

Abstract
BACKGROUND & AIMS
Primary sclerosing cholangitis (PSC) is a rare bile duct disease strongly associated with inflammatory bowel disease (IBD). Whole‐exome sequencing (WES) has contributed to understanding the molecular basis of very early‐onset IBD, but rare protein‐altering genetic variants have not been identified for early‐onset PSC. We performed WES in patients diagnosed with PSC ≤ 12 years to investigate the contribution of rare genetic variants to early‐onset PSC.
METHODS
In this multicentre study, WES was performed on 87 DNA samples from 29 patient‐parent trios with early‐onset PSC. We selected rare (minor allele frequency < 2%) coding and splice‐site variants that matched recessive (homozygous and compound heterozygous variants) and dominant (de novo) inheritance in the index patients. Variant pathogenicity was predicted by an in‐house developed algorithm (GAVIN), and PSC‐relevant variants were selected using gene expression data and gene function.
RESULTS
In 22 of 29 trios we identified at least 1 possibly pathogenic variant. We prioritized 36 genes, harbouring a total of 54 variants with predicted pathogenic effects. In 18 genes, we identified 36 compound heterozygous variants, whereas in the other 18 genes we identified 18 de novo variants. Twelve of 36 candidate risk genes are known to play a role in transmembrane transport, adaptive and innate immunity, and epithelial barrier function.
CONCLUSIONS
The 36 candidate genes for early‐onset PSC need further verification in other patient cohorts and evaluation of gene function before a causal role can be attributed to its variants.
Keywords: genetic, inflammatory bowel disease, sclerosing cholangitis
Abbreviations
- GWAS
genome‐wide association studies
- HLA
human leukocyte antigen
- IBD
inflammatory bowel disease
- MHC
major histocompatibility complex
- PSC
primary sclerosing cholangitis
- WES
whole‐exome sequencing
Key points.
It is rare to diagnose a child with primary sclerosing cholangitis (PSC) before its 13th birthday. We screened the portion of the DNA that codes for proteins in 29 young PSC children and their biological parents and identified 54 rare genetic variants in 36 genes with an assumed deleterious effect on protein function. Whether these variants play a part in the aetiology of PSC will require further verification.
1. INTRODUCTION
Primary sclerosing cholangitis (PSC) is a rare chronic cholestatic disease characterized by progressive inflammation and obliterative fibrosis of the intra‐ and extrahepatic bile ducts. 1 Disease progression is inevitable in the majority of PSC patients, with the development of biliary cirrhosis and portal hypertension requiring repeated endoscopic procedures. Liver transplantation is the only curative treatment option, but the disease recurs in 20%‐25% of transplanted patients. 2 Cholangiocarcinoma and colorectal cancer are feared complications in PSC and the most common causes of death. 3
There is a strong relation between PSC and inflammatory bowel disease (IBD). Patients who initially present with isolated PSC may go on to develop IBD years later. 4 , 5 In adult‐onset disease, approximately two thirds of patients with PSC have concurrent IBD. 1 The co‐occurrence of PSC and IBD is higher in children than in adults, varying from 76% to 97%. 2 , 6 , 7 The pathogenesis of PSC is largely unknown, but there is a strong genetic component. Genome‐wide association studies (GWAS) in adult‐onset PSC, carried out by the International PSC Study Group, identified 23 risk loci and 9 suggestive findings. 8 Not surprisingly, considering the large clinical overlap, the human leukocyte antigen (HLA) locus is by far the strongest signal in GWAS in both PSC and IBD. 9 , 10
Next to the common variants found by GWAS that so far explain <10% of PSC susceptibility, rare variants with large monogenic effect size may play a role in the onset of PSC. These variants are so rare in allele frequency (many of them private variants) that their genetic signals are hard to detect by GWAS. In contrast, whole‐exome sequencing (WES) in patients with extreme phenotypes, such as early‐onset IBD, has led to the identification of a growing number of rare monogenic disorders presenting with IBD‐like intestinal inflammation. 11 , 12 Additionally, a rare monogenic variant (a loss of function mutation in doublecortin domain containing protein 2 (DCDC2) has recently been identified in a PSC‐like disorder called neonatal sclerosing cholangitis. 13 Although neonatal sclerosing cholangitis is a different entity than PSC, histological similarities of the cholangiocytes of these patients lacking primary cilia suggest that some of the underlying pathogenic mechanisms could be shared between the two diseases. Likewise, we expect that a monogenic or oligogenic inheritance pattern may play a role in a subset of patients with early‐onset PSC as well. We therefore performed WES in a Dutch cohort of patients with early‐onset PSC and their parents to identify rare, but possibly causative genetic variants.
2. METHODS
2.1. Study design, participants and setting
In this multicentre parent‐offspring study, we collected DNA from PSC patients with disease‐onset prior to their 13th birthday and from their biological parents. PSC diagnosis was confirmed by cholangiography (presence of multifocal strictures, focal dilatation or beading of the biliary tree) or liver histology (presence of bile duct damage, onion‐skinned peri‐ductal fibrosis, inflammation, portal oedema or fibrosis, ductopenia, ductular proliferation or cholestasis), or both. Patients with sclerosing cholangitis as a result of secondary causes such as surgery, trauma, cancer or infection were excluded from participation.
Patients were recruited in five tertiary care hospitals in the Netherlands—University Medical Center Groningen (UMCG, a referral paediatric liver transplant centre), Erasmus University Medical Center–Sophia Children's Hospital, VU University Medical Center, Amsterdam University Medical Center–Emma Children's Hospital, University Medical Center Utrecht–Wilhelmina Children's Hospital—and one large general teaching hospital, the Isala Hospital. Eligible patients were those regularly attending the (paediatric) gastroenterology and hepatology clinics as part of standard care. After informed consent was given, the following information was obtained from the local patient records and entered in an online clinical registry using Castor Electronic Data Capture (Amsterdam, the Netherlands): age at PSC diagnosis, findings on cholangiography and/or histology, and follow‐up data on medication use and appearance of biliary cirrhosis, portal hypertension or liver transplantation. Between January 2017 and July 2017, blood was collected from patients and volunteering parents for genomic DNA extraction according to standard protocols.
2.2. Ethical considerations
The Medical Ethical Committee of the UMCG approved the study protocol (METC 2016/289), and secondary approval was obtained from all participating centres. All participating parents and teenagers 12‐19 years old gave informed consent prior study inclusion.
