaluate the therapeutic effect of the AAVDJ‐Alb‐2A‐MMUT vector, Mmut−/− pups (n = 4) were treated with a single dose of 8.6e11 vector genomes (VG)/pup by intrahepatic injection at birth. Untreated pups (n = 7) died by day 2 of life. Treated Mmut−/− mice displayed a significant increase in survival (mean survival of 255 days; P < 0.02; Fig. 1B) compared to untreated controls. Whereas treated Mmut−/− mice were smaller compared to WT Mmut+/− littermates, they were active and healthy in appearance. However, neither plasma methylmalonic acid levels (Fig. 1C) nor weights were improved in treated Mmut−/− mice compared to historical values from rare, untreated Mmut−/− mice that survived beyond the neonatal period( 28 ) (Supporting Fig. S1A). Because the severe lethality of the full Mmut knockout phenotype prevented the collection of bona fide control data, we performed further studies in Mmut–/–;TgINS‐MCK‐Mmut mice, also referred to as MCK MMA mice. Like Mut−/− mice, MCK MMA mice are also devoid of MMUT in the liver, and this model accurately recapitulates the early childhood form of severe isolated MMA.( 29 )
Treatment With AAV‐Alb‐2A‐MMUT Decreases Disease‐Related Circulating Metabolites in MCK MMA Mice
MCK MMA pups were intrahepatically injected at birth with doses of 8.6e11‐2.5e12 VG/pup of AAV8‐Alb‐2A‐MMUT, and longitudinally followed, in parallel with untreated MCK MMA mice, to ascertain the durability of therapy at 1, 2, and 15 months posttreatment. Treated MCK MMA mice displayed significantly lower plasma methylmalonic acid levels (P ≤ 0.04, unpaired t test) compared to untreated MCK MMA mice at all measured time points (Fig. 1D). Interestingly, plasma methylmalonic acid levels appeared to decline over time in treated MCK MMA mice. Although treated MCK MMA mice were active and appeared healthy, their weights remained similar to age‐matched untreated MCK MMA mice (Supporting Fig. S1B).
Kinetics of MMUT Expression and Transgene Integration
Liver protein extracts from treated animals were immunoblotted to determine the extent that AAV‐mediated editing resulted in increased hepatic MMUT. At 1 and 2 months posttreatment, MMUT was detected in treated MCK MMA mice at 10%‐20% of the level of endogenous MMUT in WT mice (Fig. 2A). Significantly more MMUT was detected in livers of treated MCK MMA mice at 13‐15 months posttreatment (mean = 79.0 ± 22.1% of WT protein; P = 0.01, unpaired t test; Fig. 2A,B). These highly encouraging results are in marked contrast to previous studies showing that levels of MMUT expression decreased over time after canonical AAV8 treatment of neonatal MMA mice attributable to dilution of the episomal transgene because of cellular expansion.( 32 )
FIG. 2.
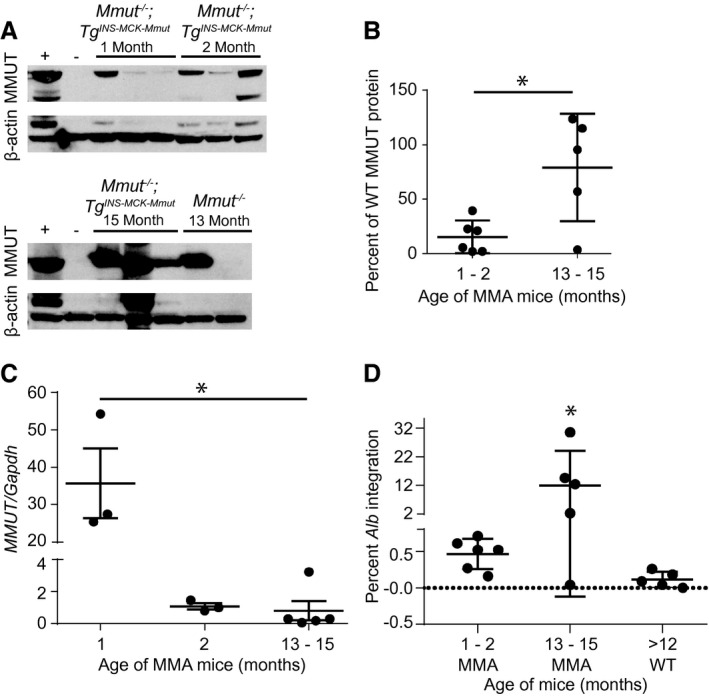
Hepatic characterization of AAV‐Alb‐2A‐MMUT transgene and MMUT expression in a pilot study. (A) Immunoblotting of hepatic MMUT protein (85 kDa) expression in WT (+), untreated MCK MMA (−), and treated MMA mice. ß‐actin (42 kDa) was used as a loading control. (B) Hepatic MMUT protein expression was quantified as a percentage of WT MMUT protein expression and normalized to ß‐actin. MMUT expression is similar between samples obtained from mice euthanized at 1 and 2 months postinjection, but is significantly elevated in samples collected 13 and 15 months postinjection (*P < 0.02, unpaired t test). (C) Number of liver viral genomes (MMUT) relative to genomic murine Gapdh copy number at 1, 2, and 13‐15 months posttreatment (n = 3, 3, and 5 respectively; *P < 0.001, one‐way ANOVA). (D) Percentage of Alb loci containing the 2A‐MMUT integration after treatment of MMA mice at 1 to 2 months posttreatment (n = 6), 13‐15 months posttreatment (n = 5), or in WT animals (n = 5) at 12‐15 months posttreatment (*P < 0.03, one‐way ANOVA).
