

Abstract
THY1773 is a novel arginine vasopressin 1B (V1B) receptor antagonist that is under development as an oral drug for the treatment of major depressive disorder (MDD). Here we report our strategy to predict a clinically effective dose of THY1773 for MDD in the preclinical stage, and discuss the important insights gained by retrospective analysis of prediction accuracy. To predict human pharmacokinetic (PK) parameters, several extrapolation methods from animal or in vitro data to humans were investigated. The fu correction intercept method and two‐species‐based allometry were used to extrapolate clearance from rats and dogs to humans. The physiologically based pharmacokinetics (PBPK)/receptor occupancy (RO) model was developed by linking free plasma concentration with pituitary V1B RO by the Emax model. As a result, the predicted clinically effective dose of THY1773 associated with 50% V1B RO was low enough (10 mg/day, or at maximum 110 mg/day) to warrant entering phase 1 clinical trials. In the phase 1 single ascending dose study, TS‐121 capsule (active ingredient: THY1773) showed favorable PKs for THY1773 as expected, and in the separately conducted phase 1 RO study using positron emission tomography, the observed pituitary V1B RO was comparable to our prediction. Retrospective analysis of the prediction accuracy suggested that the prediction methods considering plasma protein binding, and avoiding having to apply unknown scaling factors obtained in animals to humans, would lead to better prediction. Selecting mechanism‐based methods with reasonable assumptions would be critical for the successful prediction of a clinically effective dose in the preclinical stage of drug development.
Keywords: effective dose prediction, receptor occupancy, THY1773, TS‐121, V1B receptor
We evaluated preclinical pharmacokinetics of THY1773, a novel arginine vasopressin 1B (V1B) receptor antagonist, and predicted its clinically effective dose for major depressive disorder. Retrospective analysis of prediction accuracy demonstrated that selecting mechanism based methods with reasonable assumptions would be critical for the successful prediction of a clinically effective dose in the preclinical stage of drug development.
1. INTRODUCTION
Dysfunction of the hypothalamic–pituitary–adrenal (HPA) axis activity is observed in a subset of patients with major depressive disorder (MDD) (Holsboer, Haack, Gerken, & Vecsei, 1984). Arginine vasopressin (AVP) plays a crucial role in the regulation of the HPA axis in cooperation with corticotropin‐releasing factor (CRF), and excess AVP is hypothesized to be responsible for HPA axis dysfunction in depression (de Winter et al., 2003; Purba, Hoogendijk, Hofman, & Swaab, 1996). AVP regulates HPA axis activity through adrenocorticotropic hormone (ACTH) secretion from the anterior pituitary by binding to its receptor subtype vasopressin 1B (V1B) receptor (Saito, Sugimoto, Tahara, & Kawashima, 1995; Wersinger, Ginns, O’Carroll, Lolait, & Young, 2002). Therefore, the V1B receptor antagonist is expected to ameliorate the abnormalities of the HPA axis observed in depression, and consequently, to improve depressive symptoms in certain subpopulations of depressed individuals with impaired HPA axis function.
To date, several V1B receptor antagonists have been proven to exhibit antidepressant‐like effects in rodent models (Geneste et al., 2018; Griebel et al., 2002; Iijima et al., 2014). Among them, SSR149415, the first orally active V1B receptor antagonist, was tested in double‐blind, placebo‐controlled studies for patients with MDD. However, SSR149415 failed to demonstrate clear antidepressant effects in the trials (Griebel, Beeské, & Stahl, 2012). Although the reason for the failure was not fully understood, it is speculated that dose levels of SSR149415 were insufficient to show efficacy (Griebel , Beeské, & Stahl, 2012). Indeed, in a small scale depression trial (Katz, Locke, Greco, Liu, & Tracy, 2017), another V1B receptor antagonist, ABT‐436, has been reported to be associated with more favorable symptom changes at a dose that inhibits HPA function. Therefore, the therapeutic potential of V1B receptor antagonists for MDD needs to be further investigated, especially at doses that would show adequate blockade of the V1B receptor.
THY1773 is a potent and selective V1B receptor antagonist, which blocks the anterior pituitary V1B receptor and exhibits antidepressant‐like effects in rodents (Kamiya et al., 2020). In rats, THY1773 occupied the anterior pituitary V1B receptor dose‐dependently after oral administration and significantly inhibited the increase in plasma ACTH induced by CRF/desmopressin challenge at doses that showed occupancy of the anterior pituitary V1B receptor of more than 50%. Consistent with these results, THY1773 significantly improved depressive‐like behavior induced by repeated corticosterone injection, a model that induces a depressive state by impairing the HPA axis function (Kamiya et al., 2020), suggesting that THY1773 would be effective for patients with HPA axis hyperactivity, and appropriate for testing its efficacy for MDD.
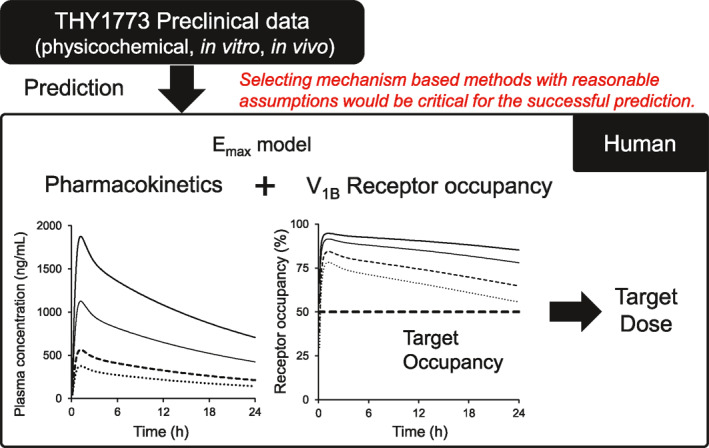
The accurate prediction of a clinically effective dose is critical for improving efficiency and the success rate in drug development (Zou et al., 2012). The pharmacokinetics/pharmacodynamics (PK/PD) modeling and simulation method is a useful tool for the quantitative decision making on the selection of drug candidates, dosing regimens, and the optimization of study designs, particularly by providing a mechanistic rationale and avoiding inaccuracy caused by interspecies differences. For human PK parameter prediction, many methods have been proposed, and the systemic evaluations of these methods using various data sets of drugs have been reported (Fagerholm, 2007; Lombardo et al., 2013a; Ring et al., 2011). However, there is no perfect method that can predict human PK of all drugs accurately. It is important to select the most appropriate prediction method for each human PK parameter of each drug based on its PK properties. For the efficacy prediction, appropriate translation of preclinical dose‐efficacy findings to human is critical. A target engaging biomarker, such as receptor occupancy (RO), which correlates with efficacy, plays an important role for this translation (Suhara et al., 2017). Establishing an appropriate quantitative model that links PK and target engaging biomarker dynamics is important for the determination of a dose that is predicted to be clinically effective with confidence.
Here, we report our strategy to predict a clinically effective dose of THY1773 for the treatment of MDD to support decision making in the preclinical stage. We applied the methods proposed by Ring et al. (2011) to predict clearance (CL). In addition, as observed human data have become available, the prediction accuracy was retrospectively evaluated, and important insights gained in selecting appropriate methods to successfully predict a clinically effective dose in the preclinical stage are also discussed.
2. MATERIALS AND METHODS
2.1. Materials
THY1773 and THY1773‐d7, deuterium‐labeled THY1773 used as internal standard (IS), were synthesized at Taisho Pharmaceutical Co., Ltd. Human liver microsomes (HLMs; pooled from 50 donors) were purchased from Sekisui XenoTech, LLC. Recombinant human cytochrome P450 (CYP) enzymes: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and control (Bactosomes, derived from Escherichia coli) were purchased from Cypex Ltd. Pooled plasma of male Crl:CD Sprague Dawley (SD) rat and male beagle dog containing EDTA‐2K as an anticoagulant, were purchased from HAMRI Co., Ltd. Male SD rat and male beagle dog liver microsomes were purchased from Corning Gentest. All other chemicals and solvents were of analytical grade and obtained from commercial sources.
2.2. Animals
Male SD rats (7‐week old) were purchased from Charles River Laboratories Japan Inc. Experiments involving male beagle dogs (approximately 3 years; LSG Corporation) were conducted at HAMRI Co., Ltd. The animals were maintained under standard laboratory conditions. All animals were maintained on a standard laboratory animal diet and fasted 16–17 h prior to dosing. Feed was provided 4–5 h postdose and water was allowed ad libitum. All of the animal experimental procedures involving animal handling were approved by the Institutional Animal Care and Use Committee of Taisho Pharmaceutical Co., Ltd (rats and dogs) and HAMRI Co., Ltd (dogs).
2.3. In vitro methods
2.3.1. Parallel artificial membrane permeability assay
The membrane permeability of THY1773 was measured using a parallel artificial membrane permeability assay (PAMPA) Evolution instrument (Pion Inc.). A “sandwich” plate consisting of a donor bottom plate with stir bars and an acceptor filter plate was used for the experiment. A donor plate was prepared by the addition of THY1773 to diluted PRISMA HT at pH 6.2 (final concentration of 12.5 μmol/L). An acceptor plate was coated with GIT‐0 lipid solution, followed by the addition of acceptor sink buffer (pH 7.4). After incubation for 4 h at room temperature, the concentrations of THY1773 in the reference, donor, and acceptor solutions were measured with a UV plate reader. The apparent permeability (Papp) was calculated using the PAMPA Evolution software (version 3.1; Pion Inc.).
2.3.2. Blood‐to‐plasma ratio
The blood‐to‐plasma ratios of THY1773 were analyzed using pooled fresh whole blood (anticoagulant: EDTA‐2K) and control plasma separated from fresh whole blood in parallel from rats, dogs, and humans. Whole blood and control plasma containing 1 μmol/L THY1773 were incubated at 37°C for 30 min in a shaking water bath. The incubated whole blood was centrifuged at 2000 × g for 10 min at 4°C to obtain plasma. An aliquot of each plasma sample (i.e., isolated plasma from incubated whole blood and control plasma) was mixed with 8 volumes of acetonitrile/methanol (9:1, vol/vol) containing the IS. After centrifugation at 3974 × g for 10 min at 4°C, the supernatant was subjected to liquid chromatography/tandem mass spectrometry (LC‐MS/MS) analysis. The blood‐to‐plasma ratio was calculated by dividing the THY1773 concentration in control plasma, which is the same as the concentration in whole blood, by that in the separated plasma from incubated whole blood.
2.3.3. Plasma protein binding
The plasma protein binding of THY1773 in rats, dogs, and humans was evaluated by an equilibrium dialysis method using a 96‐well equilibrium dialysis plate (HTDialysis) with a 12–14 kDa cut‐off dialysis membrane. THY1773 was dissolved in dimethyl sulfoxide, and spiked with rat, dog, or human plasma at a final concentration of 300 ng/ml. An aliquot of plasma containing THY1773 and sodium phosphate buffer (pH 7.4) was added into the donor side and the receiver side of each designated well, respectively. After incubation for 6 h at a rate of 50 oscillation/min at 37°C in an air incubator, an aliquot of each side of each well was collected and mixed with acetonitrile/methanol (9:1, vol/vol) containing the IS. Each sample was centrifuged at 3974 × g for 10 min at 4°C to remove the precipitated proteins, and the resultant supernatants were subjected to LC‐MS/MS analysis. The protein binding (%) was calculated using the Boudinot formula (Boudinot & Jusko, 1984).
2.3.4. Metabolic stability in animal liver microsomes
The oxidative metabolism of THY1773 was evaluated in rat and dog liver microsomes. THY1773 (1 μmol/L) was incubated with pooled liver microsomes (0.25 mg protein/ml) in sodium–potassium phosphate buffer (pH 7.4) consisting of 100 mmol/L sodium phosphate, 100 mmol/L potassium chloride, 2.5 mmol/L magnesium chloride (MgCl2), 1.5 mmol/L glucose‐6‐phosphate (G‐6‐P), and 0.18 units/ml G‐6‐P dehydrogenase (G‐6‐PDH). The reactions were initiated by the addition of β‐nicotinamide–adenine dinucleotide phosphate, oxidized form (NADP+, 0.16 mmol/L), and incubated for 10, 20, 30, 40, 50, and 60 min. The reactions were terminated by the addition of an equal volume of acetonitrile/methanol (9:1, vol/vol) containing the IS, followed by centrifugation at 3974 × g for 10 min at 4°C. An aliquot of the supernatant was subjected to LC‐MS/MS analysis. The intrinsic clearance (CLint) was calculated by the substrate depletion method described by Naritomi et al. (2001).
2.3.5. Reaction phenotyping
Reaction phenotyping using cDNA expressed recombinant human CYP isoforms, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, was conducted. Each recombinant enzyme (100 pmol/ml) in potassium phosphate buffer (pH 7.4; 100 mmol/L for CYP3A4, 50 mmol/L for the others) was preincubated with [3H]THY1773 (10 μmol/L) at 37°C for 5 min. The reactions were initiated by the addition of NADPH‐generating system (1 mmol/L NADP+, 5 mmol/L G‐6‐P, 1 unit/ml G‐6‐PDH, and 5 mmol/L MgCl2) and incubated for 0 or 60 min. The reactions were terminated by the addition of an equal volume of acetonitrile/methanol (9:1, vol/vol), followed by centrifugation at 1650 × g for 10 min at 4°C. An aliquot of the supernatant was subjected to an Agilent 1200 HPLC system (Agilent Technologies, Inc.) equipped with a radiochemical flow detector (Radiomatic 625TR; Perkin Elmer, Inc.), and formation of metabolites was evaluated. Reaction phenotyping study was also conducted using HLMs and selective chemical inhibitors for CYP2D6 and CYP3A, which were assumed to have significant roles in THY1773 metabolism from recombinant metabolic stability. Quinidine (1 μmol/L) for CYP2D6 and ketoconazole (1 μmol/L) for CYP3A were used as selective inhibitors. THY1773 (500 nmol/L) was incubated with pooled HLMs (0.5 mg protein/ml) in potassium phosphate buffer (100 mmol/L, pH 7.4) in the absence or presence of the selective inhibitors. The reactions were initiated by the addition of NADPH generating system (1.3 mmol/L β‐NADP+, 3.3 mmol/L G‐6‐P, 3.3 mmol/L MgCl2, and 0.4 units/ml G‐6‐PDH), and incubated at 37°C for 30 min. The reactions were terminated by the addition of two volumes of acetonitrile/methanol (9:1, vol/vol) containing the IS. After centrifugation at 3974 × g for 10 min at 4°C, an aliquot of the supernatant was subjected to LC‐MS/MS analysis. The CLint was calculated by the method as described previously under metabolic stability in animal liver microsomes, and the fraction metabolized (fm) was calculated by Equation (1):
| (1) |
where CLint is intrinsic clearance without specific inhibitor and CLint,with inh is intrinsic clearance in the presence of specific inhibitor.
2.4. PK in rats and dogs
The PK and urinary excretion of THY1773 in rats (1 mg/kg) and dogs (0.5 mg/kg) under fasted conditions were evaluated following intravenous or oral administration of THY1773. THY1773 was dissolved in 10% (vol/vol) hydroxypropyl‐β‐cyclodextrin at pH 4 for intravenous administration, or suspended in 0.5% (wt/vol) methyl cellulose 400 for oral administration. After a single intravenous or oral administration of THY1773, blood samples were collected into tubes containing EDTA‐2K as an anticoagulant. The scheduled time points were predose (dog only), 5 (intravenous only), 15, 30 min, 1, 2, 4, 8, 12, and 24 h postdose. Plasma was separated by centrifugation. Urine samples were collected into bottles set on ice for 24 h after intravenous administration of THY1773. Plasma and urine samples were treated with 4 volumes of acetonitrile/methanol (9:1, vol/vol) containing the IS. After centrifugation (3639 × g, 4°C, 10 min) to remove precipitated proteins, the supernatant was subjected to LC‐MS/MS analysis. The distributions of THY1773 into brain and pituitary gland in rats were investigated after a single oral administration of THY1773 (1 mg/kg). Blood was collected from the tail vein into tubes containing EDTA‐2K at 1 h after a single oral administration of THY1773, and the plasma was separated by centrifugation (12000 × g, 4°C, 3 min). After blood collection, the brain and pituitary gland were excised followed by homogenization with approximately 4 volumes of distilled water. Plasma, brain, and pituitary samples were treated with 4 volumes of acetonitrile/methanol (9:1, vol/vol) containing the IS and centrifuged (3639 × g, 4°C, 10 min). Aliquots of the supernatant were subjected to LC‐MS/MS analysis.
2.5. PK analysis
The plasma concentration–time profiles of THY1773 were analyzed by a non‐compartmental analysis with PK analysis software Phoenix WinNonlin® (version 6.1; Certara), and the PK parameters were calculated. The fraction excreted in urine was calculated by dividing the amount of THY1773 excreted in urine by the administered dose.