2.3. Whole‐exome sequencing
Libraries were prepared using the Illumina Nextera prep kit and hybrid capture (Illumina Rapid Capture Enrichment – 37 Mb target), and sequencing was performed using the Illumina HiSeq 2500 at the Broad Institute of MIT and Harvard. All raw data underwent quality control steps (https://hub.docker.com/r/broadinstitute/gatk/) without any noticeable negative features to achieve 86.06 million high‐quality reads per sample with 98.85% of reads aligned, on average, resulting in a coverage of 81% of the target region with a read depth of >30X. Sequence reads were aligned to the human reference genome using Novoalign (http://www.novocraft.com). Next, the Genome Analysis Toolkit of the Broad Institute 14 was used for calling single‐nucleotide variant and insertions/deletions.
2.3.1. Variant annotation
Variants were annotated with SNPEff 15 using publicly available data from Ensembl and Refseq, and with GAVIN, an annotation tool with an algorithm that scores the likely pathogenicity of the variants. 16 Additional annotations at the variant, exon and gene level were obtained from the 1000 Genomes Project (http://www.1000genomes.org); National Heart, Lung and Blood Institute GO Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu/EVS); PolyPhen2 17 and the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org). To facilitate further discoveries in this fairly narrow field of research, the VCF files of our study are now publicly available in The Groningen Data Catalogue ( catalogus.helpdesk@umcg.nl ) at (https://groningendatacatalogus.nl/menu/groningendatacatalogue/dataexplorer/details/umcg_collections/aaaac526ldngr6qwh2xc53aaae).
2.3.2. Variant filtering
We used variants with a sequence coverage of ten or greater. We used a Genomics Data Management System (Alissa Interpret –Agilent technologies) to create a filtering tree specifically designed for this study (see Figure 1).
FIGURE 1.
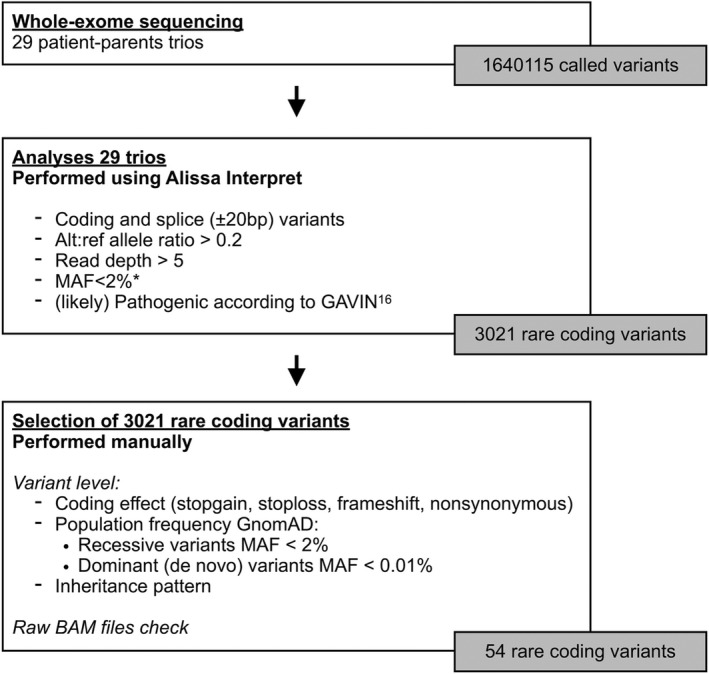
Variant selection. * Population databases used: ExAC, GnoMAD, 1000 Genomes project. Abbreviation: MAF, Minor allele frequency
We performed patient‐parent trio analyses. On the variant‐level, we selected variants matching recessive (homozygous and compound heterozygous variants) and dominant (de novo) inheritance in the index patients. HLA variants were excluded from this analysis as it is unfeasible to distinguish functional polymorphism from random variation in a patient cohort of modest size. Minor allele frequency (MAF) cut‐offs from the GnomAD database (http://gnomad.broadinstitute.org/) were <2% for recessive variants and <0.01% for dominant (de novo) variants. Variants were selected when they fulfilled the following two criteria: (a) deemed to be coding (missense‐ and nonsense mutations, frameshift insertions and deletions) or to have an effect on splicing; and (b) predicted to be (likely) pathogenic according to GAVIN. 16
2.3.3. Variant categorization
We then searched in Genecards (www.genecards.com), Reactome (www.reactome.org) and OMIM (Online Mendelian Inheritance in Man; (www.omim.org) databases for information about gene function and their involvement in diseases. We prioritized the genes into three categories (Box 1).
BOX 1. Prioritization of genes.
| Category 1 | Well‐known gene function connected with PSC or a similar phenotype (i.e., immunological, inflammatory or bile salt homeostasis). |
| Category 2 | Unknown gene function and not (or rarely) reported in literature, and therefore cannot be excluded from having a potential role in the disease pathogenesis, |
| Category 3 | Well‐known gene function not directly associated with the disease phenotype. |
2.3.4. Variant verification and validation
De novo variants were manually checked for coverage and allelic balance in the BAM files. If there was doubt about the validity of the variant, confirmatory Sanger sequencing was performed.
3. RESULTS
3.1. Patient and disease characteristics
A total of 29 patient‐parent trios were enrolled in this study (see Figure 1). Table 1 shows the characteristics of the affected persons, who were diagnosed with PSC at a median age of 10 years (range: 2‐12) and were predominantly male (72%). Twenty‐two of 29 patients (76%) had concurrent IBD, with ulcerative colitis significantly more prevalent than Crohn's disease (73% vs 27%). Other autoimmune disorders included celiac disease (n = 1), idiopathic thrombocytopenic purpura (n = 1) and vitiligo (n = 1). None of the parents was known to have liver disease, but three had IBD. The median time (range) between PSC diagnosis and study enrolment was 4 (0‐30) years. Biliary complications, including cholangitis or bile duct obstruction, had occurred in one patient (3%), and cirrhosis with portal hypertension had developed in nine (31%). Two patients underwent a liver transplantation after a disease duration of 10 and 11 years, respectively, and two other patients are currently listed for liver transplantation. One of the cirrhotic patients had experienced bleeding of oesophageal varices and required a transjugular intrahepatic portosystemic shunt procedure.
TABLE 1.
