

Abstract
Mitochondria share attributes of vesicular transport with their bacterial ancestors given their ability to form mitochondrial‐derived vesicles (MDVs). MDVs are involved in mitochondrial quality control and their formation is enhanced with stress and may, therefore, play a potential role in mitochondrial‐cellular communication. However, MDV proteomic cargo has remained mostly undefined. In this study, we strategically used an in vitro MDV budding/reconstitution assay on cardiac mitochondria, followed by graded oxidative stress, to identify and characterize the MDV proteome. Our results confirmed previously identified cardiac MDV markers, while also revealing a complete map of the MDV proteome, paving the way to a better understanding of the role of MDVs. The oxidative stress vulnerability of proteins directed the cargo loading of MDVs, which was enhanced by antimycin A (Ant‐A). Among OXPHOS complexes, complexes III and V were found to be Ant‐A‐sensitive. Proteins from metabolic pathways such as the TCA cycle and fatty acid metabolism, along with Fe‐S cluster, antioxidant response proteins, and autophagy were also found to be Ant‐A sensitive. Intriguingly, proteins containing hyper‐reactive cysteine residues, metabolic redox switches, including professional redox enzymes and those that mediate iron metabolism, were found to be components of MDV cargo with Ant‐A sensitivity. Last, we revealed a possible contribution of MDVs to the formation of extracellular vesicles, which may indicate mitochondrial stress. In conclusion, our study provides an MDV proteomics signature that delineates MDV cargo selectivity and hints at the potential for MDVs and their novel protein cargo to serve as vital biomarkers during mitochondrial stress and related pathologies.
Keywords: hyper‐reactive cysteine residues, mitochondrial stress, mitochondrial‐derived vesicle proteome, mitochondrial quality control, mitochondrial iron transport
Abbreviations
- Ant‐A
antimycin A
- ETF
electron transferring flavoprotein system
- FDR
false discovery rate
- FBS
fetal bovine serum
- FC
fold change
- GO
gene ontology
- HRCR
hyper‐reactive cysteine residues
- IMPI
integrated mitochondrial protein index
- MDV
mitochondrial‐derived vesicle
- mQC
mitochondrial quality control
- SCS
shortest common superstring
- TEM
transmission electron microscopy
1. INTRODUCTION
Vesicular transport is an ancient and highly conserved means of communication. 1 , 2 In bacteria, where this process has been extensively studied, vesicles are commonly used to exchange signaling factors for the regulation of gene expression and the coordination of collective behaviors in a population, a process known as quorum sensing. 1 Furthermore, bacteria use vesicles for the transport of proteins with numerous specific tasks, including host invasion, elimination of competing strains, and the formation of protective biofilms to ensure their survival and growth in adverse environments. 2 , 3 , 4
It is common knowledge that as endosymbiotic organelles, mitochondria share several similarities with their bacterial ancestors, including their circular plasmid‐like DNA, their double‐membrane system, and their unique translation machinery. Interestingly, emerging evidence indicates that the capacity for vesicular transport has also been conserved through evolution, as mitochondria were recently shown to generate Mitochondrial‐Derived Vesicles (MDVs) of 70‐150 nm in diameter, composed of one or two membranes, and enriched with specific mitochondrial protein markers. 5 , 6 , 7 , 8 , 9
Two different fates have been identified for MDVs. The first involves the transport of cargo to peroxisomes, 5 , 6 , 9 an organelle that shares several functions with mitochondria, including bile acid synthesis, and fatty acid metabolism. The second fate involves the delivery of mitochondrial proteins to late endosomes/multivesicular bodies, where mitochondrial cargoes entering this compartment are destined for degradation in lysosomes, 5 implying a role for MDVs in mitochondrial quality control (mQC). 10 , 11 In addition, under certain circumstances, multivesicular bodies can also be routed to the cell surface, where their limiting membranes can fuse with the plasma membrane, 12 suggesting that MDVs could also lead to the secretion of mitochondrial proteins in extracellular vesicles. 11
Studies in cell culture models indicate that MDVs destined to late endosomes/multivesicular bodies are produced at a significant rate even under baseline conditions, particularly when cells are grown in galactose‐containing media, which forces cellular dependence on oxidative metabolism, and causes wear and tear of mitochondrial proteins. 5 , 7 In addition, the production of these vesicles increases within minutes following the induction of mitochondrial oxidative stress, compared to hours‐days for mitophagy. 5 , 7 , 13 In vivo evidence supporting the importance of this process is also emerging. For instance, recent studies indicate that cardiac mitochondria constitutively bud MDVs under normal healthy conditions, while their rate of production increases by more than twofold in response to doxorubicin, a chemotherapeutic drug with well known cardiotoxic side effects. 7 Together, these observations have led to the suggestion that MDVs likely represent an essential component of the mitochondrial housekeeping process, the first line of defense against proteotoxic oxidative insults, 10 , 11 and may act as a potential source of biomarkers reflecting the presence of mitochondrial distress. However, because it is impossible to purify MDVs from tissue or cultured cells, our knowledge of cargoes incorporated into these vesicles is currently limited to a handful of proteins that were previously identified using immunofluorescence approaches. 11
In the present study, we, therefore, took advantage of an in vitro budding/reconstitution assay to characterize the proteome of MDVs generated by isolated cardiac mitochondria under oxidative stress. Our proteomics data validate previously established MDV markers and identifies novel cargoes. We provide evidence that under oxidative stress, MDVs become enriched with several metabolic enzymes with properties that make them vulnerable to dysfunction/damage. Furthermore, we report that a number of proteins released into MDVs are also found in extracellular vesicles and could be reflective of mitochondrial stress.
2. MATERIALS AND METHODS
2.1. Preparation of cytosolic and mitochondrial fractions
All experiments on animals were approved by the University of Ottawa Institutional Animal Care Committee and conducted according to the directives of the Canadian Council on Animal Care. Rats were euthanized by thoracotomy following ketamine‐xylazine anesthesia. Heart was excised and the atrium was discarded. Heart samples from four rats were pooled in order to obtain enough mitochondria and cytosolic fraction to perform several MDV budding/reconstitution assays in parallel. Experiments were repeated for three days, the pool of samples from four rats on each day being considered as a biological replicate. Tissue and fractions were kept cold on ice, and centrifugation was performed at 4°C by precooled rotors. Heart tissue was washed in cold PBS to remove blood, blotted to remove liquid, and weighed. Tissue was minced into small pieces and homogenized using a polytron (three times 5 seconds each at setting 3) in 3 mL/g wet weight of cold isolation buffer (220 mM mannitol, 20 mM HEPES, 68 mM sucrose, 76 mM potassium chloride, 4 mM potassium acetate, 2 mM magnesium chloride, and 0.5 mM EGTA at pH 7.4) supplemented with protease inhibitors (30 µg/mL of leupeptin, 5 µg/mL of aprotinin, 100 µM phenylmethylsulfonyl fluoride, and 1 mM benzamidine). Samples were then centrifuged for 10 min at 1000 g in order to pellet debris and the nuclear fraction. The supernatant was recovered in a new tube and subsequently centrifuged for 10 min at 10 000 g in order to pellet mitochondria. The supernatant of this spin was set aside for the preparation of the cytosolic fraction, while the mitochondrial pellet was resuspended in 30 mL of isolation buffer with protease inhibitors and spun again at 10 000 g to wash‐out microsomes and other non‐mitochondrial contaminants. The final mitochondrial pellet (mitochondrial fraction) obtained was then resuspended in 200 µL of isolation buffer with protease inhibitors and stored on ice until used. For the preparation of the cytosolic fraction, the supernatant from the first 10 000 g centrifugation step was ultra‐centrifuged (Optima MAX, Beckman Coulter) at 200 000 g for 90 minutes using a TLA 100.3 rotor to pellet ER vesicles and other subcellular components. The supernatant of this spin was then aliquoted and stored at −80°C until use.
2.2. In vitro MDV budding/reconstitution assay
Protein concentrations in mitochondrial and cytosolic fractions were estimated using the BCA assay. The mitochondrial fraction was washed twice in the reaction buffer (220 mM mannitol, 20 mM HEPES, 68 mM sucrose, 76 mM potassium chloride, 4 mM potassium acetate, 2 mM magnesium chloride, and 0.5 µM EGTA at pH 7.4) with no protease inhibitors by centrifuging at 10 000 g for 10 minutes in a microfuge. MDV budding assays were performed in the reaction buffer according to Soubannier et al 6 using 6 mg/mL of mitochondria and 1.5 mg/mL of the cytosolic fraction in an ATP regenerating mixture (1 mM ATP, 5 mM succinate, 80 µM ADP, and 2 mM dipotassium hydrogen phosphate at pH 7.4), in the presence or absence of 50 µM antimycin A. After 30 min, the suspension was centrifuged at 10 000 g for 10 min to pellet the mitochondria, while the supernatant containing the crude MDV fraction was recovered for further purification.
2.3. Purification of MDV fractions
The crude MDV fraction was purified by flotation using a discontinuous sucrose gradient. For this, the crude fraction was diluted twofold in an 80% w/w sucrose solution and loaded at the bottom of a 14 × 89 mm Ultra‐Clear ultracentrifuge tube (Beckman Coulter). The sucrose gradients were then established by sequentially adding equal volumes of 35%, 30%, 25%, 20%, 15%, and 10% sucrose solutions to each tube. Sucrose concentration was carefully measured using a hand‐held refractometer, and marks were made on the tube to indicate the location of the various sucrose interfaces. Fractionation was performed overnight (minimum of 12 hours) using a swinging bucket rotor (SW 41 Ti) in an ultracentrifuge (Optima L‐90K, Beckman Coulter) at 151 000 g at 4°C. Following centrifugation, equal volumes (500 µL) were sequentially recovered with a pipet starting from the top of the tube (ie, lower to higher sucrose %) and processed for western blotting. Only the fractions found to be enriched with markers of MDVs were submitted for proteomics analysis.