Next, gDNA extracted from livers of treated mice was assayed for total vector genome copy number and 2A‐MMUT integrations into the Alb locus. The ratio of MMUT transgene to genomic Gapdh alleles declined dramatically over time from a mean of 35.7 at 1 month to 1.1 at 2 months following AAV delivery in the neonatal period (Fig. 2C), consistent with the well‐established episomal loss of vector genomes during rapid liver growth.( 5 , 6 , 46 , 47 )
Integration of the MMUT transgene into the Alb locus was detected by long‐range quantitative PCR assay (Supporting Fig. S2A,B). In MCK MMA mice, 13 and 15 months posttreatment, the percentage of Alb integration events was significantly increased (P < 0.03, one‐way ANOVA) when compared to those euthanized at 1 and 2 months postinjection, suggesting an expansion of corrected hepatocytes over time in mice (Fig. 2D). Interestingly, an increase in integrations was not observed in treated WT animals, suggesting that selection for corrected hepatocytes in livers of MMA mice was operative. Additionally, the fused Alb‐2A‐MMUT mRNA was sequenced and confirmed to encode the Alb‐2A‐MMUT sequence inserted precisely 5’ to the Alb stop codon (Supporting Fig. S3A,B).
A Pattern of Selective Advantage for Corrected Hepatocytes in the Diseased Liver After AAV Alb Editing
The unique sequence of the MMUT codon‐optimized transgene allowed for the distinction between native murine Mmut and MMUT transgene mRNA expression using RISH. Livers were stained for MMUT, and the percentage of area stained was determined (Fig. 3A‐D). Only a small number of hepatocytes expressed MMUT at 2 months posttreatment in MCK MMA mice (1.24 ± 0.32%; Fig. 3A). However, MMUT expression increased significantly in treated MCK MMA mice 13‐15 months posttreatment (7.74 ± 0.88%; P < 0.005, one‐way ANOVA), and presented a markedly different appearance than at 2 months posttreatment, with large clusters of cells expressing MMUT (Fig. 3B). In stark contrast, the 15‐month posttreatment mice with WT phenotypes and with a Mmut +/+ or Mmut+/− genotypes, had minimal staining (Fig. 3C,D), corresponding to a small number of initially modified cells (0.83 ± 0.11%), consistent with a low‐frequency integration event that is expected from in vivo homologous recombination.
FIG. 3.
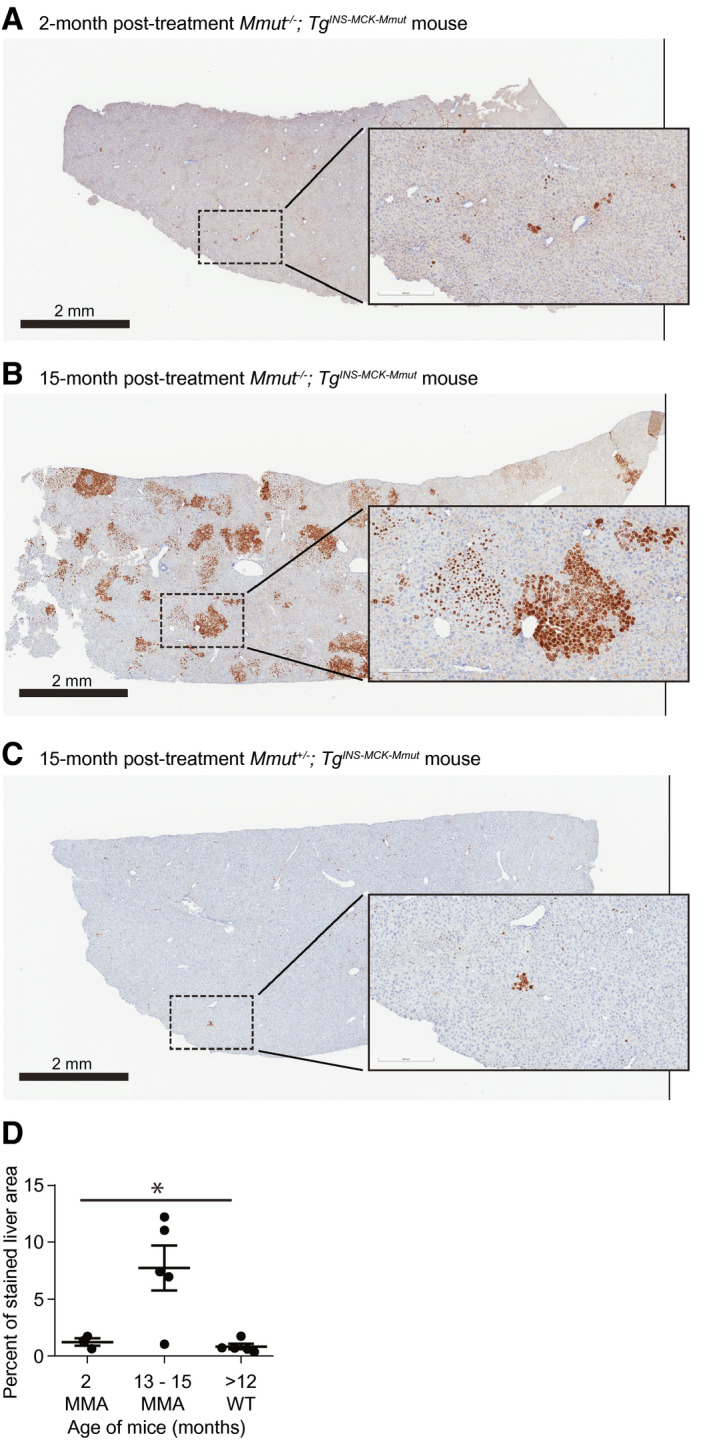
Hepatic mRNA expression of MMUT in a pilot study. (A‐C) Representative RISH images of AAV‐Alb‐2A‐MMUT–treated mice. Cells positive for MMUT mRNA are stained brown. The scale bar is 2 mm, and the insert is a 10× magnification. (A) A 2‐month treated Mmut–/–;TgINS‐MCK‐Mmut mouse liver. (B) A 15‐month treated Mmut–/–;TgINS‐MCK‐Mmut mouse liver. (C) A 13‐month treated Mmut+/− WT control mouse liver. (D) Percentage of liver area stained for MMUT mRNA in treated animals; MMA mice at 2 months posttreatment (n = 3), 13‐15 months posttreatment (n = 5), or WT mice posttreatment (n = 5; *P < 0.005, one‐way ANOVA). All error bars are ± the SEM.