2.6. Prediction of human PK parameters
2.6.1. CL
Of the many reported methods for the prediction of human CL, the top two methods reported by Ring et al. (2011) were selected based on prediction accuracy; that is, the unbound fraction corrected intercept method (FCIM) and two‐species‐based allometry (rat and dog) described by Equations (2) and (3), respectively (Tang, Hussain, Leal, Mayersohn, & Fluhler, 2007; Tang & Mayersohn, 2005):
| (2) |
| (3) |
where the allometric coefficient (a) is obtained from the intercept of the simple allometry log–log plot between rats and dogs, and Rfu is the ratio of unbound fraction in plasma between rats and humans. Human body weight was assumed to be 70 kg. The hepatic CL (CLliver) was also predicted by physiologically based in vitro–in vivo extrapolation (IVIVE) method using physiological parameters listed in Table 1 based on the well stirred model (Yang, Jamei, Yeo, Rostami‐Hodjegan, & Tucker, 2007).
TABLE 1.
Physiological parameters used for in vitro–in vivo extrapolation
Since the CLliver was underestimated in rats and dogs from CLint in liver microsomes, the ratio of observed in vivo CLint and predicted CLint was calculated as the empirical scaling factor (ESF) for the extrapolation of CLliver from in vitro data. Human CLliver was predicted from CLint in HLMs with and without applying the average of ESF derived from rats and dogs. The total plasma clearance (CLtotal) was calculated by Equation (4):
| (4) |
where QH is hepatic blood flow, fu is the free fraction in plasma, fu,mic is the fraction unbound in HLMs, Rb is blood‐to‐plasma ratio, and CLrenal is renal clearance. The fu,mic value was calculated using the Simcyp population based simulator (Version 16 R1; Simcyp Ltd.). The CLrenal was estimated by multiplying fu by glomerular filtration rate (GFR), assuming glomerular filtration only.
2.6.2. Volume of distribution
Human volume of distribution at steady state (Vdss) was predicted by single species scaling from rat assuming that the unbound Vdss is comparable across species, as described in Equation (5):
| (5) |
where fu is the unbound fraction in plasma. Human Vdss was also predicted by the two methods reported to be the best methods based on the prediction accuracy (Jones et al., 2011), which were Øie‐Tozer method (Obach et al., 1997) and rat–dog–human proportionality equation described (Wajima, Fukumura, Yano, & Oguma, 2003) by Equations (6) and (7), respectively:
| (6) |
| (7) |
where fu,t is the fraction unbound in tissues, RE/I is the ratio of binding protein in the extracellular fluid to that in plasma, Ve, Vp, and Vr is the extracellular, plasma, and remaining fluid volume, respectively. Volumes and plasma protein levels were used as described by Obach et al. (1997).
2.7. Physiologically based pharmacokinetics/RO modeling and simulation
2.7.1. PBPK/RO model development
The human PBPK/RO models were developed using Simcyp simulator following the previously reported strategy (Jones, Parrott, Jorga, & Lavé, 2006). Since Vdss in rat was underpredicted using tissue‐to‐plasma partition method as proposed by Rodgers and Rowland (2007), the tissue‐to‐plasma partition coefficient (Kp) scalar of 0.422 was applied to all human tissue compartments to match the predicted human Vdss by the Øie‐Tozer method. The CLint in HLMs were inputted into Simcyp as CYP2D6 and CYP3A4 dependent CLint based on the estimated fraction metabolized in HLMs, with (Model 1) or without (Model 2) applying ESF. The predicted CLrenal assuming glomerular filtration only was also inputted. As the relationship between drug concentration and RO typically follow the Emax model (Kim et al., 2012), the Emax model described by Equation (8) was used as a PD model for THY1773 V1B RO. As the pituitary gland is outside of the blood–brain barrier, the free concentration of THY1773 around the V1B receptor in the pituitary gland was assumed to be equal to the free concentration in plasma:
| (8) |
where Cp,u is free concentration in plasma, Emax is the maximum V1B RO, and EC50 is plasma free concentration required to produce 50% of Emax. Emax was assumed to be 100%, and EC50 was assumed to be comparable to the inhibition constant (Ki) calculated from the in vitro binding assay, which evaluated the binding of [3H]AVP to the membranes of 1321‐N1 cells expressing the human V1B receptor with or without 0.001–1000 nmol/L of THY1773 by a previously reported method (Iijima et al., 2014). The input parameters used for human PBPK/RO model development in Simcyp are listed in Table 2.
TABLE 2.
Human PBPK/RO model input parameters for THY1773
| Parameter | Value | Reference/comment | |
|---|---|---|---|
| Physicochemical properties | |||
| Molecular weight | 483.99 | ||
| Log P | 3.17 | Experimental data | |
| Compound type | Monoprotic base | ||
| pKa | 6.85 | Experimental data | |
| Fraction unbound in plasma | 0.017 | Experimental data | |
| Blood‐to‐plasma ratio | 0.55 | Theoretical limit (experimental data: 0.54) | |
| Absorption | |||
| Absorption model | Advanced dissolution, absorption, and metabolism model | ||
| Fraction absorbed | 0.998 | Simcyp predicted | |
| Absorption rate constant (1/h) | 3.10 | Simcyp predicted | |
| PAMPA permeability (10−6 cm/s) | 128.9 | Experimental data | |
| Fraction unbound in gut | 1 | Assumption | |
| Distribution | |||
| Distribution model | Full PBPK | ||
| Vdss (L/kg) | 0.405 | Adjusted to predicted value by Øie‐Tozer method with tissue to plasma partition coefficient scalar (0.422) | |
| Elimination | |||
| In vitro metabolic system | HLMs enzyme kinetics | ||
| CLint (μl/min/mg protein) | 11.2 (Model 1) | Experimental data | |
| 56.4 (Model 2) | Experimental data with empirical scaling factor | ||
| fm,CYP2D6/fm,CYP3A4 | 0.089/0.911 | Experimental data | |
| Fraction unbound in microsomes | 0.714 | Simcyp predicted | |
| Renal clearance (L/h) | 0.133 | Estimated by multiplying fu and mean body weight of “Sim‐Healthy Volunteers” population (72.4 kg) by GFR | |
| Pharmacodynamic (receptor occupancy) model | |||
| Model | Simple Emax model | ||
| Emax (%) | 100 | Assumption | |
| EC50 (nmol/L) | 3.51 | Assumption (Ki value for human V1B receptor binding inhibition) | |
GFR, glomerular filtration rate; PAMPA, parallel artificial membrane permeability assay; PBPK, physiologically based pharmacokinetics; RO, receptor occupancy.
2.7.2. Prediction of a clinically effective dose
The plasma concentration– and RO–time profiles of THY1773 after oral administration of various doses of THY1773 were simulated by the previously described PBPK/RO model in 10 virtual trials of 10 healthy volunteers (ages 20–50 years, male/female ratio of 50/50) assuming linear PK. The built‐in “Sim‐Healthy Volunteers” population was used. According to the results of a CRF/desmopressin challenge study and a forced swim test in rats, significant effects for both studies were shown at doses which showed more than 50% occupancy of the anterior pituitary V1B receptor (Kamiya et al., 2020). Therefore, the clinically effective dose was assumed to be the dose that maintains RO at more than 50% for the dosing interval (24 h after administration).
2.8. Phase 1 clinical study
A first‐in‐human, randomized, double‐blind, placebo‐controlled, adaptive, single‐ascending dose study was conducted to evaluate the safety, tolerability, and PK of single oral doses of TS‐121 capsules in the United States. This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, International Council for Harmonization Good Clinical Practice Guidelines, and the United States Food and Drug Administration Code of Federal Regulations. The study was approved by an institutional ethical review board. All subjects provided written informed consent before participating in the study. Sixty‐eight healthy male or female volunteers were enrolled and received a single oral dose of either TS‐121 capsules (0.5, 2.5, 4, 8, 15, 30, or 50 mg as THY1773/day) or placebo in a 3:1 ratio in a fasted state.
Blood samples were collected at predose and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, and 48 h postdose. Urine samples were collected before dosing and at the following time intervals after dosing: 0–6, 6–12, 12–24, 24–36, and 36–48 h. THY1773 plasma and urine concentrations were determined by validated LC‐MS/MS methods. PK parameters were evaluated following the method as described in Section 2.5 PK analysis.
2.9. Evaluation of prediction accuracy
The fold error of the difference between observed and predicted human PK parameters was calculated by dividing the predicted value by the observed value. The bioavailability of THY1773 was assumed to be 100%, and the volume of distribution at steady state was assumed to be comparable to that during the terminal phase.