2.4. Transmission electron microscopy
Following the MDV budding assay, diluted samples of the mitochondrial suspension or the purified MDV fractions were fixed with equal volumes of 3.2% glutaraldehyde in PBS at room temperature for 1 hour. Fixed mitochondria and MDV fractions were, respectively, centrifuged at 10 000 g for 10 minutes and 200 000 g for 1 hour, and the supernatants were discarded leaving around 20 µL. Using a dissection microscope, pellets were lifted using a sharp needle, and an equal volume of melted 2% agarose solution was added making sure that the pellet was suspended in the middle. Following solidification, samples were stored at 4°C. Transmission electron microscopy (TEM) was performed at the Facility for Electron Microscopy Research, McGill University, according to standard operating procedures. Following sample preparation, 90‐100 nm thick sections were mounted onto a 200 mesh copper grid (Electron Microscopy Sciences, Hatfield, PA, USA) and imaged with an FEI Tecnai 12 120 kV transmission electron microscope equipped with an AMT XR80C 8 megapixel CCD camera.
2.5. Western blot
Mitochondrial suspensions or MDV fractions were collected in SDS‐PAGE buffer (6X Laemmli buffer), vortexed, boiled at 95°C for 5 minutes, and stored at −80°C. Equal volumes of samples were loaded onto a stain‐free gel (TGX Stain‐Free FastCast, 10%, 1.5 mm thick) and electrophoresis was performed at 150 V for 1 hour. PVDF membranes were activated in 100% methanol for 20 seconds, transferred to water for 2 minutes, and left in cold transfer buffer until use. Gels were activated using UV light for 45 seconds in a ChemiDoc Touch imaging system (Bio‐Rad) at high power and 365 nm wavelength. Proteins were transferred onto the membrane using a Trans‐Blot Turbo transfer system (Bio‐Rad) at 1.3 A and up to 25 V for 10 min. Stain‐free images were taken by exposing membranes to UV light using the above‐mentioned settings. Membranes were probed with an anti‐PDHE2/E3bp antibody (ab110333, Abcam) using Snap ID 2.0. Bands were visualized via chemiluminescence using Clarity Western ECL substrate (Bio‐Rad) and imaged under a ChemiDoc Touch imaging system (Bio‐Rad).
2.6. MS sample preparation and analysis
The top and bottom adjacent layers of 35%‐40% sucrose interface, containing the MDV fraction was mixed with four times the volume of a protein precipitation buffer (50% acetone + 50% ethanol + 0.1% acetic acid), then vortexed and incubated at −20°C overnight. Samples were centrifuged for 10 minutes at 10,000 x g in a microfuge. The supernatant was removed carefully and discarded, and the residual precipitation buffer was allowed to evaporate at room temperature for 20 minutes, making sure not to over‐dry the pellet. The protein precipitate was then solubilized by adding 200 µL of 8 M urea solution prepared in 50 mM ammonium bicarbonate (pH 8.0‐8.5). Reduction buffer (1 M dithiothreitol in water, prepared fresh) was added to a final concentration of 10 mM and incubated for 1 hour at room temperature. Alkylation buffer (1M iodoacetamide in water) was added to a final concentration of 20 mM and incubated for 40 minutes at room temperature in the dark. Samples were then diluted with five volumes of 50 mM ammonium bicarbonate solution. Trypsin (Trypsin Gold, Promega) was added to the samples at a ratio of 1:100 of trypsin to protein by weight and incubated overnight at 37°C. Sep‐Pakcolumns (Waters) were activated using 1 mL of acetonitrile. Columns were then equilibrated with 1 mL of 0.1% formic acid solution in water. Trypsinized samples were acidified using 5% formic acid in water to reach a final concentration of 0.1% formic acid. Samples were then loaded onto the Sep‐Pakcolumns. Columns were washed with 1 mL of 0.1% formic acid. Samples were eluted using 0.5 mL of elution buffer (80% acetonitrile and 0.1% formic acid in water) and collected in microcentrifuge tubes. Samples were dried in a SpeedVac using no heat and stored at −20°C.
Following reconstitution in 2% acetonitrile and 0.1% formic acid final concentration: 1 µg/µL) peptide fragments were analyzed by HPLC‐ESI‐MS/MS. The system used consisted of an Eksigent Nano2D ultra HPLC (Sciex) coupled with an LTQ Velos‐Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a nano‐electrospray interface operated in positive ion mode. The mobile phases consisted of 0.1% (v/v) FA in water as buffer A and 0.1% (v/v) FA in 80% acetonitrile as buffer B. The pre‐column used was 100μm × 20 mm, packed with 5 μm 120‐Å pore size Magic C18AQ resins (Dr Maisch GmbH, Ammerbuch, Germany). The analytical column was 75 μm × 100 mm and packed in‐house with reverse‐phase Magic C18AQ resins (1.9 μm; 120‐Å pore size; Dr Maisch GmbH, Ammerbuch, Germany).
Briefly, equal amounts (4 µg) of samples were loaded on the column using 98% of buffer A at a flow rate of 1.5 µL/min for 15 min. Then, a gradient from 5% to 35% buffer B was performed over the course of 120 minutes at a flow rate of 300 nL/min. The MS method consisted of one full MS scan from 300 to 1700 m/z followed by a data‐dependent MS/MS scan of the 20 most intense ions, with a dynamic exclusion repeat count of 2, and a repeat duration of 90 s. As well, for the experiments on the Orbitrap MS the full MS was performed in the Orbitrap analyzer with R = 60 000 defined at m/z 400, while the MS/MS analyses were performed in the iontrap. To improve the mass accuracy, all the measurements in Orbitrap mass analyzer were performed with internal recalibration (“Lock Mass”). On the Orbitrap, the charge state rejection function was enabled, with single and “unassigned” charge states rejected.
2.7. Protein identification and quantification
The raw files generated by the LTQ‐Orbitrap were processed and analyzed using MaxQuant (version 1.6.6.0). 14 A target‐decoy database search was performed against Rattus norvegicus UniProtKB/Swiss‐Prot and UniProtKB/TrEMBL protein sequence databases 15 with the addition of commonly observed contaminants and their reversed sequences. The database search was performed using a precursor ion mass tolerance of 0.5 Da. Trypsin was selected as the protease and a semi‐tryptic search with at most two miscleavages was executed to allow for the identification of putative protein cleavage products, which may be incorporated into MDVs following partial breakdown by mitochondrial proteases involved in quality control. Cysteine carboxyamidomethylation was set as a fixed modification. Mono‐oxidation of methionine and hydroxylation of leucine, proline, phenylalanine and lysine, which are common modifications occurring in response to oxidative stress, were also considered. Resulting protein identifications with false discovery rates (FDR) < 0.01 were deemed of high confidence for a given MS experiment. To maximize stringency, a protein was deemed confidently detected in an experimental condition when identified in at least two biological replicates. Protein abundances were determined by label‐free quantitation using the LFQ algorithm. 16 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 17 partner repository with the dataset identifier PXD020110. Normalized protein quantification values were then imported into the Perseus 18 software platform for log transformation, data imputation, and statistical analysis. For proteins with a single missing value in one of the three experimental replicates in a particular condition, the mean LFQ value of the two other replicates was imputed. For statistical analysis, fold change of the log‐transformed LFQ value vs control was calculated for each of the paired experiments and multiple t tests were performed. For each p value obtained a corresponding False Discovery Rate (FDR) was estimated using the Benjamini and Hochberg procedure and lists of significantly affected proteins were generated using FDR thresholds of 5, 10, and 15%, corresponding to very high, high, and moderate confidence hits, respectively (Supplemental File S1). The GraphPad Prism 8 software package (version 8.4.2) was used to build basic graphical representations. To map the mitochondrial proteins recovered in the MDV fractions, the dataset was mapped against the Rattus norvegicus Integrated Mitochondrial Protein Index (IMPI) database.
2.8. Gene ontology and bioinformatics
Venn diagrams were generated using the Eulerr R Package. Hypergeometric tests were used to determine the statistical significance of the enrichments of mitochondrial proteins in MDVs that harbor hyper‐reactive cysteine residues using the mitochondrial proteome database as a reference (MitoMiner 4.0 database 19 ). Similarly, hypergeometric tests also assessed the enrichments of proteins from the MDV proteome that match mitochondrial proteins known to be detected as part of extracellular vesicles. To identify the Gene Ontology (GO) annotations of the MDV proteome, the data were uploaded into the g:Profiler 20 platform and queried using the Rattus norvegicus database. Unless otherwise indicated, only GO terms annotating 350 proteins or less were considered in order to filter out large annotations that provide limited interpretative value. 21 The g:SCS (shortest common superstring) algorithm was used for multiple hypothesis testing corrections using a default alpha threshold of 0.05. 20 The ggPlot2 R package was used to generate bubble plots in which pathway enrichments were expressed as Rich Factors, which represents the ratio of the number of proteins observed for a given GO term to the total number of proteins for this term. The Cytoscape 22 (version 3.8.0) Enrichment Map 23 and Autoannotate 24 plugins were used to cluster redundant GO terms that were significantly enriched and to automatically generate cluster labels based on word frequencies of selected attributes.