Expanded Longitudinal Dose‐Finding Study of AAV Alb Editing in MCK MMA Mice
Based on the findings of the pilot studies, we next performed an expanded longitudinal dose‐finding study to define a clinically relevant dose, verify the findings of our pilot studies, and explore the kinetics of the selective growth advantage conferred after MMUT insertion into Alb. MCK MMA mice and control littermates were treated with 2.5e11 or 2.5e12 VG/pup of AAVDJ‐Alb‐2A‐MMUT by intrahepatic injection at birth and followed for 10‐11 months. The larger cohort of MCK MMA treated with 2.5e11 VG exhibited an increased rate of survival relative to untreated MCK MMA mice (Fig. 4A).
FIG. 4.
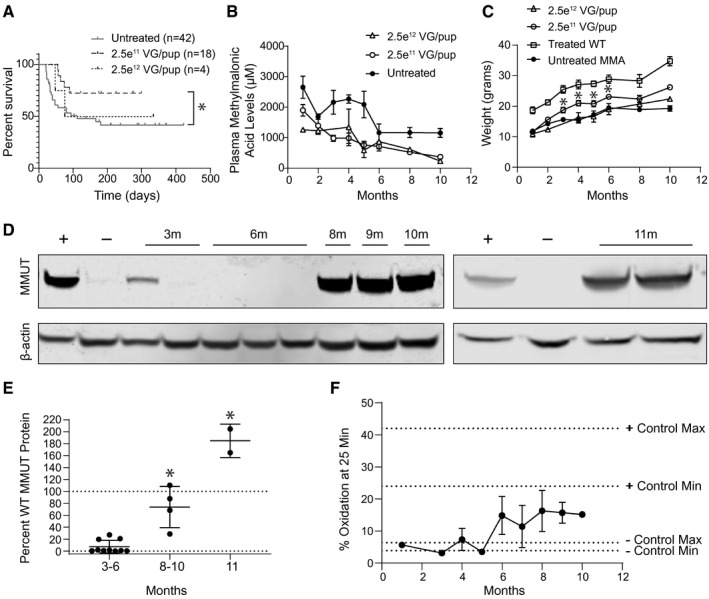
AAV‐Alb‐2A‐MMUT longitudinal dosage study in Mmut–/–;TgINS‐MCK‐Mmut mice. (A) Survival curve of Mmut–/–;TgINS‐MCK‐Mmut mice treated with 2.5e12 VG/pup (n = 4) or 2.5e11 VG/pup (n = 18) of AAVDJ‐Alb‐2A‐MMUT versus untreated Mmut–/–;TgINS‐MCK‐Mmut mice (n = 42; *P < 0.05). Some mutants treated with 2.5e11 VG/pup were sacrificed at 3 (n = 5), 6 (n = 5), 8 (n = 1), and 9 months (n = 2) for terminal assays. They are censored in the survival curve at the day of euthanization. (B) Plasma methylmalonic acid levels of Mmut–/–;TgINS‐MCK‐Mmut mice treated with 2.5e12 VG/pup (n = 4, 3, 0, 2, 2, 2, 2, and 2), with 2.5e11 VG/pup treated (n = 18, 18, 15, 7, 9, 9, 4, and 1), or untreated Mmut–/–;TgINS‐MCK‐Mmut mice (n = 5, 11, 10, 5, 7, 5, 7, and 7; P < 0.03 and 1, 2, 3, 4, 5, 8, and 10 months, one‐way ANOVA). (C) Weights of Mmut–/–;TgINS‐MCK‐Mmut mice (MMA) treated with 2.5e12 VG/pup (n = 4, 3, 0, 2, 2, 2, 2, and 2), with 2.5e11 VG/pup (n = 16, 17, 7, 9, 4, 9, 4, and 1), or untreated (n = 18, 25, 18, 8, 5, 12, 6, and 4; *P < 0.05, unpaired t test). (D) Immunoblotting of hepatic MMUT (85 kDa) and ß‐actin (42 kDa) expression in WT (+), untreated MMA (−), MMA mice treated with 2.5e11 VG/pup at 3, 6, 8, 9, and 10 months (m), or treated with 2.5e12 VG/pup at 11 months. (E) Quantification of hepatic MMUT protein in 2.5e11 VG/pup and 2.5e12 VG/pup treated MMA mice expressed as a percentage of MMUT WT expression and normalized to ß‐actin (*P < 0.0001, unpaired t test and one‐way ANOVA). (F) Propionic oxidation improves in both treatment groups beginning at 6 months compared to untreated controls, but the oxidation in treated mice never reaches WT levels. Oxidation measurements are displayed for each dose: 2.5e12 VG/pup (n = 1, 2, and 1) and 2.5e11 (n = 1, 1, 3, 1, 1, 2, 2, and 1). Positive control range was defined from 5 assayed WT mice, and negative control range was defined from 5 assayed untreated MMA mice.