Characteristics of patients with early‐onset PSC (n = 29)
| Patient identification number | Age at PSC diagnosis (in years) | PSC confirmed by | IBD present? | 1st degree relative with IBD or PSC? | Complicated disease course? |
Candidate gene identified [Prioritized genes printed in bold] |
||
|---|---|---|---|---|---|---|---|---|
| MRCP | ERCP | Liver histology | [No, UC or CD] | Biliary complications [B]; Cirrhosis with portal hypertension [C]; liver transplantation [LT] | ||||
| 1 | 11 |

|

|
|
CD | No | ‐ | ‐ |
| 2 | 10 |
|
|
|
UC | IBD (father) | ‐ | ‐ |
| 3 | 7 |
|
|
|
UC | No | ‐ | ABCB6 |
| 4 | 8 |
|
|
|
No | No | ‐ | ADAM32 , MARCH1, PTX4 , PLXDC1 |
| 5 | 11 |
|
|
|
UC | IBD (mother) | ‐ | ‐ |
| 6 | 12 |
|
|
|
No | No | ‐ | EDC4 |
| 7 | 8 |
|
|
|
No | No | C, LT | PHC2, TNRC18 |
| 8 | 11 |
|
|
|
No | No | ‐ | ‐ |
| 9 | 12 |
|
|
|
No | No | ‐ | HMCN2 |
| 10 | 10 |
|
|
|
CD | No | C | CCN4 |
| 11 | 7 |
|
|
|
No | No | C | ‐ |
| 12 | 9 |
|
|
|
CD | No | ‐ | DNAH6, EHD1 , LAMA2 |
| 13 | 7 |
|
|
|
No | No | C | DNAH7 |
| 14 | 12 |
|
|
|
UC | No | ‐ | KRTAP5‐1, MCM8 |
| 15 | 12 |
|
|
|
CD | No | ‐ | CASKIN2 |
| 16 | 10 |
|
|
|
UC | No | ‐ | PPFIA4 |
| 17 | 5 |
|
|
|
UC | No | ‐ | CDHR2 , DDX47 |
| 18 | 2 |
|
|
|
CD | No | ‐ | CALCRL, USP17L2 |
| 19 | 11 |
|
|
|
UC | No | ‐ | ANKRD36, DNAH11, NOTCH2NLA |
| 20 | 10 |
|
|
|
UC | No | ‐ | ‐ |
| 21 | 11 |
|
|
|
UC | No | C | JMJD1C, DACT1 |
| 22 | 11 |
|
|
|
UC | No | C, LT | TSPYL5 |
| 23 | 10 |
|
|
|
UC | No | ‐ | SLC9B1 |
| 24 | 5 |
|
|
|
UC | No | C, LT | TRDN |
| 25 | 8 |
|
|
|
UC | No | ‐ | FAM234B |
| 26 | 7 |
|
|
|
UC | No | B | SMCHD1, TIRAP |
| 27 | 12 |
|
|
|
UC | IBD (father) | ‐ | ‐ |
| 28 | 12 |
|
|
|
CD | No | C | CHST11 |
| 29 | 6 |
|
|
|
UC | No | C | KIF4A, ZNF30 |
3.2. Patient‐parent trio analyses of WES data
Figure 1 provides an overview of WES variant selection and prioritization. In 22 of 29 trios, we identified at least 1 candidate pathogenic variant. We identified a total of 54 candidate variants with predicted pathogenic effect, annotated to 36 genes (see Table 2). In 15 trios we identified 36 unique compound heterozygous variants, and in 12 trios we identified 18 unique de novo variants. Twelve of 36 candidate risk genes were assigned to prioritization category 1, 11 to category 2 and 13 to category 3.
TABLE 2.
Complete list of 36 candidate risk genes for early‐onset PSC from 22 patient‐parent trios
| Trio | Chr: position:alleles | rs number | Candidate risk gene | Inheritance mode (parental allele) | GnomAD allele count Population frequency | Amino Acid change | CADD‐score | Literature category | Protein function |
|---|---|---|---|---|---|---|---|---|---|
| 3 | 2:220075521:C/T | rs148211042 | ABCB6 | Compound heterozygous (mother) | 217 = 0.00077 | p.R723Q | 35.0 | 1 | Binds heme and porphyrins and functions in their ATP‐dependent uptake into the mitochondria. 20 , 21 Mutations in this gene underlie familial pseudohyperkalemia (OMIM 609 153) and dyschromatosis universalis hereditarian (OMIM 615 402). |
| 2:220078006:C/T | rs145526996 | ABCB6 | Compound heterozygous (father) | 1236 = 0.0043 | p.G588S | 32.0 | |||
| 4 | 8:39044452:G/A | rs745952927 | ADAM32 | Compound heterozygous (father) | 30 = 0 | p.A314T | 12.93 | 1 | This gene encodes a member of the disintegrin family of membrane‐anchored proteins that play a role in diverse biological processes such as brain development, fertilization, tumour development and inflammation. 28 |
| 8:39103683:A/G | rs150114293 | ADAM32 | Compound heterozygous (mother) | 192 = 0.001 | p.G634R | 29.8 | |||
| 4:164775272:C/T | Unknown | MARCH1 | De novo | Unobserved | p.W4* | 38.0 | 1 | Downregulates surface expression of major histocompatibility complex (MHC) class II molecules and other glycoproteins by directing them to the late endosomal/lysosomal compartment. 18 , 19 | |
| 16:1537911:C/T | rs775407157 | PTX4 | De novo | 3 = 0.000012 | p.V63 M | 12.5 | 1 | Pentraxins are part of the humoral arm of innate immunity and behave as functional ancestors of antibodies by mediating agglutination, complement activation and opsonization. PTX4 is a new unique member of the pentraxin superfamily, conserved in evolution. Further studies are needed to define its function. 29 | |
| 17:37234300:G/A | Unknown | PLXDC1 | De novo | Unobserved | p.A351V | 23.8 | 2 | Plays a critical role in endothelial cell capillary morphogenesis. 30 | |
| 6 | 16:67911677:G/A | rs111231628 | EDC4 | Compound heterozygous (mother) | 447 = 0.002 | p.S275G | 10.93 | 1 | Enhancer Of MRNA Decapping. 31 Diseases associated with EDC4 include Human Granulocytic Anaplasmosis and Anteroseptal Myocardial Infarction. |
| 16:67916920:C/T | rs563149577 | EDC4 | Compound heterozygous (father) | 21 = 0 | p.A1230V | 27.7 | |||
| 7 | 1:33794634:G/C | rs376869490 | PHC2 | Compound heterozygous (father) | 41 = 0 | p.I753 M | 23.2 | 2 | Component of a Polycomb group (PcG) multiprotein PRC1‐like complex, a complex class required to maintain the transcriptionally repressive state of many genes, including Hox genes, throughout development. 32 |
|
1:33820146:T/C |
rs142759750 | PHC2 | Compound heterozygous (mother) | 40 = 0 | p.D471N | 18.51 | |||
| 7:5427971:C/A | Unknown | TNRC18 | De novo | 0 | p.G495V | 14.88 | 2 |
Protein Coding gene. Diseases associated with TNRC18 include Atrial Septal Defect and Seckel Syndrome. 33 Lead CpGs at TNRC18 map to active enhancers in kidney cortex and are associated with renal fibrosis.. 34 |