To further analyze subsets of the MDV proteome, the Cytoscape stringApp plugin was used to import protein‐protein interaction data from STRING. 22 For this analysis, an interaction score of 0.7 (high confidence based on default active interaction sources) was set as minimum and GO enrichment data were used to manually cluster proteins based on mitochondrial process and location, providing a detailed map of the MDV proteome under various experimental conditions.
2.9. H9c2 myoblast culture
H9c2 myoblasts were purchased from ATCC (Manassas, VA, USA). Cells were initially cultured in high glucose DMEM (319‐020‐CL; Wisent Inc, Saint‐Jean‐Baptiste, QC, Canada) supplemented with sodium pyruvate (1 mM, 600‐110‐EL; Wisent), penicillin/streptomycin (450‐201‐EL; Wisent) and 10% fetal bovine serum (FBS, 37°C, 5% CO2). To force cultured cells to rely on mitochondrial oxidative phosphorylation, glucose was replaced with galactose as the major energy substrate. For this, cells were cultured in glucose‐free DMEM (319‐060‐CL; Wisent), supplemented with galactose (10 mM), sodium pyruvate (1 mM, 600‐110‐EL; Wisent), HEPES (10 mM, 330‐050‐EL; Wisent), 1 × penicillin/streptomycin (100 IU/100 μg/ml, 450‐201‐EL; Wisent) and 10% FBS. Cells were maintained in galactose media for 1 week prior to experiments. Cells were then exposed to doxorubicin (25 µM) during 30 min, and an appropriate vehicle‐treated time‐matched control was employed at each time point. All treatments were performed in 24‐well plates with cells adhered to deposited coverslips. Each 24‐well plate included at least two time‐matched control wells. Experiments were repeated on three separate occasions.
2.10. Immunofluorescence
Immunofluorescence was performed as previously described 7 with all solutions made in PBS (in mM: 136 NaCl, 2.6 KCl, 10 Na2HPO4, 1.8 KH2PO4, pH 7.4). Briefly, at the end of the treatments, cells were first fixed at 37°C in 5% paraformaldehyde, then quenched in NH4Cl (50 mM), and permeabilized with 0.25% Triton X‐100. Cells were then blocked with 10% FBS for 1 hour prior to primary antibody incubation (5% FBS, 1 hour) and subsequently incubated with a secondary antibody (5% FBS, 1 hour, in the dark). The coverslips were then mounted on glass slides using ProLong Gold without DAPI (P36935, respectively; Life Technologies, Carlsbad, CA, USA).
Slides were imaged by confocal microscopy using a 100X, 1.4 NA objective on an LSM 510 Meta confocal microscope (Zeiss, Oberkochen, Germany). Excitation/emission wavelengths were set at 561/640 nm (red) to measure the secondary antibody fluorescence of Translocase of the Outer Membrane (TOM20) and 488/520 nm (green) for that of Oxoglutarate Dehydrogenase (OGDH). Acquired images were analyzed using ImageJ (NIH, Bethesda, MD, USA). Vesicles were identified visually, based on apparent size and cargo selectivity (OGDH positive or TOM20 positive, but not both) after the application of a co‐localization mask to discriminate them from mitochondria (which are positive for both OGDH and TOM20). Apparent vesicle size was ~300 nm, which represents an estimated actual size of ~50 nm considering the lateral resolution limit of confocal microscopy. For the analysis, overlay TIF images were opened on ImageJ, color channels were split, and the co‐localization mask plugin was applied using only the green and red channels (ratio: 50%, Thresholds 1 and 2:50, Display value: 255). After this transformation, the mitochondrial network or any small mitochondria containing both OGDH and TOM20 appeared white and were visually excluded from the analysis. On the resulting image, the color balance was then adjusted in each channel to a minimum signal (30‐60 to eliminate background) and a maximum of 121 to enhance contrast. The resulting image was used to visually identify circular structures (>9 pixels 2 ) positive for either OGDH and negative for TOM20 as previously described. 7 The number of MDVs per cell was quantified in 5‐6 different fields of view from three independent experiments.
3. RESULTS
3.1. Isolated mitochondria actively bud vesicles when exposed to graded oxidative stress in vitro
To analyze the cargo composition of MDVs in response to graded mitochondrial stress, a budding/reconstitution assay that enables generation and purification of cardiac mitochondrial MDVs under well‐controlled conditions was used (Figure 1A). Cardiac mitochondria were incubated with substrates (succinate, ADP, ATP, Pi) eliciting constant submaximal rates of oxidative phosphorylation in buffers equilibrated with ambient PO2 (20.9% O2, room air), as this relatively hyperoxic state, in comparison to PO2 in cellulo (0.1%‐3% O2), promotes the formation of superoxide anions even under conditions of unrestricted electron flow through the respiratory chain. 25 To further increase oxidative stress, experiments were also performed in presence of the complex III inhibitor antimycin A, which in presence of succinate, enhances superoxide formation both at the matrix and intermembrane space through mechanisms that are well established. 26 , 27 This approach achieves moderate and maximal levels of oxidative stress as confirmed by measuring rates of H2O2 release by mitochondria at baseline and after the addition of antimycin A (Figure 1B). MDVs generated by mitochondria were collected after 30 min of treatment during ambient PO2 respiration conditions and purified by flotation ultracentrifugation on a discontinuous sucrose gradient. This short 30 min incubation, when compared to 60 and 120 min, combined with conditions unfavorable to the opening of the permeability transition pore (ie, no Ca, 2 + presence of EGTA, adenine nucleotides, and MgCl2), were used to rule out a contribution of pore opening and subsequent mitochondrial disruption to the release of proteins into the MDV fraction. Oximetric tests confirmed the bioenergetic integrity of mitochondria after 30 min of incubation (Figure 1C).
FIGURE 1.
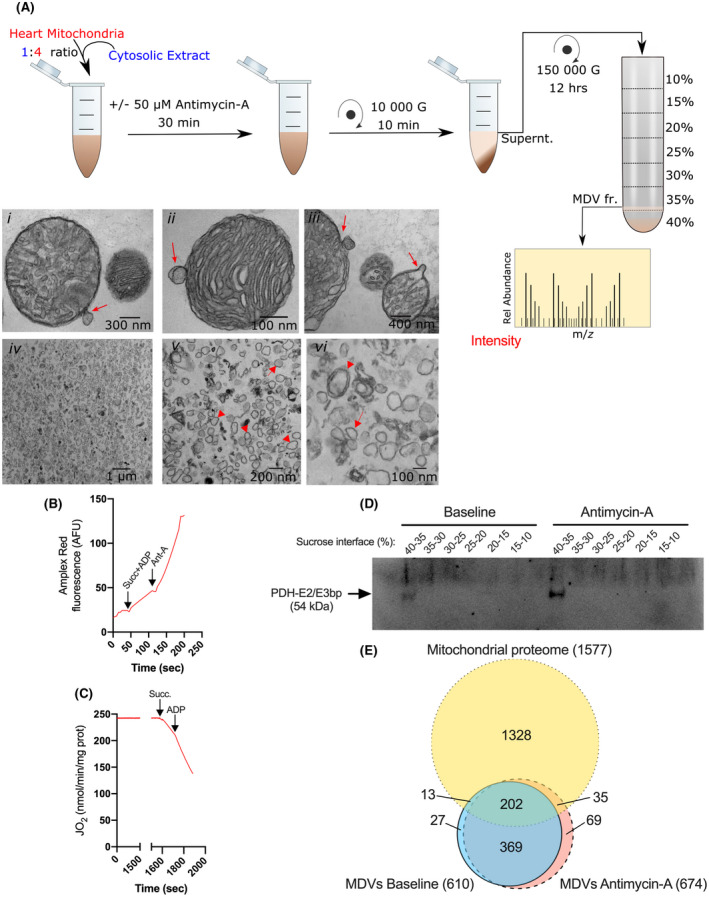
Overview of the in vitro budding assay employed to survey the MDV proteome following graded oxidative stress. A, 5 mg of freshly isolated cardiac mitochondria were incubated at 37°C and ambient PO2, in a budding medium supplemented with a cytosolic extract as per the indicated proportion. Parallel experiments were performed in the absence or presence of the complex III inhibitor antimycin A (Ant‐A: 50 µM), which allowed comparison to be made at two levels of oxidative stress (ie, baseline and antimycin A). The cytosolic extract used for the budding assay was prepared in large quantities on a separate day and kept frozen in aliquots, which allowed to use the same extract across all experimental replicates and conditions. Following incubation, mitochondria were rapidly sedimented and the supernatant was processed for the isolation of MDV fractions as indicated. Images i‐iii show TEM micrographs of individual organelles actively budding single or double‐membrane vesicles of 60‐150 nm in diameter (Direct Magnification: 49 000×). Images iv‐vi show the accumulation of vesicular structures of the same size range at the 35%‐40% sucrose interface (Direct Magnification: 1900 and 30 000×). Arrowheads point to double‐membrane MDVs, while arrows point to single membrane MDVs (B): Amplex Red fluorescence tracing showing differences in the rate of H2O2 release achieved when mitochondria are incubated under baseline (ie, in presence of succinate and ADP) or in presence of antimycin A. C, Oxygraph trace of mitochondria incubated for 30 min at room temperature. The capacity to respond to the addition of succinate and ADP confirms that bioenergetic integrity is well maintained. D, Equal volume of the various sucrose interfaces recovered were submitted to SDS‐PAGE and probed with an antibody directed against the MDV marker PDHE2/E3bp. PDH immunoreactivity was observed only at the 35%‐40% interface, which was subsequently used for MS/MS analysis. E, Venn diagram representing the overlap between the Rattus norvegicus mitochondrial proteome, and proteins identified in the MDV fractions following incubation under baseline or antimycin A conditions. Proteins in the mitochondrial proteome included 1127 known mitochondrial proteins and 431 proteins with a predicated mitochondrial location based on the MitoMiner 4.0 database 19 (http://mitominer.mrc‐mbu.cam.ac.uk/release‐4.0/impi.do). For the MDV fractions, proteins detected in at least two‐thirds of experimental replicates (FDR < 0.01) within any experimental condition are considered. Number in parentheses indicates the total number of proteins in each set of data
To confirm that mitochondria actively produced MDVs in the experimental conditions used, mitochondrial suspensions were fixed in glutaraldehyde, embedded in low‐melting agarose, and visualized using TEM. Vesicular structures of 60‐150 nm in diameter and attached to mitochondria were observed both under baseline conditions and following exposure to antimycin A (Figure 1A i‐iii). Vesicles were either composed of a single layer derived from the outer mitochondrial membrane or two layers derived from the inner and outer membranes. Constrictions at the base of the vesicular structures and regions of high membrane curvature were often evident, consistent with the occurrence of a protein‐mediated budding process.
