

Abstract
Alcaligenes faecalis is the predominant Gram‐negative bacterium inhabiting gut‐associated lymphoid tissues, Peyer's patches. We previously reported that an A. faecalis lipopolysaccharide (LPS) acted as a weak agonist for Toll‐like receptor 4 (TLR4)/myeloid differentiation factor‐2 (MD‐2) receptor as well as a potent inducer of IgA without excessive inflammation, thus suggesting that A. faecalis LPS might be used as a safe adjuvant. In this study, we characterized the structure of both the lipooligosaccharide (LOS) and LPS from A. faecalis. We synthesized three lipid A molecules with different degrees of acylation by an efficient route involving the simultaneous introduction of 1‐ and 4′‐phosphates. Hexaacylated A. faecalis lipid A showed moderate agonistic activity towards TLR4‐mediated signaling and the ability to elicit a discrete interleukin‐6 release in human cell lines and mice. It was thus found to be the active principle of the LOS/LPS and a promising vaccine adjuvant candidate.
Keywords: glycolipids, lipid A, lipopolysaccharides, oligosaccharides, vaccines
A host–microbe chemical ecology study revealed an effective and safe immunomodulator from Alcaligenes faecalis resident in gut‐associated lymphoid tissues, Peyer's patches (see picture). The complete structures of both the lipooligosaccharide and the lipopolysaccharide from A. faecalis were characterized. Furthermore, A. faecalis lipid A molecules were synthesized with varying degrees of acylation and found to be a promising vaccine adjuvant candidate.
Introduction
Over 100 trillion microorganisms living in the gastrointestinal tract, known as the gut microbiota, play a fundamental role in human physiology, disease, and maintenance of homeostasis through complex interactions, including immunomodulation of microbial components and metabolites. [1] In healthy individuals, Bacteroidetes, Firmicutes, and Proteobacteria are major phyla among gut microbes, [2] with the former as one of the most commonly found Gram‐negative phylum in the human intestine. [2] The main component of the Gram‐negative bacterial outer membrane and, therefore, in constant contact with the host environment, is its lipopolysaccharide (LPS), which is known as a potent stimulator of the host immune system. [3] LPS typically consists of a glycolipid portion termed lipid A, a core oligosaccharide (OS), and an optional polysaccharide O‐antigen region. [3] If the O‐antigen moiety is absent, the terminology employed is a lipooligosaccharide (LOS or R‐LPS). The lipid A portion generally acts as the immunoactive entity of LPS through specific recognition, primarily by the host innate immune receptorial complex built up of Toll‐like receptor 4 (TLR4)/myeloid differentiation factor‐2 (MD‐2). [3] Importantly, depending on the fine structure of such a glycolipid moiety, LPS can fine‐tune the degree of the TLR4/MD‐2‐mediated inflammatory response. [3]
LPS has been extensively investigated for its negative impact on the development and exacerbation of diseases caused by Gram‐negative bacterial infections. Nevertheless, recent studies have reported the role of specific gut microbiota LPS and lipid A in immune homeostasis and autoimmunity.[ 1a , 1b , 1c , 1d , 1e , 1f ] However, these studies have been conducted on bacteria inhabiting the intestinal surfaces such as Bacteroides vulgatus. We recently determined the chemical and immunological properties of its LPS. Its O‐antigen was also synthesized and tested for affinity with defined lectins to better understand its beneficial role in the intestine.[ 1 , 4 ] Herein, we focus on bacteria that colonize gut‐associated lymphoid tissues (GALT). Peyer's patches (PPs) are the major GALT immediately underlying the intestinal epithelium over the entire length of the small intestine. [5] PPs are also known as the largest sites for the initiation and regulation of intestinal IgA responses by crosstalk via cytokines and cell–cell interactions comprising dendritic, T, and B cells. [6] It is widely accepted that microbial stimuli are required for the development and maintenance of intestinal IgA production. Within this framework, we proposed that such stimuli in PPs are represented by Alcaligenes spp., which are unique Gram‐negative bacteria inhabiting these GALT and crucially involved in the regulation of dendritic cells (DCs) for the efficient production of intestinal IgA.[ 5a , 7 ] Alcaligenes spp. were known as opportunistic bacteria but, focusing on the LPS, we recently revealed that these bacteria create and maintain a homeostatic environment in PPs without triggering any harmful responses. [8] Furthermore, we demonstrated that Alcaligenes spp. LPS acts as a weak TLR4 agonist and it could promote interleukin (IL)‐6 release from DCs, which, in turn, enhanced IgA production. [8] Therefore, we hypothesized that Alcaligenes spp. LPS tends to favor bacterial persistence in PPs by promoting homeostasis rather than inflammation. Acting as such, Alcaligenes spp. LPS would take part in the host immune system vigilance through the production of IgA, which, in turn, might favor commensal persistence in PPs. [8]
These remarkable TLR4‐mediated immunomodulating properties of Alcaligenes spp. LPS must, of course, be attributed to the lipid A chemical structure, which is expected to be different from that of canonical Escherichia coli. This, in turn, opens the intriguing possibility of elucidating novel synthetic compounds inspired by gut commensal‐derived lipid A as a potential vaccine adjuvant. It is well known that lipid A from canonical E. coli LPS, which is a potent inducer of inflammation, [9] cannot be applied as an adjuvant. We identified that the immune functions of E. coli lipid A decrease if the phosphate group is removed, yielding the so called monophosphoryl lipid A (MPL). [10] Furthermore, GlaxoSmithKline developed and commercialized 3D‐MPL [11] as a novel lipid A derivative adjuvant for its selective antiviral effect. As the response of TLR4/MD‐2 is species‐specific, [12] this complicates structure–activity relationship studies of lipid A to control signaling pathways and toxicity. On the other hand, focusing on lipid A from human symbiotic bacteria to elucidate host–bacterial chemical ecology, we revealed a close connection between bacterial characteristics and lipid A activity. [13] We synthesized lipid A moieties from two parasitic bacteria (Helicobacter pylori[ 13a , 13c ] and Porphyromonas gingivalis [13c] ) which showed antagonistic activity in pro‐inflammatory cytokine induction, such as IL‐6, 8, 1β, and TNF‐α. This could be counterbalanced by a selective induction of IL‐18, which is thought to be related to chronic inflammatory diseases. [14] This observation suggested that parasitic bacteria, to allow a permanent relationship with the host, tend to induce chronic inflammation, avoiding the antibacterial effects derived from the induction of pro‐inflammatory cytokine release. [13]
A similar behavior was hypothesized for lipid A from GALT‐specific intracellular symbiotic bacteria, such as in the case of Alcaligenes spp. To shed light on this, we aimed to establish the complete chemical structure of LPS and its lipid A.