2.10. LC‐MS/MS conditions
The LC‐MS/MS system consisted of a Shimadzu LC‐10ADvp, LC‐20AD, LC‐30AD, or Nexera (Shimadzu) and an API 3000, API 4000, or Triple Quad 5500 mass spectrometer (AB Sciex). The data were collected and processed using Analyst software (version 1.4 or later; AB Sciex). THY1773 was analyzed using a Shim‐pack XR‐ODS column (3.0 mm I.D. × 30 mm, 2.2 μm particle size; Shimadzu) or a ZORBAX Eclipse Plus C18 column (4.6 mm I.D. × 50 mm, 3.5 μm particle size; Agilent Technologies) with water/formic acid (1000:1, vol/vol) and acetonitrile or water/formic acid (95:5, vol/vol) and acetonitrile/methanol (95:5, vol/vol) as the mobile phase under a gradient condition. The selected reaction monitoring transitions were as follows: THY1773, m/z 484 → m/z 397; and THY1773‐d7 (IS), m/z 491 → m/z 404.
3. RESULTS
3.1. Permeability, plasma protein binding, and blood‐to‐plasma ratio
The passive membrane permeability of THY1773 was measured using PAMPA. The Papp of THY1773 in PAMPA at pH 6.2 was 128.9 (10−6 cm/s), suggesting high permeability in the gut. Plasma protein binding was determined by equilibrium dialysis at a THY1773 concentration of 300 ng/ml, which was roughly estimated as an average concentration in human plasma at an effective dose. The mean plasma protein binding of THY1773 at 300 ng/ml was 89.2% in rats, 89.6% in dogs, and 98.3% in humans. The blood‐to‐plasma ratio of THY1773 was evaluated in rat, dog, and human pooled whole blood at 1 μmol/L (484 ng/ml). The mean blood‐to‐plasma ratio was 0.822 in rats, 0.846 in dogs, and 0.540 in humans. THY1773 showed higher plasma protein binding and lower blood‐to‐plasma ratio in humans than in animals.
3.2. Metabolic stability in animal liver microsomes
The oxidative metabolism of THY1773 in rat and dog liver microsomes was evaluated at 1 μmol/L, at which the reaction was assumed to be linear. The CLint (in units of μL/min/mg protein) in liver microsomes was 22.0 for rats and 32.2 for dogs.
3.3. Reaction phenotyping
Reaction phenotyping studies were conducted using cDNA expressed recombinant human CYP isoforms and HLMs. In the reaction phenotyping study using recombinant human CYP isoforms, [3H]THY1773 metabolite formation was evaluated at 10 μmol/L. [3H]THY1773 was metabolized by CYP2D6 and CYP3A4, and to a minor extent by CYP2C8. In the reaction phenotyping study using HLMs and selective chemical inhibitors, the disappearance of THY1773 at 500 nmol/L (242 ng/ml) was evaluated. The CLint of THY1773 was significantly reduced by ketoconazole, a CYP3A inhibitor, whereas it was partially reduced by quinidine, a CYP2D6 inhibitor. The fractions metabolized were estimated to be 0.914 by CYP3A and 0.0893 by CYP2D6 (Table 3).
TABLE 3.
Effects of specific inhibitor of CYP3A and CYP2D6 on CLint of THY1773 in human liver microsomes and estimated fraction metabolized
| CYP isoforms | Selective inhibitor (concentration) | CLint (μl/min/mg) | fm |
|---|---|---|---|
| – | None | 11.2 ± 0.898 | N.A. |
| CYP3A | Ketoconazole (1 μmol/L) | 0.961 a | 0.914 |
| CYP2D6 | Quinidine (1 μmol/L) | 10.2 ± 0.509 | 0.0893 |
Values are presented as the mean ± SD of triplicate determinations.
N.A., not applicable.
Value is presented as the mean of duplicate determinations.
3.4. PK in rats and dogs
The concentration–time profiles in rats and dogs after a single intravenous or oral administration of THY1773 under fasted conditions are shown in Figure 1, and the PK parameters are summarized in Table 4. In rats and dogs, the CLtotal was moderate (approximately 30% of hepatic blood flow) after single intravenous administration. The urinary excretion of unchanged THY1773 within 24 h postintravenous dosing was 4.6% in rats and 2.0% in dogs. In both species, the CLrenal of THY1773 was comparable to fu × GFR (within twofold), which suggested that active transport was not likely to play an important role in the CLrenal. Following oral administration, THY1773 was absorbed rapidly, and oral bioavailability reached 46.4% in rats and 45.0% in dogs. The concentration ratio of brain and pituitary gland to plasma in rats at 1 h after oral administration was 0.2 and 4.7, respectively.
FIGURE 1.
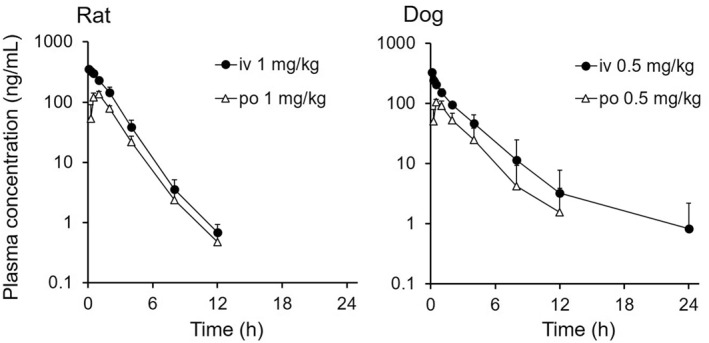
THY1773 plasma concentration–time profiles (mean ± SD, n = 3) in male rats (left) and dogs (right) after a single intravenous (iv) or oral (po) administration. The plasma concentrations declined below the lower limit of quantification (<0.1 ng/ml) at 24 h except for after intravenous dose to dogs.
TABLE 4.
Pharmacokinetic parameters of THY1773 after a single intravenous or oral administration to male rats (1 mg/kg) and dogs (0.5 mg/kg)
| Species | Route | CLtotal (ml/h/kg) | Vdss (ml/kg) | t1/2 (h) | Cmax (ng/ml) | tmax (h) | AUC0‐∞ (h·ng/ml) | F (%) |
|---|---|---|---|---|---|---|---|---|
| Rat | iv | 1330 (13.7) | 2220 (5.7) | 1.38 (2.3) | N.C. | N.C. | 762 (14.9) | |
| po | N.C. | N.C. | 1.45 (5.5) | 136 (11.8) | 0.833 (34.6) | 355 (13.5) | 46.6 | |
| Dog | iv | 785 (26.4) | 2020 (37.6) | 3.07 (59.3) | N.C. | N.C. | 664 (23.7) | |
| po | N.C. | N.C. | 2.40 (81.2) | 105 (12.3) | 0.500 (0.0) | 307 (37.7) | 45.0 (16.6) |
Values are presented as the mean (percent coefficient of variation) of three animals.
N.C., not calculated.
3.5. Prediction of human PK parameters
Of many reported methods for the prediction of human PK parameters, the methods reported to be relatively accurate among various methods using data sets containing lipophilic and highly plasma protein‐bound drugs (Jones et al., 2011; Ring et al., 2011) were selected, as THY1773 was also lipophilic and highly plasma protein‐bound. The human CLtotal was predicted by FCIM, two‐species‐based allometry, or IVIVE method (Table 5). Before conducting IVIVE, the discrepancy between in vitro and in vivo CLint was evaluated in rats and dogs. The in vivo CLint was higher than in vitro CLint in both species, and the ESF for IVIVE was calculated to be 6.5 in rats and 3.6 in dogs. The predicted human CLtotal values varied greatly depending on the prediction methods, and the difference was over 14‐fold. The human Vdss was predicted by rat single species scaling with Equation (5), Øie‐Tozer method, or rat–dog–human proportionality equation (Table 6). The predicted Vdss values varied depending on the prediction methods, and the difference was over threefold.
TABLE 5.
Predicted and observed human CLtotal of THY1773 and fold error
| Prediction method | Predicted human CLtotal (ml/h/kg) | Observed human CLtotal/F (ml/h/kg) | Fold error a |
|---|---|---|---|
| FCIM | 64.1 |
17.2 |
3.7 |
| Two species allometry | 224 | 13 | |
| IVIVE method without ESF | 15.0 | 0.87 | |
| IVIVE method with ESF | 63.4 | 3.7 |
ESF, empirical scaling factor; FCIM, fraction corrected intercept method; IVIVE, in vitro–in vivo extrapolation.
Fold error was calculated by dividing predicted CLtotal by observed oral CLtotal (CLtotal/F) assuming bioavailability (F) was 100%.
TABLE 6.
Predicted and observed human Vdss of THY1773 and fold error
| Prediction method | Predicted human Vdss (ml/kg) | Observed human Vdz/F (ml/kg) | Fold error a |
|---|---|---|---|
| Single species scaling (rat) | 349 |
528 |
0.66 |
| Rat–dog–human proportionality equation | 1380 | 2.6 | |
| Øie‐Tozer method | 404 | 0.77 |
Fold error was calculated by dividing predicted Vdss by observed oral apparent volume of distribution during terminal phase (Vdz/F) assuming bioavailability (F) was 100% and the volume of distribution at steady state was comparable to that during terminal phase.
3.6. Prediction of a clinically effective dose
The plasma concentration– and RO–time profiles in humans were simulated by two PBPK/RO models considering best and worst case scenarios for human liver CLint. The simulated plasma concentration– and RO–time profiles in humans at various doses of THY1773 using PBPK/RO model (Model 1 as best and Model 2 as worst case scenario) are shown in Figure 2. As described in Section 2.7.2, preclinical studies indicated that THY1773 exhibits antidepressant‐like effects in several animal models at doses that occupied more than 50% of the pituitary V1B receptor; consequently, we assumed that significant efficacy would be shown at doses that maintain more than 50% occupancy of the V1B receptor for the dosing interval (24 h after administration). Based on our assumption, the clinically effective dose was predicted to be 10 mg by Model 1 and 110 mg by Model 2.
FIGURE 2.
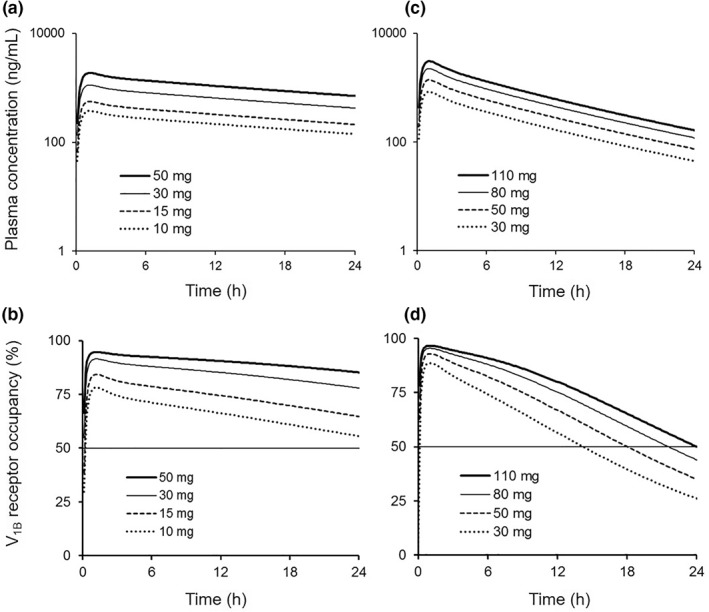
The simulated plasma THY1773 concentration–time (a and c) and V1B RO–time (b and d) profiles in humans after a single oral administration of THY1773 at various doses (10, 15, 30, and 50 mg for Model 1; 30, 50, 80, and 110 mg for Model 2) using the human PBPK/RO model. The PBPK/RO model was established using CLint in HLMs without ESF (Model 1; a and b) or with ESF (Model 2; c and d). The solid horizontal lines represent 50% RO. ESF, empirical scaling factors; HLM, human liver microsome; PBPK, physiologically based pharmacokinetics; RO, receptor occupancy
3.7. Phase 1 clinical study
Following a single oral administration of TS‐121 capsules in healthy male or female volunteers, THY1773 was absorbed rapidly, reached maximum plasma concentration (Cmax) at 1.3 h, and eliminated with a half‐life of 21 h at 30 mg/day as THY1773 (Figure 3 and Table 7). THY1773 exposures (Cmax and area under the concentration–time curve) increased nearly dose‐proportionally (Figure 3). Oral CLtotal and apparent volume of distribution during the terminal phase were stable over the dose range of 0.5 to 50 mg, and averaged 1.32 L/h and 40.6 L, respectively. The urinary excretion of unchanged THY1773 within 48 h postoral dosing was also stable over the dose range and averaged 4.2%.
FIGURE 3.
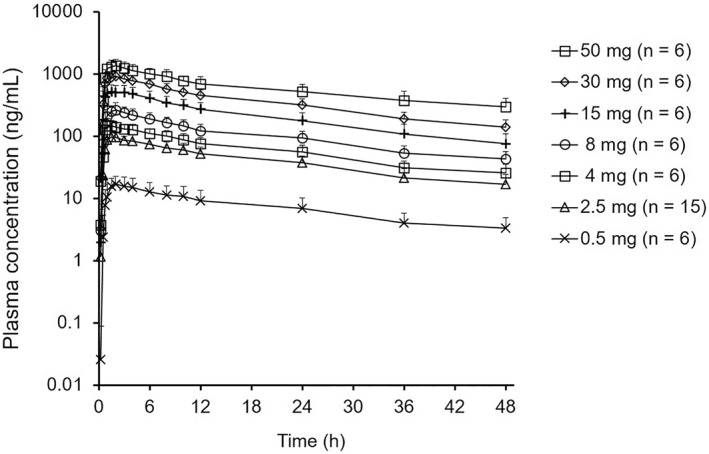
The plasma concentration–time profiles (mean ± SD) of THY1773 after oral administration in the phase 1 single ascending dose study. Healthy male or female volunteers received a single oral dose of either TS‐121 capsules or placebo in a fasted state.
TABLE 7.
Observed human PK parameters of THY1773 after a single oral administration (30 mg)
| Parameter | AUC0‐∞ (h·ng/ml) | Cmax (ng/ml) | tmax a (h) | t1/2 (h) | CLpo (L/h) | Vdz/F (L) |
|---|---|---|---|---|---|---|
| Mean | 21340 | 949 | 1.27 | 20.5 | 1.52 | 43.6 |
| (% CV) | (24.2) | (22.4) | (1.00, 2.10) | (14.4) | (37) | (27.3) |
Median (minimum, maximum) values are presented.
3.8. Evaluation of prediction accuracy
The observed oral blood CL was only 2.6% of the hepatic blood flow, which indicated that hepatic metabolism of THY1773 was not extensive in humans. As THY1773 was mainly metabolized by CYP3A in HLMs, we assumed that gut metabolism, which was known to be mainly mediated by CYP3A, was not extensive in humans either. Then we assumed that the human bioavailability of THY1773 was 100% to compare the predicted human PK parameters with the observed values obtained after oral administration. The fold errors of the difference between observed and predicted human PK parameters are shown in Table 5 (CLtotal) and Table 6 (Vdss). The simulated concentration–time profiles of THY1773 using PBPK model (Model 1) overlaid with observed plasma concentrations after a single oral administration of THY1773 at a dose of 30 mg and at doses of 0.5–50 mg are shown in Figure 4. Overall, concentrations of THY1773 were accurately predicted by Model 1, whereas they were underestimated by Model 2.
FIGURE 4.
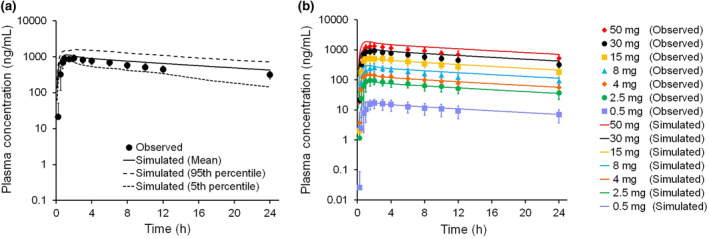
The simulated versus observed plasma concentration–time profiles of THY1773 in humans after a single oral administration of THY1773 at a dose of 30 mg (a) and at doses of 0.5–50 mg (b) using the PBPK model. The PBPK model was established using CLint in HLMs without ESF (Model 1). Points, observed values (mean ± SD) in clinical study; solid line, simulated mean; dotted lines, 5th and 95th percentiles. ESF, empirical scaling factors; HLM, human liver microsome; PBPK, physiologically based pharmacokinetics
4. DISCUSSION AND CONCLUSION
The development of drugs for central nervous system (CNS) diseases is challenging relative to other therapeutic areas (Harrison, 2016; Suhara et al., 2017). Modeling and simulation technology has been used widely as an initiative to overcome the difficulty and to improve success rates of drug discovery and development (Visser, De Alwis, Kerbusch, Stone, & Allerheiligen, 2014). We developed THY1773, a novel selective V1B receptor antagonist that exhibited antidepressant‐like effects in animal models (Kamiya et al., 2020). In the present studies, we established PBPK/RO models of THY1773 fully based on the preclinical data and predicted a clinically effective dose for MDD to support decision making in the preclinical stage of drug development.