3.2. Budding/reconstitution assays allow relative purification of MDVs for proteomic analysis
Upon ultracentrifugation, western blot analysis using an antibody against PDHE2/E3bp, a common marker of cardiac MDVs, was performed on the various recovered sucrose fractions (Figure 1D). Isolated MDVs were found to float at the 35%‐40% sucrose interface. Visual inspection of the biological material recovered from this fraction revealed the presence of single or double‐membrane vesicular structures of 60‐150 nm in diameter (Figure 1A iv‐vi). Broken mitochondrial membranes were occasionally observed, but this represented a small proportion of the material recovered.
A total of 714 proteins (610 and 674 in baseline and antimycin A, respectively) proteins were identified in the MDV fractions (Supplemental File S1). As shown in Figure 1E, A total of 250 MDV proteins were identified as being mitochondrial, based on the MitoMiner database (215 and 237 at baseline and antimycin A, respectively). A large proportion of the mitochondrial proteins in MDVs (ie, 202) were present both under baseline and after exposure to antimycin A, albeit at different abundances, while only a few were unique to one of the two conditions (ie, 13 and 35 in baseline and antimycin A, respectively).
Because a cytosolic extract was added to the reaction buffer to promote the budding process, a significant number of non‐mitochondrial proteins were recovered in the MDV fraction (ie, 465 proteins total, 65% of the proteins identified). As expected, a large proportion (80%; 369 out of 465) of these non‐mitochondrial proteins were common to baseline and antimycin A, since the same cytosolic extract was added to the budding mixture in both conditions.
In order to decipher the nature of proteins recovered in the MDV fractions, functional enrichment analysis was performed on the pooled dataset (ie, baseline and antimycin A) using the GO database. GO enrichments of non‐mitochondrial proteins within the MDV fractions were not observed for vesicular budding and trafficking (Figure S1 and File S2). However, the non‐mitochondrial proteins identified were enriched for cardiac muscle development/contraction, cytoskeleton organization/regulation, haptoglobin/hemoglobin, serine‐type endopeptidase/chaperones, and lipoprotein and fatty acid binding (complete list of proteins provided in Supplemental File S1).
We next focused on characterizing the nature of the 250 mitochondrial proteins identified in the MDV dataset. Based on GO cellular components terms, the main submitochondrial compartments from which the proteins originated were the inner membrane and matrix, while the outer membrane contributed to a lesser extent (Figure 2A and Supplemental File S3). Proteins with a dual association to mitochondria and peroxisomes or secretory vesicles were also present. As shown in Figure 2B‐D and Supplemental Files S2‐S3, MDVs were enriched with proteins associated with OXPHOS complexes, TCA cycle, pyruvate metabolism, and fatty acid oxidation, which are all prominent metabolic pathways in cardiac mitochondria. Interestingly, iron‐sulfur cluster binding proteins and proteins involved in redox homeostasis / antioxidant activity, mitochondrial protein import and processing, and the transfer of phosphogroups were also significantly enriched in the MDV fractions. Proteins involved in aldehyde and amino acid metabolic processes, including branched‐chain amino acid catabolism and transamination reactions, were also enriched. Last, small GTPases of the RAB sub‐family that are predicted or known to be localized to the mitochondria and are known to play a role in vesicle‐mediated transport were detected in MDV fractions.
FIGURE 2.
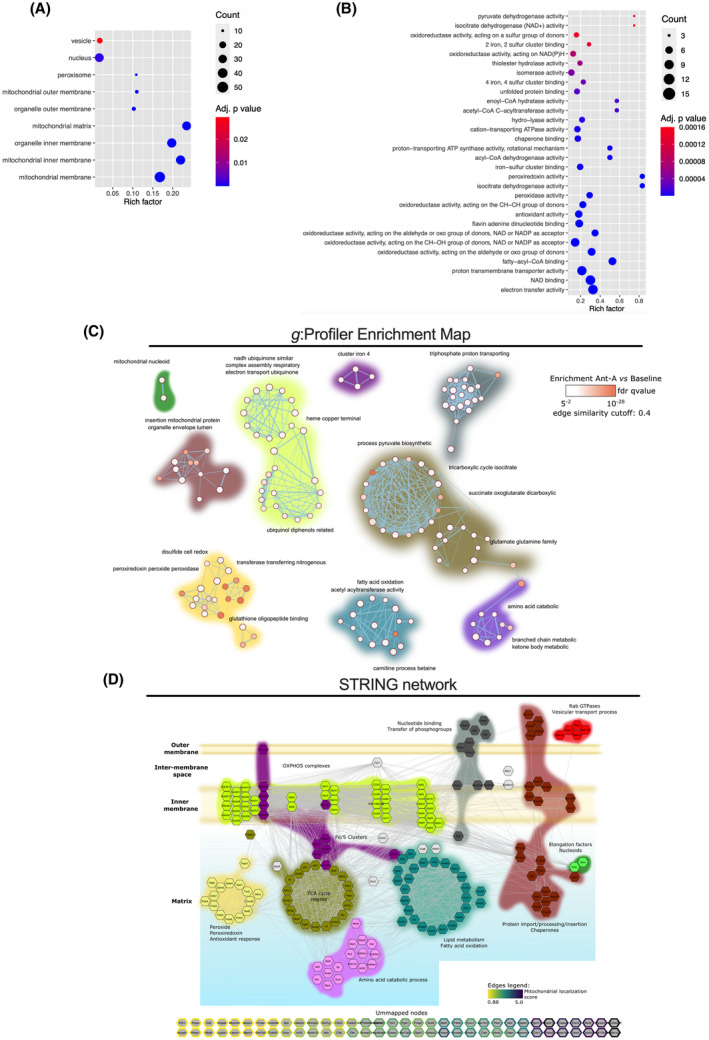
Characterization of the mitochondrial proteins identified in the combined MDV dataset. Functional Enrichment analysis of over‐represented GO cellular component (A) and biological process/molecular function (B) in the combined MDV dataset (baseline and antimycin A) performed using g:Profiler. Proteins with a known predicted mitochondrial localization were included in the analysis. Maximum size of functional categories was set at 90 to filter out large annotations that provide limited interpretative value. 21 The g:SCS algorithm was used for multiple hypothesis testing corrections using a default alpha threshold of 0.05 for significance. Enrichment is expressed as a rich factor, which represents the ratio of the number of proteins observed for a given GO term to the total number of proteins for this term. Circle size reflects the number of proteins per GO term, while color indicates the level of significance. C, g:Profiler enrichment map illustrating the main mitochondrial processes represented in the MDV proteome. Nodes, edges, and node color represent individual GO terms, mutual overlap, and the level of significance of enrichment, respectively. The auto‐annotation tool was used on Cytoscape to automatically generate cluster labels. D, High confidence (interaction score > 0.7 based on default active interaction sources) STRING network of mitochondrial proteins identified in the MDV fractions. GO enrichment data were used to manually cluster proteins based on biological process and location, providing a detailed map of the MDV proteome. The color code used to identify clusters is the same for panels C and D
Importantly, the few proteins currently known to be incorporated in cardiac MDVs were confirmed in the proteomics analysis. This includes the TCA cycle enzyme PDH, which has been used to track MDV production in cardiac myoblasts, 7 as well as CIII subunit UQCRC2 and VDAC, which were previously identified by immunoblotting in MDV budding/reconstitution assays performed in heart mitochondria 6 (Figure 2D). Furthermore, OGDH, a recently identified protein in vesicular cargo in immune cells, 28 was also identified in the MDV fraction, suggesting that cardiac mitochondria also release this TCA cycle enzyme. This was further verified by confocal imaging in H9c2 cardioblasts. OGDH‐positive MDVs were actively released by mitochondria under normal cell culture conditions, and their release was nearly doubled within 30 minutes of exposure to the anthracycline doxorubicin, a well‐known inducer of superoxide production in mitochondria (Figure 3).
FIGURE 3.