|
| 9 | 9:133047587:C/G | Unknown | HMCN2 | Compound heterozygous (father) | 8 = 0 | p.Q161E | 22.8 | 2 |
Protein Coding gene. Diseases associated with HMCN2 include Posterior Myocardial Infarction. 35 |
| 9:133305895:A/G | rs559374161 | HMCN2 | Compound heterozygous (mother) | 63 = 0 | p.G4293E | 3.171 | |||
| 10 | 8:134225273:G/A | Unknown | CCN4 | De novo | Unobserved | p.C79Y | 27.2 | 3 | Mediates diverse developmental processes, such as control of cell proliferation, adhesion, cell polarity and establishment of cell fates. 36 |
| 12 | 2:84816032:T/A | Unknown | DNAH6 | De novo | Unobserved | p.L855H | 22.8 | 3 | Force generating protein of respiratory cilia. Produces force towards the minus ends of microtubules. Diseases associated include Primary Ciliary Dyskinesia and Anomalous Left Coronary Artery From The Pulmonary Artery. 37 : |
| 11:64645648:G/A | rs747258453 | EHD1 | Compound heterozygous (mother) | Unobserved |
p.F97L |
27.8 | 1 | Important motif in proteins involved in protein‐protein interactions and in intracellular sorting. The protein encoded by this gene is thought to play a role in the endocytosis of IGF1 receptors. 38 | |
| 11:64645841:C/T | Unknown | EHD1 | Compound heterozygous (father) | 2 = 0 | p.= | 15.68 | |||
| 6:129621952:A/T | Unknown | LAMA2 | Compound heterozygous (father) | Unobserved | p.I1037F | 18.85 | 3 | It is thought to mediate the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components. Diseases associated with LAMA2 include Muscular Dystrophy. 39 | |
| 6:129824345:T/C | rs151334775 | LAMA2 | Compound heterozygous (mother) | 22 = 0 | p.P2823S | 25.3 | |||
| 13 | 2:196720589:C/A | rs749776504 | DNAH7 | Compound heterozygous (father) | 6 = 0 | p.R2847S | 12.06 | 3 | Force generating protein of respiratory cilia. Produces force towards the minus ends of microtubules. 40 Diseases associated with DNAH7 include Situs Inversus and Dextrocardia With Situs Inversus. |
| 2:196889160:A/G | rs182086316 | DNAH7 | Compound heterozygous (mother) | 439 = 0.002 | p.R246C | 29.2 | |||
| 14 | 11:1606144: ACAAGAGCCACAGCCCCCCTTGG/. | rs761147271 | KRTAP5‐1 | De novo | Unobserved | p.S105Wfs*115 | ‐ | 3 | In the hair cortex, hair keratin intermediate filaments are embedded in an interfilamentous matrix, consisting of hair keratin‐associated protein (KRTAP), which are essential for the formation of a rigid and resistant hair shaft through their extensive disulphide bond cross‐linking with abundant cysteine residues of hair keratins. 41 : |
| 20:5935281:A/G | rs377435486 | MCM8 | De novo | 14 = 0 | p.E94G | 23.1 | 3 | The protein encoded by this gene is one of the highly conserved mini‐chromosome maintenance proteins (MCM) that are essential for the initiation of eukaryotic genome replication. Diseases associated with MCM8 include Premature Ovarian Failure 10 and Amenorrhea. 42 | |
| 15 | 17:73499287:T/C | rs200947487 | CASKIN2 | Compound heterozygous (mother) | 150 = 0.001 | p.R623Q | 18.14 | 2 | This gene encodes a large protein that contains six ankyrin repeats, as well as a Src homology 3 (SH3) domain and two sterile alpha motif (SAM) domains, which may be involved in protein‐protein interactions. 43 |
| 17:73500515:G/A | rs201521912 | CASKIN2 | Compound heterozygous (father) | 83 = 0 | p.P454L | 17.33 | |||
| 16 | 1:203018043:G/C | rs61756414 | PPFIA4 | Compound heterozygous (mother) | 43 = 0 | ‐ | 15.66 | 2 | May regulate the disassembly of focal adhesions. May localize receptor‐like tyrosine phosphatases type 2A at specific sites on the plasma membrane, possibly regulating their interaction with the extracellular environment and their association with substrates. 44 |
| 1:203036894:G/T | rs528573275 | PPFIA4 | Compound heterozygous (father) | 9 = 0 | p.R1041L | 33 | |||
| 17 | 5:176002840:G/A | rs780769740 | CDHR2 | De novo | 5 = 0.00003 | p.=SPLICE_SITE DONOR | 16.9 | 1 | Intermicrovillar adhesion molecule that controls the packing of microvilli at the apical membrane of epithelial cells. 23 , 24 |
| 12:12974228:C/T | rs780873695 | DDX47 | De novo | 2 = 0 | p.P90S | 24.5 | 1 | Involved in apoptosis. May have a role in rRNA processing and mRNA splicing. Associates with pre‐rRNA precursors. 45 : | |
| 18 | 2:188228104:G/A | Unknown | CALCRL | De novo | Unobserved | p.P209L | 29.6 | 2 | Receptor for calcitonin gene‐related peptide (CGRP) and adrenomedullin. 46 |
| 8:11996026:T/G | rs201734663 | USP17L2 | Compound heterozygous (mother) | 51 = 0 | p.L82I | ‐ | 1 | Has deubiquitinating activity. Also regulates cell proliferation and apoptosis through deubiquitination of SUDS3 a regulator of histone deacetylation. 47 | |
| 8:11996122:C/T | rs199985479 | USP17L2 | Compound heterozygous (father) | 76 = 0 | p.D50N | 0.034 | |||
| 19 | 2:97790321:A/G | rs770051110 | ANKRD36 | Compound heterozygous (mother) | 16 = 0 | p.A240T | 11.81 | 2 | Protein Coding gene. Diseases associated with ANKRD36 include Giant Axonal Neuropathy. 48 : |
| 2:97823866:C/T | rs567234399 | ANKRD36 | Compound heterozygous (father) | 12 = 0 | p.T428 M | 18.47 | |||
| 7:21805095:A/G | rs35865357 | DNAH11 | Compound heterozygous (mother) | 3087 = 0.011 | p.R2997Q | 35 | 3 | Force generating protein of respiratory cilia. Produces force towards the minus ends of microtubules. 49 Diseases associated with DNAH11 include Ciliary Dyskinesia, Primary and Primary Ciliary Dyskinesia. | |