Plasma methylmalonic acid and weights were evaluated monthly in treated animals. As with the pilot studies, a trend of decreasing plasma methylmalonic acid levels over time was observed in both dose cohorts (Fig. 4B). A significant weight improvement was observed starting at 3 months posttreatment and persisted for the remainder of the study in the 2.5e11 VG/pup group. However, treated MCK MMA mice never achieved the weights of their age‐matched WT littermates (Fig. 4C).
Hepatic MMUT protein levels in treated MCK MMA mice were determined by immunoblotting and normalized to the percentage of WT murine MMUT protein (Fig. 4D,E). Low levels of MMUT protein were detected in MMA mice treated with 2.5e11 VG/pup dose at 3 (mean = 10.0 ± 5.6%) and 6 months (mean = 4.6 ± 4.1%) posttreatment. However, MMUT protein was significantly higher at 8‐10 months posttreatment with the 2.5e11 VG/pup dose (mean = 73.7 ± 17.3%; P < 0.0001, unpaired t test). The highest levels of hepatic MMUT protein were found in the 11‐month‐old mice treated with the 2.5e12 VG/pup dose (mean = 184.8 ± 20.0%; P < 0.0001, one‐way ANOVA). Immunoblotting of isolated mitochondria showed that the MMUT protein from the therapeutic transgene was properly localized (Supporting Fig. S4).
To determine the functionality of hepatic MMUT produced following genome editing, we assayed enzymatic function by measuring whole‐animal in vivo 1C13 propionate oxidative capacity, disease‐associated biomarkers, and peptide emanating from the edited locus. Untreated MCK MMA mice oxidized between 3.9% and 6.4% of the labeled propionate( 29 ) (Fig. 4F) whereas treated MCK MMA mice displayed a gradual increase in 1C13 propionate oxidative capacity, with older treated mutant mice metabolizing more label than younger mice.
Recent studies have identified plasma Fgf21, a metabolic regulator and marker of mitochondrial stress, as an important biomarker in the MMA patient population.( 29 ) In agreement with the previous observation that liver‐directed canonical AAV8 gene therapy resulted in a dramatic lowering of Fgf21 levels in treated MCK MMA mice, AAV‐mediated editing of MMUT into Alb likewise resulted in a 22‐ fold reduction of Fgf21 plasma levels (Fig. 5A; P < 0.001, unpaired t test). This reduction was observed beginning at 1 month posttreatment and was maintained for the duration of the study (Supporting Fig. S2C).
FIG. 5.
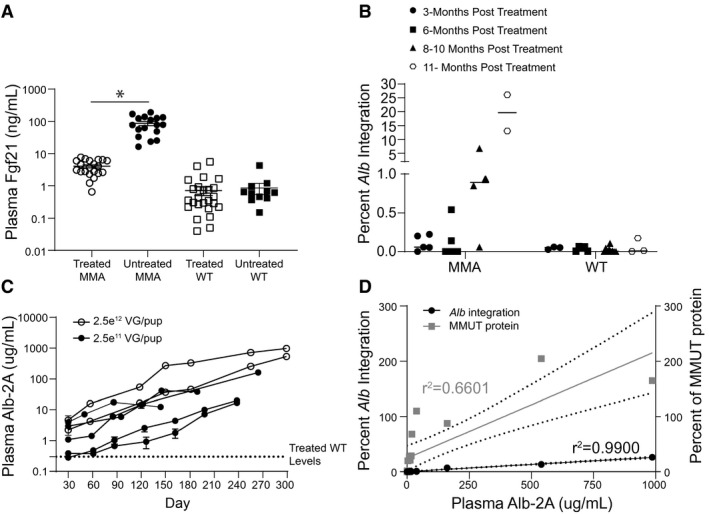
Plasma biomarkers in a longitudinal dosage study in Mmut–/–;TgINS‐MCK‐Mmut mice. (A) Reduction of Fgf21 plasma levels in treated MMA mice relative to untreated MMA mice (P < 0.0001, unpaired t test). Error bars are ± the SEM. (B) Percentage of Alb loci containing the 2A‐MMUT integration in treated MMA mice (n = 5, 5, 4, and 2) and treated WT mice (n = 3, 5, 9, and 3). (C) Increase in plasma 2A protein levels in individual treated mice over time. (D) A linear regression analysis shows the correlation between plasma Alb‐2A levels and Alb‐2A‐MMUT integrations (r 2 = 0.99; P < 0.0001) and hepatic MMUT protein levels (r 2 = 0.66; P = 0.0001).
Because AAV‐Alb‐2A‐MMUT–mediated genome‐edited hepatocytes produce a 21‐amino‐acid P2A peptide fused to 3′ terminus of Alb protein (Fig. 1A), we developed an assay to detect Alb‐2A in plasma and measured the concentration after mice were treated with AAV‐Alb‐2A‐MMUT. Most treated MMA mice showed a trend of continuously increasing integrations (Fig. 5B) and plasma Alb‐2A protein with age (Fig. 5C). A linear regression analysis revealed that plasma Alb‐2A was highly correlated with both Alb‐2A‐MMUT integrations (r = 0.995; P < 0.0001) and MMUT protein levels (r = 0.812; P = 0.0001; Fig. 5D). In most treated mice, Alb‐2A plasma levels represented <1% of the total plasma albumin except in the animal with the highest Alb‐2A where it measured 3.4%.
To further define the kinetics of correction following Alb editing, livers from AAV‐Alb‐2A‐MMUT–treated mice were stained for MMUT mRNA and MMUT protein by immunohistochemistry. Expression of the MMUT transgene at 3 and 6 months in MCK MMA livers (n = 5) was ~1%‐2%, and displayed predominantly single‐cell staining with rare small clusters of positively stained cells (Fig. 6A). However, at 8‐10 months posttreatment, expression of the transgene RNA in MCK MMA livers (n = 3) increased to ~5%, with multiple clusters of MMUT mRNA and MMUT protein‐positive cells dispersed throughout the liver (Fig. 6B,C). After 11 months, the corrected cells had further expanded into larger distinct clusters, with ~9% hepatocytes expressing MMUT (Fig. 6D). Conversely, treated WT control livers only had small numbers of cells with positive mRNA staining at both early and late time points (Fig. 6E). The relatively higher level of MMUT protein in comparison to MMUT RNA staining in Fig. 6E is attributable to the presence of endogenous MMUT protein in the WT animals. In all the treated MMA mice, the intensity and location of MMUT mRNA staining corresponded with MMUT protein staining.
FIG. 6.
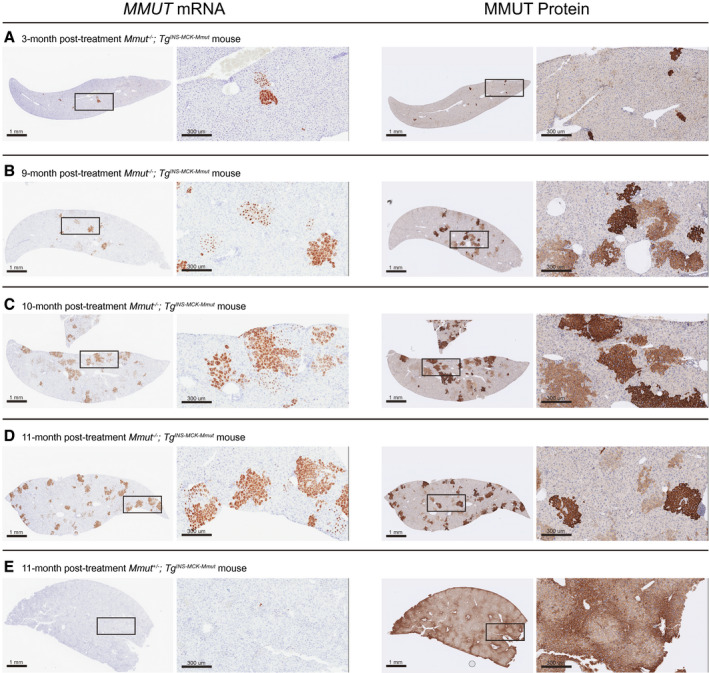
MMUT mRNA and MMUT protein expression in livers of treated mice. Representative MMUT RISH and MMUT protein immunohistochemistry staining of serial liver sections in treated mice. Cells positive for mRNA or protein expression are stained brown. The scale bar is 1 mm, and the insert is a 10× magnification. (A) A Mmut–/–;TgINS‐MCK‐Mmut mouse 3 months posttreatment (2.5e11 VG). (B) A Mmut–/–;TgINS‐MCK‐Mmut mouse 9 months posttreatment (2.5e11 VG). (C) A Mmut–/–;TgINS‐MCK‐Mmut mouse 10 months posttreatment (2.5e11 VG). (D) A Mmut–/–;TgINS‐MCK‐Mmut mouse 11 months posttreatment (2.5e12 VG). (E) A Mmut+/–;TgINS‐MCK‐Mmut (WT phenotype, 2.5e12 VG) mouse 11 months posttreatment.
No HCC Observed After AAV‐Alb‐2A‐MMUT Treatment
To validate the safety of AAV‐mediated genome editing, 56 livers from treated MMA (n = 21) and WT (n = 35) mice that ranged in age from 3 to 26 months were evaluated for hereditary hemochromatosis, grossly by visual dissection, and histologically by a veterinary pathologist. None of the AAV‐treated livers showed gross liver abnormalities or abnormal hepatocellular morphology other than what is associated with MMA (Fig. 7A‐E). Additionally, a subset of livers harvested from AAV‐treated mice (ranged in age from 18 to 26 months) were screened for increased expression of Afp, an established HCC biomarker.( 48 ) Afp levels were not increased in treated animals compared to the levels observed in untreated controls (Supporting Fig. S2D).
FIG. 7.