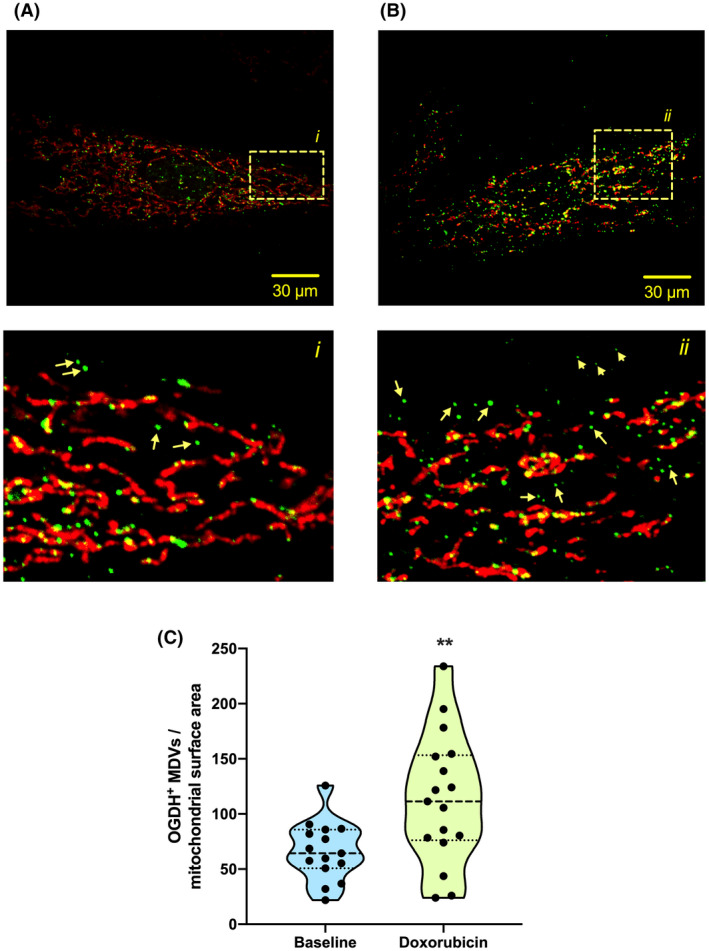
Generation of OGDH‐positive MDV in cardiac myoblasts. H9c2 cardiac myoblasts cultured in galactose‐containing media and treated with vehicle (A) or 25 μM doxorubicin (B) for 30 min. Cells were immunolabeled for the TOM20 (red) and the OGDH (green). Numbered panels below are the magnifications of the respective boxed areas shown in the top panels. C, Quantification of OGDH+/TOM20− MDVs in control and doxorubicin‐treated cells at 30 min (three independent experiments per group, 5‐6 random fields of view). The number of vesicles in each cell is expressed per unit of mitochondrial surface area. Statistical comparison was made between doxorubicin and control using a two‐tailed nonparametric Mann‐Whitney test: a, P < .01
3.3. Proteomics analysis identifies vulnerable proteins incorporated into MDVs under oxidative stress
To further delineate the impact of oxidative stress on the incorporation of mitochondrial proteins into the MDV fraction, we focused on proteins that were altered following incubation in the presence or absence of antimycin A. As shown in Figure 4A, the number of proteins that were unique (Antimycin A: 34 vs Baseline: 13) or more abundant, based on LFQ intensity (Antimycin A: 58, vs Baseline: 12, with 37%, 32% and 28% corresponding to very high, high, and moderate confidence hits, respectively, Supplemental File S1), was 3.8‐fold higher in the MDV fraction following antimycin A treatment when compared to the baseline group. In addition to these differentially abundant proteins, 132 other mitochondrial proteins were also detected in the MDV fractions, but their abundance did not differ significantly between the two experimental groups.
FIGURE 4.
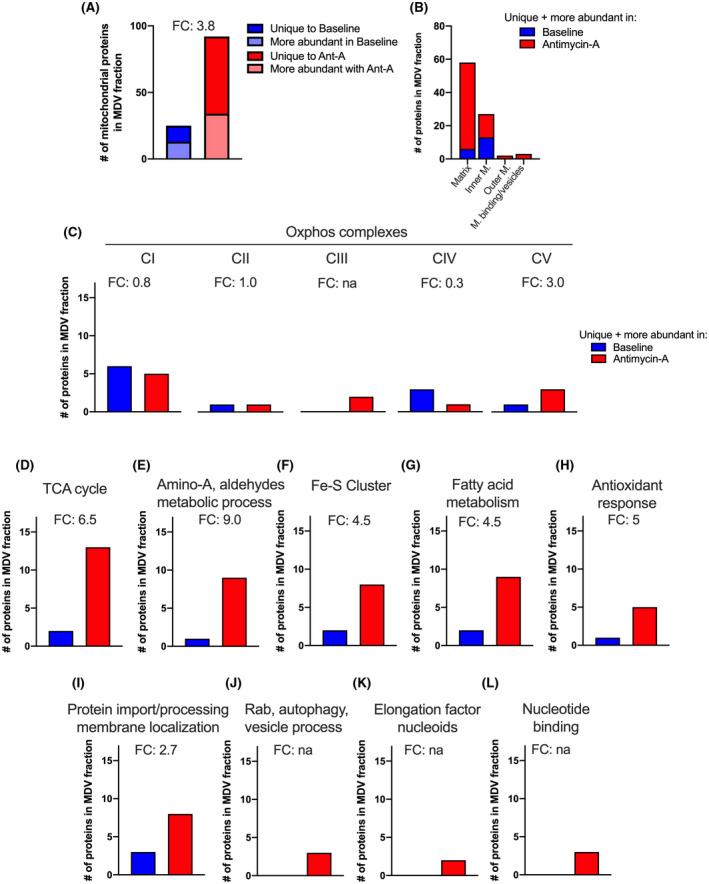
Differential analysis of the MDV proteome at baseline and following exposure to antimycin A. A, Total number of mitochondrial proteins enriched in the MDV fraction in one experimental condition vs the other. This includes proteins that were uniquely detected in one experimental condition and proteins that were significantly (FDR < 15%) more abundant based on LFQ values. B, Submitochondrial localization of mitochondrial proteins detected as unique or more abundant in each of the experimental condition. C‐L, Number of enriched (ie, unique and more abundant) mitochondrial proteins associated with specific mitochondrial processes and molecular functions under baseline and antimycin A conditions. Fold change (FC) is indicated for each process/molecular function
Closer investigation of differentially abundant proteins based on GO terms and network mapping revealed that proteins that were more abundant following antimycin A exposure originated mainly from the mitochondrial matrix. In comparison, inner membrane proteins contributed half as much, while proteins from the inter‐membrane space (not shown) and outer mitochondrial membrane were present in much smaller numbers in both MDV populations (Figures 4C and 6).
FIGURE 6.
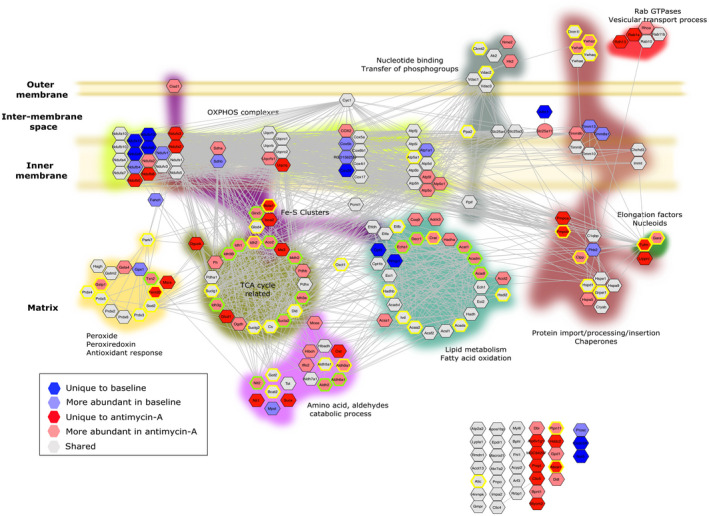
Differential analysis of the MDV proteome at baseline and after exposure to antimycin A. The STRING network of mitochondrial proteins identified in the MDVs (see Figure 2 for details) was used to map proteins that were differentially enriched under baseline or antimycin A conditions, and to identify those reported to harbor hyper‐reactive cysteine residues. 29 Proteins labeled in dark blue or red were uniquely detected in one of the two experimental conditions. Proteins labeled in pale blue or red were identified in both experimental conditions, but their abundance differed significantly (FDR < 15%). Proteins labeled in grey were present in both experimental conditions and did not show any significant difference. Proteins with hyper‐reactive cysteine residues identified in the murine heart and cancer cell lines are labeled with green and yellow borders, respectively
In line with these results, OXPHOS complexes, which are located in the inner membrane and contain over 100 different subunits, only contributed to 17% of the differentially abundant proteins found within MDVs in response to antimycin A (Figures 4D and 6). Among the five OXPHOS complexes, only complex V and complex III, the latter being the direct target of antimycin A, and an important site of ROS production, presented greater differential enrichment following exposure to antimycin A (Figures 4D and 6).
Proteins associated with the TCA cycle, amino‐acid and aldehyde metabolic processes, fatty acid metabolism, iron‐sulfur clusters, and antioxidant systems were 4.5 to 9‐fold more numerous following exposure to antimycin A when compared to baseline (Figures 4E‐I and 6). Together, proteins associated with these metabolic and redox processes represented over 65% of the proteins enriched in the MDV fraction in response to antimycin A. As for the remaining 16%, they were related to protein import/processing, Rab‐GTPases, elongation factors linked to nucleoids, and proteins involved in the transfer of phosphogroups (Figures 4J‐M and 6).