| 7:21901605:G/C | rs751035617 | DNAH11 | Compound heterozygous (father) | Unobserved | p.K3779N | 19.07 | |||
| 1:145273295:C/T | rs782819394 | NOTCH2NLA | De novo | 4 = 0 | p.T50 M | 24.1 | 1 | Human‐specific protein that promotes neural progenitor proliferation and evolutionary expansion of the brain neocortex by regulating the Notch signalling pathway via direct interaction with NOTCH2. 50 NOTCH2 variants are associated with Alagille syndrome, including cholestasis phenotypes (www.omim.org/entry/600275). | |
| 21 | 10:64927837:C/T | rs71508957 | JMJD1C | Compound heterozygous (father) | 1162 = 0.0041 | p.E2531K | 26.5 | 3 | A candidate histone demethylase thought to be a co‐activator for key transcription factors. Plays a role in the DNA‐damage response pathway. 51 |
| 10:64974807:C/G | rs200016210 | JMJD1C | Compound heterozygous (mother) | 132 = 0.00047 | p.D374H | 26.2 | |||
| 14:59104943:C/T | Unknown | DACT1 | Compound heterozygous (mother) | 5 = 0 | p.T8 M | 23.6 | 3 |
Interacts with, and positively regulates, dishevelled‐mediated signalling pathways during development. 52 Associated with Townes‐Brocks syndrome‐2 (OMIM 617 466). |
|
| 14:59113376:T/C | rs200977826 | DACT1 | Compound heterozygous (father) | 166 = 0.001 | p.W679R | 25.5 | |||
| 22 | 8:98289238:T/C | rs151015596 | TSPYL5 | Compound heterozygous (father) | 1053 = 0.004 | p.S279G | 17.38 | 3 | Involved in modulation of cell growth and cellular response to gamma radiation probably via regulation of the Akt signalling pathway. Involved in regulation of p53/TP53. 53 : |
| 8:98290012:A/C | rs79679520 | TSPYL5 | Compound heterozygous (mother) | 485 = 0.003 | p.A21S | 23.3 | |||
| 23 | 4:103832611:G/A | rs75599926 | SLC9B1 | De novo | 2 = 0.000011 | p.R305* | 36. | 1 | Sodium/hydrogen exchanger and transmembrane protein. 54 |
| 24 | 6:123786033:./A | rs201431159 | TRDN | De novo | Unobserved | p.S297Ffs*32 | n.a. | 2 | Contributes to regulation of luminal Ca2 + release via the sarcoplasmic reticulum calcium release channels. 55 Associated with ventricular tachycardia (OMIM 615 441) |
| 25 | 12:13219604:T/C | rs367547952 | FAM234B | Compound heterozygous (mother) | 23 = 0 | p.R295W | 35 | 2 | Protein Coding gene. Diseases associated with FAM234B include Temtamy Syndrome and Autosomal Dominant Non‐Syndromic Intellectual Disability. 56 : |
| 12:13221607:T/A | rs140271825 | FAM234B | Compound heterozygous (father) | 150 = 0.001 | p.F444I | 25.6 | |||
| 26 | 18:2722603:G/A | Unknown | SMCHD1 | De novo | Unobserved | p.D849N | 31 | 3 | Involved in DNA management and plays an essential role in X chromosome inactivation. 57 |
| 11:126162948:G/A | rs185114125 | TIRAP | De novo | 49 = 0 | p.R215H | 23.6 | 1 | Adapter involved in TLR2 and TLR4 signalling pathways in the innate immune response. Acts via IRAK2 and TRAF‐6, leading to the activation of NF‐kappa‐B, MAPK1, MAPK3 and JNK, and resulting in cytokine secretion and the inflammatory response. 58 | |
| 28 | 12:105151159:G/A | Unknown | CHST11 | De novo | Unobserved | p.G213S | 32. | 3 | Catalyses the transfer of sulphate in chondroitin. 59 Diseases associated with CHST11 include Mucinoses and Costello Syndrome (OMIM 618 167). |
| 29 | X:69623814:A/G | Unknown | KIF4A | De novo | Unobserved | p.N907S | 10.11 | 3 | Iron‐sulphur (Fe‐S) cluster binding motor protein that has a role in chromosome segregation during mitosis. 60 |
| 19:35434411:C/T | rs367651957 | ZNF30 | Compound heterozygous (mother) | 43 = 0 | p.C182R | 23.2 | 2 | May be involved in transcriptional regulation. 61 Diseases associated with ZNF30 include Chromosome 19Q13.11 Deletion Syndrome and Brugada Syndrome. | |
| 19:35435632:C/T | rs140215760 | ZNF30 | Compound heterozygous (father) | 542 = 0.002 | p.R589W | 25.8 |
Abbreviations: CADD‐score, Combined Annotation‐Dependent Depletion scoreChr, Chromosome.
The category 1 genes are depicted in Figure 2. The genes SLC9B1 and ABCB6 encode for membrane transporter proteins and relate to the ‘Transport of glucose and other sugars, bile salts and organic acids, metal ions and amine compounds’ pathway (www.pathcards.genecards.org). The gene NOTCH2NLA regulates the ‘Notch Signaling’ pathway (www.pathcards.genecards.org) and NOTCH2 variants are related to syndromes with cholestatic phenotypes (www.omim.org/entry/600275). The genes MARCH1, PTX4, TIRAP, ADAM32, DDX47, USP17L2, EHD1 and EDC4 are associated with defects of the immune system. The CDHR2 gene is involved in the epithelial barrier function.
FIGURE 2.
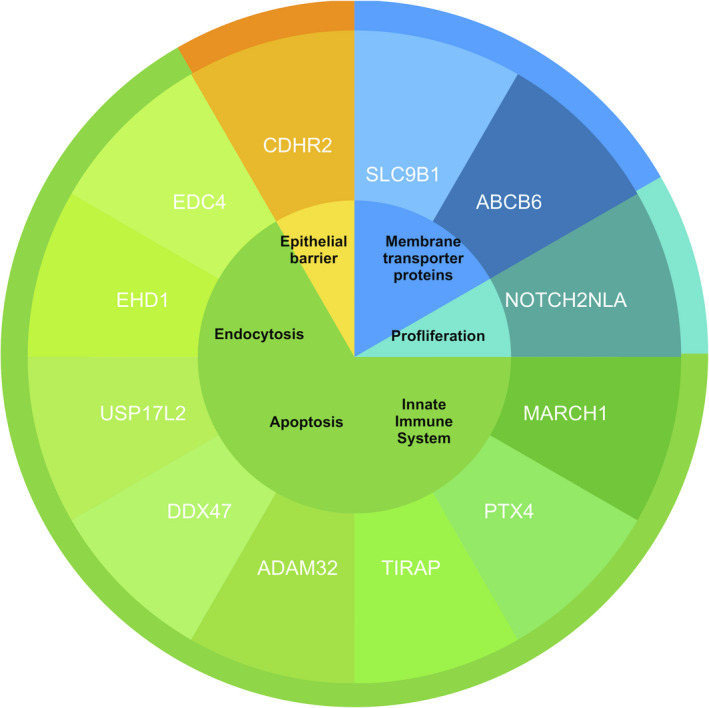
Function of the 12 prioritized candidate risk genes for early‐onset PSC. The core indicates the protein functions and the inner ring shows the 12 candidate risk genes. Information on genetic functions comes from multiple databases including Genecards (www.genecards.com), Reactome (www.reactome.org) and OMIM (Online Mendelian Inheritance in Man; www.omim.org)
4. DISCUSSION
4.1. Key results
In this study we examined the exomes of patients with early‐onset PSC and their biological parents. With this trio‐analysis approach we identified 54 rare variants with predicted large effects on protein function in 36 genes. We prioritized 12 candidate risk genes that are most likely to contribute to the development of PSC in patients with early‐onset disease based on their presumed role in (auto) immunity pathways, membrane transport (including bile salt homeostasis) and epithelial barrier functioning.
4.2. Prioritized genes and their possible role in PSC pathogenesis
Previous GWAS studies on the genetics of PSC suggested the autoimmune origin of the disease, with strong associations with the genes encoding for the HLA complex on chromosome 6, along with several susceptibility genes that are critically involved in T‐cell function. 1 , 8 Liver tissue from biopsied patients with PSC showed mainly T cells and, to a lesser degree, macrophages and neutrophils in the infiltrates. 1 We found a de novo stop‐gain variant located at the very beginning of the MARCH1‐gene (transcript position 4) in patient #4, a boy of 8 with PSC–autoimmune hepatitis overlap syndrome, also called autoimmune sclerosing cholangitis. Such an early stop‐gain results in loss of the corresponding protein from this allele and is predicted to be highly pathogenic. As the inheritance mode of this gene is still unknown, we cannot predict the biological effect of this variant. Functional studies of MARCH1 suggested that this gene mediates the immunosuppressive effect of the anti‐inflammatory cytokine interleukin 10 (IL10) on antigen presentation in monocytes. 18 , 19 Knockdown of MARCH1 strongly inhibited IL‐10–dependent down‐regulation of cell surface HLA‐DR. 19 The exact contribution of the MARCH1 gene regulation to immunopathology remains to be explored.
We identified variants in genes ABCB6 and SLC9B1, which are (according to www.pathcards.genecards.org) related to the ‘transport of glucose and other sugars, bile salts and organic acids, metal ions and amine compounds’ pathway. Patient #3, a girl with both PSC and IBD diagnosed at age 7, had compound heterozygous ABCB6 variants. The gene encodes a member of the ATP‐binding cassette (ABC) transporter superfamily and is known to bind heme and porphyrins and function in their ATP‐dependent uptake in the mitochondria. 20 , 21 Both variants are predicted to be damaging and to disrupt the highly conserved ABC transport and ABC transmembrane regions of the protein, respectively. However, whether this genetic variant is truly involved in alteration of bile salt homeostasis has to be investigated further.
Another possible cause of development of PSC is the epithelial cell lining of the bile ducts. Bile salts are toxic in high concentrations. 22 Damage of the epithelial border may result in leakage of bile and could be an important driver of toxicity. In patient #17 we identified one de novo intronic variant positioned exactly at a splice‐donor consensus sequence site in CDHR2, predicted to disrupt splicing of the transcript and causing loss of function. CDHR2 plays a central role in the integrity of epithelial tissues such as the bile duct epithelium. 23 , 24
Uncovering the functional consequences of the newly discovered candidate variants, in particular for the genes without a known function, and the pathways involved in the onset of PSC will require detailed functional experiments involving different functional read‐outs, given the broad nature of the identified genes, and further verification of our findings in independent cohorts.
4.3. From theory to definitive proof
This is the first explorative study of high‐impact rare coding variants in young patients with PSC which could help to identify causative genes. To prove that our set of candidate genes contains a causative gene, replication in an independent patient cohort is a first step. One method to replicate low‐frequency and rare variants in complex immune diseases is by targeted genotyping using the Illumina exome chip array (HumanExome BeadChips, Illumina, Inc, San Diego, CA). This next‐generation genotyping array includes several regions on the genome that are known to play a role in immune mediated disease based on the results from existing re‐sequencing datasets. In a first attempt to further study the contribution of rare variants in PSC, our research group is in the process of replication genotyping of identified variants in a large international exome array case‐control study and identified several genetic loci containing rare variants that are associated with PSC. Adding our new candidate risk genes and variants to such arrays may contribute to efficient replication.
Furthermore, replication in other early‐onset PSC cohorts is needed. In 2013, the BROAD Institute partnered with researchers worldwide to develop a collaborative exome sequencing network in IBD, and this initiative is currently ongoing. A similar project is now up and running in PSC with the aim to meta‐analyse the exomes of more than 1000 patients of European ancestry. This will enable modelling of the combined contribution of polygenic and oligogenic variants to the inheritance of PSC.
Convincing evidence to prove that a gene is causal in a disease is identifying a similar genetic variant in the same gene in another patient. In monogenic diseases, a freely accessible Web‐based tool called GeneMatcher (www.genematcher.org) is used to identify additional individuals with rare phenotypes who have variants in the same candidate gene. 25 A similar tool does not yet exist for complex genetic diseases but data sharing such as GeneMatcher might be useful here as well.
4.4. Implications for clinical practice
At this moment, there is no curative therapy available for PSC. Quality‐of‐life undermining complications may eventually justify a liver transplantation. The ultimate goal of investigating the genetic basis of this disease is to help reveal mechanisms of disease pathology and guide the selection of new targets for drug discovery. Each genetic risk locus can be seen as a potential drug target and the starting point of new treatment opportunities. This has successfully been demonstrated in the field of IBD, in which small‐molecule inhibitors were used to recapitulate the anti‐inflammatory function of CARD9 variants associated with protection from IBD. 26 Scientists now recognize that genes with evidence for causality in disease are more promising for identification of new drug targets, and this has led to an increased interest in disease‐associated genes with variants that reduce gene function, such as nonsense, frameshift or essential splice‐site variants. 27 Our study is one of the first steps on this road to drug discovery for patients with PSC.
5. CONCLUSION
We identified 54 rare protein‐altering genetic variants in 36 genes, of which 12 were prioritized as candidate risk genes. The functional consequences of these variants and their causality to PSC will need to be studied in replication cohorts and with functional testing.
CONFLICTS OF INTEREST
None.
AUTHOR CONTRIBUTIONS
SMH, RKW, PFvR and CCvD contributed to study concept and design, interpretation of data and drafting of the manuscript. BAEdK, MEJ, BGPK, TdM, VMW and ON contributed to the collection of patient data. SMH obtained the data. Whole‐exome sequencing and quality control were performed by MJD, CS, RJX and MAR. SMH and CCvD analysed the data. HJV, RB, DBHJ, NF and MV contributed important intellectual content. PFvR and CCvD had full responsibility for the study.
ETHICS APPROVAL STATEMENT
The Medical Ethical Committee of the UMCG approved the study protocol (METC 2016/289), and secondary approval was obtained from all participating centres.
PATIENT CONSENT STATEMENT
All participating parents and teenagers 12‐19 years old gave informed consent prior study inclusion.
CONFERENCE PRESENTATION
This study was selected for a lecture presentation during the Digestive Disease Week ® in May 2019 in San Diego.
ACKNOWLEDGEMENTS
We thank Krista van Dijk‐Bos and Kristin Abbott for assistance with variant filtering and interpretation. We thank Kate Mc Intyre for editorial assistance.
Haisma S‐M, Weersma RK, Joosse ME, et al. Exome sequencing in patient‐parent trios suggests new candidate genes for early‐onset primary sclerosing cholangitis. Liver Int. 2021;41:1044–1057. 10.1111/liv.14831
Patrick F van Rheenen, Cleo C van Diemen are shared last authors.
Handling Editor: Ana Lleo
FUNDING INFORMATION
This work was supported by the European Crohn's and Colitis Organization (ECCO) [grant number Grant_2017/ECCO/PatrickvanRheenen].
REFERENCES
- 1. Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis ‐ a comprehensive review. J Hepatol. 2017;67(6):1298‐1323. [DOI] [PubMed] [Google Scholar]
- 2. Deneau MR, El‐Matary W, Valentino PL, et al. The natural history of primary sclerosing cholangitis in 781 children: A multicenter, international collaboration. Hepatol. 2017;66(2):518‐527. [DOI] [PubMed] [Google Scholar]
- 3. Boonstra K, Weersma RK, van Erpecum KJ, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58(6):2045‐2055. [DOI] [PubMed] [Google Scholar]
- 4. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237‐267. [DOI] [PubMed] [Google Scholar]
- 5. Saich R, Chapman R. Primary sclerosing cholangitis, autoimmune hepatitis and overlap syndromes in inflammatory bowel disease. World J Gastroenterol. 2008;14(3):331‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary sclerosing cholangitis, autoimmune hepatitis, and overlap in Utah children: epidemiology and natural history. Hepatology. 2013;58(4):1392‐1400. [DOI] [PubMed] [Google Scholar]
- 7. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single‐center longitudinal cohort study. J Pediatr Gastroenterol Nutr. 2016;63(6):603‐609. [DOI] [PubMed] [Google Scholar]
- 8. Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol. 2017;14(5):279‐295. [DOI] [PubMed] [Google Scholar]
- 9. Ji SG, Juran BD, Mucha S, et al. Genome‐wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. 2017;49(2):269‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease‐specific patterns at shared loci. Nat Genet. 2016;48(5):510‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ouahed J, Spencer E, Kotlarz D, et al. Very early onset inflammatory bowel disease: a clinical approach with a focus on the role of genetics and underlying immune deficiencies. Inflamm Bowel Diseases. 2020;26(6):820‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelsen JR, Baldassano RN. The role of monogenic disease in children with very early onset inflammatory bowel disease. Curr Opin Pediatr. 2017;29(5):566‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grammatikopoulos T, Sambrotta M, Strautnieks S, et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol. 2016;65(6):1179‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cingolani P, Platts A, le Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 2012;6(2):80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Velde KJ, de Boer EN, van Diemen CC, et al. GAVIN: Gene‐aware variant INterpretation for medical sequencing. Genome Biol. 2017;18(1):6‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013. 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thibodeau J, Bourgeois‐Daigneault MC, Huppe G, et al. Interleukin‐10‐induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol. 2008;38(5):1225‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Gassart A, Camosseto V, Thibodeau J, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation‐dependent MARCH I down‐regulation. Proc Natl Acad Sci U S A. 2008;105(9):3491‐3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Helias V, Saison C, Ballif BA, et al. ABCB6 is dispensable for erythropoiesis and specifies the new blood group system Langereis. Nat Genet. 2012;44(2):170‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitsuhashi N, Miki T, Senbongi H, et al. MTABC3, a novel mitochondrial ATP‐binding cassette protein involved in iron homeostasis. J Biol Chem. 2000;275(23):17536‐17540. [DOI] [PubMed] [Google Scholar]
- 22. Shah A, Macdonald GA, Morrison M, Holtmann G. Targeting the gut microbiome as a treatment for primary sclerosing cholangitis: a conceptional framework. Am J Gastroenterol. 2020;115(6):814‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pinette JA, Mao S, Millis BA, Krystofiak ES, Faust JJ, Tyska MJ. Brush border protocadherin CDHR2 promotes the elongation and maximized packing of microvilli in vivo. Mol Biol Cell. 2019;30(1):108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crawley SW, Shifrin DA, Grega‐Larson NE, et al. Intestinal brush border assembly driven by protocadherin‐based intermicrovillar adhesion. Cell. 2014;157(2):433‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leshchiner ES, Rush JS, Durney MA, et al. Small‐molecule inhibitors directly target CARD9 and mimic its protective variant in inflammatory bowel disease. Proc Natl Acad Sci U S A. 2017;114(43):11392‐11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vallabh ME, Karczewski Konrad J, Martin Hilary C, et al. Evaluating potential drug targets through human loss‐of‐ function genetic variation. Nature. 2020;581:459‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi I, Woo JM, Hong S, Jung YK, Kim DH, Cho C. Identification and characterization of ADAM32 with testis‐predominant gene expression. Gene. 2003;30(304):151‐162. [DOI] [PubMed] [Google Scholar]
- 29. Martinez de la Torre Y, Fabbri M, Jaillon S, et al. Evolution of the pentraxin family: the new entry PTX4. J Immunol. 2010;184(9):5055‐5064. [DOI] [PubMed] [Google Scholar]
- 30. St Croix B, Rago C, Velculescu V, et al. Genes expressed in human tumor endothelium. Science. 2000;289(5482):1197‐1202. [DOI] [PubMed] [Google Scholar]
- 31. D'Lima NG, Ma J, Winkler L, et al. A human microprotein that interacts with the mRNA decapping complex. Nat Chem Biol. 2017;13(2):174‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levine SS, Weiss A, Erdjument‐Bromage H, Shao Z, Tempst P, Kingston RE. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol. 2002;22(17):6070‐6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiao M, Li J, Zhang Q, et al. Identification of four potential biomarkers associated with coronary artery disease in non‐diabetic patients by gene co‐expression network analysis. Front Genet. 2020;24(11):542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chu AY, Tin A, Schlosser P, et al. Epigenome‐wide association studies identify DNA methylation associated with kidney function. Nat Commun. 2017;8(1):1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tereshchenko LG, Sotoodehnia N, Sitlani CM, et al. Genome‐Wide Associations of Global Electrical Heterogeneity ECG Phenotype: The ARIC (Atherosclerosis Risk in Communities) Study and CHS (Cardiovascular Health Study). J Am Heart Assoc. 2018;7(8):e008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP‐1 is a Wnt‐1‐ and beta‐catenin‐responsive oncogene. Genes Dev. 2000;14(5):585‐595. [PMC free article] [PubMed] [Google Scholar]
- 37. Oltean A, Schaffer AJ, Bayly PV, Brody SL. Quantifying ciliary dynamics during assembly reveals stepwise waveform maturation in airway cells. Am J Respir Cell Mol Biol. 2018;59(4):511‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rotem‐Yehudar R, Galperin E, Horowitz M. Association of insulin‐like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem. 2001;276(35):33054‐33060. [DOI] [PubMed] [Google Scholar]
- 39. Tezak Z, Prandini P, Boscaro M, et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin alpha 2 (LAMA2) deficiency. Hum Mutat. 2003;21(2):103‐111. [DOI] [PubMed] [Google Scholar]
- 40. Zhang YJ, O'Neal WK, Randell SH, et al. Identification of dynein heavy chain 7 as an inner arm component of human cilia that is synthesized but not assembled in a case of primary ciliary dyskinesia. J Biol Chem. 2002;277(20):17906‐17915. [DOI] [PubMed] [Google Scholar]
- 41. Yahagi S, Shibuya K, Obayashi I, et al. Identification of two novel clusters of ultrahigh‐sulfur keratin‐associated protein genes on human chromosome 11. Biochem Biophys Res Commun. 2004;318(3):655‐664. [DOI] [PubMed] [Google Scholar]
- 42. Volkening M, Hoffmann I. Involvement of human MCM8 in prereplication complex assembly by recruiting hcdc6 to chromatin. Mol Cell Biol. 2005;25(4):1560‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabuchi K, Biederer T, Butz S, Sudhof TC. CASK participates in alternative tripartite complexes in which Mint 1 competes for binding with caskin 1, a novel CASK‐binding protein. J Neurosci. 2002;22(11):4264‐4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Katoh M, Katoh M. Identification and characterization of human PPFIA4 gene in silico. Int J Mol Med. 2003;12(6):1009‐1014. [PubMed] [Google Scholar]
- 45. Lee JH, Rho SB, Chun T. GABAA receptor‐associated protein (GABARAP) induces apoptosis by interacting with DEAD (Asp‐Glu‐Ala‐Asp/His) box polypeptide 47 (DDX 47). Biotechnol Lett. 2005;27(9):623‐628. [DOI] [PubMed] [Google Scholar]
- 46. McLatchie LM, Fraser NJ, Main MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin‐receptor‐like receptor. Nature. 1998;393(6683):333‐339. [DOI] [PubMed] [Google Scholar]
- 47. McFarlane C, Kelvin AA, de la Vega M, et al. The deubiquitinating enzyme USP17 is highly expressed in tumor biopsies, is cell cycle regulated, and is required for G1‐S progression. Cancer Res. 2010;70(8):3329‐3339. [DOI] [PubMed] [Google Scholar]
- 48. Johnson‐Kerner BL, Garcia Diaz A, Ekins S, Wichterle H. Kelch Domain of Gigaxonin Interacts with Intermediate Filament Proteins Affected in Giant Axonal Neuropathy. PLoS One. 2015;10(10):e0140157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bartoloni L, Blouin JL, Pan Y, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A. 2002;99(16):10282‐10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fiddes IT, Lodewijk GA, Mooring M, et al. Human‐specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell. 2018;173(6):1356‐1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saez MA, Fernandez‐Rodriguez J, Moutinho C, et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet Med. 2016;18(4):378‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shi Y, Ding Y, Lei YP, et al. Identification of novel rare mutations of DACT1 in human neural tube defects. Hum Mutat. 2012;33(10):1450‐1455. [DOI] [PubMed] [Google Scholar]
- 53. Epping MT, Meijer LA, Krijgsman O, Bos JL, Pandolfi PP, Bernards R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat Cell Biol. 2011;13(1):102‐108. [DOI] [PubMed] [Google Scholar]
- 54. Holmes RS, Spradling‐Reeves KD, Cox LA. Evolution of vertebrate solute carrier family 9B genes and Proteins (SLC9B): Evidence for a marsupial origin for testis specific SLC9B1 from an ancestral vertebrate SLC9B2 gene. J Phylogenetics Evol Biol. 2016;4(3): 10.4172/2329-9002.1000167. Epub 2016 Jun 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Oddoux S, Brocard J, Schweitzer A, et al. Triadin deletion induces impaired skeletal muscle function. J Biol Chem. 2009;284(50):34918‐34929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagase T, Kikuno R, Ishikawa K, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7(2):143‐150. [DOI] [PubMed] [Google Scholar]
- 57. Blewitt ME, Gendrel AV, Pang Z, et al. SmcHD1, containing a structural‐maintenance‐of‐chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;40(5):663‐669. [DOI] [PubMed] [Google Scholar]
- 58. Nagpal K, Plantinga TS, Wong J, et al. A TIR domain variant of MyD88 adapter‐like (Mal)/TIRAP results in loss of MyD88 binding and reduced TLR2/TLR4 signaling. J Biol Chem. 2009;284(38):25742‐25748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamauchi S, Mita S, Matsubara T, et al. Molecular cloning and expression of chondroitin 4‐sulfotransferase. J Biol Chem. 2000;275(12):8975‐8981. [DOI] [PubMed] [Google Scholar]
- 60. Ben‐Shimon L, Paul VD, David‐Kadoch G, et al. Fe‐S cluster coordination of the chromokinesin KIF4A alters its subcellular localization during mitosis. J Cell Sci. 2018;131(12):jcs211433. [DOI] [PubMed] [Google Scholar]
- 61. Thiesen HJ. Multiple genes encoding zinc finger domains are expressed in human T cells. New Biol. 1990;2(4):363‐374.PMID: 2288909. [PubMed] [Google Scholar]