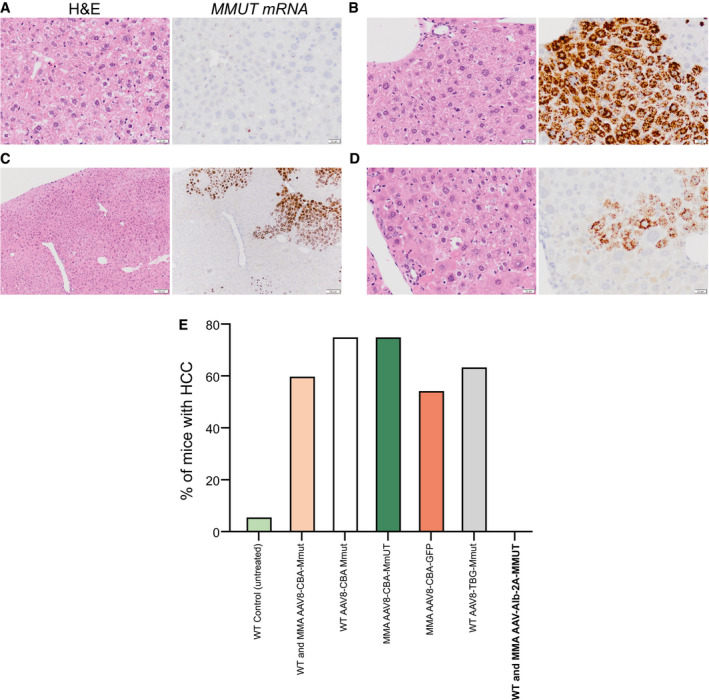
No signs of HCC detected following AAV treatment. Representative H&E and MMUT RISH staining of serial liver sections in treated mice. (A) A Mmut–/– mouse 13 months posttreatment (8.58e11 VG). Areas with little MMUT RNA staining show a normal MMA hepatocyte phenotype; no sign of HCC present. Both images taken at 40×, scale bar is 20 µm. (B) A Mmut–/–;TgINS‐MCK‐Mmut mouse 15 months posttreatment (1.9e12 VG). Areas with significant MMUT RNA staining show a normal microscopic appearance. Both images taken at 40×, scale bar is 20 µm. (C) A Mmut–/–;TgINS‐MCK‐Mmut mouse 15 months posttreatment (8.58e11 VG). Areas that border regions of corrected and uncorrected hepatocytes again show normal hepatocyte phenotypes for MMA or healthy tissues. At these borders, there is no sign of HCC. Both images taken at 10×, scale bar is 100 µm. (D) A magnification of (C). Both images taken at 40×, scale bar is 20 µm. (E) Incidence of HCC in WT and MMA mice treated with AAV‐Alb‐2A‐MMUT versus historical HCC samples.( 9 ) Abbreviations: CBA, chicken beta actin; GFP, green fluorescent protein; TBG, thyroxine‐binding globulin.
Discussion
With the implementation of newborn screening for lethal disorders comes an accompanying need to develop effective treatments. Indeed, the successful gene therapy for cerebral adrenoleukodystrophy,( 48 ) and the devastating neuromuscular disorder, spinal muscular atrophy type 1,( 49 ) has helped establish a rationale for population detection of previously fatal genetic disorders during infancy. It is therefore pressing to address the treatment options for patients with IMDs that are routinely detected yet remain recalcitrant to medical management, such as organic acidemias and urea cycle disorders. Based on proof‐of‐concept efficacy in small and large animal models, systemic AAV gene therapy has emerged as a promising possibility; however, durable transgene expression needs to be achieved and long‐term toxicity requires further investigation.
There is an evolving concern that exposure to high doses of AAV, both WT and perhaps recombinant, might confer an increased risk to develop HCC. Fundamentally, the genotoxicity associated with AAV exposure derives from random integration. However, numerous studies with varied metabolic mouse models, treated with canonical promoter‐driven nonintegrating AAV vectors, have revealed a significantly increased risk of HCC formation as a long‐term complication of neonatal gene therapy.( 8 , 9 ) Although the earliest observations describing an AAV HCC association in mice were controversial, more recent studies have been documenting that a small subset of HCCs in humans contained clonal and genotoxic insertions of WT AAV2,( 12 ) and almost always contain a small hepatic enhancer embedded immediately adjacent to the AAV inverted terminal repeats.( 50 ) The renewed concerns surrounding AAV genotoxicity have stimulated alternative approaches to develop genomic therapies suitable for young patients with severe metabolic liver diseases.
We therefore sought to develop a genome editing strategy that would be applicable for patients with MMA. After a single neonatal treatment with a promoterless, integrating AAV‐Alb‐2A‐MMUT vector in mouse models of severe MMA, durable therapeutic benefits, with effects measurable as long as 15 months posttreatment, were noted. Treated MMA mice had improved survival, dramatic increases in hepatic MMUT mRNA and protein expression, increased whole‐body enzyme activity, decreased disease‐related plasma metabolites, and were free of HCC. Expression of MMUT from the Alb locus in MMA mice, but not treated controls, was accompanied by a pattern of increased Alb integrations, clusters of MMUT staining cells that increased in size over time, and a steady increase in Alb‐2A, a pattern that demonstrates a selective advantage for corrected hepatocytes in the context of the disease state. Although we do not define a mechanism for the growth effects we observed after MMUT editing into albumin, we posit that corrected hepatocytes manifest improved mitochondrial function and decreased oxidative stress, giving them an in vivo selective advantage in the abnormal metabolic milieu of an MMA hepatic environment.( 28 , 29 ) Given that the edited hepatocytes are permanently corrected, they transmit the therapeutic integration to their daughter cells, which propagate as the liver grows, and account for the observed in vivo expansion. Treated MMA mice do not develop HCC, despite exposure to AAV doses that were uniformly genotoxic in previous studies, and have grossly normal liver histology, specifically in the sections that correspond to the area where the in vivo expansion of corrected cells was noted by RISH, which provides a strong argument against a post‐AAV selective advantage originating from an independent driver mutation(s) as the cause of subsequent clonal proliferation.
Severe medical complications of MMA, and related IMDs, can arise early in life, and therefore immediate gene therapy could allow for the greatest potential benefit. Canonical AAV gene therapy, particularly when administered in the neonatal period, however, is associated with rapid and substantial loss of transgene expression and, possibly, genotoxicity. This raises both theoretical and practical concerns for translation to human subjects. The studies detailed here demonstrate that a promotor‐less, nuclease‐free integrating AAV designed to treat MMUT MMA is therapeutic, durable, safe, and improves with time. Unlike conventional AAV gene therapy, AAV‐mediated editing should be especially beneficial when administered early in life, to capitalize on the physiological peak of maximal hepatic growth and proliferation experienced by neonatal and pediatric patients. In aggregate, our results support clinical translation of AAV‐mediated editing into Alb as a genomic therapeutic approach to treat patients with MMA and related IMDs.
Author Contributions
R.J.C., M.A.K., A.B., and C.P.V. developed the experimental ideas and design of the study. Experiments were performed by L.E.V., R.J.C., J.L., S.G., S.Z., A.B., N.C., J.L.S., C.W.K., and B.T.H. Data analysis was performed by L.E.V., R.J.C., J.L., S.G., N.C., K.C., B.T.H., and V.H. Figures were produced by L.E.V., R.J.C., and C.P.V. The text was written by L.E.V., R.J.C., and C.P.V. with input from all authors. N.C., R.J.C, and C.P.V. supervised the study. L.E.V and R.J.C. contributed equally to this article. M.A.K. received support from NIH‐R01HL064274.
Supporting information
Supplementary Material
Acknowledgment
L.E.V., R.J.C., J.L.S., B.T.H., and C.P.V. were supported by the Intramural Research Program of the National Human Genome Research Institute. B.T.H. and J.L.S. were also supported by the Angels for Alyssa MMA Research Fund. J.L., S.G., S.Z., N.C., and K.C. are employees of, and supported by, LogicBio Therapeutics. M.A.K was supported by NIH‐R01HL064274. This work was completed with assistance from the National Human Genome Research Institute Cytogenetics and Microscopy Core and the National Cancer Institute Pathology/Histotechnology Laboratory. We owe special thanks to Andrew Warner, Elijah Edmondson, and Stephen Wincovitch for assistance with RNAscope studies and analysis. We also acknowledge Darwin Romero for animal husbandry, the tracking of mice, and data entry. Figures were created with the aid of Julia Fekecs.
Supported by the Intramural Research Program of the National Human Genome Research Institute, the Angels for Alyssa MMA Research Fund, and LogicBio Therapeutics, Inc.
Potential conflict of interest: Dr. Gordo is employed by and owns stock in LogicBio. Dr. Zang is employed by LogicBio. Dr. Kay consults for, advises for, and owns stock in LogicBio. Dr. Chiang is employed by and owns stock in LogicBio. Dr. Ko is employed by LogicBio. Dr. Chau is employed by and owns stock in LogicBio. Dr. Barzel consults for, advises for, and owns stock in LogicBio. Dr. Venditti received grants from LogicBio.
References
Author names in bold designate shared co‐first authorship.
- 1. Baruteau J, Waddington SN, Alexander IE, Gissen P. Gene therapy for monogenic liver diseases: clinical successes, current challenges and future prospects. J Inherit Metab Dis 2017;40:497‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med 2020;382:29‐40. [DOI] [PubMed] [Google Scholar]
- 3. Nienhuis AW, Nathwani AC, Davidoff AM. Gene therapy for hemophilia. Mol Ther 2017;25:1163‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus‐associated virus vector‐mediated gene transfer in hemophilia B. N Engl J Med 2011;365:2357‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno‐associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol 2001;75:6969‐6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L, Bell P, Lin J, Calcedo R, Tarantal AF, Wilson JM. AAV8‐mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta). Mol Ther 2011;19:2012‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cunningham SC, Spinoulas A, Carpenter KH, Wilcken B, Kuchel PW, Alexander IE. AAV2/8‐mediated correction of OTC deficiency is robust in adult but not neonatal Spf(ash) mice. Mol Ther 2009;17:1340‐1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chandler RJ, LaFave MC, Varshney GK, Burgess SM, Venditti CP. Genotoxicity in mice following AAV gene delivery: a safety concern for human gene therapy? Mol Ther 2016;24:198‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo‐Carrasco N, Senac JS, et al. Vector design influences hepatic genotoxicity after adeno‐associated virus gene therapy. J Clin Invest 2015;125:870‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007;317:477. [DOI] [PubMed] [Google Scholar]
- 11. Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, et al. Observed incidence of tumorigenesis in long‐term rodent studies of rAAV vectors. Gene Ther 2001;8:1343‐1346. [DOI] [PubMed] [Google Scholar]
- 12. Nault JC, Datta S, Imbeaud S, Franconi A, Mallet M, Couchy G, et al. Recurrent AAV2‐related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet 2015;47:1187‐1193. [DOI] [PubMed] [Google Scholar]
- 13. La Bella T, Imbeaud S, Peneau C, Mami I, Datta S, Bayard Q, et al. Adeno‐associated virus in the liver: natural history and consequences in tumour development. Gut 2020;69:737‐747. [DOI] [PubMed] [Google Scholar]
- 14. Manoli I, Sloan JL, Venditti CP. Isolated methylmalonic acidemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. 2005. Aug 16 [updated 2016 Dec 1]. [Google Scholar]
- 15. Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med 1983;308:857‐861. [DOI] [PubMed] [Google Scholar]
- 16. Baumgartner MR, Horster F, Dionisi‐Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis 2014;9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr 2016;28:682‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hauser NS, Manoli I, Graf JC, Sloan J, Venditti CP. Variable dietary management of methylmalonic acidemia: metabolic and energetic correlations. Am J Clin Nutr 2011;93:47‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manoli I, Myles JG, Sloan JL, Shchelochkov OA, Venditti CP. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: isolated methylmalonic acidemias. Genet Med 2016;18:386‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sloan JL, Manoli I, Venditti CP. Liver or combined liver‐kidney transplantation for patients with isolated methylmalonic acidemia: who and when? J Pediatr 2015;166:1346‐1350. [DOI] [PubMed] [Google Scholar]
- 21. Spada M, Calvo PL, Brunati A, Peruzzi L, Dell’Olio D, Romagnoli R, et al. Liver transplantation in severe methylmalonic acidemia: the sooner, the better. J Pediatr 2015;167:1173. [DOI] [PubMed] [Google Scholar]
- 22. Niemi AK, Kim IK, Krueger CE, Cowan TM, Baugh N, Farrell R, et al. Treatment of methylmalonic acidemia by liver or combined liver‐kidney transplantation. J Pediatr 2015;166:1455‐1461.e1. [DOI] [PubMed] [Google Scholar]
- 23. Chen PW, Hwu WL, Ho MC, Lee NC, Chien YH, Ni YH, et al. Stabilization of blood methylmalonic acid level in methylmalonic acidemia after liver transplantation. Pediatr Transplant 2010;14:337‐341. [DOI] [PubMed] [Google Scholar]
- 24. Pillai NR, Stroup BM, Poliner A, Rossetti L, Rawls B, Shayota BJ, et al. Liver transplantation in propionic and methylmalonic acidemia: a single center study with literature review. Mol Genet Metab 2019;128:431‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sakamoto R, Nakamura K, Kido J, Matsumoto S, Mitsubuchi H, Inomata Y, et al. Improvement in the prognosis and development of patients with methylmalonic acidemia after living donor liver transplant. Pediatr Transplant 2016;20:1081‐1086. [DOI] [PubMed] [Google Scholar]
- 26. Chu TH, Chien YH, Lin HY, Liao HC, Ho HJ, Lai CJ, et al. Methylmalonic acidemia/propionic acidemia—the biochemical presentation and comparing the outcome between liver transplantation versus non‐liver transplantation groups. Orphanet J Rare Dis 2019;14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vernon HJ, Sperati CJ, King JD, Poretti A, Miller NR, Sloan JL, et al. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. J Inherit Metab Dis 2014;37:899‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chandler RJ, Zerfas PM, Shanske S, Sloan J, Hoffmann V, DiMauro S, et al. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J 2009;23:1252‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manoli I, Sysol JR, Epping MW, Li L, Wang C, Sloan JL, et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight 2018;3:e124351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chandler RJ, Sloan J, Fu H, Tsai M, Stabler S, Allen R, et al. Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle. BMC Med Genet 2007;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carrillo‐Carrasco N, Chandler RJ, Chandrasekaran S, Venditti CP. Liver‐directed recombinant adeno‐associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long‐term phenotypic correction. Hum Gene Ther 2010;21:1147‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chandler RJ, Venditti CP. Long‐term rescue of a lethal murine model of methylmalonic acidemia using adeno‐associated viral gene therapy. Mol Ther 2010;18:11‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chandler RJ, Venditti CP. Pre‐clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl‐CoA mutase in a murine model of methylmalonic acidemia (MMA). Mol Genet Metab 2012;107:617‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Senac JS, Chandler RJ, Sysol JR, Li L, Venditti CP. Gene therapy in a murine model of methylmalonic acidemia using rAAV9‐mediated gene delivery. Gene Ther 2012;19:385‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barzel A, Paulk NK, Shi Y, Huang Y, Chu K, Zhang F, et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature 2015;517:360‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Porro F, Bortolussi G, Barzel A, De Caneva A, Iaconcig A, Vodret S, et al. Promoterless gene targeting without nucleases rescues lethality of a Crigler‐Najjar syndrome mouse model. EMBO Mol Med 2017;9:1346‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borel F, Tang Q, Gernoux G, Greer C, Wang Z, Barzel A, et al. Survival advantage of both human hepatocyte xenografts and genome‐edited hepatocytes for treatment of alpha‐1 antitrypsin deficiency. Mol Ther 2017;25:2477‐2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nygaard S, Barzel A, Haft A, Major A, Finegold M, Kay MA, et al. A universal system to select gene‐modified hepatocytes in vivo. Sci Transl Med 2016;8:342ra379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donnelly MLL, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, et al. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol 2001;82:1013‐1025. [DOI] [PubMed] [Google Scholar]
- 40. Sharma P, Yan F, Doronina VA, Escuin‐Ordinas H, Ryan MD, Brown JD. 2A peptides provide distinct solutions to driving stop‐carry on translational recoding. Nucleic Acids Res 2012;40:3143‐3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T‐cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114:535‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus‐1 in human cell lines, zebrafish and mice. PLoS One 2011;6:e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno‐associated viruses. J Virol 2008;82:5887‐5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nakai H, Thomas CE, Storm TA, Fuess S, Powell S, Wright JF, et al. A limited number of transducible hepatocytes restricts a wide‐range linear vector dose response in recombinant adeno‐associated virus‐mediated liver transduction. J Virol 2002;76:11343‐11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marcell PD, Stabler SP, Podell ER, Allen RH. Quantitation of methylmalonic acid and other dicarboxylic acids in normal serum and urine using capillary gas chromatography‐mass spectrometry. Anal Biochem 1985;150:58‐66. [DOI] [PubMed] [Google Scholar]
- 46. Cunningham SC, Dane AP, Spinoulas A, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther 2008;16:1081‐1088. [DOI] [PubMed] [Google Scholar]
- 47. Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 2011;12:341‐355. [DOI] [PubMed] [Google Scholar]
- 48. Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, et al. Hematopoietic stem‐cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med 2017;377:1630‐1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mendell JR, Al‐Zaidy S, Shell R, Arnold WD, Rodino‐Klapac LR, Prior TW, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713‐1722. [DOI] [PubMed] [Google Scholar]
- 50. Logan GJ, Dane AP, Hallwirth CV, Smyth CM, Wilkie EE, Amaya AK, et al. Identification of liver‐specific enhancer‐promoter activity in the 3’ untranslated region of the wild‐type AAV2 genome. Nat Genet 2017;49:1267‐1273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material