The fact that metabolic and redox processes were strongly over‐represented in MDVs, and more so in presence of antimycin A, led us to examine potential commonalities that could underlie their preferential incorporation into MDVs. In this regard, recent proteome‐wide studies have shown that metabolic and redox enzymes in mitochondria account for a significant proportion of cellular proteins harboring oxidation‐prone cysteine residues. 29 This includes “professional” redox enzymes involved in H2O2 metabolism, as well as multiple metabolic enzymes. 30
We, therefore, compared our MDV proteome to the dataset published by Weerapana et al 29 in which a chemoproteomic method was used to quantify intrinsic cysteine reactivity in the native proteome of murine heart and cancer cells lines. We found that 44 of the 47 mitochondrial proteins harboring hyper‐reactive cysteine residues (HRCRs) in the heart were present in our dataset (Figure 5A). Almost all of these proteins (ie, 43) were found in MDVs even under baseline conditions, and 18 (41%) of them became significantly more abundant when oxidative stress was further enhanced by blocking the respiratory chain with antimycin A (hypergeometric test p value 1.7e‐31; Figure 5C). Similar observations were made when the MDV dataset was compared to the hyper‐reactive cysteine proteome of human cancer cell lines (Figure 5B), which further suggested a very strong preferential incorporation of proteins containing HRCRs in MDVs when mitochondria are exposed to oxidative stress (hypergeometric test p value: 1.0e‐21; Figure 5C).
FIGURE 5.
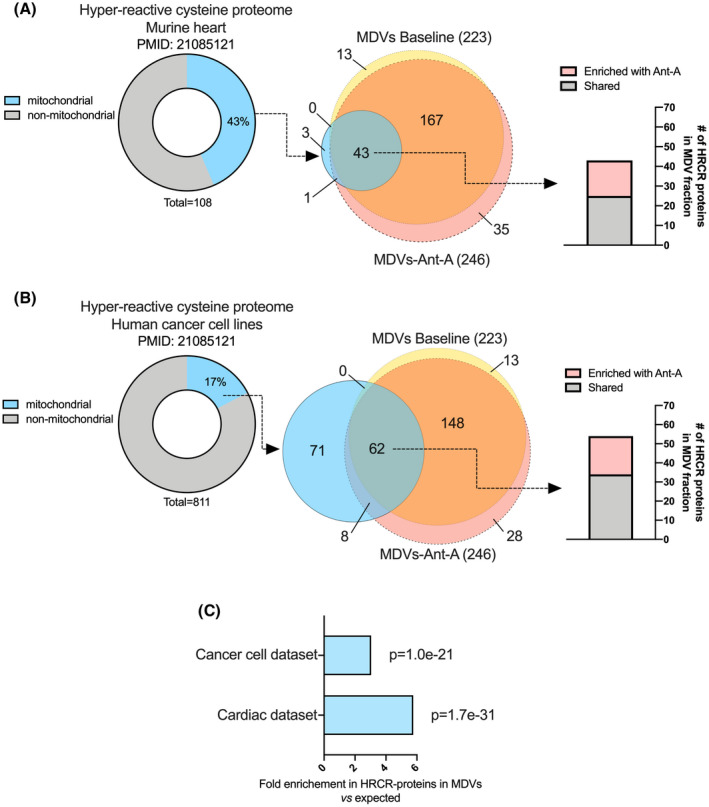
Enrichment of proteins containing hyper‐reactive cysteine residues in MDVs. Two proteomics datasets published by Weerapana et al 29 were used to examine the potential enrichment of MDVs with proteins harboring hyper‐reactive cysteine residues (HRCRs). The first dataset contained 108 HRCR proteins identified in the soluble fraction of murine heart (A), while the second dataset contained 811 HRCR proteins identified in various human cancer cell lines (MCF7, MDA‐MB‐231, and Jurkat) (B). For each comparison, the percentage of mitochondrial proteins in the HRCR dataset was determined using the MitoMiner 4.0 database. Venn diagrams were then used to determine the number of mitochondrial proteins harboring HRCRs present in MDVs under baseline and antimycin A conditions. Mitochondrial proteins known to harbor HRCRs were strongly enriched in MDVs. C, Fold enrichments in mitochondrial HRCR proteins in MDVs vs the mitochondrial proteome along with their hypergeometric test p values
As shown in Figure 6, several of the TCA cycle‐related enzymes released in MDVs contained HRCRs (ACO2, IDH, MDH2, DLD, SUCLA2, CS, and SUCLG2), and the majority of those were present in larger amounts following exposure to antimycin A. Similarly, most of the fatty acid metabolism enzymes released in MDVs contained HRCRs, including mitochondrial thiolases (ACAT1, HADHB, ACAA2), which are among the most reactive protein thiols in mammalian cells. 29 Several aldehyde dehydrogenases, which contain redox‐sensitive thiol groups were also among the proteins incorporated into MDVs (ALDH2, ALDH5A1, ALDH6A1, ALDH9A1), including ALDH2, which is listed as the protein containing the most reactive cysteine residue in the Weerapana et al dataset. 29 Professional redox enzymes with highly reactive cysteine residues were also present in MDVs, including SOD2, peroxiredoxin isoforms (PRDX3, PRDX5, PRDX6), and thioredoxin/thioredoxin reductase (TRX2, TXRD2).
Interestingly, several MDV proteins containing HRCRs were related to iron metabolism (Figure 6). This includes proteins involved in the maturation (ISCA2), handling (GLXR5, CISD1), and insertion (BOLA1) of Fe‐S clusters in mitochondrial proteins, as well as the Fe‐S cluster containing TCA cycle enzyme ACO2. In addition, several other proteins containing Fe‐S clusters were also incorporated into MDVs (NDUFS1‐3, NDUFV1, SDHB, UQCRFS1, ETFDH, ETFA, ETFB), with some of them displaying sensitivity to antimycin A (Figure 6).
3.4. Comparison of proteomic datasets suggests that MDVs deliver a number of mitochondrial proteins to extracellular vesicles
MDVs destined to lysosomes were previously shown to transit through the late endosome/multivesicular body compartment. 5 Because some multivesicular bodies can be routed to the cell surface, where their limiting membranes can fuse with the plasma membrane, 12 we investigated the overlap between mitochondrial proteins released into MDVs during our budding assay and those present in the proteome of extracellular vesicles (EVs). As shown in Figure 7, 24% (191/800) of the mitochondrial proteins listed in the Exocarta database were present in our MDV dataset and more than a third of those displayed sensitivity to antimycin A. To refine comparisons, publicly available proteomic datasets from EVs released under stress were also examined. This included EVs released by normal cells exposed to metabolic stress such as nutrient deprivation or severe hypoxia as well as cancer cells. 31 , 32 , 33 , 34 , 35 Strikingly, we found that more than 50% (58/110) of the mitochondrial proteins released by cardiac fibroblasts were present in MDVs with about half displaying sensitivity to antimycin A (Figure 7). Overlaps ranging between 30%‐40% were also observed between MDV and EV proteins released under serum deprivation or by various cancer cells (Figure 7). Mitochondrial proteins present in MDVs that consistently overlapped across EV datasets included HSPD1 (ie, mitochondrial HSP60), TCA‐cycle related enzymes (PDH, OGDH, and their E3 component DLD, MDH, IDH, CS, GLUD1, and GOT2), fatty acid oxidation enzymes (HADHA, HADHB), and enzymes involved in peroxide and glutathione metabolism (PRDX5, PRDX6, GSTP1) (Supplemental Figures [Link], [Link]). Interestingly, several mitochondrial proteins present in our MDV dataset were also found in circulating EVs isolated from human blood, 36 , 37 , 38 , 39 with some being enriched in patients with diseases associated with perturbed mitochondrial function and oxidative stress, namely Parkinson's, HTLV‐1 infection, lung cancer, and myocardial infarction (Figure S4).
FIGURE 7.
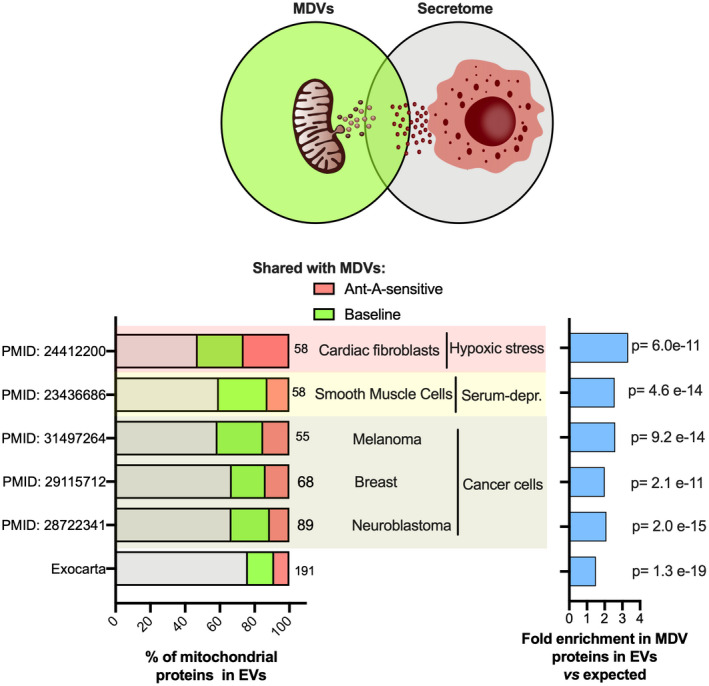
Shared proteomic signature among MDVs and extracellular vesicles. The proportion of mitochondrial proteins that appear in both MDVs and EVs, which might predict the MDVs fate of contributing to the extracellular vesicles, was estimated by comparing the MDV proteome with the ExoCarta database and the proteome of six other publicly available EV datasets obtained under various stress conditions. A, A depiction of shared mitochondrial proteomic profile between MDVs and EVs. B, The total number of mitochondrial proteins in the EV datasets derived from the ExoCarta database, cardiac fibroblasts under hypoxia (PMID—24412200 35 ]), serum‐starved smooth muscle cells (PMID—23436686 31 ), and cancerous cells (PMIDs – 31 497 264, 29 115 712 & 28722341 32 , 33 , 34 ) was determined using the MitoMiner 4.0 database. The percentage of mitochondrial proteins from each EV dataset that was common to MDV proteome and among those that were sensitive to antimycin A were indicated. C, Fold change of actual vs expected percentage of MDV mitochondrial proteins observed in EVs with respect to mitochondrial proteome along with their hypergeometric test p values
4. DISCUSSION
While the budding of MDVs is increasingly viewed as a mitochondrial housekeeping process and is hypothesized to defend against mitochondrial proteotoxic stress, the repertoire of proteins carried by these vesicles remains largely unknown. The present study provides the first proteomic survey, to our knowledge, of MDV cargoes released by mitochondria under oxidative stress, a major pathogenic factor underlying several human diseases with mitochondrial involvement. Our proteomic data validate previously established protein markers of MDVs and identifies several novel cargoes. Importantly, we provide evidence that under oxidative stress, MDVs are preferentially enriched with proteins containing ROS emitting sites, several enzymes harboring highly reactive cysteine residues and proteins that bind or contain iron, which likely makes them vulnerable to oxidative damage. Furthermore, our data show that a strong overlap exists between the proteome of MDVs and that of EVs secreted under a variety of stress conditions, which suggests that MDVs might account for a significant proportion of mitochondrial proteins commonly reported in extracellular vesicles.
4.1. Susceptibility to damage/toxic oxidative modifications underlies the cargo selectivity of MDVs under oxidative stress
Beyond their shape and size, MDVs have been defined by their cargo selectivity. 5 , 11 , 13 For instance, MDVs carrying markers, such as PDH or OGDH, were systematically reported to lack TOM20 and vice versa. 5 , 7 , 8 , 13 However, it remains unknown whether this selectivity applies more broadly, and what factors could influence the incorporation of protein subsets in vesicles.
Our data indicate that proximity to a source of free radicals is likely a key factor promoting the incorporation of selective proteins into MDVs. The main sources of free radicals in mitochondria are the flavin (NDUFV1‐3) and iron‐sulfur (NDUFS1‐8) containing subunits of complex I, 40 and the quinol‐oxidizing site of complex III (UQCRFS1, UQCRC1, UQCRC2). 26 , 40 In addition, several other sources of ROS exist, namely the flavin site of complex II (SDHA), 41 mitochondrial glycerol‐3‐phosphate dehydrogenase, the electron transferring flavoprotein system (ETFs) of fatty acid β‐oxidation, 42 and the dihydrolipoamide dehydrogenase (DLD) of three distinct 2‐oxoacid dehydrogenase complexes, namely OGDH, BCKDH, and PDH. 43 The fact that almost all of these proteins were preferentially present under moderate levels of oxidative stress in MDVs collected at ambient PO2, and that many of the proteins became enriched in presence of antimycin A (Figure 6), strongly suggests that they are among the most susceptible to damage when ROS emission is elevated. This also emphasizes the likely role that MDVs play the in cellular response to proteotoxic stress.
In addition to proximity to a source of radicals, susceptibility to oxidative modifications is also likely to promote selective incorporation of proteins into MDVs. Due to its relative stability, H2O2 generated by the dismutation of superoxide is one of the main reactive oxidants interacting with mitochondrial proteins, particularly protein thiols. 30 While the oxidation of most protein thiols occurs slowly, 44 some notable exceptions exist. This is the case for abundantly expressed redox enzymes such as SOD2, GPXs, PRXs, and TXN/TXNRDs which display accessible cysteine residues that are extremely reactive with their targets. 30 In addition, several metabolic enzymes including thiolases, aldehyde dehydrogenases, and TCA cycle dehydrogenases, harbor HRCRs, 29 most often in their catalytic site, which allow them to act as thiol switches for redox‐dependent regulation of metabolism. 30 However, under conditions of oxidative stress, H2O2 can cause irreversible sulfinic and sulfonic acid modifications that often lead to the catalytic inactivation of these enzymes. 30 Similarly, the activity or function of redox enzymes are also altered in presence of oxidative stress. Exposure of GPX1 to increasing concentrations of H2O2 is known to convert the selenocysteine residue at its active site to dehydroalanine (DHA) causing its inactivation. 45 , 46 PRX2 is inactivated by the hyperoxidation of its catalytic cysteine to cysteine sulfinic acid. 47 , 48 Moreover, the interaction of TRX with its target proteins is profoundly disrupted by the formation of disulfide bridges between two of their three non‐catalytic cysteine residues. 49 , 50 , 51 Our data thus suggest that irreversible cysteine oxidation could be a factor underlying the enrichment of proteins containing HRCRs in MDVs. Chemoproteomic methods should be used in future studies to directly quantify changes in HRCRs abundance in MDVs in response to graded oxidative stress.
Because of the well‐known susceptibility of Fe‐S clusters to oxidation, proteins harboring Fe‐S clusters and enzymes involved in cluster synthesis may also be particularly vulnerable to dysfunction and damage. For instance, it is well established that the TCA enzyme ACO2 harbors labile Fe‐S clusters that are released under oxidative stress, leading to catalytic inactivation. 30 , 52 Furthermore, during their synthesis, Fe‐S clusters are bound loosely to scaffold and transfer proteins, which facilitate their release during assembly. 53 However, under conditions of oxidative stress, the release of these labile Fe‐S clusters is likely to promote the accumulation of free iron in mitochondria, leading to further ROS production through the reaction between free iron and superoxide anion. 52 , 54 Our data thus raises the intriguing possibility that Fe‐S cluster containing/handling proteins in mitochondria are incorporated into MDVs under conditions of oxidative stress to eliminate irreversibly damaged proteins and prevent mitochondrial iron overload. While our data provide a rationale for the incorporation of specific sets of proteins into MDVs, the molecular mechanisms underlying their segregation and packaging into vesicles, however, remains to be determined.
4.2. MDV cargoes in extracellular vesicles
Over the last two decades, extracellular vesicles have emerged as important players in conveying and disseminating biological information between cells and across organs. Extracellular vesicles result either from the fusion of multivesicular bodies with the cellular membrane or from the protrusion and scission of the plasma membrane, which causes the release of either small vesicles (eg, exosomes) or large vesicles (eg, microvesicles) into the extracellular milieu. 12 Due to their different subcellular origins, extracellular vesicles can carry different subsets of proteins, lipids, and nucleic acids and the pathways leading to their specific cargo enrichment remain largely unknown. Results from the present study suggest that MDVs contribute to the presence of mitochondrial proteins in EVs. The efflux of mitochondrial proteins into EVs under stress conditions could reflect enhanced mitochondrial damage and/or the incapacity of cells to clear MDVs destined for lysosomal degradation. Alternately, an increasing amount of evidence suggests that mitochondria or mitochondrial components released into EVs may contribute to cell‐cell signaling. 55 , 56 , 57
In conclusion, our data provide an overall blueprint of mitochondrial proteins incorporated into MDVs under oxidative stress and provide a rationale for the incorporation of specific sets of proteins. Furthermore, the extensive overlap observed between the proteome of MDVs generated under oxidative stress and that of EVs generated by cells exposed to a variety of stressors suggests that vesicles are a potential source of biomarkers reflecting the presence of mitochondrial stress.
CONFLICT OF INTEREST
The authors report no conflicts of interest in this work.
AUTHOR CONTRIBUTIONS
Y. Burelle concepted the hypothesis; G. Vasam, KJ Menzies, and Y. Burelle designed the work; G. Vasam and VJJ Cadete performed the research and acquired the data; G. Vasam, Y. Burelle, and KJ Menzies drafted the manuscript; and all authors analyzed and interpreted the data and approved the final version of the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Supplemental File S1
Supplemental File S2
Supplemental File S3
ACKNOWLEDGMENTS
The authors are grateful for the technical support of LC‐MS sample preparation and processing from the Proteomic Resource Center of the Ottawa Institute of Systems Biology (OISB), University of Ottawa, especially Dr Zhibin Ning from the facility. TEM images were obtained with the help of the Facility for Electron Microscopy Research (FEMR), McGill University. This work was funded by grants from the Canadian Institutes of Health Research (MOP 136999, MOP 159455), and the Natural Sciences and Engineering Research Council of Canada (RGPIN 2016‐09932, RGPIN 2018‐06838, DGECR 2018‐00012) to YB and KJM. YB is a University of Ottawa Chair in Integrative Mitochondrial Biology. MLA holds an NSERC Discovery Grant and Rachel Nadeau holds an Ontario Graduate Scholarship as well as an NSERC Canada Graduate Scholarship.
Vasam G, Nadeau R, Cadete VJJ, Lavallée‐Adam M, Menzies KJ, Burelle Y. Proteomics characterization of mitochondrial‐derived vesicles under oxidative stress. The FASEB Journal.2021;35:e21278. 10.1096/fj.202002151R
REFERENCES
- 1. Mashburn LM, Whiteley M. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature. 2005;437(7057):422‐425. [DOI] [PubMed] [Google Scholar]
- 2. Deatherage BL, Cookson BT. Membrane Vesicle Release in Bacteria, Eukaryotes, and Archaea: a Conserved yet Underappreciated Aspect of Microbial Life. Infection and Immunity. 2012;80(6):1948‐1957. 10.1128/IAI.06014-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li Z, Clarke AJ, Beveridge TJ. Gram‐negative bacteria produce membrane vesicles which are capable of killing other bacteria. J Bacteriol. 1998;180(20):5478‐5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kadurugamuwa JL, Beveridge TJ. Membrane vesicles derived from Pseudomonas aeruginosa and Shigella flexneri can be integrated into the surfaces of other Gram‐negative bacteria. Microbiology. 1999;145(8):2051‐2060. [DOI] [PubMed] [Google Scholar]
- 5. Soubannier V, McLelland G‐L, Zunino R, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22(2):135‐141. [DOI] [PubMed] [Google Scholar]
- 6. Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. 2012;7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cadete VJJ, Deschênes S, Cuillerier A, et al. Formation of mitochondrial‐derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol. 2016;594(18):5343‐5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin‐17 delivers PINK1/parkin‐dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214(3):275‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neuspiel M, Schauss AC, Braschi E et al. Cargo‐selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18(2):102‐108. [DOI] [PubMed] [Google Scholar]
- 10. Cadete VJJ, Vasam G, Menzies KJ, Burelle Y. Mitochondrial quality control in the cardiac system: an integrative view. Biochim Biophys Acta—Mol Basis Dis. 2019;1865(4):782‐796. [DOI] [PubMed] [Google Scholar]
- 11. Sugiura A, McLelland G, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial‐derived vesicles. EMBO J. 2014;33(19):2142‐2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33(4):282‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol. 2008;26(12):1367‐1372. [DOI] [PubMed] [Google Scholar]
- 15. D506‐D515 . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506‐D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asara JM, Christofk HR, Freimark LM, Cantley LC. A label‐free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics. 2008;8(5):994‐999. [DOI] [PubMed] [Google Scholar]
- 17. Perez‐Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442‐D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731‐740. [DOI] [PubMed] [Google Scholar]
- 19. Smith AC, Robinson AJ. MitoMiner v3.1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 2016;44(D1):D1258‐D1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler—a web‐based toolset for functional profiling of gene lists from large‐scale experiments. Nucleic Acids Res. 2007;35(suppl. 2):W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reimand J, Isserlin R, Voisin V, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA. Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. 2019;18(2):623‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Merico D, Isserlin R, Bader GD. Visualizing gene‐set enrichment results using the cytoscape plug‐in enrichment map. Methods Mol Biol. 2011;781:257‐277. [DOI] [PubMed] [Google Scholar]
- 24. Kucera M, Isserlin R, Arkhangorodsky A, AutoAnnotate BGD. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Research. 2016;5:1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nanadikar MS, Vergel Leon AM, Borowik S, et al. O2 affects mitochondrial functionality ex vivo. Redox Biol. 2019;22:101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin‐inhibited mitochondrial Q‐cycle. J Biol Chem. 2011;286(36):31361‐31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. St‐Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277(47):44784‐44790. [DOI] [PubMed] [Google Scholar]
- 28. Matheoud D, Sugiura A, Bellemare‐Pelletier A, et al. Parkinson’s disease‐related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166(2):314‐327. [DOI] [PubMed] [Google Scholar]
- 29. Weerapana E, Wang C, Simon GM, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468(7325):790‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riemer J, Schwarzländer M, Conrad M, Herrmann JM. Thiol switches in mitochondria: operation and physiological relevance. Biol Chem. 2015;396(5):465‐482. [DOI] [PubMed] [Google Scholar]
- 31. Pallet N, Sirois I, Bell C, et al. A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics. 2013;13(7):1108‐1120. [DOI] [PubMed] [Google Scholar]
- 32. Jang SC, Crescitelli R, Cvjetkovic A, et al. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J Extracell Vesicles. 2019;8(1):1635420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gangoda L, Liem M, Ang C‐S, et al. Proteomic profiling of exosomes secreted by breast cancer cells with varying metastatic potential. Proteomics. 2017;17(23–24):1600370‐1600375. [DOI] [PubMed] [Google Scholar]
- 34. Colletti M, Petretto A, Galardi A et al. Proteomic analysis of neuroblastoma‐derived exosomes: new insights into a metastatic signature. Proteomics. 2017;17(23–24):1600430‐1600511. [DOI] [PubMed] [Google Scholar]
- 35. Cosme J, Guo H, Hadipour‐Lakmehsari S, Emili A, Gramolini AO. Hypoxia‐induced changes in the fibroblast secretome, exosome, and whole‐cell proteome using cultured, cardiac‐derived cells isolated from neonatal mice. J Proteome Res. 2017;16(8):2836‐2847. [DOI] [PubMed] [Google Scholar]
- 36. Jeannin P, Chaze T, Giai Gianetto Q et al. Proteomic analysis of plasma extracellular vesicles reveals mitochondrial stress upon HTLV‐1 infection. Sci Rep. 2018;8(1):5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang R, Rong C, Ke R et al. Differential proteomic analysis of serum exosomes reveals alterations in progression of Parkinson disease. Medicine (Baltimore). 2019;98(41):e17478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuang M, Tao X, Peng Y et al. Proteomic analysis of plasma exosomes to differentiate malignant from benign pulmonary nodules. Clin Proteomics. 2019;16(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheow ESH, Cheng WC, Lee CN, de Kleijn D, Sorokin V, Sze SK. Plasma‐derived extracellular vesicles contain predictive biomarkers and potential therapeutic targets for myocardial ischemic (MI) injury. Mol Cell Proteomics. 2016;15(8):2628‐2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH‐ubiquinone oxidoreductase (Complex I). J Biol Chem. 2011;286(31):27103‐27110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial Complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287(32):27255‐27264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perevoshchikova IV, Quinlan CL, Orr AL, Gerencser AA, Brand MD. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic Biol Med. 2013;61:298‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quinlan CL, Goncalves RLS, Hey‐Mogensen M, Yadava N, Bunik VI, Brand MD. The 2‐oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014;289(12):8312‐8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549‐561. [DOI] [PubMed] [Google Scholar]
- 45. Pigeolet E, Corbisier P, Houbion A, et al. Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev. 1990;51(3):283‐297. [DOI] [PubMed] [Google Scholar]
- 46. Blum J, Fridovich I. Inactivation of glutathione peroxidase by superoxide radical. Arch Biochem Biophys. 1985;240(2):500‐508. [DOI] [PubMed] [Google Scholar]
- 47. Cho C‐S, Lee S, Lee GT, Woo HA, Choi E‐J, Rhee SG. Irreversible Inactivation of Glutathione Peroxidase 1 and Reversible Inactivation of Peroxiredoxin II by H2O2 in Red Blood Cells. Antioxid Redox Signal. 2010;12(11):1235‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang K‐S, Kang SW, Woo HA et al. Inactivation of human peroxiredoxin i during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine‐sulfinic acid. J Biol Chem. 2002;277(41):38029‐38036. [DOI] [PubMed] [Google Scholar]
- 49. Du Y, Zhang H, Zhang X, Lu J, Holmgren A. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J Biol Chem. 2013;288(45):32241‐32247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Watson WH, Pohl J, Montfort WR et al. Redox potential of human thioredoxin 1 and Identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278(35):33408‐33415. [DOI] [PubMed] [Google Scholar]
- 51. Hashemy SI, Holmgren A. regulation of the catalytic activity and structure of human thioredoxin 1 via oxidation and S ‐nitrosylation of cysteine residues. J Biol Chem. 2008;283(32):21890‐21898. [DOI] [PubMed] [Google Scholar]
- 52. Cantu D, Schaack J, Patel M. Oxidative Inactivation of Mitochondrial Aconitase Results in Iron and H2O2‐Mediated Neurotoxicity in Rat Primary Mesencephalic Cultures. Rutherford J, ed. PLoS One. 2009;4(9):e7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boyd ES, Thomas KM, Dai Y, Boyd JM, Outten FW. Interplay between oxygen and Fe–S cluster biogenesis: insights from the Suf pathway. Biochemistry. 2014;53(37):5834‐5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ibrahim WH, Habib HM, Kamal H, St. Clair DK, Chow CK. Mitochondrial superoxide mediates labile iron level: evidence from Mn‐SOD‐transgenic mice and heterozygous knockout mice and isolated rat liver mitochondria. Free Radic Biol Med. 2013;65:143‐149. [DOI] [PubMed] [Google Scholar]
- 55. Puhm F, Afonyushkin T, Resch U et al. Mitochondria are a subset of extracellular vesicles released by activated monocytes and induce type I IFN and TNF responses in endothelial cells. Circ Res. 2019;125(1):43‐52. [DOI] [PubMed] [Google Scholar]
- 56. Zhang Q, Raoof M, Chen Y et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221‐225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
Supplemental File S1
Supplemental File S2
Supplemental File S3