In rats and dogs, THY1773 was rapidly and well absorbed with moderate bioavailability, and exhibited moderate CLtotal and Vdss. The renal excretion ratio was less than 5%. As THY1773 had high permeability in PAMPA (a class 2 compound in the extended CL classification system; Varma, Steyn, Allerton, & El‐Kattan, 2015), THY1773 was assumed to be primarily eliminated by hepatic metabolism. THY1773 showed species differences in plasma protein binding and blood‐to‐plasma ratio. THY1773 is a CYP3A4 substrate, and mainly metabolized by CYP3A in HLMs. The CLint in HLMs was lower than that in rats and dogs. From these preclinical PK properties, THY1773 was expected to be well absorbed with good bioavailability and eliminated with moderate CL mainly mediated by CYP3A4 metabolism in humans. Based on these preclinical data, a PBPK/RO model of THY1773 in humans was established for the prediction of a clinically effective dose for MDD.
First, human CL and Vdss were predicted by several methods. Human CL was predicted by two allometric and two IVIVE methods, and we found that widely different values were obtained depending on the methods. Of these methods, we selected IVIVE methods for the following reasons: (1) the plasma protein binding of THY1773 was higher in humans than in animals, and it has been reported that allometric methods that did not account for protein binding tended to overpredict human CL (Ring et al., 2011); and (2) as THY1773 is a CYP3A4 substrate, the CLliver, or intestinal first pass, was expected to be relatively accurately predicted using CLint in HLMs (Bowman & Benet, 2019b; Gertz, Harrison, Houston, & Galetin, 2010). Since IVIVE methods using HLMs reportedly have a tendency to underpredict human CLliver (Wood, Houston, & Hallifax, 2017), the IVIVE method, with or without ESF obtained from rats and dogs, was used for THY1773 CLliver prediction as the worst or best case scenario, respectively. To predict CLtotal, CLrenal was predicted assuming glomerular filtration only as in animals, and added to the predicted CLliver. The human Vdss was predicted by two empirical and one semi‐mechanistic method. The predicted values were different depending on whether or not the plasma protein binding was considered. The unbound Vdss was comparable between rats and dogs, suggesting that the assumption in rat single species scaling with Equation (5), (i.e., the unbound Vdss is comparable across species), was valid, and the predicted human Vdss would therefore be confident. We ultimately selected the semi‐mechanistic Øie‐Tozer method as it was reported to be most accurate for many compounds (Jones et al., 2011), and the predicted Vdss was comparable to that predicted by rat single species scaling with Equation (5).
Then, the PBPK model was established using preclinical data, adjusting the Vdss to fit the predicted value by the Øie‐Tozer method, and linked to the RO. The following two assumptions were made: (1) the pituitary V1B RO of THY1773 was correlated to the plasma free concentration, and the relationship follows Emax model, and (2) the in vivo EC50 value for pituitary V1B RO was comparable to the in vitro Ki value for human V1B receptor binding inhibition.
Finally, a clinically effective dose was predicted by the PBPK/RO model assuming that significant efficacy would be shown at a dose that shows more than 50% occupancy of the V1B receptor for the dosing interval. As a result, the predicted effective dose of THY1773 was relatively low (10 mg/day, or at maximum, 110 mg/day), and was thought to be optimal for further clinical development. These results supported our decision to enter phase 1 clinical trials.
In the phase 1 single ascending dose study, THY1773 was rapidly absorbed, and eliminated with a terminal half‐life of approximately 21 h, which was long enough to support once daily dosing. THY1773 demonstrated linear PK over the dose range of 0.5–50 mg. Together with the additional information that THY1773 did not have any apparent in vitro inhibition or induction potential on major CYP isoforms (data not shown), THY1773 demonstrated ideal PK features as a CNS drug. Plasma concentration–time profiles of THY1773 after a single oral administration were consistent with the simulated profiles by PBPK model without applying ESF. Based on the phase 1 RO study using PET (ClinicalTrials.gov Identifier: NCT02448212), V1B RO at steady state was estimated to be 55.3% at 10 mg and 75.1% at 50 mg (unpublished data), which were comparable to our prediction (68.2% at 10 mg and 90.3% at 50 mg) and expected to be enough to show efficacy. In our phase 2 study conducted in patients with MDD (ClinicalTrials.gov Identifier: NCT03093025), statistically nonsignificant greater improvements in depressive symptoms were observed with TS‐121 in MDD patients at doses of 10 and 50 mg, which would achieve more than 50% RO at steady state (Kamiya et al., 2020). Although further investigation of the dose‐efficacy relationship is required, these findings support our assumption that a clinically effective dose that maintains RO at more than 50% for the dosing interval is achievable.
A retrospective analysis of the prediction accuracy was conducted after obtaining the observed data. For CLtotal, only the IVIVE method without ESF could predict human CLtotal within twofold. This finding was consistent with the previous report that the IVIVE method using human microsomal data provided better prediction than the other methods for compounds eliminated primarily by CYP‐mediated mechanisms (Hosea et al., 2009). Two species allometry for Vdss without a protein binding correction showed a large discrepancy (13‐fold overprediction) between predicted and observed human oral CLtotal. Although the predicted human CLtotal by the IVIVE method with ESF was comparable to the value predicted by FCIM, both methods overpredicted human oral CLtotal by approximately fourfold. These results suggest that the similarity of predicted values by different prediction methods does not always indicate higher reliability. Even though FCIM was reported to yield the best predictions among various methods using larger data sets (Lombardo et al., 2013b), it failed to predict human CLtotal of THY1773, which was possibly due to the lack of monkey data. The observed CLrenal was lower than the predicted value, suggesting the potential of renal reabsorption of THY1773 in humans, and indicating that the overprediction of human CLtotal by IVIVE method with ESF was due to the overprediction of CLliver. The calculated ESF was not comparable across species, and similar results were obtained when the CLint in hepatocytes were used for IVIVE (data not shown). The reasons for the discrepancy between in vitro and in vivo CLint and species difference of ESF are not clear. These results indicate, however, the limitations of extrapolation of animal data to humans; that is, the unknown scaling factor obtained in animals should not be applied to human prediction without a reasonable hypothesis, especially when the value is not comparable across species. Recently, it has been reported that the IVIVE method using liver microsomes is highly predictive for CYP3A4 substrates than for other metabolic enzymes, and that the IVIVE method tends to underpredict CLliver but the underprediction is CLint‐ and CLliver‐dependent (Bowman & Benet, 2019a; Hallifax, Foster, & Houston, 2010). Taken together with these reports, our results suggested that the IVIVE method would be the best method for the prediction of human CLliver of a compound that is assumed to be metabolized primarily by CYP3A4 with low CLint.
Based on the retrospective analysis of the prediction accuracy of Vdss, the rat–dog–human proportionality equation, a method without plasma protein binding correction, showed discrepancy (approximately threefold overprediction) between predicted and observed human oral Vdss. The Øie‐Tozer method and rat single species scaling with Equation (5), both of which consider plasma protein binding, predicted human Vdss accurately (within twofold). Our results support the previous report that concluded the semi‐mechanistic Øie‐Tozer method as the most accurate method verified with more compounds (Lombardo et al., 2013a; Petersson, Papasouliotis, Lecomte, Badolo, & Dolgos, 2020).
The PBPK model using human liver microsomal CLint without ESF (Model 1) described well the human plasma concentration–time profiles (Figure 4). In the PBPK model, as it was reported that CLint in human intestinal microsomes and HLMs were not significantly different after normalization for tissue‐specific CYP3A abundance (Gertz, Harrison, Houston, & Galetin, 2010), CYP3A4 metabolism in the gut was estimated from CYP3A4 CLint in HLMs, accounting for the differences in enzyme abundance between the liver and intestine. The fraction escaping gut first‐pass metabolism was predicted to be 0.97, and the bioavailability was predicted to be 95% by Model 1, which was almost consistent with our assumption for the evaluation of prediction accuracy of PK parameters.
Retrospective analysis of the PBPK/RO model also revealed the limitation of extrapolation of animal data to humans. According to the results of the phase 1 RO study using PET, the in vivo EC50 value for V1B RO was comparable (within twofold) to the in vitro Ki value for human V1B receptor binding inhibition (unpublished data). Based on the results of a rat V1B RO study using a tritium labeled V1B receptor ligand (Kamiya et al., 2020), the EC50 value for V1B RO of THY1773 in the rat anterior pituitary was estimated to be 4.8 nmol/L, which was much lower than the in vitro Ki value of 80.1 nmol/L for rat anterior pituitary membrane V1B receptor (unpublished data). This discrepancy (17‐fold higher affinity in vivo) was not likely to be explained by the target concentration because, even if the observed concentration ratio of the pituitary gland to plasma (4.7) was assumed to be applicable for free concentration, it would be much lower than the observed discrepancy. Also, hysteresis (time delay between target concentration and RO) was not likely to explain the discrepancy because hysteresis would result in a higher in vivo EC50 value than the in vitro Ki value, as the RO was evaluated at approximately tmax. The discrepancy was possibly due to a more complex biological mechanism in vivo, but such a complex mechanism was difficult to include in the RO model and translate to humans based on the limited data obtained in the preclinical stage. For the prediction of a human effective dose, the discrepancy between in vitro Ki and in vivo EC50 observed in rats was not considered because there was no reasonable hypothesis that the discrepancy would be the same in humans, and we aimed to avoid the risk of lowering the probability of success of clinical trials by underpredicting a clinically effective dose. In addition, in the PET imaging study in rhesus monkeys, as previously reported (Koga et al., 2017), the monkey in vivo EC50 value of THY1773 for V1B RO was similar to the in vitro Ki value for human recombinant V1B receptor binding inhibition (unpublished data). Even though the in vitro Ki value for monkey V1B receptor was not evaluated, we assumed that the monkey in vivo EC50 value for V1B RO was comparable to the in vitro Ki value based on the high degree of V1B receptor protein homology (96%) between human and monkey, according to the HomoloGene database (https://www.ncbi.nlm.nih.gov/homologene/22678). The aforementioned results provided the rationale for our approach. Retrospective analysis revealed that the discrepancy observed in rats was not observed in humans, and that the unknown scaling factor obtained in preclinical animal studies should not be applied to humans unless the mechanism is clear.
In summary, we established a human PBPK/RO model of THY1773, and succeeded in nearly accurately predicting a clinically effective dose for the treatment of MDD in the preclinical stage. To predict human PK parameters and a clinically effective dose more accurately in the preclinical stage, it is important to identify the most promising prediction method of various available methods. From our studies, it is suggested that mechanism based methods should be selected with reasonable assumptions for the successful predictions. Our studies also showed the limitations of extrapolation of animal data to humans. Caution should be exercised when applying an in vitro–in vivo scaling factor obtained in the preclinical animal studies to humans, especially when the reason for the in vitro–in vivo discrepancy is not clear.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors sincerely thank Dr. Shigeyuki Chaki for reviewing this manuscript. The authors greatly thank Yasunori Kawakita for his support of the clinical and nonclinical studies. The clinical studies and the preclinical experiments were supported by Taisho Pharmaceutical R&D Inc. and Taisho Pharmaceutical Co., Ltd., respectively.
REFERENCES
- Boudinot, F. D. , & Jusko, W. J. (1984). Fluid shifts and other factors affecting plasma protein binding of prednisolone by equilibrium dialysis. Journal of Pharmaceutical Sciences, 73(6), 774–780. 10.1002/jps.2600730617 [DOI] [PubMed] [Google Scholar]
- Bowman, C. M. , & Benet, L. Z. (2019a). In vitro‐in vivo extrapolation and hepatic clearance‐dependent underprediction. Journal of Pharmaceutical Sciences, 108(7), 2500–2504. 10.1016/j.xphs.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman, C. M. , & Benet, L. Z. (2019b). In vitro‐in vivo inaccuracy: The CYP3A4 anomaly. Drug Metabolism & Disposition, 47(12), 1368–1371. 10.1124/dmd.119.088427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, H. S. , Griffin, M. , & Houston, J. B. (2007). Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metabolism & Disposition, 35(2), 293–301. 10.1124/dmd.106.011569 [DOI] [PubMed] [Google Scholar]
- de Winter, R. F. P. , Van Hemert, A. M. , Derijk, R. H. , Zwinderman, K. H. , Frankhuijzen‐Sierevogel, A. C. , Wiegant, V. M. , & Goekoop, J. G. (2003). Anxious‐retarded depression: Relation with plasma vasopressin and cortisol. Neuropsychopharmacology, 28(1), 140–147. 10.1038/sj.npp.1300002 [DOI] [PubMed] [Google Scholar]
- Fagerholm, U. (2007). Prediction of human pharmacokinetics – evaluation of methods for prediction of hepatic metabolic clearance. Journal of Pharmacy and Pharmacology, 59(6), 803–828. 10.1211/jpp.59.6.0007 [DOI] [PubMed] [Google Scholar]
- Geneste, H. , Bhowmik, S. , van Gaalen, M. M. , Hornberger, W. , Hutchins, C. W. , Netz, A. , … Unger, L. (2018). Novel, potent, selective and brain penetrant vasopressin 1b receptor antagonists. Bioorganic & Medicinal Chemistry Letters, 28(19), 3260–3264. 10.1016/j.bmcl.2018.07.043 [DOI] [PubMed] [Google Scholar]
- Gertz, M. , Harrison, A. , Houston, J. B. , & Galetin, A. (2010). Prediction of human intestinal first‐pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metabolism & Disposition, 38(7), 1147–1158. 10.1124/dmd.110.032649 [DOI] [PubMed] [Google Scholar]
- Griebel, G. , Beeské, S. , & Stahl, S. M. (2012). The vasopressin V1b receptor antagonist SSR149415 in the treatment of major depressive and generalized anxiety disorders: Results From 4 Randomized, Double‐Blind, Placebo‐Controlled Studies. Journal of Clinical Psychiatry, 73(11), 1403–1411. 10.4088/JCP.12m07804 [DOI] [PubMed] [Google Scholar]
- Griebel, G. , Simiand, J. , Serradeil‐Le Gal, C. , Wagnon, J. , Pascal, M. , Scatton, B. , … Soubrié, P. (2002). Anxiolytic‐ and antidepressant‐like effects of the non‐peptide vasopressin V1b receptor antagonist, SSR149415, suggest an innovative approach for the treatment of stress‐related disorders. Proceedings of the National Academy of Sciences of the United States of America, 99(9), 6370–6375. 10.1073/pnas.092012099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallifax, D. , Foster, J. A. , & Houston, J. B. (2010). Prediction of human metabolic clearance from in vitro systems: Retrospective analysis and prospective view. Pharmaceutical Research, 27(10), 2150–2161. 10.1007/s11095-010-0218-3 [DOI] [PubMed] [Google Scholar]
- Harrison, R. K. (2016). Phase II and phase III failures: 2013‐2015. Nature Reviews Drug Discovery, 15(12), 817–818. 10.1038/nrd.2016.184 [DOI] [PubMed] [Google Scholar]
- Holsboer, F. , Haack, D. , Gerken, A. , & Vecsei, P. (1984). Plasma dexamethasone concentrations and differential suppression response of cortisol and corticosterone in depressives and controls. Biological Psychiatry, 19(3), 281–291 [PubMed] [Google Scholar]
- Hosea, N. A. , Collard, W. T. , Cole, S. , Maurer, T. S. , Fang, R. X. , Jones, H. , … Beaumont, K. (2009). Prediction of human pharmacokinetics from preclinical information: Comparative accuracy of quantitative prediction approaches. The Journal of Clinical Pharmacology, 49(5), 513–533. 10.1177/0091270009333209 [DOI] [PubMed] [Google Scholar]
- Iijima, M. , Yoshimizu, T. , Shimazaki, T. , Tokugawa, K. , Fukumoto, K. , Kurosu, S. , … Chaki, S. (2014). Antidepressant and anxiolytic profiles of newly synthesized arginine vasopressin V1B receptor antagonists: TASP0233278 and TASP0390325. British Journal of Pharmacology, 171(14), 3511–3525. 10.1111/bph.12699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, H. M. , Parrott, N. , Jorga, K. , & Lavé, T. (2006). A novel strategy for physiologically based predictions of human pharmacokinetics. Clinical Pharmacokinetics, 45(5), 511–542. 10.2165/00003088-200645050-00006 [DOI] [PubMed] [Google Scholar]
- Jones, R. D. , Jones, H. M. , Rowland, M. , Gibson, C. R. , Yates, J. W. T. , Chien, J. Y. , … Poulin, P. (2011). PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 2: Comparative assessment of prediction methods of human volume of distribution. Journal of Pharmaceutical Sciences, 100(10), 4074–4089. 10.1002/jps.22553 [DOI] [PubMed] [Google Scholar]
- Kamiya, M. , Sabia, H. D. , Marella, J. , Fava, M. , Nemeroff, C. B. , Umeuchi, H. , … Nishino, I. (2020). Efficacy and safety of TS‐121, a novel vasopressin V1B receptor antagonist, as adjunctive treatment for patients with major depressive disorder: A randomized, double‐blind, placebo‐controlled study. Journal of Psychiatric Research, 128, 43–51. 10.1016/j.jpsychires.2020.05.017 [DOI] [PubMed] [Google Scholar]
- Katz, D. A. , Locke, C. , Greco, N. , Liu, W. , & Tracy, K. A. (2017). Hypothalamic‐pituitary‐adrenal axis and depression symptom effects of an arginine vasopressin type 1B receptor antagonist in a one‐week randomized Phase 1b trial. Brain and Behavior, 7(3), e00628. 10.1002/brb3.628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E. , Howes, O. D. , Kim, B.‐H. , Jeong, J. M. , Lee, J. S. , Jang, I.‐J. , … Kwon, J. S. (2012). Predicting brain occupancy from plasma levels using PET: Superiority of combining pharmacokinetics with pharmacodynamics while modeling the relationship. Journal of Cerebral Blood Flow and Metabolism, 32(4), 759–768. 10.1038/jcbfm.2011.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga, K. , Nagai, Y. , Hanyu, M. , Yoshinaga, M. , Chaki, S. , Ohtake, N. , … Higuchi, M. (2017). High‐contrast PET imaging of vasopressin V1B receptors with a novel Radioligand,11C‐TASP699. Journal of Nuclear Medicine, 58(10), 1652–1658. 10.2967/jnumed.116.188698 [DOI] [PubMed] [Google Scholar]
- Lombardo, F. , Waters, N. J. , Argikar, U. A. , Dennehy, M. K. , Zhan, J. , Gunduz, M. , … Obach, R. S. (2013b). Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 2: Clearance. The Journal of Clinical Pharmacology, 53(2), 178–191. 10.1177/0091270012440282 [DOI] [PubMed] [Google Scholar]
- Lombardo, F. , Waters, N. J. , Argikar, U. A. , Dennehy, M. K. , Zhan, J. , Gunduz, M. , … Obach, R. S. (2013a). Comprehensive assessment of human pharmacokinetic prediction based on in vivo animal pharmacokinetic data, part 1: Volume of distribution at steady state. The Journal of Clinical Pharmacology, 53(2), 167–177. 10.1177/0091270012440281 [DOI] [PubMed] [Google Scholar]
- Naritomi, Y. , Terashita, S. , Kimura, S. , Suzuki, A. , Kagayama, A. , & Sugiyama, Y. (2001). Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metabolism & Disposition, 29(10), 1316–1324. [PubMed] [Google Scholar]
- Obach, R. S. , Baxter, J. G. , Liston, T. E. , Silber, B. M. , Jones, B. C. , Macintyre, F. , & Wastall, P. (1997). The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. Journal of Pharmacology and Experimental Therapeutics, 283(1), 46–58. [PubMed] [Google Scholar]
- Paine, S. W. , Ménochet, K. , Denton, R. , McGinnity, D. F. , & Riley, R. J. (2011). Prediction of human renal clearance from preclinical species for a diverse set of drugs that exhibit both active secretion and net reabsorption. Drug Metabolism & Disposition, 39(6), 1008–1013. 10.1124/dmd.110.037267 [DOI] [PubMed] [Google Scholar]
- Petersson, C. , Papasouliotis, O. , Lecomte, M. , Badolo, L. , & Dolgos, H. (2020). Prediction of volume of distribution in humans: Analysis of eight methods and their application in drug discovery. Xenobiotica, 50(3), 270–279. 10.1080/00498254.2019.1625084 [DOI] [PubMed] [Google Scholar]
- Purba, J. S. , Hoogendijk, W. J. G. , Hofman, M. A. , & Swaab, D. F. (1996). Increased number of vasopressin‐ and oxytocin‐expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Archives of General Psychiatry, 53(2), 137–143. 10.1001/archpsyc.1996.01830020055007 [DOI] [PubMed] [Google Scholar]
- Ring, B. J. , Chien, J. Y. , Adkison, K. K. , Jones, H. M. , Rowland, M. , Jones, R. D. , … Poulin, P. (2011). PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: Comparative assessment of prediction methods of human clearance. Journal of Pharmaceutical Sciences, 100(10), 4090–4110. 10.1002/jps.22552 [DOI] [PubMed] [Google Scholar]
- Rodgers, T. , & Rowland, M. (2007). Mechanistic approaches to volume of distribution predictions: Understanding the processes. Pharmaceutical Research, 24(5), 918–933. 10.1007/s11095-006-9210-3 [DOI] [PubMed] [Google Scholar]
- Saito, M. , Sugimoto, T. , Tahara, A. , & Kawashima, H. (1995). Molecular cloning and characterization of rat V1B vasopressin receptor: Evidence for its expression in extrapituitary tissues. Biochemical and Biophysical Research Communications, 212(3), 751–757. 10.1006/bbrc.1995.2033 [DOI] [PubMed] [Google Scholar]
- Suhara, T. , Chaki, S. , Kimura, H. , Furusawa, M. , Matsumoto, M. , Ogura, H. , & Yamawaki, S. (2017). Strategies for utilizing neuroimaging biomarkers in CNS drug discovery and development: CINP/JSNP working group report. International Journal of Neuropsychopharmacology, 20(4), 285–294. 10.1093/ijnp/pyw111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Hussain, A. , Leal, M. , Mayersohn, M. , & Fluhler, E. (2007). Interspecies prediction of human drug clearance based on scaling data from one or two animal species. Drug Metabolism & Disposition, 35(10), 1886–1893. 10.1124/dmd.107.016188 [DOI] [PubMed] [Google Scholar]
- Tang, H. , & Mayersohn, M. (2005). A novel model for prediction of human drug clearance by allometric scaling. Drug Metabolism & Disposition, 33(9), 1297–1303. 10.1124/dmd.105.004143 [DOI] [PubMed] [Google Scholar]
- Varma, M. V. , Steyn, S. J. , Allerton, C. , & El‐Kattan, A. F. (2015). Predicting clearance mechanism in drug discovery: Extended clearance classification system (ECCS). Pharmaceutical Research, 32(12), 3785–3802. 10.1007/s11095-015-1749-4 [DOI] [PubMed] [Google Scholar]
- Visser, S. A. G. , De Alwis, D. P. , Kerbusch, T. , Stone, J. A. , & Allerheiligen, S. R. B. (2014). Implementation of quantitative and systems pharmacology in large pharma. CPT: Pharmacometrics & Systems Pharmacology, 3(10), e142. 10.1038/psp.2014.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajima, T. , Fukumura, K. , Yano, Y. , & Oguma, T. (2003). Prediction of human pharmacokinetics from animal data and molecular structural parameters using multivariate regression analysis: Volume of distribution at steady state. Journal of Pharmacy and Pharmacology, 55(7), 939–949. 10.1211/0022357021477 [DOI] [PubMed] [Google Scholar]
- Wersinger, S. R. , Ginns, E. I. , O’Carroll, A.‐M. , Lolait, S. J. , & Young III, W. S. (2002). Vasopressin V1b receptor knockout reduces aggressive behavior in male mice. Molecular Psychiatry, 7(9), 975–984. 10.1038/sj.mp.4001195 [DOI] [PubMed] [Google Scholar]
- Wood, F. L. , Houston, J. B. , & Hallifax, D. (2017). Clearance prediction methodology needs fundamental improvement: Trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metabolism & Disposition, 45(11), 1178–1188. 10.1124/dmd.117.077040 [DOI] [PubMed] [Google Scholar]
- Yang, J. , Jamei, M. , Yeo, K. R. , Rostami‐Hodjegan, A. , & Tucker, G. T. (2007). Misuse of the well‐stirred model of hepatic drug clearance. Drug Metabolism & Disposition, 35(3), 501–502. 10.1124/dmd.106.013359 [DOI] [PubMed] [Google Scholar]
- Zou, P. , Yu, Y. , Zheng, N. , Yang, Y. , Paholak, H. J. , Yu, L. X. , & Sun, D. (2012). Applications of human pharmacokinetic prediction in first‐in‐human dose estimation. The AAPS Journal, 14(2), 262–281. 10.1208/s12248-012-9332-y [DOI] [PMC free article] [PubMed] [Google Scholar]