Therefore, we defined the complete structure of the LOS and LPS isolated from the predominant Alcaligenes spp. component in human and murine PPs, that is, Alcaligenes faecalis. This structural study revealed an uncommon chemical structure with interesting peculiarities both in the glycolipid and the saccharide domain. Furthermore, we synthesized and tested the immunological properties of its lipid A components with different degrees of acylation. The overall aim of this study is to propose a new chemical structure to work on and develop a novel strategy for potential vaccine adjuvants based on the structural characteristics of gut commensal‐derived LPS.
Results
Structural Determination of LOS and LPS from A. faecalis
Briefly (see the Supporting Information for a full description), LOS and LPS material were obtained from dried bacterial cell extract, [15] and the nature and degree of purity of LPS and LOS fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE). The convergent information from compositional analyses via gas liquid chromatography–mass spectrometry (GLC–MS), matrix‐assisted laser desorption/ionization MS (MALDI MS), and NMR spectroscopy allowed us to define the complete structure of A. faecalis LOS and LPS. Monosaccharide analysis of LOS revealed the presence of terminal D‐galactose (Gal), 6‐substituted D‐glucose (Glc), terminal D‐glucosamine (GlcN), 6‐substituted D‐GlcN, terminal D‐galactosamine (GalN), 4‐substituted D‐GalN, 3,4‐disubstituted L,D‐heptose (Hep), 2,7‐disubstituted L,D‐Hep, and 5‐substituted 3‐deoxy‐D‐manno‐oct‐2‐ulosonic (Kdo). In contrast, the monosaccharide analysis of LPS highlighted the presence mainly of 2‐substituted, 3‐substituted, and 2,3,4‐trisubstituted D‐rhamnose (Rha) and terminal D‐xylose (Xyl). All sugars were in pyranose form. Fatty acid content, identical to that of LPS and LOS, revealed the presence of (R)‐3‐hydroxytetradecanoic acid (14:0(3‐OH)), either in both ester and amide linkages, and of ester‐linked (R)‐3‐hydroxydodecanoic acid (12:0(3‐OH)), decanoic (10:0), dodecanoic (12:0), and tetradecanoic acids (14:0).
The core OS structure was primarily determined on the fully deacylated LOS after alkaline treatment (Figures 1 a,b and S1–S4), as assessed by compositional analyses and NMR spectroscopy. Further information of the core OS was obtained after mild acid hydrolysis of the LOS by NMR investigation (1H NMR spectrum in Figure S5). This soft chemical degradation selectively cleaves the mild linkage between Kdo and the non‐reducing GlcN of the lipid A portion, thus enabling the isolation of both lipid A and core OS moieties.
Figure 1.
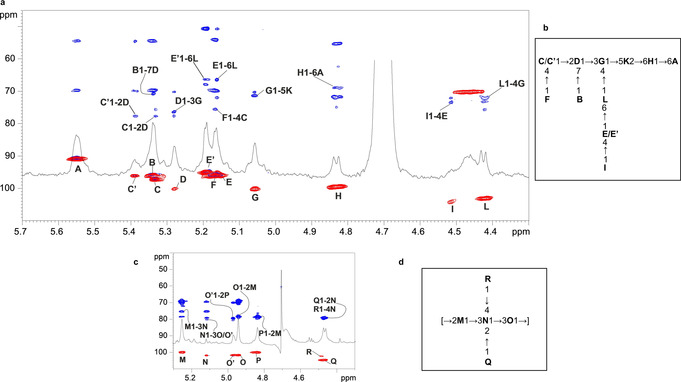
Structural characterization of A. faecalis core OS and O‐antigen. a) Zoom of the superimposition of 1H, 1H,13C HSQC and 1H,13C HMBC NMR spectra of the fully deacylated LOS from A. faecalis. Key inter‐residue long‐range correlations are indicated. Numbering of sugar residues is as reported in Table S1 and Figure 2. b) Structure of the core OS elucidated by NMR spectroscopy, reported using letters as indicated in Table S1 and Figure 2. c) Zoom of the superimposition of proton, HSQC and HMBC NMR spectra after mild acid hydrolysis performed on the LPS fraction. Key inter‐residue long‐range correlations are indicated as in Table S2. d) Structure of the main pentasaccharide repeating unit comprising the O‐antigen as deduced by NMR spectroscopy and reported using the letters in Table S2 and Figure 2.
The 1H and 1H,13C heteronuclear single quantum coherence (HSQC) NMR spectra of the fully deacylated product revealed the occurrence of twelve anomeric signals, indicative of twelve spin systems (A–L, Figure 1 b; Table S1), which were fully assigned (see the Supporting Information for complete chemical and spectroscopic analyses and full structural discussion; Figures S1–S4; Table S1). Moreover, the upfield‐shifted signals clearly visible in the spectra were attributed to the H‐3 methylene resonances of the Kdo unit (K). The HSQC spectrum revealed the presence of four amino sugars, as evident from the correlation of four H‐2 signals to nitrogen‐bearing carbon atoms resonating in the range 50.8–55.4 ppm (Table S1). As deduced from the NMR analysis of the core OS product after acid treatment (Figure S5), all the four amino sugar bear N‐acetyl groups. Finally, analysis of the 31P,1H HSQC spectrum (not shown) disclosed that the Kdo was phosphorylated at position O‐4, as also proven by the downfield displacement of C‐4/H‐4 (δ C/δ H=70.2/4.46 ppm) observed in the HSQC spectrum (Figure S1). Therefore, we concluded that the core OS was a monophosphorylated nonasaccharide made up of one α‐Kdo (K), two α‐L,D‐Hep (D and G), two α‐D‐GalNAc (E/E′ and C/C′), two α‐D‐GlcNAc (B and F), one β‐D‐Gal(I), and one β‐D‐Glc (L). The primary sequence of the core OS was inferred by using NOE contacts of the nuclear Overhauser effect spectroscopy (NOESY) spectrum (Figure S3), and by long‐range correlations visible in the 1H,13C heteronuclear multiple bond correlation (HMBC) (Figure 1 a, full spectrum is shown in Figure S4). Briefly, starting from the lipid A sugar backbone, built up of GlcN residues A and H, the latter was found to be substituted at position O‐6 by Kdo unit (K). This was, in turn, substituted at O‐5 by α‐Hep G as proven by the inter‐residual NOE correlation of H‐1 G with H‐5 K (Figure S3), and by the corresponding long‐range correlation observed in the HMBC spectrum (Figure 1 a). α‐Hep G was, in turn, substituted at positions O‐3 and O‐4 by α‐Hep D and β‐Glc L, respectively. The latter β‐Glc L was, in turn, substituted at its O‐6 by terminal α‐GalN unit E/E′. Residue E was found to be non‐stoichiometrically substituted at its position O‐4 by β‐Gal I. Moreover, the α‐Hep D was substituted at O‐2 by α‐GalN C/C′ and at O‐7 by α‐GlcN B. Finally, residue C was found to be non‐stoichiometrically substituted at its position O‐4 by α‐GlcN residue F, as shown in Scheme S1 and Figure 2 a).
Figure 2.
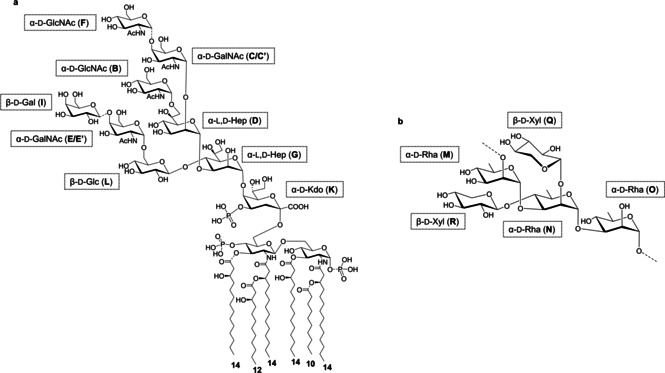
Chemical structure of a) the LOS and b) the O‐antigen from A. faecalis. The letter labels used for NMR analysis are as reported in Tables S1 and S2 and Figure 1. N‐Acetyl groups were deduced by NMR spectroscopic investigation of the core OS product after mild acid hydrolysis.
Mild acid hydrolysis was also performed on the LPS fraction, yielding the O‐antigen moiety of the A. faecalis LPS. NMR investigation of the obtained product (Figures 1 c,d and S6–S9) enabled the identification of the O‐antigen moiety as a xylosylated rhamnan chain (Scheme S2 and Figure 2 b), as previously reported. [16]
Detailed MALDI‐TOF MS and MS2 analyses were performed on lipid A isolated through mild acid hydrolysis of LOS/LPS to define its structure. MALDI‐TOF MS and MS2 investigations (Figures S10–S14) revealed that lipid A was a mixture of species differing in acylation and phosphorylation patterns. Mono‐ and bisphosphorylated tetra‐ to hexaacylated lipid A species were identified, with the latter form presenting a 3+3 distribution of the acyl chains with respect to the GlcN disaccharide backbone. A discrete heterogeneity in each lipid A cluster (tetra‐ to hexa‐) was visible due to the presence of mass differences of 28 atomic mass units (amu; ‐(CH2)2‐ unit), indicative of the existence of species differing in acyl chain length. In addition, a difference of 16 amu was also detected, indicating the absence or presence of hydroxylated acyl chains of lipid A species (Figure S10). The relative abundance of lipid A species was unfortunately not measurable due to both the high heterogeneity of lipid A moieties and the intrinsic inaccuracy of MALDI MS approach. The MS2 analysis (Figures S11–S14) of the main ion peak of each cluster demonstrated that a 12:0 (3‐OH) moiety was present as a secondary acyl substituent of the nonreducing GlcN unit (Figure 2 a). This was further unequivocally confirmed by a positive‐ion MALDI‐TOF MS and MS2 analysis (Figures S13–S14). In particular, positive‐ion MS2 analysis provided key information about the oxonium ion, that is, a fragment ion that arises from the cleavage of the glycosydic linkage and that gave indication of the 12:0 (3‐OH) moiety linked as a secondary acyl moiety to the primary amide bound 14:0 (3‐OH) of the nonreducing glucosamine (Figure 2 b).
Therefore, the combination of structural data on both lipid A and core OS revealed an unprecedented LOS structure (Figure 2 a), whereas the O‐antigen was as previously reported (Figure 2 b). [15a]
With the perspective of a structure‐to‐function relationship study and the immunological properties observed in the LPS fraction, [8] we then performed the chemical synthesis of A. faecalis lipid A, which is typically the main component of the TLR4‐mediated immune elicitation function of LPS. [3a]
Chemical Synthesis of Hexaacylated A. faecalis lipid A
Taking into account that the bisphosphorylated hexaacylated lipid A from E. coli is the most active form, [3a] we synthesized bisphosphorylated hexaacylated A. faecalis lipid A (hexa‐AfLA, Scheme 1) and then the tetra‐ and pentaacyl patterns present in the MALDI MS as well (penta‐AfLA, tetra‐AfLA, Scheme S4).
Scheme 1.
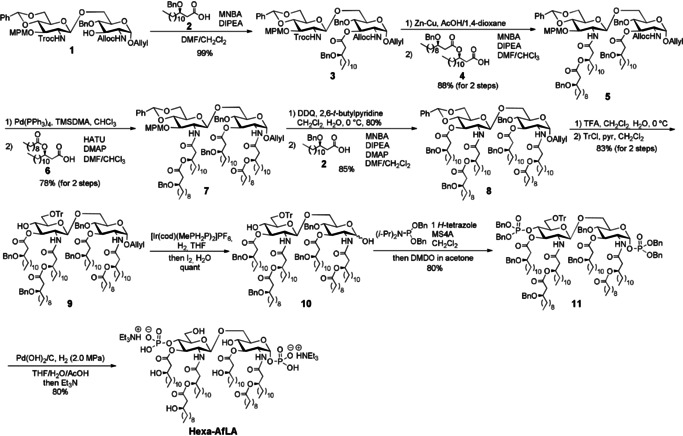
Synthesis of the hexaacylated Alcaligenes faecalis lipid A (hexa‐AfLA).
We previously developed a diversity‐oriented synthetic strategy for MPL based on a key disaccharide, intermediate 1, [13] whose appropriate protection allowed the efficient chemical synthesis of hypoacylated bacterial MPLs derived from H. pylori[ 13a , 13b ] and P. gingivalis. [13c] Although the availability of such a strategy for MPLs has already been demonstrated, our synthetic target was here bisphosphorylated. Therefore, in this study, we applied intermediate 1 to the synthesis of bisphosphorylated lipid A. [17]
Chemical synthesis of hexa‐AfLA was accomplished as illustrated in Scheme 1 (see also the Supporting Information, Scheme S3). Each protecting group, that is, 1‐O‐allyl, 2‐N‐allyloxycarbonyl (Alloc), 2′‐N‐2,2,2‐trichloroethyloxycarbonyl (Troc), 3′‐O‐p‐methoxybenzyl (MPM), and 4′,6′‐O‐benzylidene groups of 1 could be selectively removed to enable the sequential introduction of acyl or phosphate) groups to the proper position.
2‐Methyl‐6‐nitrobenzoic anhydride (MNBA) [18] was employed in our strategy since we have previously shown that it is effective in the acylation of this class of compounds, [13a] whereas other coupling reagents tend to give considerable amounts of the symmetrical acid anhydride, which showed low reactivity in acylation. Benzyl‐protected β‐hydroxy fatty acid 2 [19] was introduced to the 3‐position of 1 in the presence of MNBA reactivity in acylation. Benzyl‐protected β‐hydroxy fatty acid 2 [19] was introduced to the 3‐position of 1 in the presence of MNBA and N,N‐diisopropylethylamine (DIPEA) to obtain 3 with 99 % yield. Then, the 2′‐N‐Troc group was removed by a Zn–Cu couple and the resulting amino group was acylated with fatty acid 4 (Scheme S3) using MNBA with 88 % overall yield. Subsequently, the 2‐N‐Alloc group was removed by Pd(PPh3)4 and (dimethylamino)trimethylsilane (TMSDMA), [20] and the introduction of acyloxycarboxylic acid 6 (Scheme S3) to the free 2‐amino group using O‐(7‐azabenzotriazol‐1‐yl)‐N,N,N′,N′‐tetramethyluronium hexafluorophosphate (HATU) and 4‐dimethylaminopyridine (DMAP) gave 7 with 78 % yield. In this case, we used HATU instead of MNBA to avoid the formation of 2‐methyl‐6‐nitrobenzoyled by‐product. After the cleavage of the MPM group at 3‐position by 2,3‐dichloro‐5,6‐dicyanobenzoquinone (DDQ) oxidation, β‐hydroxy fatty acid 2 was introduced using MNBA to obtain 8 with 85 % yield. Next, the 4′,6′‐O‐benzylidene group was cleaved by trifluoroacetic acid (TFA), and then the trityl (Tr) group was selectively introduced to the primary hydroxyl group. The anomeric allyl group was then isomerized to the 1‐propenyl group with the Ir complex [21] and the resulting 1‐propenyl group was then removed by iodine and water to yield the 1,4′‐dihydroxy disaccharide 10 quantitatively. The simultaneous phosphitylation of 1 and 4′‐position using phosphoramidite in the presence of 1H‐tetrazole and MS4A gave the desired 1,4′‐O‐diphosphite, which was then oxidized to 1,4′‐O‐diphosphorylated 11 using dimethyldioxirane (DMDO) [22] with 80 % yield. After hydrogenolysis of 11 with Pd(OH)2/C under H2 (2.0 MPa) in THF/H2O/AcOH, the first chemical synthesis of hexa‐AfLA was accomplished with 80 % yield. Furthermore, penta‐AfLA and tetra‐AfLA were also synthesized according to similar synthetic schemes (Scheme S4).
Immunological Properties of AfLAs and Isolated A. faecalis LOS
The immunomodulating activities of synthesized AfLAs were evaluated and compared to the strong immunostimulatory LPS from E. coli strain O111 (Figures 3 and S15–S17).
Figure 3.
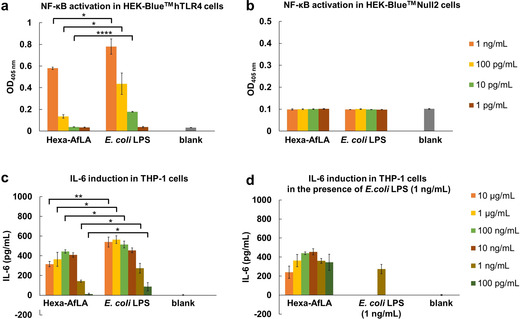
Hexaacylated A. faecalis lipid A (hexa‐AfLA) mediated NF‐κB activation in a) HEK‐Blue™ hTLR4 cells and b) HEK‐Blue™ Null2 cells was evaluated by a SEAP reporter assay. Hexa‐AfLA‐mediated cytokine induction activity in PMA‐differentiated THP‐1 cells was evaluated by ELISA: c) release of IL‐6, d) release of IL‐6 in the presence of E. coli LPS O111 (1 ng mL−1). Results in (a–d) represent the mean±standard deviation of three independent experiments. *p<0.05, **p<0.005, ****p<0.0001 by Student's t‐test.
Nuclear factor (NF)‐κB activation was analyzed by a secretory alkaline phosphatase (SEAP) reporter assay in HEK‐Blue™ hTLR4 and HEK‐Blue™ Null2 cells to evaluate the effective immunoactivation of hexa‐AfLA through TLR4. As previously shown for the whole LPS, [8] hexa‐AfLA induced a weaker NF‐κB activation than E. coli LPS in HEK‐Blue™ hTLR4 cells (p<0.05 for hexa‐AfLA vs. E. coli LPS, both at 1 ng mL−1 and 100 pg mL−1; p<0.0001 for hexa‐AfLA vs. E. coli LPS at 10 pg mL−1; Figure 3 a). The lack of any NF‐κB activation in HEK‐Blue™ Null2 cells, that is, without TLR4/MD‐2/CD14 expression (Figure 3 b), indicated that NF‐κB activation by hexa‐AfLA was TLR4‐dependent. On the other hand, penta‐AfLA and tetra‐AfLA did not show any immunostimulatory effect (Figure S15).
Next, we evaluated the ability of hexa‐AfLA to induce cytokine release, such as IL‐6, in phorbol 12‐myristate 13‐acetate (PMA)‐differentiated THP‐1 human monocytic cells [23] (Figure 3 c,d) using an enzyme‐linked immunosorbent assay (ELISA). Hexa‐AfLA induced IL‐6 release, although the inducing activity was significantly weaker than that observed for E. coli LPS (p<0.005 for hexa‐AfLA vs. E. coli LPS at 10 μg mL−1; p<0.05 for hexa‐AfLA vs. E. coli LPS, at 1 μg mL−1, 10 ng mL−1, 1 ng mL−1 and 100 pg mL−1; Figure 3 c). Furthermore, its capacity to induce the release of other cytokines such as tumor necrosis factor‐α (TNF‐α), IL‐1β, and IL‐18 in PMA‐differentiated THP‐1 cells [23] was investigated (Figure S16). In all cases, hexa‐AfLA exhibited low immunostimulant activity compared to E. coli LPS. A competition assay of hexa‐AfLA against E. coli LPS activity was also performed, and the cytokine levels in the supernatants of the incubated mixtures were measured using ELISA (Figure 3 d). Hexa‐AfLA exhibited almost no inhibitory and/or synergistic effect in the presence of E. coli LPS (1 ng mL−1).
Finally, we evaluated the IL‐6‐inducing ability of hexa‐AfLA in mice. As shown in Figure S17, serum cytokines significantly increased upon subcutaneous hexa‐AfLA administration. Interestingly, the levels of induced IL‐6 after administration of hexa‐AfLA were comparable to those previously observed after administration of A. faecalis LPS. [8] These results, although preliminary, suggested that hexa‐AfLA is potentially able to induce IL‐6‐mediated IgA production, that is, it is the LPS moiety responsible to induce IL‐6 in mice.
Discussion
Comprehension of the host–gut microbe chemical ecology, which is the molecular mechanism underlying host‐microbe interactions at the gut level, is at the crossroads of many fields of science. The extent and the manner in which the molecular recognition of microbes is achieved by the host is not clearly defined yet. Specifically, how and why commensal LPS does not cause an inflammatory response remains underexplored.
Even more fascinating is the case of Alcaligenes spp., the predominant Gram‐negative bacteria inhabiting the gut lymphoid tissues PPs.[ 5 , 8 ] We have previously demonstrated that Alcaligenes spp. can establish symbiotic relationships with the host in PPs despite continuous contact with immune cells. [8] We showed that Alcaligenes spp. LPS is directly involved in the maintenance of homeostatic immunological conditions within PPs by induction of IgA production through an IL‐6‐dependent mechanism. [8]
Given that the immunological functions of LPS are strictly related to its chemical structure, it was expected that Alcaligenes spp. express their LPS, and in particular the lipid A moiety, with a particular chemical architecture enabling its permanence in PPs, avoiding immune vigilance and simultaneously participating in surveillance by enhancing IgA production.
In this study, we have taken a step forward at the molecular level by establishing the complete chemical structure of Alcaligenes spp. LPS/LOS and synthesizing its bisphosphorylated lipid A portions with different acylation patterns, whose immunological properties have also been investigated.
Our structural studies revealed that the monophosphorylated core OS of A. faecalis is composed of nine monosaccharide units comprising Kdo, two L‐glycero‐D‐manno‐heptoses, two N‐acetyl D‐glucosamine, two N‐acetyl D‐galactosamine, one D‐glucose, and one D‐galactose residue (Figure 2 a). The O‐antigen is made up of a xylosylated rhamnan chain, with a double‐branched pentasaccharide repeating unit, where two xylose residues on a linear rhamnan chain are substituted with a rhamnose residue (Figure 2 b). [ 15a] As for the lipid A component, a mixture of tetra‐ to hexaacylated species was identified. The lipid A species with the highest acylation degree was composed of a bisphosphorylated glucosamine disaccharide backbone carrying 14:0 (3‐OH) as primary and 12:0 (3‐OH) and 10:0 as secondary fatty acids, which were distributed in a 3+3 fashion with respect to the disaccharide backbone (Figure 2 a). A. faecalis lipid A moieties in its tetra‐ to hexaacylated forms were completely synthesized in this study (hexa‐AfLA, penta‐AfLA, tetra‐AfLA) and immunological investigation demonstrated that, similar to what we previously observed for the A. faecalis LPS, [8] only hexa‐AfLA induces weaker NF‐κB activation than E. coli LPS in TLR4‐expressing cells. Moreover, no activation was observed in the absence of TLR4 expression (Figure 3 a,b); this observation concomitantly confirmed that the active principle of A. faecalis LOS/LPS is hexa‐AfLA. Hexa‐AfLA was able to induce IL‐6 release in THP‐1 human monocytic cells (Figure 3 c) and mice (Figure S17).
We previously proposed that Alcaligenes‐derived LPS acts as a weak agonist and creates a homeostatic inflammatory condition within PPs without causing pathological inflammation. [8] Such low immunopotency is definitively related to the fine structure of the A. faecalis lipid A moiety. Indeed, although in the hexaacylated form, the presence of two phosphates, the number and nature of most of the acyl chains is identical to that of E. coli, several different structural features are likely responsible for the diverse immunological functions observed. In this frame, i) the symmetrical distribution of the acyl chains in the A. faecalis lipid A (3+3 distribution, in its hexaacylated form, vs. 4+2 symmetry for the E. coli lipid A), ii) the presence of a shorter secondary fatty acid (10:0 in place of 14:0), which is also in a different position compared to E. coli, iii) the presence of a hydroxylated secondary acyl moiety, and iv) the occurrence of hypoacylated and monophosphorylated lipid A species, are notable. These chemical features, in fact, could globally result in a weak elicitation of the TLR4‐mediated immune response. For most of the above structural characteristics, the impact on the lipid A binding mode to the TLR4/MD‐2 complex has been reported, limiting its proper dimerization/activation, resulting in poor immune response.[ 3a , 3b , 10b , 24 ]
We previously synthesized hexaacylated lipid A with shorter acyl groups (3+3 symmetry with four 10:0 β‐hydroxy and two 12:0 fatty acids) and its carboxymethyl (CM) analogues, in which 1‐phosphate was replaced with a CM group, with a different distribution of the acyl groups. The immunological properties of these compounds varied from potent antagonistic, moderate agonistic, to no activity depending on the acyl group distribution as well as on the structure of the acidic functional groups. [25] Moreover, we have shown that the synthesized hypoacylated and monophosphorylated lipid A molecules act as weak TLR4 agonists or antagonists. [13]
Similarly, LPS from Bacteroides spp., the predominant Gram‐negative bacteria in human gut microbiota, are monophosphorylated and hypoacylated[ 1a , 1e ] and show antagonistic [1a] or extremely weak cytokine‐inducing activity compared to E. coli LPS.[ 1e , 4a ] Such lipid A monophosphorylation and expression of less than six acyl chains are considered to be a trick adopted by some bacteria to prevent the TLR4 elicitation either in the case of commensal bacteria (avoiding their elimination), and in the case of parasitic bacteria (enabling the infection to become chronic). In this context, due to its physiological location and persistence, a monophosphorylated hypoacylated lipid A was also expected for A. faecalis LPS/LOS. However, we revealed that together with hypoacylated and monophosphorylated species comprising the lipid A moiety of A. faecalis LPS/LOS, hexaacylated bisphosphorylated forms are also present. In this study, we synthesized this hexaacylated lipid A, taking into account that hexaacylated E. coli lipid A represents the TLR4‐immunoactive portion of E. coli LPS, [26] and then also synthesized tetra‐ and pentaacylated lipid A species. We previously revealed that a homogeneous mixture of hexaacylated E. coli lipid A and tetraacylated antagonistic lipid IVa still exhibits immunostimulating activity comparable to or higher than that of E. coli lipid A itself. [27] Tests for a mixed lipid A species system are in progress.
Conclusion
Despite the fact that only a limited number of LPSs from gut commensals have been structurally defined, the capability to only poorly engage the TLR4‐mediated signaling seems to be a leitmotiv adopted by bacteria inhabiting the intestinal surfaces to avoid host immune system detection. We have recently demonstrated that LPS from Bacteroides also functions via the synergistic action of TLR4/MD‐2 and TLR2‐mediated signaling pathways. [4a]
Nonetheless, this is the first case of complete structural description of an LPS/LOS from a Gram‐negative mutualistic bacterium inhabiting GALT. We identified hexa‐AfLA as the active moiety of A. faecalis LPS/LOS through the immunological characterization of its chemically synthesized tetra‐ to hexaacylated lipid A forms (hexa‐AfLA, penta‐AfLA, tetra‐AfLA). Future studies will be devoted to the definition of the role of the carbohydrate portion of A. faecalis LOS/LPS in the previously observed immunological functions. [8] Finally, it should be noted that the capability to induce only weak activation of TLR4 signaling in combination with potent IgA production‐inducing activity are of great importance in the evaluation of a molecule as a potential adjuvant candidate in vaccine production. From this perspective, a detailed structure–function study is currently ongoing to shed light on the molecular bases of the A. faecalis LOS/LPS and lipid A immunological functions, aiding in the evaluation of potential use of synthetic derivatives as efficient and safe adjuvants in vaccine production. These structure‐related immunological functions also demonstrate that hexa‐AfLA is a promising adjuvant candidate with the intrinsic fascinating feature to not derive from detoxification of useful but toxic immunomodulators, but rather on host–microbe chemical ecology research. In other words, LPS from GALT‐resident mutualistic bacteria can inspire the synthesis of a pool of safer and effective adjuvants. Within this frame, we recently also demonstrated that hexa‐AfLA promotes antigen‐specific IgA and IgG production and enhances Th17 response in the vaccination of female BALB/c mice without excessive inflammation. [28]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported in part by JSPS KAKENHI Grant Number JP15H05836 (K.F.) in Middle Molecular Strategy, JP20H04776 (A.Sh.), JP18H04620 (A.Sh.) in Frontier Research on Chemical Communications, JP20K05749 (A.Sh.), JP19KK0145 (K.F.), JP16H01885 (K.F.), JP16K01914 (A.Sh.), JP18K17997 (K.H.), JP18H02674 (J.K.), JP17K17686 (N.S.), JP18H05280 (H.K.), AMED Grant Number JP20ak0101068h0004 (J.K.), JP20fk0108122h0001 (H.K.), AMED ACT‐MS Grant Number JP20im0210623h0002 (H.K.), AMED Seeds B Grant Number JP19lm0203082h0001 (H.K.), AMED CiCLE Grant Number JP17pc0101001h0001 (H.K.), AMED SATREPS Grant Number JP20jm0110012h0006 (H.K.), JST ACT‐X Grant Number JPMJAX1917 (N.S.), Cross‐ministerial Strategic Innovation Promotion Program: SIP (J.K.), the Ministry of Health and Welfare of Japan and Public/Private R&D Investment Strategic Expansion Program: PRISM (J.K.) and MEXT Program for Leading Graduate Schools: Interactive Materials Science Cadet (K.M.). We also acknowledge Dr. Naoya Inazumi and Dr. Yasuto Todokoro (Osaka University) for their support with NMR measurements. This research was carried out also in the frame of Programme STAR, financially supported by UniNA and Compagnia di San Paolo, as acknowledged by F.D.L.; A.M. is supported by the Progetto POR SATIN POR‐FESR 2014–2020 grant n. B61C17000070007 (OR3) and Progetto POR Campania Oncoterapia 2014–2020 grant n. B61G18000470007; F.D.L., M.D.P., A.Si. and A.M. acknowledge the H2020‐MSCA‐814102–Sweet Crosstalk project; A.M. and A.Si. acknowledge COST (European Cooperation in Science and Technology) Action CA16231 “ENOVA” and Action CA18103 “INNOGLY”, respectively.
A. Shimoyama, F. Di Lorenzo, H. Yamaura, K. Mizote, A. Palmigiano, M. D. Pither, I. Speciale, T. Uto, S. Masui, L. Sturiale, D. Garozzo, K. Hosomi, N. Shibata, K. Kabayama, Y. Fujimoto, A. Silipo, J. Kunisawa, H. Kiyono, A. Molinaro, K. Fukase, Angew. Chem. Int. Ed. 2021, 60, 10023.
Contributor Information
Prof. Antonio Molinaro, Email: molinaro@unina.it.
Prof. Koichi Fukase, Email: koichi@chem.sci.osaka-u.ac.jp.
References
- 1.
- 1a. Vatanen T., Kostic A. D., d'Hennezel E., Siljander H., Franzosa E. A., Yassour M., Kolde R., Vlamakis H., Arthur T. D., Hamalainen A. M., Peet A., Tillmann V., Uibo R., Mokurov S., Dorshakova N., Ilonen J., Virtanen S. M., Szabo S. J., Porter J. A., Lahdesmaki H., Huttenhower C., Gevers D., Cullen T. W., Knip M., Group D. S., Xavier R. J., Cell 2016, 165, 842–853; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. d'Hennezel E., Abubucker S., Murphy L. O., Cullen T. W., mSystems 2017, 2, e00046-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Naito T., Mulet C., De Castro C., Molinaro A., Saffarian A., Nigro G., Berard M., Clerc M., Pedersen A. B., Sansonetti P. J., Pedron T., mBio 2017, 8, e01680-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Di Lorenzo F., De Castro C., Silipo A., Molinaro A., FEMS Microbiol. Rev. 2019, 43, 257–272; [DOI] [PubMed] [Google Scholar]
- 1e. Steimle A., Michaelis L., Di Lorenzo F., Kliem T., Munzner T., Maerz J. K., Schafer A., Lange A., Parusel R., Gronbach K., Fuchs K., Silipo A., Oz H. H., Pichler B. J., Autenrieth I. B., Molinaro A., Frick J. S., Mol. Ther. 2019, 27, 1974–1991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Erturk-Hasdemir D., Oh S. F., Okan N. A., Stefanetti G., Gazzaniga F. S., Seeberger P. H., Plevy S. E., Kasper D. L., Proc. Natl. Acad. Sci. USA 2019, 116, 26157–26166; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Durack J., Lynch S. V., J. Exp. Med. 2019, 216, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Human Microbiome Project C., Nature 2012, 486, 207–214; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Kamada N., Chen G. Y., Inohara N., Nunez G., Nat. Immunol. 2013, 14, 685–690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Hillman E. T., Lu H., Yao T., Nakatsu C. H., Microbes Environ. 2017, 32, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Molinaro A., Holst O., Di Lorenzo F., Callaghan M., Nurisso A., D'Errico G., Zamyatina A., Peri F., Berisio R., Jerala R., Jimenez-Barbero J., Silipo A., Martin-Santamaria S., Chem. Eur. J. 2015, 21, 500–519; [DOI] [PubMed] [Google Scholar]
- 3b. Di Lorenzo F., Billod J. M., Martin-Santamaria S., Silipo A., Molinaro A., Eur. J. Org. Chem. 2017, 4055–4073; [Google Scholar]
- 3c. Di Lorenzo F., De Castro C., Lanzetta R., Parrilli M., Silipo A., Molinaro A., RSC Drug Discovery 2015, 43, 38–63. [Google Scholar]
- 4.
- 4a. Di Lorenzo F., Pither M., Martufi M., Scarinci I., Guzman Caldentey J., Łakomiec E., Jachymek W., Bruijns S., Martin-Santamaria S., Frick J.-S., Van Kooyk Y., Chiodo F., Silipo A., Bernardini M. L., Molinaro A., ACS Cent. Sci. 2020, 6, 1602–1616; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Zhu Q., Shen Z., Chiodo F., Nicolardi S., Molinaro A., Silipo A., Yu B., Nat. Commun. 2020, 11, 4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Obata T., Goto Y., Kunisawa J., Sato S., Sakamoto M., Setoyama H., Matsuki T., Nonaka K., Shibata N., Gohda M., Kagiyama Y., Nochi T., Yuki Y., Fukuyama Y., Mukai A., Shinzaki S., Fujihashi K., Sasakawa C., Iijima H., Goto M., Umesaki Y., Benno Y., Kiyono H., Proc. Natl. Acad. Sci. USA 2010, 107, 7419–7424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Kunisawa J., Kiyono H., Front. Immunol. 2012, 3, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Kunisawa J., Nochi T., Kiyono H., Trends Immunol. 2008, 29, 505–513; [DOI] [PubMed] [Google Scholar]
- 6b. Fagarasan S., Kawamoto S., Kanagawa O., Suzuki K., Annu. Rev. Immunol. 2010, 28, 243–273. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Fung T. C., Bessman N. J., Hepworth M. R., Kumar N., Shibata N., Kobuley D., Wang K., Ziegler C. G. K., Goc J., Shima T., Umesaki Y., Sartor R. B., Sullivan K. V., Lawley T. D., Kunisawa J., Kiyono H., Sonnenberg G. F., Immunity 2016, 44, 634–646; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Sonnenberg G. F., Monticelli L. A., Alenghat T., Fung T. C., Hutnick N. A., Kunisawa J., Shibata N., Grunberg S., Sinha R., Zahm A. M., Tardif M. R., Sathaliyawala T., Kubota M., Farber D. L., Collman R. G., Shaked A., Fouser L. A., Weiner D. B., Tessier P. A., Friedman J. R., Kiyono H., Bushman F. D., Chang K. M., Artis D., Science 2012, 336, 1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shibata N., Kunisawa J., Hosomi K., Fujimoto Y., Mizote K., Kitayama N., Shimoyama A., Mimuro H., Sato S., Kishishita N., Ishii K. J., Fukase K., Kiyono H., Mucosal Immunol. 2018, 11, 693–702. [DOI] [PubMed] [Google Scholar]
- 9. Beutler B., Rietschel E. T., Nat. Rev. Immunol. 2003, 3, 169–176. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Brade L., Brandenburg K., Kuhn H. M., Kusumoto S., Macher I., Rietschel E. T., Brade H., Infect. Immun. 1987, 55, 2636–2644; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Tanimura N., Saitoh S., Ohto U., Akashi-Takamura S., Fujimoto Y., Fukase K., Shimizu T., Miyake K., Int. Immunol. 2014, 26, 307–314. [DOI] [PubMed] [Google Scholar]
- 11. Mata-Haro V., Cekic C., Martin M., Chilton P. M., Casella C. R., Mitchell T. C., Science 2007, 316, 1628–1632. [DOI] [PubMed] [Google Scholar]
- 12. Akashi S., Nagai Y., Ogata H., Oikawa M., Fukase K., Kusumoto S., Kawasaki K., Nishijima M., Hayashi S., Kimoto M., Miyake K., Int. Immunol. 2001, 13, 1595–1599. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Shimoyama A., Saeki A., Tanimura N., Tsutsui H., Miyake K., Suda Y., Fujimoto Y., Fukase K., Chem. Eur. J. 2011, 17, 14464–14474; [DOI] [PubMed] [Google Scholar]
- 13b. Fujimoto Y., Shimoyama A., Suda Y., Fukase K., Carbohydr. Res. 2012, 356, 37–43; [DOI] [PubMed] [Google Scholar]
- 13c. Fujimoto Y., Shimoyama A., Saeki A., Kitayama N., Kasamatsu C., Tsutsui H., Fukase K., Mol. Biosyst. 2013, 9, 987–996. [DOI] [PubMed] [Google Scholar]
- 14. Trøseid M., Seljeflot I., Arnesen H., Cardiovasc. Diabetol. 2010, 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Galanos C., Luderitz O., Westphal O., Eur. J. Biochem. 1969, 9, 245–249; [DOI] [PubMed] [Google Scholar]
- 15b. Westphal O., Jann K. in Methods in Carbohydrate Chemistry (Ed.: Whistler R. L.), Academic Press, New York, 1965, pp. 83–91. [Google Scholar]
- 16. Knirel'Iu A., Zdorovenko G. M., Shashkov A. S., Zakharova I., Kochetkov N. K., Bioorg. Khim. 1986, 12, 1530–1539. [PubMed] [Google Scholar]
- 17. Fukase K., Kunisawa J., Kiyono H., Lipid A containing Complex of Glucosamine Disaccharide Chain and Fatty Acid Chains and Adjuvant Using It, WO 2018155051 A1, 2018.
- 18. Shiina I., Ibuka R., Kubota M., Chem. Lett. 2002, 31, 286–287. [Google Scholar]
- 19. Fukase K., Fukase Y., Oikawa M., Liu W. C., Suda Y., Kusumoto S., Tetrahedron 1998, 54, 4033–4050. [Google Scholar]
- 20. Merzouk A., Guibe F., Loffet A., Tetrahedron Lett. 1992, 33, 477–480. [Google Scholar]
- 21. Baudry D., Ephritikhine M., Felkin H., J. Chem. Soc. Chem. Commun. 1978, 694–695. [Google Scholar]
- 22. Murray R. W., Jeyaraman R., J. Org. Chem. 1985, 50, 2847–2853. [Google Scholar]
- 23. Budai M. M., Varga A., Milesz S., Tozser J., Benko S., Mol. Immunol. 2013, 56, 471–479. [DOI] [PubMed] [Google Scholar]
- 24. Ohto U., Fukase K., Miyake K., Shimizu T., Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukase Y., Fujimoto Y., Adachi Y., Suda Y., Kusumoto S., Fulkase K., Bull. Chem. Soc. Jpn. 2008, 81, 796–819. [Google Scholar]
- 26. Kusumoto S., Fukase K., Chem. Rec. 2006, 6, 333–343. [DOI] [PubMed] [Google Scholar]
- 27. Mueller M., Lindner B., Kusumoto S., Fukase K., Schromm A. B., Seydel U., J. Biol. Chem. 2004, 279, 26307–26313. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Yoshii K., Hosomi K., Shimoyama A., Wang Y., Yamaura H., Nagatake T., Suzuki H., Lan H., Kiyono H., Fukase K., Kunisawa J., Microorganisms 2020, 8, 1102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Wang Y., Hosomi K., Shimoyama A., Yoshii K., Yamaura H., Nagatake T., Nishino T., Kiyono H., Fukase K., Kunisawa J., Vaccines 2020, 8, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary