

Abstract
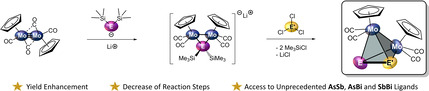
In order to improve and extend the rare class of tetrahedral mixed main group transition metal compounds, a new synthetic route for the complexes [{CpMo(CO)2}2(μ,η2:η2‐PE)] (E=As (1), Sb (2)) is described leading to higher yields and a decrease in reaction steps. Via this route, also the so far unknown heavier analogues containing AsSb (3 a), AsBi (4) and SbBi (5) ligands, respectively, are accessible. Single crystal X‐ray diffraction experiments and DFT calculations reveal that they represent very rare examples of compounds comprising covalent bonds between two different heavy pnictogen atoms, which show multiple bond character and are stabilised without any organic substituents. A simple one‐pot reaction of [CpMo(CO)2]2 with ME(SiMe3)2 (M=Li, K; E=P, As, Sb, Bi) and the subsequent addition of PCl3, AsCl3, SbCl3 or BiCl3, respectively, give the complexes 1–5. This synthesis is also transferable to the already known homo‐dipnictogen complexes [{CpMo(CO)2}2(μ,η2:η2‐E2)] (E=P, As, Sb, Bi) resulting in higher yields comparable to those in the literature reported procedures and allows the introduction of the bulkier and better soluble Cp′ (Cp′=tert butylcyclopentadienyl) ligand.
Keywords: pnictogens, bismuth, antimony, molybdenum, mixed main group elements
New synthetic pathway towards organometallic tetrahedrane derivatives [{CpMo(CO)2}2(μ,η2:η2‐EE′)] (E≠E′=P, As, Sb, Bi) involving a hetero‐dipnictogen ligand is reported, leading to dramatic yield enhancements for already known compounds, the reduction of reaction steps and access to so far unknown AsSb, AsBi and SbBi ligand complexes, which feature unseen covalent bonds between two different heavy group 15 elements without organic substituents.
Introduction
Tetrahedral molecules such as the most simple organic representative, the parent tetrahedrane (tricyclo[1.1.0.02,4]butane), have always been of great scientific interest, not only due to their aesthetical attraction as a chemical equivalent of a platonic body, but also because of their unusual bonding situation, high ring strain and reactivity. [1] These properties point already to their mostly challenging syntheses and low stability. [1] The probably most prominent inorganic example of this class of compounds is white phosphorus (P4), which can be prepared by sublimation of red phosphorus and is stable under exclusion of air. In contrast, its heavier counterpart, yellow arsenic (As4), is highly unstable and undergoes rapid autocatalytic degradation under light exposure already. [2] Furthermore, the heavier homologue Sb4 has been observed in the solid state only within thin antimony films received by the evaporation of Sb4 molecules in ultra‐high vacuum, [3] while Bi4 is only known in the gas phase. [4] Heteroatomic tetrahedranes built from p‐block elements are extremely scarce. Only three examples, namely AsP3 (I) and the tetrahedrane derivatives P(CtBu)3 (II) and P2(CtBu)2 (III), have been reported to date (Scheme 1b). [5] They are, however, highly sensitive to air. While I and III are pyrophoric, compound II degrades after 30 mins at room temperature.
Scheme 1.
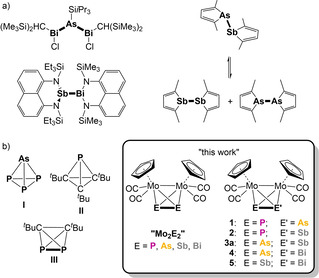
a) Rare examples of compounds featuring covalent As−Bi and Sb−Bi bonds (left), as well as a covalent As−Sb bond, which is in equilibrium with its respective homo‐dipnictogen complexes (right); b) heteroatomic main‐group tetrahedranes. This work: tetrahedral [{CpMo(CO)2}2(μ,η2:η2‐EE′)] compounds containing scarce covalent hetero‐dipnictogen bonds.
By formal substitution of one or two atoms of the E4 (E=P, As, Sb, Bi) tetrahedra with isolobal 15‐valence‐electron transition metal fragments, such as [CpMo(CO)2], stable tetrahedra such as the air‐stable (in the solid state) polypnictogen ligand complexes [{CpMo(CO)2}(η3‐E3)] (“MoE3”; E=P, As, Sb) [6] and [{CpMo(CO)2}2(μ,η2:η2‐E2)] (“Mo2E2”; E=P, As, Sb, Bi)[ 6a , 6c , 7 ] are received (Scheme 1). They can easily be synthesised, characterised and handled and are therefore well‐suited starting materials for further investigations and have already proved to be excellent precursors for the formation of extended polypnictogen frameworks upon reduction, [8] coordination [9] and oxidation. [10] While for Mo2E2 complexes all representatives of E (E=P, As, Sb, Bi; except E=N) are known, the respective hetero‐dipnictogen complexes Mo2EE′ have been far less investigated. Until now, only two compounds of this class, [{CpMo(CO)2}2(μ,η2:η2‐PE)] (E=As (1), Sb (2)), could be synthesised by Mays et al. in 1998 (Scheme 2). [11] Their approach based on the deprotonation of [{CpMo(CO)2}2(μ‐H)(μ‐PH2)] (C), which yields the lithium salt of the anion [{CpMo(CO)2}2(μ‐PH2)]− (D). Further reaction with AsCl3 or SbCl3, respectively, gives access to 1 and 2 by elimination of one equivalent of LiCl and two equivalents of HCl. However, the latter can also protonate the anion D back to C, which leads to just moderate yields of 33 % (1) and 39 % (2), respectively. Another disadvantage of this synthetic route is the elaborate preparation of C, where first Mo2P2 (B) is generated by thermolysis of [CpMo(CO)2]2 (A) with white phosphorus to be further hydrolysed with NaOH in THF/H2O. Following this route, C can only be obtained in a yield of just 15 %, which leads to an overall yield of 5.0 % for 1 and 5.9 % for 2, when starting from A. Moreover, a direct route affording C from A was developed by our group in 2003. [12] Additionally, only recently, we were able to show that D can be directly synthesised from B by a nucleophilic attack of OH−. [13] Mays et al. also tried to synthesise the respective PBi ligand complex by adding BiCl3 to D, but failed, which is not overly surprising as complexes containing a P−Bi bond are very rare and only of a dative bonding nature and/or stabilised by bulky substituents. [14] The number of examples containing a covalent E−E′ bond dramatically decreases by moving on to the heavier pnictogen elements. [15] While three compounds for covalent As−Sb bonds were reported, [16] only two are known for As−Bi and Sb−Bi (Scheme 1a), [17] only half of which were characterised crystallographically. Between As and Sb, only single bonds were observed, whereas the other ones mentioned also form double bonds. However, all of them can only be stabilised by very bulky organic substituents or they have a distinct tendency to disproportionate and form homonuclear bonds (Scheme 1a; right). [16a]
Scheme 2.
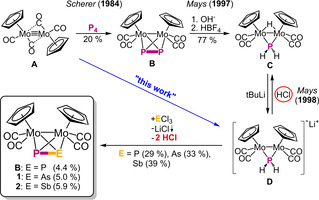
Synthetic route for the tetrahedral molybdenum pnictogen complexes of the type {[Mo]2(μ,η2:η2‐PE)} (E=P (B), As (1), Sb (2); [Mo]=CpMo(CO)2) by Mays et al. in 1998. Yields for B, 1 and 2 are calculated for the overall reaction starting from A. This work: direct synthesis of D from A and avoidance of HCl elimination by replacing the protons with SiMe3 groups.
These disadvantages and the perspective of making tetrahedral compounds with heavier hetero‐dipnictogen ligands accessible prompted us to develop a new synthetic route, which reduces the number of reaction steps and avoids the elimination of HCl to give much higher yields. In the following, we report a facile two‐step one‐pot synthesis for this type of compounds starting directly from A (Scheme 2), which is also transferable to the complexes Mo2E2, containing different CpR ligands such as the tert‐butyl substituted cyclopentadienyl ligand (Cp′=η5‐C5H4 tBu) and gives access to the so far unknown heavier hetero‐dipnictogen derivatives of 1 and 2 such as [{CpMo(CO)2}2(μ,η2:η2‐AsSb)] (3 a), [{CpMo(CO)2}2(μ,η2:η2‐AsBi)] (4) and [{CpMo(CO)2}2(μ,η2:η2‐SbBi)] (5) in higher yields. The latter represent the first examples of compounds containing covalent bonds between mixed heavier group 15 elements that do not bear any bulky organic substituents and are only stabilised by transition metal moieties.
Results and Discussion
When A is reacted with a solution of Li[P(SiMe3)2] (E1) in THF, an immediate colour change from orange‐brown to greenish red occurs suggesting the formation of the intermediate Li[{CpMo(CO)2}2{μ‐P(SiMe3)2}] (6 a) in solution (Scheme 3). Compound 6 a is a derivative of D in which the P‐bound hydrogen atoms are replaced by SiMe3 groups. Further addition of AsCl3 or SbCl3, respectively, leads to the generation of the tetrahedral molybdenum pnictogen complexes [{CpMo(CO)2}2(μ,η2:η2‐PE′)] (E′=As (1), Sb (2)) in 69 % and 59 % isolated yields (Scheme 3), respectively, after chromatographic work‐up. In the last step, two equivalents of Me3SiCl are released avoiding the disadvantageous elimination of HCl. This method not only avoids elaborate reaction steps, but also increases the yield of the compounds 1 and 2 dramatically, compared to the literature (5.0 % and 5.9 %; Table 1).
Scheme 3.
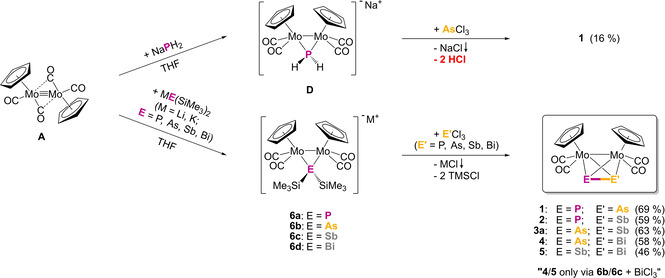
One‐pot synthesis of the tetrahedral hetero‐pnictogen complexes 1–5. Top: using NaPH2 for the synthesis of 1 leading to HCl evolution in the second reaction step; bottom: using ME(SiMe3)2 (M=Li, K; E=P, As, Sb, Bi) for the synthesis of 1–5, which avoids HCl elimination resulting in higher yields.
Table 1.
All possible combinations of the intermediates 6 a–d with E′Cl3 (E′=P, As, Sb, Bi) and the resulting products [{CpMo(CO)2}2(μ,η2:η2‐E E′)]. Yield improvements compared to the literature syntheses are given subjacent (referred to A).
|
|
6 a (“P”) |
6 b (“As”) |
6 c (“Sb”) |
6 d (“Bi”) |
|---|---|---|---|---|
|
PCl3 |
P2 20 %→57 % |
P As (1) 5 %→50 % |
P Sb (2) 6 %→18 % |
unsuccessful |
|
AsCl3 |
P As (1) 5 %→69 % |
As2 35 %→52 % |
As Sb (3 a) 0 %→63 % |
unsuccessful |
|
SbCl3 |
P Sb (2) 6 %→59 % |
As Sb (3 a) 0 %→51 % |
Sb2 76 %→37 % |
unsuccessful |
|
BiCl3 |
unsuccessful |
As Bi (4) 0 %→58 % |
Sb Bi (5) 0 %→46 % |
Bi2 14 %→25 % |
The purity of 1 and 2 was proven by 31P and 1H NMR spectroscopy as well as by elemental analysis and mass spectrometry. It has to be mentioned that compound 1 can also be synthesised by reacting A with NaPH2 (yielding the anion D immediately in solution) and the subsequent addition of AsCl3. But, here again, HCl evolves in the second reaction step leading to side reactions and a relatively low yield of 16 %, which, however, still exceeds three times the yield reported by Mays et al.
Compound Mo2P2 (B) can be obtained in the same manner by reacting the solution of the intermediate 6 a with PCl3. Here, the yield can be increased from 20 % to 57 % (Table 1). However, the synthesis of an analogous complex with a PBi ligand by adding BiCl3 to 6 a was not successful, as as it was also reported by themethod of Mays et al. Only small traces of Mo2P2 could be detected by NMR spectroscopy and mass spectrometry, which might suggest that the P−Bi bond is too unstable and the tetrahedral product is either not formed or decomposes rapidly.
Our synthetic route clearly shows that the number of reaction steps could be reduced and that the yield is considerably increased by an easy one‐pot reaction, especially for the hetero‐dipnictogen complexes 1 and 2. But the more challenging question arises whether the yet unknown hetero‐dipnictogen ligands, which only contain the heavier elements As, Sb and Bi, are also accessible. The reaction of A with either Li[As(SiMe3)2] (E2), K[Sb(SiMe3)2] (E3) or K[Bi(SiMe3)2] (E4) in THF leads to an immediate colour change from orange‐brown to greenish red (E2), greenish brown (E3) or bronze‐coloured (E4) suggesting the formation of the respective intermediates M[{CpMo(CO)2}2{μ‐E(SiMe3)2}] (M=Li, K; E=As (6 b), Sb (6 c), Bi (6 d)) in solution. The subsequent addition of AsCl3, SbCl3 or BiCl3, respectively, and easy purification by column chromatography lead to the unprecedented complexes [{CpMo(CO)2}2(μ,η2:η2‐AsSb)] (3 a), [{CpMo(CO)2}2(μ,η2:η2‐AsBi)] (4) and [{CpMo(CO)2}2(μ,η2:η2‐SbBi)] (5) in remarkable isolated yields of 63 %, 58 % and 46 % (Scheme 3, Table 1).
Thus, all combinations of dipnictogen ligands can be synthesised by reacting the respective intermediate 6 a‐d with the appropriate pnictogen‐trihalide ECl3 (except Mo2PBi). While compound 3 a can be obtained in two ways, either by combining 6 b with SbCl3 or 6 c with AsCl3, 4 and 5 are only formed by the reaction of 6 b or 6 c, respectively, with BiCl3, not by reacting 6 d with AsCl3 or SbCl3 (Table 1 or Scheme 3). This suggests that either the formation of 6 d is less favoured or that 6 d is less stable than its lighter counterparts.
The incorporated EE′ ligands feature very rare bonds between different heavier pnictogen atoms, especially within the compounds 3 a, 4 and 5. Compound 3 a represents the fourth, 4 and 5 only the third example of compounds with covalent As−Sb, [16] As‐Bi [17a] or Sb‐Bi[ 17b , 17c ] bonds, respectively. Moreover, they are the first examples of such covalent E−E′ bonds that are only stabilised by transition metals and do not bear any organic substituents. Therefore, these compounds can be regarded as complexes of the exotic diatomic As≡Sb, As≡Bi and Sb≡Bi molecules, respectively, which are the heaviest hetero‐pnictogen congeners of N2.
Analogously to the synthesis of Mo2P2, the homo‐dipnictogen complexes Mo2As2, Mo2Sb2 and Mo2Bi2 can be synthesised via this method too, again resulting in a remarkable yield enhancement compared to their hitherto existing preparations (except for the antimony complex; Table 1). Additionally, this synthesis could be scaled‐up to a multigram scale, which enables the systematic investigation of the reactivity of this class of compounds. All the tetrahedral complexes are well soluble in dichloromethane and toluene and moderately soluble in ortho‐difluorobenzene and nonpolar solvents such as n‐hexane or n‐pentane. Overall, the heavier the pnictogen atoms are, the more the solubility decreases.
The products 1–5 can be crystallised from saturated CH2Cl2 solutions at −30 °C leading to dark red blocks suitable for single crystal X‐ray diffraction. Since 1 and 2 are already described in the literature, only the solid‐state structures of 3 a‐5 (Figure 1) will be discussed. They are isostructural with a distorted Mo2EE′ tetrahedron as the central structural motif and crystallise in the monoclinic space groups P2/n (3 a), P21 (4) or I2/a (5), respectively, with two half molecules (3 a), one (4) or one half molecule (5) in the asymmetric unit. The AsSb ligand of 3 a exhibits a 50 : 50 disorder over the two sites with an average bond length of 2.515(1) Å, [18] which is in between those of the diarsenic (2.311(3) Å) [7a] and the diantimony complexes (2.678(1) Å). [19] Additionally, it is just slightly longer than the sum of the covalent radii for an As−Sb double bond (2.47 Å). [20] Therefore, it shows a structural similarity to all other existing complexes of the type [{CpMo(CO)2}2(μ,η2:η2‐EE′)]. It represents, however, the first example of an As−Sb bond with a multiple bond character. The same accounts for the compounds 4 and 5. The former bears an AsBi ligand with a bond length of 2.64(2) Å, [18] the latter an SbBi ligand with a bond length of 2.7916(2) Å. Both are again disordered over the two sites in a ratio of 50 : 50 and the bond distances are in between the sum of the respective single (As−Bi: 2.72 Å; Sb−Bi: 2.91 Å) and double bond (As=Bi: 2.55 Å; Sb=Bi: 2.77 Å) covalent radii, while 5 nears a double bond. [20] Additionally, the bond length of the SbBi ligand in 5 is again between those of the diantimony (2.678(1) Å) [19] and the dibismuth (2.838(1) Å) complexes. The same behaviour is observed for the Mo−Mo distances of 3 a‐5, which are slightly elongated in comparison to the respective lighter homo‐dipnictogen complex Mo2E2, but shortened in comparison to the heavier analogue. The Cp ligands in 3 a‐5 are arranged in an eclipsed manner to each other. Overall, the newly formed complexes 3 a‐5 can, on the one hand, be described as tetrahedra, isolobal to P4 and As4 and, on the other hand, as EE′ dumbbells stabilised by an Mo2 unit, each featuring a formal triple bond. The rather short E−E′ distances are nicely reproduced by DFT calculations (c.f. Supporting Information). For example, the As−Sb distance in the optimized geometry of 3 a is with 2.515 Å in excellent agreement with the experimental value (vide supra). Moreover, the As−Sb multiple bond character is reflected in the Wiberg bond order with a Löwdin orthogonalized basis (see Supporting Information for details) of 1.40. The corresponding Wiberg bond orders of the As−Bi and Sb−Bi bonds in 4 and 5 are 1.35 and 1.36, respectively.
Figure 1.
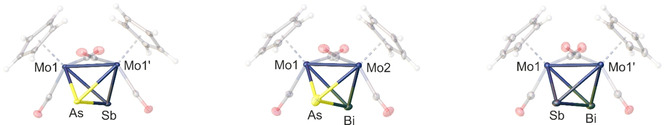
Molecular structure of the unprecedented products 3 a (left), 4 (middle) and 5 (right). Anisotropic displacement is set to the 50 % probability level. The Cp ligands as well as the CO substituents are drawn translucent and the disorder is omitted for clarity. Selected bond lengths [Å]: 3 a: As−Sb 2.515(1), Mo1‐Mo1′ 3.0656(6); 4: As−Bi 2.64(2), Mo1‐Mo2 3.084(2); 5: Sb−Bi 2.7916(2), Mo1‐Mo1′ 3.1255(6).
The 1H NMR spectra of 1–5 all feature one singlet in the characteristic region for Cp ligands. [21] Likewise, one singlet is observed in the 13C{1H} NMR spectra as well as characteristic signals for the CO ligands. The phosphorus‐containing complexes 1 and 2 were characterised by 31P NMR spectroscopy, revealing the expected signals at δ=34.5 ppm (1 in C6D6) [22] and 98.8 ppm (2 in C6D6) [22] as reported in literature. [11] However, in some cases, both solutions show an additional small signal at −44.5 ppm, which can be attributed to trace impurity of Mo2P2. This indicates that a slight intermolecular exchange between the pnictogen atoms can take place in these syntheses and, therefore, small amounts of the homo‐dipnictogen complexes are generated which cannot be separated by column chromatography. The amount of the produced Mo2P2 is very little (ratio of 20 : 1 or higher in favour of 1 or 2, respectively) though. Small amounts of homo‐dipnictogen complexes can also be observed in the 1H NMR spectra of 3 a–5 in some reactions. The identity of 1–5 has been unambiguously proven by mass spectrometry, where the molecular ion peak has been detected. In addition, in some cases, peaks of low intensity corresponding to the homo‐dipnictogen species have been also detected. The composition of 1–5 is further supported by the elemental analysis. All the synthesised tetrahedral compounds, including 3 b, 7 and 8 (vide infra), are stable in air, in contrast to other heteroatomic tetrahedranes.[ 5a , 5b , 5c ]
It was also possible to isolate crystals of the intermediates 6 a–c (regardless of several attempts, no crystals for 6 d could be obtained), which were suitable for single‐crystal X‐ray diffraction by adding the respective crown ethers (12‐crown‐4 for lithium, 18‐crown‐6 for potassium) within the first reaction step and layering with n‐hexane at −30 °C (Figure 2). The anions of the intermediates 6 a–c are isostructural to each other as well as to D, which is a derivative of 6 a where the SiMe3 groups are substituted by protons. They crystallise in the space groups P2/n (6 a), P21/n (6 b) and P −1(6 c), respectively. In the newly formed anions, the pnictogenido [E(SiMe3)2]− units bridge the Mo−Mo bond with E−Mo distances in the range between a single and a double bond (P−Mo: 2.4304(6) Å; As−Mo: 2.5170(3) Å; Sb−Mo: 2.6897(7) Å). The pnictogen atom features a distorted tetrahedral geometry. The Mo−Mo distances (6 a: 3.1890(5) Å; 6 b: 3.2445(3) Å; 6 c: 3.2598(8) Å) are dramatically increased compared to the starting material A (2.4477(12) Å) [23] reasoning that the original triple bond is completely degraded and only an elongated single bond between the molybdenum atoms is left. Additionally, the Mo−Mo distances increase from the lighter to the heavier pnictogen elements as expected. Compared to its derivative D, compound 6 a shows a similar Mo−Mo distance, but slightly elongated Mo−P bonds (D: 2.375(2)‐2.378(2) Å). This is caused by the sterically more demanding SiMe3 groups in contrast to the P‐bound hydrogen atoms of D. A similar derivative of the arsenic compound 6 b is not known yet, but a related structural motif can be found in the neutral compound [{CpMo(CO)2}2(μ‐H)(μ‐AsH2)] with a bridging [AsH2]− unit instead of [As(SiMe3)2]−. Interestingly, 6 c is the first crystallographically characterized compound with a stibenido unit bridging a dimolybdenum fragment. And, although the solid‐state structure of the bismuth derivative 6 d could not be determined, it probably reveals the same constitution, which is unprecedented for bismuth.
Figure 2.
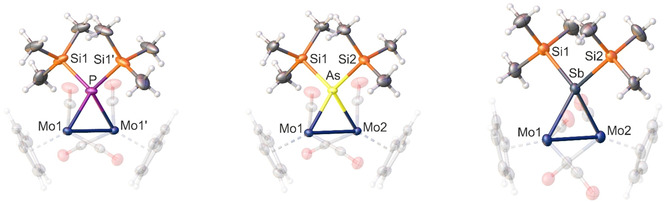
Molecular structures of the intermediates 6 a (left), 6 b (middle) and 6 c (right). Anisotropic displacement is set to the 50 % probability level. Only the anionic part is shown. The cations and the crown ethers are omitted, and Cp as well as CO substituents are drawn translucent for clarity. Selected bond lengths [Å] and angles [°]: 6 a: P−Mo1/P−Mo1′ 2.4304(6), P−Si1/P−Si1′ 2.2574(8), Mo1‐Mo1′ 3.1889(5), Si1‐P−Si1′ 103.85(5); 6 b: As−Mo1 2.5213(3), As−Mo2 2.5128(3), As−Si1 2.3410(8), As−Si2 2.3540(8), Mo1‐Mo2 3.2445(3), Si1‐As−Si2 104.39(3); 6 c: Sb−Mo1 2.6887(8), Sb−Mo2 2.6907(7), Sb−Si1 2.561(2), Sb−Si2 2.559(2), Mo1‐Mo2 3.2597(8), Si1‐Sb−Si2 105.27(7).
The electronic structures of 6 a–d have been investigated by DFT calculations, which are in excellent agreement with the experimentally determined geometric parameters, especially the elongated Mo−Mo distances. Although the Mo−Mo distances in 6 a–d are longer than in the corresponding Mo2EE’ derivatives 1–5, the Wiberg bond orders in Löwdin orthogonalized basis are only slightly lower (for example: Mo−Mo 3.199 Å, WBI 0.45 in 6 a vs. Mo−Mo 3.048 Å, WBI 0.53 in 1), indicating the presence of an elongated Mo−Mo single bond. This is also substantiated by the Intrinsic Bonding Orbitals, which show the presence of a Mo−Mo bond, although with additional orbital contribution from the CO groups (see Fig. S34 in the Supporting Information).
Besides making the novel hetero‐dipnictogen complexes 3 a–5 accessible, the synthetic strategy reported herein also allows to introduce substituted Cp ligands, such as tert‐butylcyclopentadienyl (Cp′), to vary the solubility and the electronic as well as the steric properties of the complexes. This might be crucial for further reactivity studies. The following complexes were synthesised by using [Cp′Mo(CO)2]2 as starting material exemplifying [{Cp′Mo(CO)2}2(μ,η2:η2‐As2)] (7), [{Cp′Mo(CO)2}2(μ,η2:η2‐Sb2)] (8) and [{Cp′Mo(CO)2}2(μ,η2:η2‐AsSb)] (3 b).
The complexes 7 and 8 had already been synthesised by our group in the past, [8b] even though with a remarkably low yield hampering the investigation of further reactivity. Nonetheless, they had already proved to be suitable starting materials for building different anionic pnictogen frameworks upon reduction. [8b] This shows the necessity of making these compounds available in a larger scale, which can be achieved via the new synthetic route. That way, the yields of the complexes 7 (5.4 %→55 %) and 8 (6.0 %→29 %) can be dramatically increased and also the unprecedented compound 3 b can be obtained in a remarkably good yield of 49 %.
Interestingly, the 1H NMR spectrum of 3 b (in contrast to the spectra of 7 and 8) shows three multiplets in a ratio of 1 : 2 : 1 for the aromatic protons instead of the expected two triplets, which indicates that the protons in 2,5 and 3,4 position, respectively, are not chemical or magnetical equivalent in 3 b. This is confirmed by 13C{1H} NMR spectroscopy, where four singlets instead of two for the proton bound C atoms of the Cp′ ring are observed.
Furthermore, orange red crystals suitable for X‐ray diffraction of the complexes 3 b, 7 and 8 could be obtained by cooling saturated CH2Cl2 solutions from room temperature to −30 °C. Their solid‐state structures (cf. Figure 3 for 3 b) show tetrahedral Mo2 EE′ cores, which are almost identical to their Cp congeners.
Figure 3.
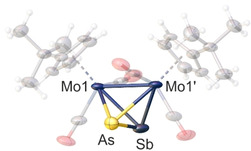
Molecular structure of the Cp′ derivative 3 b, exemplifying the isostructural compounds 3 b, 7 and 8. Anisotropic displacement is set to the 50 % probability level. The Cp′ ligands as well as the CO substituents are drawn translucent and the disorder is omitted for clarity.
Conclusions
We developed a new and easy one‐pot synthesis of air‐stable tetrahedral dimolybdenum dipnictogen complexes, which enables not only an impressive yield improvement (see Table 1) for the already known hetero‐dipnictogen complexes [{CpMo(CO)2}2(μ,η2:η2‐PE)] (E=As (1), Sb (2)) and the homo‐dipnictogen complexes Mo2E2 (E=P, As, (Sb), Bi), but, more importantly, also gives access to unprecedented complexes containing the heavier hetero‐dipnictogen ligands AsSb (3), AsBi (4) and SbBi (5). Now, these syntheses can also be carried out in a multigram scale. The complexes 3–5 extend the very rare class of En ligand complexes of the heavy pnictogen elements and the exotic tetrahedrane analogues, with all of them featuring very rare covalent bonds between two different heavy pnictogen atoms as well as representing the first ever examples in which these bonds can be stabilised without any organic substituents. Therefore, these compounds can be understood as the complexes of the exotic diatomic As≡Sb, As≡Bi and Sb≡Bi molecules with reduced E−E′ bond order, which are the heavy hetero‐pnictogen congeners of N2. Within that, compound 3 contains the very first As−Sb bond with a multiple bond character.
The intermediates M[{CpMo(CO)2}2{μ‐E(SiMe3)2}] (M=Li, K; E=P (6 a), As (6 b), Sb (6 c)) could also be crystallographically characterized revealing anionic [E(SiMe3)2]− units bridging a dimolybdenum fragment. The salt 6 c contains the first crystallographically characterized single stibenido unit of this kind.
Moreover, the new synthesis allowed to introduce the bulkier tert‐butylcyclopentadienyl (Cp′) ligand to vary the steric and electronic properties and enhance the solubility, which expands the possibilities of further reactivity studies. Overall, the newly synthesised En ligand complexes in large scale can be used as promising precursors for the synthesis of extended En ligand frameworks upon oxidation or reduction. Furthermore, due to the lack of Sbn and Bin ligand complexes, the almost unexplored coordination behaviour of these compounds towards coinage and other transition metals can now be investigated.
Experimental Section
For experimental details see the Supporting Information.
Deposition numbers 2061901 (for 2), 2061902 (3 a), 2061903 (3 b), 2061904 (4), 2061905 (5), 2061906 (6 a), 2061907 (6 b), 2061908 (6 c), 2061909 (7), 2061910 (8), and 2072587 ([Cp′Mo(CO)2(η3‐As3)] contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/36‐1. We thank Anna Garbagnati, Matthias Hautmann and Lisa Zimmermann for their support in the preparation of the compounds 7 (A. G.), E3, E4 (M. H.) and 4 (L. Z.). Open access funding enabled and organized by Projekt DEAL.
L. Dütsch, C. Riesinger, G. Balázs, M. Scheer, Chem. Eur. J. 2021, 27, 8804.
Dedicated to Christian Bruneau.
References
- 1. Maier G., Angew. Chem. Int. Ed. Engl. 1988, 27, 309; [Google Scholar]; Angew. Chem. 1988, 100, 317–332. [Google Scholar]
- 2. Seidl M., Balázs G., Scheer M., Chem. Rev. 2019, 119, 8406–8434. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Bernhardt T. M., Stegemann B., Kaiser B., Rademann K., Angew. Chem. Int. Ed. 2003, 42, 199–202; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 209–202; [Google Scholar]
- 3b.A. Stock, O. Guttmann, Berichte der deutschen chemischen Gesellschaft 1904, 37, 885–900: Stock and Guttmann reported on a possible synthesis of yellow antimony by reacting liquid stibane (antimony trihydride) with oxygen at 183 K in 1904. However, an anlytical evidence could not be provided.
- 4. Karttunen A. J., Linnolahti M., Pakkanen T. A., Theor. Chem. Acc. 2011, 129, 413–422. [Google Scholar]
- 5.
- 5a. Cossairt B. M., Diawara M.-C., Cummins C. C., Science 2009, 323, 602–602; [DOI] [PubMed] [Google Scholar]
- 5b. Riu M.-L. Y., Jones R. L., Transue W. J., Müller P., Cummins C. C., Sci. Adv. 2020, 6, eaaz3168; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Hierlmeier G., Coburger P., Bodensteiner M., Wolf R., Angew. Chem. Int. Ed. 2019, 58, 16918–16922; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17074–16922; [Google Scholar]
- 5d.G. A. Ozin, J. Chem. Soc. A 1970, 2307–2310, also the existence of the molecules As2P2, As3P and SbP3 within mixtures of phosphorus and arsenic or phosphorus and antimony vapours, respectively, has been detected by high-temperature gas-phase RAMAN spectroscpy.
- 6.
- 6a. Scherer O. J., Sitzmann H., Wolmershäuser G., J. Organomet. Chem. 1984, 268, C9-C12; [Google Scholar]
- 6b. Gorzellik M., Bock H., Gang L., Nuber B., Ziegler M. L., J. Organomet. Chem. 1991, 412, 95–120; [Google Scholar]
- 6c. Breunig H. J., Rösler R., Lork E., Angew. Chem. Int. Ed. Engl. 1997, 36, 2819; [Google Scholar]; Angew. Chem. 1997, 109, 2941–2821. [Google Scholar]
- 7.
- 7a. Sullivan P. J., Rheingold A. L., Organometallics 1982, 1, 1547–1549; [Google Scholar]
- 7b. Clegg W., Compton N. A., Errington R. J., Norman N. C., Polyhedron 1988, 7, 2239–2241. [Google Scholar]
- 8.
- 8a. Arleth N., Gamer M. T., Koppe R., Pushkarevsky N. A., Konchenko S. N., Fleischmann M., Bodensteiner M., Scheer M., Roesky P. W., Chem. Sci. 2015, 6, 7179–7184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Roesky P. W., Reinfandt N., Schoo C., Dütsch L., Köppe R., Konchenko S. N., Scheer M., Chem. Eur. J. 2021, 27, 10.1002/chem.202003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Fleischmann M., Jones J. S., Balazs G., Gabbai F. P., Scheer M., Dalton Trans. 2016, 45, 13742–13749; [DOI] [PubMed] [Google Scholar]
- 9b. Ly H. V., Parvez M., Roesler R., Inorg. Chem. 2006, 45, 345–351; [DOI] [PubMed] [Google Scholar]
- 9c. Fleischmann M., Dütsch L., Moussa M. E., Schindler A., Balázs G., Lescop C., Scheer M., Chem. Commun. 2015, 51, 2893–2895; [DOI] [PubMed] [Google Scholar]
- 9d. Fleischmann M., Dütsch L., Elsayed Moussa M., Balázs G., Kremer W., Lescop C., Scheer M., Inorg. Chem. 2016, 55, 2840–2854; [DOI] [PubMed] [Google Scholar]
- 9e. Moussa M. E., Schiller J., Peresypkina E., Seidl M., Balázs G., Shelyganov P., Scheer M., Chem. Eur. J. 2020, 26, 14315–14319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Schiller J., Schreiner A., Seidl M., Balázs G., Scheer M., Chem. Eur. J. 2020, 26, 14570–14574; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Scherer O. J., Sitzmann H., Wolmershäuser G., Angew. Chem. Int. Ed. Engl. 1984, 23, 968; [Google Scholar]; Angew. Chem. 1984, 96, 979–969; [Google Scholar]
- 9h. Bai J., Leiner E., Scheer M., Angew. Chem. Int. Ed. 2002, 41, 783–786; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 820–786; [Google Scholar]
- 9i. Scheer M., Gregoriades L., Bai J., Sierka M., Brunklaus G., Eckert H., Chem. Eur. J. 2005, 11, 2163–2169; [DOI] [PubMed] [Google Scholar]
- 9j. Welsch S., Gregoriades L. J., Sierka M., Zabel M., Virovets A. V., Scheer M., Angew. Chem. Int. Ed. 2007, 46, 9323–9326; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9483–9326; [Google Scholar]
- 9k. Scheer M., Gregoriades L. J., Zabel M., Bai J., Krossing I., Brunklaus G., Eckert H., Chem. Eur. J. 2008, 14, 282–295; [DOI] [PubMed] [Google Scholar]
- 9l. Welsch S., Bodensteiner M., Dušek M., Sierka M., Scheer M., Chem. Eur. J. 2010, 16, 13041–13045; [DOI] [PubMed] [Google Scholar]
- 9m. Fleischmann M., Welsch S., Gregoriades L. J., Gröger C., Scheer M., Zeitschr. Naturforschung B 2014, 69, 1348–1356; [Google Scholar]
- 9n. Elsayed Moussa M., Attenberger B., Peresypkina E. V., Fleischmann M., Balázs G., Scheer M., Chem. Commun. 2016, 52, 10004–10007. [DOI] [PubMed] [Google Scholar]
- 10. Dütsch L., Fleischmann M., Welsch S., Balázs G., Kremer W., Scheer M., Angew. Chem. Int. Ed. 2018, 57, 3256–3261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3311–3261. [Google Scholar]
- 11. Davies J. E., Kerr L. C., Mays M. J., Raithby P. R., Tompkin P. K., Woods A. D., Angew. Chem. Int. Ed. 1998, 37, 1428–1429; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1473–1429. [Google Scholar]
- 12. Vogel U., Scheer M., Z. Anorg. Allg. Chem. 2003, 629, 1491–1495. [Google Scholar]
- 13. Riedlberger F., Seidl M., Scheer M., Chem. Commun. 2020, 56, 13836–13839. [DOI] [PubMed] [Google Scholar]
- 14. Nejman P. S., Curzon T. E., Bühl M., McKay D., Woollins J. D., Ashbrook S. E., Cordes D. B., Slawin A. M. Z., Kilian P., Inorg. Chem. 2020, 59, 5616–5625. [DOI] [PubMed] [Google Scholar]
- 15. Conrad E., Burford N., McDonald R., Ferguson M. J., J. Am. Chem. Soc. 2009, 131, 5066–5067. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Ashe A. J., Ludwig E. G., J. Organomet. Chem. 1986, 303, 197–204; [Google Scholar]
- 16b. Nikolova D., von Hänisch C., Eur. J. Inorg. Chem. 2005, 378–382; [Google Scholar]
- 16c. Stevens J. G., Trooster J. M., Martens H. F., Meinema H. A., Inorg. Chim. Acta 1986, 115, 197–201. [Google Scholar]
- 17.
- 17a. Traut S., Hähnel A. P., von Hänisch C., Dalton Trans. 2011, 40, 1365–1371; [DOI] [PubMed] [Google Scholar]
- 17b. Marczenko K. M., Chitnis S. S., Chem. Commun. 2020, 56, 8015–8018; [DOI] [PubMed] [Google Scholar]
- 17c. Sasamori T., Takeda N., Tokitoh N., Chem. Commun. 2000, 1353–1354. [Google Scholar]
- 18. 3 a: Two half molecules in the asymmetric unit of 3 a. The given bond lengths are the average bond lengths of both. 4: The AsBi ligand is diordered over the two sites with a ratio of 50 : 50. The given bond length is the average of both As−Bi distances. For details see Supporting Information.
- 19. Harper J. R., Rheingold A. L., J. Organomet. Chem. 1990, 390, c36-c38. [Google Scholar]
- 20. Pyykkö P., J. Phys. Chem. A 2015, 119, 2326–2337. [DOI] [PubMed] [Google Scholar]
- 21.For details see Supporting Informations.
- 22.For chemical shifts in other deuterated solvents see Supporting Information.
- 23. Klingler R. J., Butler W. M., Curtis M. D., J. Am. Chem. Soc. 1978, 100, 5034–5039. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary