

Abstract
Introduction
Vitamin K antagonists (VKA) and non‐vitamin K oral antagonist anticoagulants (NOAC) are used in the clinic to reduce risk of thrombosis. However, they also exhibit vascular off‐target effects. The aim of this study is to compare VKA and NOAC on atherosclerosis progression and calcification in an experimental setup.
Material and methods
Female Apoe −/− mice (age 12 weeks) were fed Western‐type diet as control or supplemented with dabigatran etexilate or warfarin for 6 or 18 weeks. Vascular calcification was measured in whole aortic arches using µCT and [18F]‐NaF. Atherosclerotic burden was assessed by (immuno)histochemistry. Additionally, in vitro effects of warfarin, thrombin, and dabigatran on primary vascular smooth muscle cells (VSMC) were assessed.
Results
Short‐term treatment with warfarin promoted formation of atherosclerotic lesions with a pro‐inflammatory phenotype, and more rapid plaque progression compared with control and dabigatran. In contrast, dabigatran significantly reduced plaque progression compared with control. Long‐term warfarin treatment significantly increased both presence and activity of plaque calcification compared with control and dabigatran. Calcification induced by warfarin treatment was accompanied by increased presence of uncarboxylated matrix Gla protein. In vitro, both warfarin and thrombin significantly increased VSMC oxidative stress and extracellular vesicle release, which was prevented by dabigatran.
Conclusion
Warfarin aggravates atherosclerotic disease activity, increasing plaque inflammation, active calcification, and plaque progression. Dabigatran lacks undesired vascular side effects and reveals beneficial effects on atherosclerosis progression and calcification. The choice of anticoagulation impacts atherosclerotic disease by differential off target effect. Future clinical studies should test whether this beneficial effect also applies to patients.
Keywords: atherosclerosis, NOAC, oxidative stress, vascular calcification, vascular smooth muscle cells, VKA
Essentials.
Although lowering coagulation tendency to the same level, VKA and NOAC have vascular off‐target effects impacting atherosclerosis and calcification.
However, the precise mechanism underlying the effect on atherosclerosis is not known.
VKA aggravate atherosclerosis and vascular calcification whereas NOAC inhibit atherosclerotic plaque progression.
The results confirm that choice of venous and arterial thrombosis treatment impacts cardiovascular disease.
1. INTRODUCTION
Upon atherosclerotic plaque rupture or erosion, atherothrombosis may cause the occlusion of arteries, which remains the main cause of morbidity and mortality worldwide. 1 , 2 For decades, vitamin K antagonists (VKAs) have been the most widely used anticoagulant drugs for treatment of patients at risk of arterial and venous thrombosis. Because of unfavorable pharmacokinetics of VKAs, direct thrombin and factor Xa (FXa) inhibitors (non‐vitamin K antagonist oral anticoagulants, NOACs) have been introduced to the clinic as alternative anticoagulants.
Both VKAs and NOACs are proven effective in reducing thrombotic risk; however, they also influence nonhemostatic activities of coagulation factors. VKAs exhibit their side effects on the vessel wall via vitamin K–dependent proteins. 1 , 2 , 3 , 4 , 5 In the vasculature, vascular smooth muscle cells (VSMCs) synthesize the vitamin K–dependent matrix Gla protein (MGP), which acts as a local vascular calcification inhibitor. 6 Inhibition of MGP activity by VKA treatment is associated with increased vascular calcification. 7 VKA treatment increases intimal calcification in patients and experimental animal models of atherosclerosis. 8 , 9 The consequences of vascular calcification on plaque stability are debated and dependent on the extent of calcification, size, localization, and density. 10 Macrocalcification, or sheet‐like calcification, and dense calcification are suggested to stabilize the atherosclerotic plaque. 11 In contrast, microcalcification or spotty calcification as well as increased calcification volume have been implicated in promoting plaque instability and subsequently increasing risk of plaque rupture and risk for stroke and myocardial infarction. 12 , 13 , 14 Microcalcifications are metabolically active and can be detected with the positron emission tomography tracer [18F]‐NaF, which identifies high‐risk coronary atherosclerotic plaques 15 and disease progression in patients with aortic stenosis. 16 Recently, it was shown that VKA use was associated with increased spotty coronary artery calcification. 17 VKAs have been used for decades in patients at risk for thrombosis. In vivo studies showed that VKAs induce valvular and arterial calcification in a dose‐ and time‐dependent manner. 9 , 18 Supplementation with vitamin K was shown to prevent and even reverse the calcifying effects of VKAs in rats. 19
The NOAC dabigatran etexilate (DE) was one of the first non‐vitamin K oral antagonists on the market. DE is an oral prodrug that is rapidly converted by esterases to dabigatran, which is a direct, competitive inhibitor of thrombin (FIIa). Thrombin is a key enzyme in the coagulation cascade, ensuring the conversion of fibrinogen into fibrin. Thrombin has also a wide spectrum of effects directly on the vessel wall, which contribute to atherogenesis. 20 , 21 Increased thrombin formation has been shown to aggravate atherogenesis. 22 Inhibition of thrombin by dabigatran reduced plaque size and atherogenesis. 22 , 23 Because dabigatran exhibits these beneficial effects on reducing plaque size, we addressed the question whether dabigatran also has an effect on plaque calcification. Using a preclinical experimental animal model of atherosclerosis, we assessed the short‐ and the long‐term effects of the VKA warfarin and the NOAC dabigatran on atherosclerotic plaque calcification and atherogenesis.
2. METHODS
2.1. Experimental animals
All animal studies were performed under an approved protocol by the Ethics Committee for animal experiments of Maastricht University. Twelve‐week‐old female C57/BL6 Apoe −/− mice were purchased from Maastricht University and housed in climate‐controlled spaces under 12‐hour day/night cycle with ad libitum access to food and water. In our model, we used female mice because they develop more advanced atherosclerotic lesions. 24 Mice were fed an irradiated (0.9 Mrad) vitamin K–deficient Western type diet (WTD: 0.25% cholesterol and 15% cocoa butter, derived from Altromin, Lage, Germany). WTD was supplemented with corn oil–dissolved vitamin K1 (5 mg/kg, Merck KGaA, Darmstadt, Germany) and was used as control diet or supplemented with DE, 7.5 mg/g (~22.5 mg/mouse/day), Boehringer Ingelheim, Biberach, Germany) for NOAC treatment. Additionally, WTD was supplemented with warfarin (3 mg/g warfarin (~9 mg/mouse/day); Merck KGaA, Darmstadt, Germany) and vitamin K1 (1.5 mg/g K1, Merck KGaA) for VKA treatment. Vitamin K1, which counteracts warfarin's effects predominantly in the liver and less in extrahepatic tissue, was additionally added to the warfarin diet to prevent bleeding. 9 , 25 Animals were scored every day for well‐being and bleedings because of the potential bleeding effects of warfarin and dabigatran.
Mice were randomly divided to receive WTD (control), DE, or warfarin (warfarin) supplemented food for 6 or 18 weeks (n = 12 per treatment per time point; Figure 1A). Thereafter, mice were sacrificed, and blood was collected in 105 mM trisodium citrate via the portal vein. Plasma was prepared and frozen in aliquots at −80oC until analysis. Next, the vasculature was washed with a sterile binding buffer (40 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 2.5 mM CaCl2, pH 7.3) via the left ventricle. Aortas were harvested, dissected, and fixed in 1% (v/v) HEPES‐buffered formaldehyde containing 150 mM saline, overnight at 4°C, before they were embedded in paraffin. Before sections were cut, paraffin embedded aortas were analyzed ex vivo for calcification using µCT.
FIGURE 1.
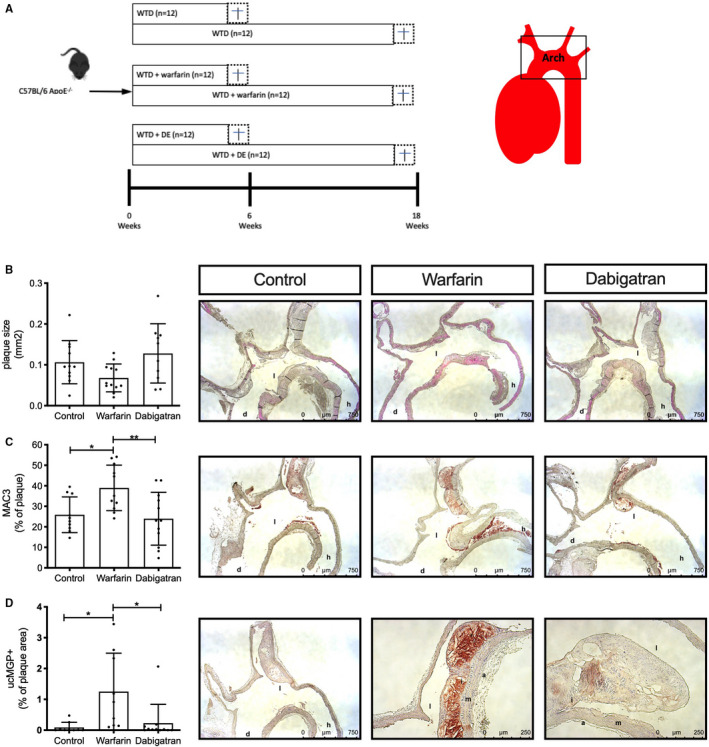
Short‐term oral anticoagulant effects on atherosclerotic lesions. (A) Schematic overview of experimental setup of the animal experiment. (B) Apoe −/− mice were treated for 6 weeks with WTD without or with warfarin or dabigatran, after which animals were sacrificed. Aortic arches of Apoe −/− mice on WTD without or with warfarin or dabigatran showed no significant difference on plaque size at 6 weeks of treatment. (C) MAC3+ cells were significantly increased in lesions of warfarin‐treated mice compared with control (p < .05) and dabigatran (p < .01). (D) Six weeks of warfarin treatment significantly increased ucMGP in atherosclerosis lesions compared to control and dabigatran. a, adventitia; d, aorta descendens; h, heart; i, intima; l, lumen; m, media; ucMGP, uncarboxylated matrix Gla protein; WTD, Western type diet
2.2. Blood analysis
Warfarin and dabigatran plasma levels were determined using high‐performance liquid chromatography and Hemoclot thrombin inhibitor assay (Hyphen Biomed, Neuville‐sur‐Oise, France), respectively. The prothrombin time of plasma, a measure for clotting tendency, was determined according to standard laboratory methods using the appropriate tests. Total plasma cholesterol and triglyceride levels were assessed with the Synchron LX20 (Beckman Coulter, Brea, CA) according to the manufacturer's instructions.
2.3. [18F]‐NaF
Mice (n = 7, 6, and 8 for control, warfarin, and DE, respectively) were fasted for 2 hours before intravenous injection with [18F]‐NaF. Thirty minutes postinjection, the animals were killed by an overdose of pentobarbital and the vasculature was washed with a sterile binding buffer (40 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 2.5 mM CaCl2, pH 7.3) via the left ventricle. Aortic arch was dissected and [18F]‐NaF uptake was measured ex vivo using autoradiography and with a gamma counter (Wallac Wizard, Turku, Finland). Uptake was corrected for injected dose and time.
2.4. µCT analysis of aortic calcification
Ex vivo µCT scans were performed to quantify calcification in the whole aortic tissue (n = 11 for control and DE; n = 12 for warfarin treatment). The paraffin‐embedded aortas were scanned for 40/50 minutes by µCT (NanoSPECT/CT, Bioscan, USA) using 45 keV energy, and 360 projections with a resolution of 0.1 mm. Scan data were assessed for Hounsfield units (HU), with HU >1000 considered as positive calcification.
2.5. Immunohistochemistry
Embedded aortic sections were cut at 4‐µm thickness. Plaque size and volume were determined using hematoxylin and eosin staining (n = 10 for control and DE; n = 11 for warfarin treatment). Additionally, plaque characteristics were determined using histochemical analysis for calcification (Alizarin Red S), and elastin fiber breaks (Elastica von Giesson). Stained sections were analyzed by ImageJ Software. Elastin breaks found beneath the atherosclerotic plaque were scored in different sections for every mouse, using a scoring system ranging from 0 to 4; 0, no breaks; 1, one to three breaks; 2, four to six breaks; 3, seven to nine breaks; and 4, >9 breaks. All immunohistochemical scoring were performed blinded to the treatment groups.
The following antibodies were used: anti‐uncarboxylated matrix Gla‐protein (1:400; IDS, Boldon, UK), anti‐bone morphogenetic protein 4 (BMP4; 1:50; Santa Cruz Biotechnology, Dallas, TX), anti‐8‐hydroxyguanosine (8OHdG; 1:300; Meridian Life Science, TN), anti‐MAC3 (1:50, Bioscience, Merck KGaA), and anti‐integrin ß 3 (ITGB3 1:100, Sigma‐Aldrich). Horseradish peroxidase–conjugated secondary antibodies were visualized with Nova‐RED substrate (Vector Labs, Amsterdam, Netherlands). Two‐dimensional analysis quantification was performed using Leica Application Suite X (Leica Microsystems, Wetzlar, Germany). Data are expressed as the percentage of positive plaque area unless otherwise stated.
2.6. Cell culture
Human primary VSMCs were derived from tissue explants from males and females aged between 18 and 65 years of age and cultured as described previously. 26 In brief, VSMCs were cultured in DMEM medium (Gibco, Thermo Fisher Scientific Inc, Paisley, Scotland, UK) supplemented with 20% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were used between passage 4 and 10.
2.7. Calcification assays
VSMCs were placed in calcifying medium consisting of DMEM supplemented with 2.5% FBS and 1% PS in the presence of an additional 1.8 mmol/l calcium. After incubation with stimulus, calcium deposits were assessed by the O‐cresolphthalein method and calcium content was normalized for protein content.
2.8. VSMC proliferation
xCELLigence VSMCs were seeded in 96‐xCELLigence‐well. After 24 hours, stimulus was added directly in the well. The slope of impudence over 3 days was determined as an indication of proliferation. EdU and VSMCs were seeded in a 24‐well plate. The cells were allowed to adhere overnight, then the medium was replaced with stimulus and EdU (5‐Ethynyl‐2'‐deoxyuridine; 10 µM, Life Technologies) containing 0.5% FBS. After 24 hours, cells were washed twice and fixed using 4% paraformaldehyde (Merck KGaA). Cells were then incubated with staining solution (0.1 M Tris, 1 mM CuSO4, 10.5 µM azide‐F488, and 0.1 M ascorbic acid) for 1 hour in the dark. Cells were counterstained with Hoechst (1 µg/ml) for 15 min. Green fluorescent protein and DAPI images were obtained using Cytation 3 Cell Imaging Multi‐Mode Reader (BioTek Instruments Inc., Winooski, VT) and % of green fluorescent protein–positive cells over all cells were calculated.
2.9. VSMC migration
VSMC migration was determined using xCELLigence using the Boyden chamber principal. First, VSMCs were starved overnight in 0.5% FBS. The next day, bottom and upper chambers were coated with collagen (0.1 mg/ml collagen G, Biochrome MERCK). Thereafter, 40,000 VSMC were placed in the upper chamber and migration was analyzed over the following 4 hours.
2.10. Quantitative polymerase chain reaction
RNA extraction, complementary DNA synthesis, and quantitative polymerase chain reaction were performed as described previously. 27 The following primers were used: monocyte chemoattractant protein 1 (MCP1) forward; 5′‐CCCCAGTCACCTGCTGTTAT‐3′, MCP1 reverse; 5′‐TGGAATCCTGAACCCACTTC‐3′, interferon (IFN)‐γ forward; 5′‐CCAACGCAAAGCAATACATGA‐3′, IFN‐γ reverse; 5′‐CCTTTTTCGCTTCCCTGTTTTA‐3′, interleukin (IL)‐1ß forward; 5′‐AAACCTCTTCGAGGCACAAG‐3′, IL‐1ß reverse; 5′‐GTTTAGGGCCATCAGCTTCA‐3, tumor necrosis factor (TNF)‐α forward; 5′‐TGCACTTTGGAGTGATCGGC‐3′, TNF‐α reverse; 5′‐AGCTTGAGGGTTTGCTACAACA‐3′; and for normalization: GAPDH forward; 5′‐AACGGATTTGGTCGTATTGGGC‐3′, GAPDH reverse; 5′‐CTTGACGGTGCCATGGAATTTG‐3′.
2.11. Messenger RNA isolation for micro‐assay
VSMCs were fasted overnight and stimulated with thrombin, warfarin, or control for 4 hours. We have chosen to compare thrombin and warfarin because both are known to affect VSMC phenotypes relevant for calcification. After 4 hours, VSMCs were washed in ice‐cold phosphate‐buffered saline before RNA was isolated using the miRNeasy Mini Kit (Qiagen, The Netherlands) including a DNase treatment, according to the manufacturer's protocols. The concentration of total RNA was measured on a Nanodrop ND‐1000 spectrophotometer (Thermofisher, The Netherlands). The integrity of total RNA was checked using RNA Nanochips on a 2100 Bioanalyzer (Agilent Technologies, The Netherlands). Only samples with an RNA Integrity Number > 8.9 were used for microarray analysis.
2.12. Microarray preparation and data validation
A total amount of 200 μg RNA of each sample was used as input material for synthesizing dye (cyanine 3) labeled complementary RNA according to the Agilent one‐color Quick‐Amp labeling protocol (Agilent Technologies, Amstelveen, The Netherlands). Individual samples were hybridized to an Agilent SurePrint G3 Human Gene Expression 8 × 60 K v2 (Agilent Technologies, Santa Clara, CA). The microarrays were scanned using the Agilent Microarray Scanner (Agilent Technologies, Amstelveen, The Netherlands). Pixel intensities were extracted as raw data from the scan images using Agilent Feature Extraction Software (Agilent). Quantile normalization and data processing were performed using ArrayQC (https://github.com/BiGCAT‐UM/arrayQC_Module/), a quality control pipeline in R (version 2.10.1; The R Foundation for Statistical Computing, Vienna, Austria). For each spot, the following steps were taken: local background correction, flagging of bad spots, controls, and spots with too low of an intensity, log2 transformation, and quantile normalization. Probes with no flagged bad spots were selected and repeated identifiers merged, resulting in 21,532 transcripts, representing 15,163 unique known genes, and were used for statistical analysis.
2.13. Selection of differentially expressed genes
Differentially expressed genes were selected using the linear model for microarrays approach. 28 The resulting p values were FWER‐corrected using the false discovery rate method. The following criteria were applied: (1) a false discovery rate–corrected p value <.05 obtained through a moderated t‐test, and (2) for the biological replicates an average absolute fold change of 1.5 or higher. Selected gene data sets used “inflammation” and “atherosclerosis” are available on disgenet.
2.14. Intracellular generation of oxidative stress
To identify intracellular reactive oxygen species (ROS), we used the cell‐permeable fluorogenic substrate 2,7‐dichlorofluorescein diacetate (Merck KGaA), which is oxidized to 2,7‐dichlorofluorescein in the presence of oxidants.
VSMCs were seeded in a 96‐well plate and left to adhere overnight. Next, cells were incubated with 20 µM 2,7‐dichlorofluorescein diacetate for 60 min in the dark at 37°C and 5% CO. After washing, cells were incubated with stimulus in combination with Hoechst (1 μg/ml) and fluorescence intensity was measured (Excitation 485, Emission 529) with Cytation 3 Cell Imaging Multi‐Mode Reader (BioTek Instruments Inc., Winooski, VT) for a total of 30 min. Fluorescence intensity was corrected for background measurement and normalized to cell count.
2.15. Extracellular vesicles quantification
VSMCs were seeded in a 12‐well plate and allowed to adhere overnight. Next, VSMCs were stimulated in DMEM 31966, supplemented with 0.5% FBS and 1% PS for 24 hours, after which medium was collected. Medium was centrifuged at 3000g and incubated with CD‐63 coupled beads overnight. After washing with phosphate‐buffered saline 2% bovine serum albumin, beads were incubated with secondary antibody CD81‐APC (1:50; BD Biosciences) and incubated for 60 min in the dark. After washing, exosomes were detected by flow cytometry (BD Accuri C6). Exosome secretion is expressed as arbitrary units, which were calculated as follows: median fluorescence was multiplied by percentage positive beads and this was normalized for cell number.
2.16. Statistical analysis
All in vitro data were obtained in three or more independent experiments in triplicate (or more) wells. Data are expressed as mean with standard deviation. Data were analyzed using the Mann‐Whitney t‐test or Kruskal‐Wallis one‐way analysis of variance using PRISM software (GraphPad 8.2.1, San Diego, CA). Values of p < .05 were considered statistically significant.
3. RESULTS
Female BL6/C57‐Apoe −/− mice were fed a WTD (control) or WTD supplemented with VKA (warfarin) or DE for 6 or 18 weeks (Figure 1A). After 18 weeks of treatment, no difference was observed in weight, cholesterol, and triglyceride levels between the groups (Table 1). Increased warfarin and dabigatran plasma levels confirmed respective treatment. Moreover, warfarin significantly increased prothrombin time compared with control, confirming anticoagulant effect (Table 1).
TABLE 1.
Model validation of control, warfarin, and dabigatran after 18 weeks of treatment
| Control | Warfarin | Dabigatran | |
|---|---|---|---|
| Weight (g) | 23.82 ± 1.60 | 22.50 ± 1.68 | 24.33 ± 2.46 |
| Cholesterol (mM) | 21.27 ± 3.34 | 21.54 ± 3.07 | 18.93 ± 1.74 |
| Triglycerides (mM) | 2.77 ± 0.20 | 2.96 ± 0.28 | 2.70 ± 0.17 |
| Dabigatran (ng/ml) | — | — | 370.3 ± 175.9 |
| Warfarin (mg/l) | — | 11,52 ± 5,81 | — |
| Prothrombin time (s) | 8.73 ± 0.83 | 54.2 ± 22.7* | 12.5 ± 4.28 |
p < .05
3.1. Effect of short‐term oral anticoagulation on atherosclerotic development
Serial sections of the aortic arch were used to visualize atherosclerotic plaques. First, we compared two methods to express plaque size: (1) plaque size based on the section with the largest measured plaque area in mm2 or (2) plaque size based on volume (mm3) determined by reconstructing every fifth section of serially cut specimen. The two methods showed a very strong correlation (r = 0.911, p < .0001; Figure S1A); thus, because of time, we selected the first method and expressed plaque size in mm2. Short‐term (6 weeks) treatment did not significantly alter plaque size between groups, although warfarin showed a trend toward smaller plaques compared with control and dabigatran treatment (p = .24 and p = .09 for warfarin vs control and DE, respectively; Figure 1B). Next, we measured the inflammatory status of plaques by staining macrophage and macrophage‐like cells using anti‐MAC3 antibodies. Short‐term warfarin treatment significantly increased MAC3 positive staining (38.9% of plaque area) compared with both control (25.8% of plaque area) and dabigatran treatment (23.9% of plaque area) (Figure 1C). At 6 weeks of treatment, Alizarin Red S stain did not detect any calcification in any of the groups (data not shown). However, warfarin treatment significantly increased uncarboxylated MGP (ucMGP) in atherosclerotic lesions as compared to both control and dabigatran (p < .05; Figure 1D). Furthermore, a trend of increased integrin ß3 expression was observed after warfarin treatment compared with dabigatran (p = .09; Figure S1B). No significant difference was observed in smooth muscle cell content and oxidative stress levels (8‐OHdG) between the plaques of short‐term treatment groups (data not shown).
3.2. Long‐term VKA treatment increases active vascular calcification
We next determined long‐term (18 weeks of treatment) effects of warfarin treatment on plaque progression and calcification and compared those with control and dabigatran treatment. Warfarin treatment significantly increased atherosclerotic plaque size compared with dabigatran treatment by some 1.7‐fold after 18 weeks (0.54 mm2 and 0.31 mm2 lesion size, respectively; Figure 2A). Next, the amount of vascular calcification was detected by μCT in whole mouse aortic arches. Warfarin treatment for 18 weeks significantly increased vascular calcification compared to control (30.8 ± 31.9 and 6.22 ± 4.59 total HU for warfarin and control, respectively; p < .05) and dabigatran (3.77 ± 6.57 total HU; p < .01) (Figure 2B). Dabigatran treatment showed a borderline significant reduction of vascular calcification compared with control (p = .07). Second, vascular calcification at 18 weeks was assessed histologically by Alizarin Red S staining of serial sections. Warfarin treatment showed a significant increase in plaque calcification compared with both control and dabigatran treatment (61%, 33%, and 21% of plaque area for warfarin, control, and DE, respectively; Figure 2D). Detection of calcification by μCT and Alizarin Red S showed a strong and significant correlation (r = .72, p < .05; Figure 2C). To determine whether warfarin induces active calcification, calcification was investigated using the positron emission tomography tracer [18F]‐NaF. 15 After 18 weeks of treatment, [18F]‐NaF uptake by aortas, as measured ex vivo, was significantly increased by warfarin treatment compared with control and dabigatran treatments (Figure 2E). Autoradiography showed that [18F]‐NaF activity predominantly localizes to the aortic arch where the atherosclerotic burden is highest (Figure 2F).
FIGURE 2.
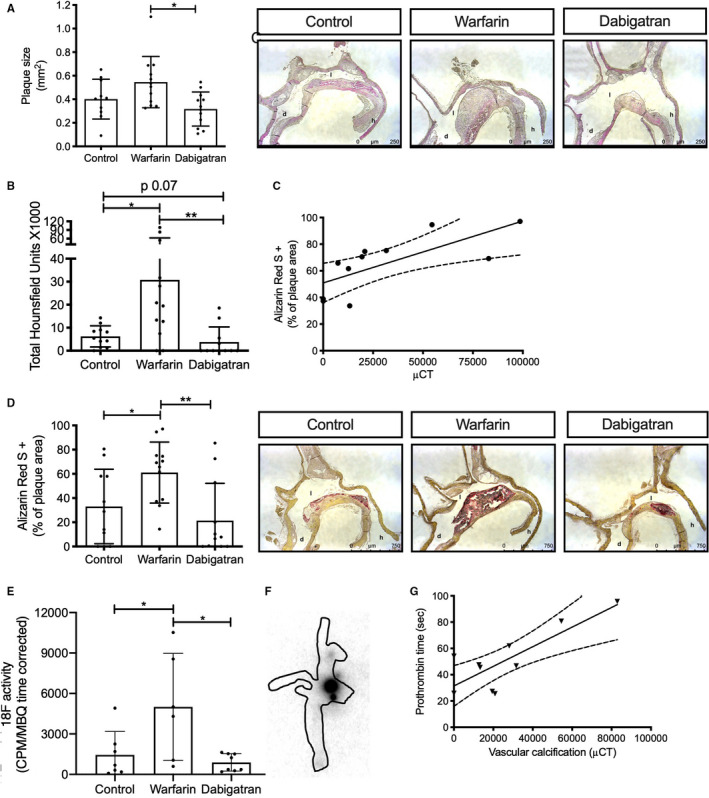
Long‐term oral anticoagulant treatment has a differential effect on vascular calcification. Apoe −/− mice were treated for 18 weeks with WTD without or with warfarin or dabigatran, after which animals were sacrificed. (A) Aortic plaque size was significantly increased after 18 weeks of warfarin treatment compared with dabigatran treatment (p < .05). (B) Vascular calcification detected by µCT on whole aortic arches showed significant increased calcification after 18 weeks of warfarin treatment compared with control (p < .05) and dabigatran (p < .01). (C) Calcification detected by µCT and Alizarin Red S strongly correlate (r = 0.72, p < .05). (D) Calcification in atherosclerotic lesions was visualized by Alizarin Red S and was significantly increased after warfarin treatment compared with control (p < .05) and dabigatran (p < .01). (E) [18F]‐NaF activity in whole aortic arches was significantly increased after 18 weeks warfarin treatment compared with control and dabigatran (p < .05). (F) Visualization of [18F]‐NaF in an aortic arch by autoradiography. (G) In warfarin‐treated mice, amount of vascular calcification correlates significantly with anticoagulation as measured by elevated prothrombin time (r = 0.81, p < .01). d, aorta descendens; h, heart; l, lumen; WTD, Western type diet
To investigate whether calcification was associated with vitamin K deficiency, we determined the correlation between anticoagulation (prothrombin time) and vascular calcification. Increased vascular calcification in the warfarin treated group correlated strongly with prothrombin time after 18 weeks of treatment (r = 0.81, p < .01; Figure 2G).
3.3. Differential effects of oral anticoagulation on atherogenesis
Vascular calcification is also linked to plaque progression and used in the clinic to determine cardiovascular risk. 29 Therefore, we calculated the increase in plaque progression as an x‐fold increase in plaque size from 6 to 18 weeks of treatment. Warfarin treatment showed a significant 7.2‐fold increase in plaque progression compared with a 3.7‐fold increase in control (p < .05) and a 1.9‐fold increase in dabigatran treatment (p < .001). Moreover, dabigatran treatment significantly reduced plaque progression compared with control (p < .01; Figure 3A). To further test whether calcification is related to vascular vitamin K deficiency, ucMGP positivity in the plaques was analyzed. Warfarin significantly increased ucMGP in atherosclerotic plaques (0.89% of plaque area) compared with control (0.09% of plaque area; p < .001) and dabigatran (0.38% of plaque area; p < .05) (Figure 3B). UcMGP positivity was also significantly increased in dabigatran compared with control treatment (Figure 3B). To examine the contribution of MGP in the process of calcification we correlated ucMGP presence with vascular calcification detected with Alizarin Red S. UcMGP significantly correlates with vascular calcification in atherosclerotic plaques (r = 0.58, p < .01; Figure S1C). Moreover, effects of increased ucMGP in atherosclerotic plaques was determined by assessing BMP4, a known downstream effector and promoter of osteochondrogenic differentiation. 30 Similar to ucMGP, warfarin treatment significantly increased BMP4 in the atherosclerotic plaque compared with both control and dabigatran (Figure 3C). In contrast to ucMGP, no difference was observed between dabigatran and control.
FIGURE 3.
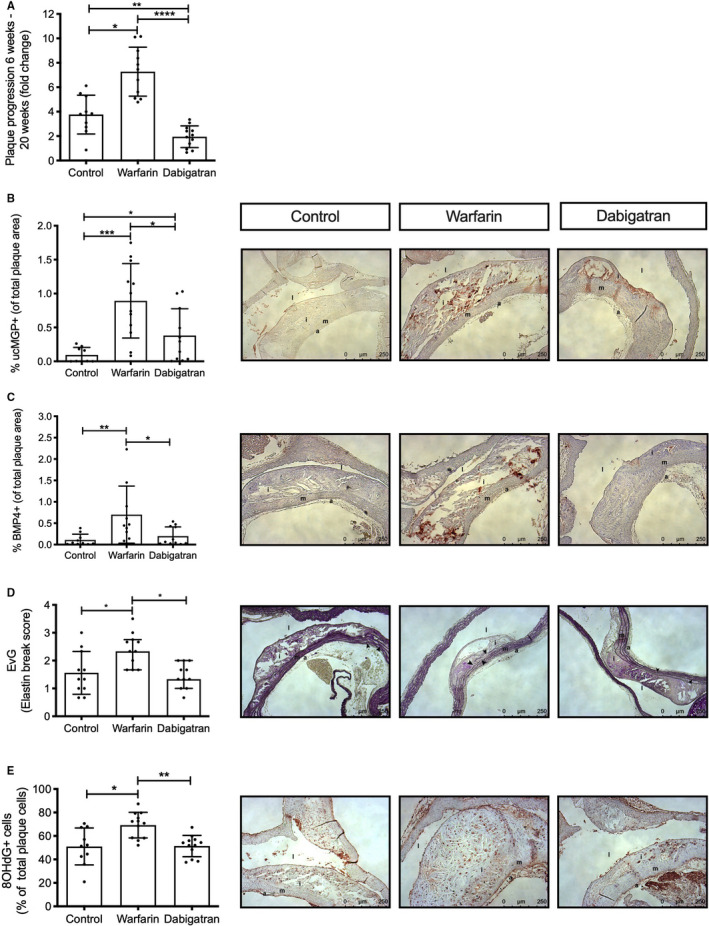
Long‐term warfarin and not dabigatran promote plaque vulnerability. Apoe −/− mice were treated for 18 weeks with WTD without or with warfarin or dabigatran, after which animals were sacrificed. (A) Calculating plaque progression based on the fold change of plaque size measured at 6 and 18 weeks of treatment demonstrated significantly higher plaque progression after warfarin treatment compared with control (p < .05) and dabigatran (p < .0001). Moreover, dabigatran treatment significantly reduced plaque progression compared with control (p < .01). (B) Vascular vitamin K deficiency was analyzed by uncarboxylated matrix Gla protein (ucMGP) presence in atherosclerotic lesions. The ucMGP was significantly increased in warfarin treated mice compared with control (p < .001) and dabigatran (p < .05). Moreover, dabigatran treatment significantly increased ucMGP presence compared with control (p < .05). (C) Presence of bone morphogenetic protein 4 (BMP4), a marker for bone formation, in plaques was only elevated in warfarin‐treated mice (p < .01 and p < .05 for control and dabigatran, respectively). (D) Warfarin treatment resulted in significant more elastin breaks under atherosclerotic lesions compared with control and dabigatran (p < .05). (E) Oxidative stress, measured by presence of 8OHdG, a marker of oxidative stress to DNA and a risk factor for atherosclerosis, was significantly increased after warfarin treatment compared with control (p < .05) and dabigatran (p < .01). a, adventitia; d, aorta descendens; h, heart; i, intima; l, lumen; m, media; WTD, Western type diet
3.4. Long‐term VKA treatment induces a vulnerable atherosclerotic plaque phenotype
Next, plaque composition was determined to gain insight into plaque vulnerability. Elastin breaks in the tunica media underneath the atherosclerotic plaque were significantly more abundant after warfarin treatment (score 2.3) compared with control (score 1.5) or dabigatran treatment (score 1.3) (p < .05; Figure 3D). Oxidative stress is considered a major cause of vascular calcification. 31 , 32 Oxidative stress, measured by 8OHdG+ cells, was significantly increased after 18 weeks warfarin treatment compared with control and dabigatran (Figure 3E). No difference was observed in smooth muscle cell content, macrophages (MAC3), matrix metalloproteinases 2 and 9, or necrotic core size between any of the groups (data not shown).
3.5. Systemic inflammation is not affected by warfarin or dabigatran treatment
After 18 weeks of treatment, cytokine levels were determined in plasma to investigate whether systemic inflammation in involved in OAC effects on atherosclerosis. No difference in cytokines levels were found between any of the treatment regimens (Table S1).
4. WARFARIN PROMOTES AN INFLAMMATORY RESPONSE IN VASCULAR SMOOTH MUSCLE CELLS
Because warfarin treatment induced a proinflammatory phenotype at 6 weeks of treatment, we stimulated VSMC with warfarin. Proliferation and migration of VSMC is a key feature in the genesis of atherosclerosis. Unexpectedly, warfarin treatment reduced proliferation to 68% compared with control as measured by xCELLigence (Figure 4A). Reduced VSMC proliferation by warfarin was confirmed by lower EdU incorporation in a dose‐dependent manner (p < .001; Figure 4B). Moreover, warfarin significantly reduced VSMC migration compared with control (Figure 4C). Because warfarin treatment for 6 weeks increased MAC3+ cells in the atherosclerotic plaque, we measured effects of warfarin on VSMC inflammation. VSMC treated with warfarin for 4 hours significantly increased MCP1 (1.6‐fold; Figure 4D), IFN‐γ (4.3‐fold; Figure 4E), and IL‐1b (2‐fold; Figure 4F) messenger RNA levels. Although not significant, warfarin treatment also increased TNF‐α messenger RNA levels (Figure 4G).
FIGURE 4.
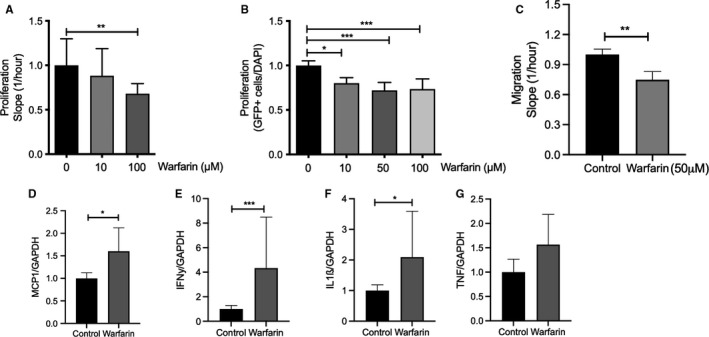
Differential effect of warfarin in vascular smooth muscle cells. Primary human vascular smooth muscle cells (VSMCs) were cultured in the presence of warfarin or vehicle. (A, B) Warfarin treatment significantly reduced VSMC proliferation and migration (C). Warfarin (50 µM) significantly promoted inflammatory messenger RNA levels of (D) MCP1, (E) IFN‐γ, (F) IL‐1ß, and (G) elevated TNF‐α. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor
4.1. Interaction of warfarin but not dabigatran with oxidized low‐density lipoprotein
The experimental animal model used to investigate the effect of oral anticoagulation was the BL6/C57‐Apoe −/−, which is known for its increased cholesterol levels. We tested potential synergistic effects of warfarin or dabigatran in combination with oxidized low‐density lipoprotein (oxLDL). Although oxLDL alone was a significant inducer of VSMC calcification (4.5‐ to 6.4‐fold compared with control), no effect of additional dabigatran on VSMC calcification was observed (Figure 5A). Moreover, dabigatran alone did not influence VSMC calcification (Figure 5A). On the other hand, warfarin treatment alone significantly increased VSMC calcification (2‐fold; Figure 5B). Co‐stimulation of warfarin with oxLDL significantly increased VSMC calcification compared with either warfarin or oxLDL alone (2.2‐fold compared with oxLDL alone; Figure 5B).
FIGURE 5.
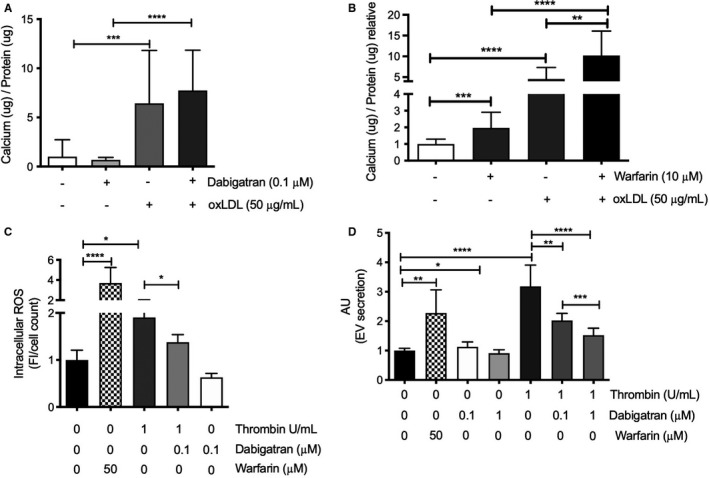
Interaction of warfarin and thrombin in vascular smooth muscle cells. Primary human vascular smooth muscle cells (VSMCs) were cultured in the presence of (A) warfarin, oxidized LDL (oxLDL), thrombin, dabigatran, or vehicle. oxLDL significantly increased calcification on VSMCs. Moreover, dabigatran did not influence oxLDL‐mediated calcification. (B) Both warfarin and oxLDL significantly increased calcification compared to control. Co‐stimulation of warfarin with oxLDL resulted in significantly more calcification compared with warfarin or oxLDL alone, indicating an additive effect. (C) Both warfarin and thrombin significantly increased intracellular ROS compared with control. Thrombin‐induced intracellular ROS was significantly attenuated when thrombin was combined with dabigatran, whereas dabigatran alone had no significant effect on ROS. (D) Extracellular vesicles, a hallmark of vascular calcification, were significantly increased after both warfarin and thrombin stimulation. In contrast, high dosage of dabigatran showed a significant reduction in extracellular vesicles. Elevated extracellular vesicles after thrombin stimulation were significantly and dose dependently reduced with dabigatran. ROS, reactive oxygen species
4.2. Thrombin increased oxidative stress and secretion of extracellular vesicles
Because warfarin increased oxidative stress in Apoe −/− mice, we tested whether warfarin and dabigatran also induce oxidative stress in VSMCs in vitro. Intracellular oxidative stress in VSMCs was determined over 30 minutes, directly after stimulation with either warfarin, thrombin, or thrombin combined with dabigatran. Warfarin significantly increased oxidative stress compared with control (50 µM warfarin, 3.7‐fold increase; Figure 5C). Thrombin significantly increased oxidative stress in VSMCs compared with vehicle treatment (1.9‐fold; Figure 5C). Co‐stimulation of dabigatran with thrombin significantly reduced thrombin induced oxidative stress (p < .05; Figure 5C). Dabigatran stimulation alone had no effect on oxidative stress. Oxidative stress is an inducer of extracellular vesicle secretions, which are known to induce VSMC calcification. 27 , 33 Therefore, we analyzed the number of secreted extracellular vesicles from VSMCs. Warfarin stimulation significantly increased extracellular vesicles compared with control (2.2‐fold, p < .01; Figure 5D). Thrombin also increased extracellular vesicle release significantly (3.1‐fold), which was dose dependently reduced after co‐stimulation with dabigatran (2‐fold for 0.1 μM dabigatran and 1.5‐fold for 1 μM dabigatran compared with control; Figure 5D). Moreover, dabigatran stimulation alone significantly reduced the number of extracellular vesicles compared with control (0.9‐fold of control for 1 μM dabigatran; Figure 5D).
4.3. Differently expressed genes after thrombin and warfarin stimulation in VSMCs
To further analyze the effects of warfarin and thrombin on VSMCs, we performed transcriptomic analysis after 4 hours of stimulation of VSMCs with warfarin and thrombin compared with vehicle control. Although warfarin affected 44 transcripts (27 genes increased expression and 17 reduced compared with control), thrombin altered a total of 1582 transcripts (expression of 979 genes were increased and expression of 603 genes were reduced compared with control). Altered genes were compared with known datasets associated with atherosclerosis development (1133 genes; Table 2). Thrombin significantly changed expression of 107 genes in this transcriptome whereas warfarin only significantly altered expression of four genes. FABP5 and IL1b were significantly altered for both warfarin and thrombin. Transcriptomic analysis was also compared with a known inflammatory gene dataset (428 genes; Table 3) because of the important role of inflammation in atherosclerosis together with the observed inflammatory effect of warfarin both in vivo and in vitro. Here, thrombin altered 43 genes, of which only three were also altered by warfarin stimulation (IL‐1b, leukemia inhibitory factor, CXCL2).
TABLE 2.
Altered gene transcripts of an atherosclerotic dataset of genes
| Atherosclerosis | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Thrombin | Warfarin | Thrombin | Warfarin | Gene | Thrombin | Warfarin | |
| LogFC | logFC | Gene | LogFC | logFC | LogFC | logFC | ||
| ABCA1 | −1.29 | 0.02 | GREM1 | 2.15 | 0.20 | PLA2G4A | 0.67 | 0.11 |
| ABR | 0.64 | 0.18 | HAS2 | 2.00 | 0.44 | PLAT | 0.67 | 0.14 |
| ADAM8 | −0.75 | −0.08 | HILPDA | 0.88 | −0.06 | PLAUR | 1.52 | 0.05 |
| ADAMTS1 | 0.66 | −0.18 | HMGA1 | 0.62 | 0.09 | PNP | 0.96 | 0.04 |
| ADAMTS5 | −2.08 | −0.08 | HS3ST1 | 1.28 | 0.12 | PPARG | 1.28 | 0.09 |
| AHRR | 0.01 (NS) | 0.78 * | HSD11B1 | 0.62 | 0.12 | PTGS1 | 0.62 | 0.19 |
| APOL6 | −0.83 | −0.03 | HSPA2 | 1.95 | −0.16 | PTK2B | −0.88 | −0.12 |
| BDNF | −0.60 | −0.34 | ICAM1 | 0.83 | 0.24 | RGS2 | 0.73 | −0.14 |
| BMP4 | −0.74 | −0.16 | IL11 | 0.60 | −0.04 | SAMD4A | 1.45 | 0.01 |
| CASP1 | −0.71 | 0.18 | IL1B | 1.30 | 0.76 * | SAMD9 | 0.79 | 0.05 |
| CASP3 | 0.73 | −0.06 | IL33 | 0.83 | 0.05 | SERPINE1 | 1.69 | −0.12 |
| CAV1 | 0.85 | 0.06 | IL6 | 0.76 | −0.25 | SLC16A3 | 0.94 | −0.16 |
| CCL2 | 1.47 | 0.24 | IRS2 | −0.81 | −0.04 | SLC17A5 | 0.79 | −0.14 |
| CD44 | 0.67 | 0.17 | ISG20 | 1.46 | 0.09 | SLC6A4 | −0.67 | 0.09 |
| CDKN1C | −0.82 | 0.15 | ITIH4 | −0.70 | 0.13 | SMAD3 | 0.78 | 0.00 |
| CHDH | 1.53 | −0.12 | KLF4 | 0.88 | −0.22 | SOCS1 | 0.65 | 0.37 |
| CREB3 | 0.60 | −0.09 | KLF5 | −0.72 | −0.32 | SOCS3 | 0.78 | 0.10 |
| CRY1 | 0.99 | −0.32 | LDB2 | −1.19 | 0.06 | SOD2 | 0.99 | 0.04 |
| CTF1 | −0.83 | 0.08 | LIPE | −0.70 | 0.06 | ST8SIA1 | −1.56 | 0.08 |
| CXCL1 | 2.49 | 0.41 | LMNA | 0.95 | −0.08 | TCF7L2 | −0.91 | −0.38 |
| DDAH1 | 1.45 | −0.25 | LRP8 | 0.67 | 0.26 | TFPI2 | 0.97 | 0.08 |
| DUSP1 | 0.76 | −0.29 | MAP2 K1 | 0.89 | −0.05 | THBD | 2.71 | 0.19 |
| ECE2 | 0.71 | −0.09 | MAP3 K5 | −1.17 | 0.16 | TNFRSF11B | 0.76 | −0.23 |
| EDN1 | −1.25 | −0.20 | MPRIP | 0.89 | −0.19 | TNFRSF12A | 1.03 | −0.39 |
| EDNRA | −0.99 | 0.32 | MTSS1 | 1.81 | 0.23 | TNFRSF14 | −0.72 | 0.14 |
| EFNB1 | −0.77 | 0.17 | MYH9 | 0.63 | −0.28 | TNFRSF4 | 0.29 (NS) | 1.22 * |
| EGFR | 0.78 | 0.08 | NCEH1 | 0.71 | 0.21 | TNFSF4 | 1.10 | −0.05 |
| ELK1 | 0.61 | 0.14 | NOTCH1 | 0.78 | 0.21 | TNFSF9 | 0.66 | 0.01 |
| ENPP1 | 0.63 | −0.12 | NOV | 0.69 | −0.09 | TRAF3IP2 | 0.88 | 0.24 |
| ETS1 | 0.68 | 0.02 | NR1D1 | −0.82 | 0.10 | TRIB3 | −1.57 | −0.05 |
| FABP5 | −0.70 | 0.60 * | NR4A3 | 3.41 | 0.18 | UNC5B | −0.76 | −0.05 |
| FGF2 | −0.96 | −0.25 | NRG1 | −1.47 | −0.16 | VCL | 1.05 | −0.15 |
| FGFR1 | 0.60 | 0.00 | NUP62 | 0.69 | −0.13 | VKORC1 | 0.61 | 0.01 |
| FOS | −1.00 | −0.23 | PDGFA | 1.50 | −0.09 | VWF | 1.72 | 0.21 |
| FST | 0.95 | −0.28 | PECAM1 | 1.12 | 0.15 | WNT5A | −0.69 | 0.08 |
| FURIN | 0.87 | 0.20 | PIK3CD | 1.15 | 0.00 | XPR1 | 0.94 | −0.20 |
| GDF15 | −0.71 | −0.05 | ||||||
Increased (red) and reduction (blue) compared with control. All thrombin, except for (NS), altered gene expressions were significant different compared with control and *present significant difference of warfarin compared with control. FC, fold change; NS, non significant
TABLE 3.
Altered gene transcripts of an inflammatory dataset
| Inflammation | |||||
|---|---|---|---|---|---|
| Thrombin | Warfarin | Thrombin | Warfarin | ||
| Gene | LogFC | logFC | Gene | LogFC | logFC |
| AQP3 | 0.90 | 0.08 | NRG1 | −1.47 | −0.16 |
| BDKRB1 | 0.64 | 0.20 | PDGFA | 1.50 | −0.09 |
| BDNF | −0.60 | −0.34 | PECAM1 | 1.12 | 0.15 |
| BMP4 | −0.74 | −0.16 | PLAT | 0.67 | 0.14 |
| CASP1 | −0.71 | 0.18 | PLAUR | 1.52 | 0.05 |
| CCL2 | 1.47 | 0.24 | PPARG | 1.28 | 0.09 |
| CD44 | 0.67 | 0.17 | SERPINE1 | 1.69 | −0.12 |
| CD83 | 0.63 | 0.40 | SETD7 | 0.66 | 0.01 |
| CXCL1 | 2.49 | 0.41 | SMAD3 | 0.78 | 0.00 |
| CXCL2 | 1.68 | 1.04 * | SOCS1 | 0.65 | 0.37 |
| CXCL6 | 1.55 | 0.47 | SOCS3 | 0.78 | 0.10 |
| EDN1 | −1.25 | −0.20 | TFPI2 | 0.97 | 0.08 |
| EFNB1 | −0.77 | 0.17 | THBD | 2.71 | 0.19 |
| FGF2 | −0.96 | −0.25 | TIMP4 | 0.65 | 0.12 |
| ICAM1 | 0.83 | 0.24 | TNFAIP3 | 1.70 | −0.03 |
| IL1B | 1.30 | 0.76 * | TNFRSF11B | 0.76 | −0.23 |
| IL1RAP | −1.19 | 0.13 | TNFRSF12A | 1.03 | −0.39 |
| IL33 | 0.83 | 0.05 | UCN | −1.04 | −0.01 |
| IL4R | 0.74 | 0.12 | VWF | 1.72 | 0.21 |
| IL6 | 0.76 | −0.25 | WDR1 | 0.90 | −0.08 |
| LIAS | −0.61 | −0.02 | YWHAH | 0.73 | −0.06 |
| LIF | 1.65 | 0.75 * | |||
Increased (red) and reduction (blue) compared with control. All thrombin altered gene expressions were significant different compared with control and *present significant difference of warfarin compared with control. FC, fold change
5. DISCUSSION
In the presented study, we demonstrate that warfarin and dabigatran have differential effects on atherosclerosis. Warfarin treatment accelerated plaque inflammation, oxidative stress, calcification activity, and plaque progression. By contrast dabigatran had no effect on vascular inflammation or calcification and reduced plaque progression in vivo. These data suggest that the choice of oral anticoagulant treatment in patients at risk of venous or arterial thrombosis may have important effects on atherosclerotic disease, indicating that further detailed clinical studies are now required.
Warfarin promotes atherosclerotic disease activity by increased presence of calcification and calcification activity. Recently, greater calcification volume—but not higher density—was associated with increased cardiovascular disease risk. 34 Our data show that warfarin increases calcification volume in atherosclerotic plaques, suggesting that warfarin increases plaque vulnerability. Dabigatran had no effect on calcification and even reduced plaque progression, implying that local effects play key roles in cardiovascular disease development. It is known that warfarin, as with all VKAs, inhibits carboxylation of vitamin K–dependent proteins thereby reducing coagulation tendency at the expense of MGP activity. 35 MGP is one of the strongest vascular calcification inhibitors 36 and reduced activation of MGP by warfarin appears to tip the balance towards calcification. Carboxylated MGP binds to BMP4, thereby reducing osteochondrogenic differentiation. 29 We confirm that warfarin treatment increased the presence of BMP4 in the atherosclerotic plaque. Unexpectedly, dabigatran increased ucMGP presence in atherosclerotic plaque, but in contrast to warfarin did not affect BMP4 presence in atherosclerotic plaque. This suggests that in dabigatran‐treated animals elevated levels of ucMGP point toward a coping mechanism for elevated calcification pressure being part of increased total MGP 2 , 37
Carboxylated MGP binds to calcium crystals and extracellular vesicles, blocking further growth of calcification. 38 Warfarin treatment increased ucMGP already in early atherosclerotic lesions, even before calcification could be visualized. Active calcification, detected by [18F]‐NaF is associated with increased plaque vulnerability. 39 We show that mice treated with warfarin displayed higher [18F]‐NaF positivity in the vasculature compared with dabigatran and control, indicating that VKA induces active mineralization. Our data are in line with recently published preclinical studies 40 and clinical studies comparing VKA, NOAC, or no oral anticoagulant treatment on vascular calcification. VKA treatment showed higher prevalence of vascular and valvular calcification, which was not seen in NOAC‐treated patients. 8 Moreover, plaque burden and prevalence of high‐risk plaques have been shown to be increased in patients using VKA compared with NOAC treatment, 17 although caution should be taken with generalizing NOAC treatments.
Oxidative stress has been shown to increase vascular calcification development. 32 Warfarin increases oxidative stress in vivo and in vitro, whereas dabigatran reduced thrombin‐induced oxidative stress. VKAs have been put forward to induce oxidative stress via inhibition of the vitamin K cycle. 41 VKAs are also known to interact with cytochrome P450 (CYP) and our transcriptomic data show elevated CYP1B1 transcript after warfarin treatment. CYP1B1 has been shown to increase oxidative stress in VSMCs. 42 Dabigatran, on the other hand, has been shown to block oxidative stress in mice thereby improving nitric oxide synthesis. 43 , 44 , 45 , 46 Extracellular vesicles from VSMCs play an important role in the initiation of calcification, 27 and both warfarin and thrombin increased extracellular vesicle secretion. Taken together, warfarin induces impaired carboxylation of MGP, and increases oxidative stress and extracellular vesicle release, thereby supporting vascular calcification. In contrast, dabigatran inhibits the detrimental side effects of thrombin on VSMCs.
Both thrombin and warfarin increased IL‐1ß transcripts, which has been associated with exacerbation of atherosclerosis. 47 Microcalcification can initiate inflammation 48 and inflammation can promote calcification. 49 We show increased presence of macrophages in early atherosclerotic lesions and elevated inflammatory markers in VSMCs after warfarin treatment. Moreover, we demonstrate that warfarin treatment increases [18F]‐NaF uptake, which is indicative of presence of microcalcification. Warfarin has previously been shown to have immunomodulatory effects, including promoting an inflammatory environment. 50 , 51 It has been demonstrated that calcified VSMCs initiate macrophage migration and promote a pro‐inflammatory environment. 48 In early atherosclerotic lesions, hydroxyapatite nanocrystals are present, associated with initial mineralization. 49 , 52 Additionally, calcium crystals activate macrophages thereby promoting a pro‐inflammatory environment 53 and gaining osteogenic activity. 49 Consequently, inflammatory macrophages promote calcification via release of TNF‐α. In line with these data, we confirm that mice treated with warfarin had more macrophages in early atherosclerotic lesions. In conclusion, macrophage rich plaques display increased osteogenic activity, further enhancing the strong correlation between inflammation and vascular calcification.
Thrombin can affect atherosclerosis development by promoting inflammation via cytokine production, proliferation and cytoskeletal rearrangement of VSMC, fibroblast stimulation and nitric oxide synthesis. 44 , 45 , 46 , 47 In atherosclerosis, thrombin also promotes leukocyte recruitment, whereas reduction of thrombin by approximately 50% reduced macrophages content. 22 Here, we show that dabigatran reduced atherogenesis and that thrombin inhibition induced oxidative stress and extracellular vesicle secretion in vitro. Moreover, thrombin stimulation in VSMCs increased a wide range of transcript genes associated with atherosclerosis and inflammation. Thrombin binds and cleaves the N‐terminal exodomain of PAR1, thereby initiating PAR1 signaling. Dabigatran inhibits thrombin, thereby attenuating thrombin‐induced PAR signaling resulting in diminished or altered atherosclerosis. Indeed, dabigatran has been shown to reduce thrombin induced inflammatory response. 54 , 55 Moreover, it has been shown that dabigatran increases bone turnover and reduces bone mineralization compared with warfarin treatment. 56 Finally, edoxaban, a direct FXa inhibitor, has recently been shown to inhibit maladaptive vascular remodeling compared with warfarin treatment, 57 in line with our findings.
This study has several limitations. The atherosclerotic Apoe −/− mice model is a widely accepted animal model for studying atherosclerosis. Although there are great similarities in morphology of plaques compared with humans, atherosclerotic plaques in mice will not rupture. Therefore, potential consequences of myocardial infarction or stroke are absent in this model. In this study, we achieved dabigatran plasma levels of 370 ng/ml (Table 1), which is higher compared with patients with concentrations varying between 50 and 180 (trough‐peak). However, murine thrombin has lower affinity to dabigatran than human thrombin. 21 Thus, increased dabigatran concentration in our model is roughly comparable to patient concentrations. Moreover, our dosage of warfarin in mice (e.g., 3 mg/g and mice eat approximately 3 g per day, daily intake of 9 mg per day) exceeded dose in humans (5 mg per day/70 kg). 58 Nevertheless, warfarin was combined with elevated vitamin K1 to prevent bleeding and was used to study VKA effects on the vessel wall. In the present study, we determined the differential effects of warfarin and dabigatran on the vessel wall and possible consequence in atherosclerosis development.
Our data are of clinical importance because many patients are treated with cholesterol‐lowering drugs such as statins, known to also impact the vasculature. Statins have been shown to reduce atherosclerotic plaque vulnerability and subsequent cardiovascular morbidity and mortality. 59 Combination therapy of statin treatment with either warfarin or NOAC might impact treatment efficacy and interactions between medication needs to be investigated in future clinical trials.
In conclusion, VKA aggravates atherogenesis via its direct effects on VSMCs. Our data show that VKA increases atherosclerotic disease activity, increasing plaque inflammation, oxidative stress, and calcification activity; effects that translate into faster plaque progression. In contrast, these effects are not observed with dabigatran, which appeared to have a beneficial effect on disease progression. Our findings suggest that the choice of anticoagulant in patients at risk of arterial of venous thrombosis may have important off‐target effects on bystander atherosclerotic disease, with further studies in humans now required.
CONFLICT OF INTEREST
Rich H. van Gorp is employed by NattoPharma. Joanne van Ryn is employed by Boehringer Ingelheim. Leon J. Schurgers received research funding from Boehringer Ingelheim related to this work and from NattoPharma, Bayer, Daichii‐Sankyo, and IDS not related to this work. Felix Mottaghy is on the advisory board of Advanced Accelerator Applications GmbH and has received institutional grants from GE Healthcare, Siemens, and Nano‐Map not related to this work. Vanessa Bröker received lecture fees and research funding from Bayer, Daichii‐Sankyo, and BMS‐Pfizer not related to this work. Herni M. H. Spronk received funding for research from Bayer and Pfizer outside this work. Henri M. H. Spronk and Leon J. Schurgers are stockholders in Coagulation Profile. All other authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Rick H. van Gorp, Vanessa Bröker, Matthis Bauwens, and Peter Leenders conducted the animal experiments. Peter Leenders performed the experimental surgery. Rick H. van Gorp and Peter Leenders performed the histological analysis. Danyel Jennen and Jacco J. Briedé performed microarrays and data validation. Rick H. van Gorp, Joanne van Ryn, Henri M. H. Spronk, Chris P. Reutelingsperger, and Leon J. Schurgers designed and prepared the study. Ingrid Dijkgraaf, Marc R. Dweck, Jan Bucerius, Vincent Brandenberg, and Felix Mottaghy analyzed and interpreted vascular calcification data. Rick H. van Gorp and Vanessa Bröker performed in vitro experiments. Rick H. van Gorp, Chris P. Reutelingsperger, and Leon J. Schurgers wrote the manuscript. Joanne van Ryn and Henri M. H. Spronk proofread the manuscript. Chris P. Reutelingsperger, and Leon J. Schurgers performed final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy and integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Fig S1
{kind=link}
Table S1
ACKNOWLEDGMENTS
This work was financed by Dutch Thrombosis Society (2014.02), the Norwegian Research Council, Nattopharma, Boehringer Ingelheim, and the European Union’s Horizon 2020 research and innovation programmes under the Marie Sklodowska‐Curie grant agreement No 813409. Dabigatran etexilate was kindly provided by Boehringer‐Ingelheim.
Handled and decision: Saskia Middeldorp, 02 March 2021
REFERENCES
- 1. Kapustin AN, Schoppet M, Schurgers LJ, et al. Prothrombin loading of vascular smooth muscle cell–derived exosomes regulates coagulation and calcification highlights. Arterioscler Thromb Vasc Biol. American Heart Association, Inc; 2017;37(3):e22‐e32. [DOI] [PubMed] [Google Scholar]
- 2. Schurgers LJ, Teunissen KJF, Knapen MHJ, et al. Novel conformation‐specific antibodies against matrix gamma‐carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25(8):1629‐1633. [DOI] [PubMed] [Google Scholar]
- 3. Hasific S, Øvrehus KA, Gerke O, et al. Extent of arterial calcification by conventional vitamin K antagonist treatment. PLoS One. 2020;15(10):e0241450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alappan HR, Kaur G, Manzoor S, Navarrete J, O'Neill WC. Warfarin accelerates medial Arterial calcification in humans. Arterioscler Thromb Vasc Biol. 2020;40(5):1413‐1419. [DOI] [PubMed] [Google Scholar]
- 5. Andrews J, Psaltis PJ, Bayturan O, et al. Warfarin use is associated with progressive coronary arterial calcification: insights from serial intravascular ultrasound. JACC Cardiovasc Imaging. 2018;11(9):1315‐1323. [DOI] [PubMed] [Google Scholar]
- 6. Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two Gla‐containing proteins. J Cell Biol. 2004;165(5):625‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schurgers LJ, Cranenburg ECM, Vermeer C. Matrix Gla‐protein: the calcification inhibitor in need of vitamin K. Thromb Haemost. 2008;100(4):593‐603. [PubMed] [Google Scholar]
- 8. Peeters F, Dudink E, Kimenai D, et al. Vitamin K antagonists, non–vitamin K antagonist oral anticoagulants, and vascular calcification in patients with atrial fibrillation. TH Open. Georg Thieme Verlag KG. 2018;02(04):e391‐e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schurgers LJ, Joosen IA, Laufer EM, et al. Vitamin K‐antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS One. 2012;7(8):e43229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shioi A, Ikari Y. Plaque calcification during atherosclerosis progression and regression. JAT. Japan Atherosclerosis Society. 2018;25(4):294‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin TC, Tintut Y, Lyman A, Mack W, Demer LL, Hsiai TK. Mechanical response of a calcified plaque model to fluid shear force. Ann Biomed Eng. 2006;34(10):1535‐1541. [DOI] [PubMed] [Google Scholar]
- 12. Ehara S, Kobayashi Y, Yoshiyama M, et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110(22):3424‐3429. [DOI] [PubMed] [Google Scholar]
- 13. Vengrenyuk Y, Carlier S, Xanthos S, et al. A hypothesis for vulnerable plaque rupture due to stress‐induced debonding around cellular microcalcifications in thin fibrous caps. Proc Natl Acad Sci USA. 2006;103(40):14678‐14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Criqui MH, Denenberg JO, Ix JH, et al. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA. American Medical Association. 2014;311(3):271‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Joshi NV, Vesey AT, Williams MC, et al. 18F‐fluoride positron emission tomography for identification of ruptured and high‐risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet. 2014;383(9918):705‐713. [DOI] [PubMed] [Google Scholar]
- 16. Dweck MR, Jenkins WSA, Vesey AT, et al. 18F‐sodium fluoride uptake is a marker of active calcification and disease progression in patients with aortic stenosis. Circ Cardiovasc Imaging. 2014;7(2):371‐378. [DOI] [PubMed] [Google Scholar]
- 17. Plank F, Beyer C, Friedrich G, et al. Influence of vitamin K antagonists and direct oral anticoagulation on coronary artery disease: a CTA analysis. Int J Cardiol. Elsevier B.V. 2018;260(C):11‐15. [DOI] [PubMed] [Google Scholar]
- 18. Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. 1998;18(9):1400‐1407. [DOI] [PubMed] [Google Scholar]
- 19. Schurgers LJ, Spronk HM, Soute BA, Schiffers PM, DeMey JG, Vermeer C. Regression of warfarin‐induced medial elastocalcinosis by high intake of vitamin K in rats. Blood. 2007;109(7):2823‐2831. [DOI] [PubMed] [Google Scholar]
- 20. Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vascul Pharmacol. 2014;62(2):72‐81. [DOI] [PubMed] [Google Scholar]
- 21. Borissoff JI, Spronk HMH, Heeneman S, ten Cate H. Is thrombin a key player in the “coagulation‐atherogenesis” maze? Cardiovasc Res. 2009;82(3):392‐403. [DOI] [PubMed] [Google Scholar]
- 22. Borissoff JI, Otten JJT, Heeneman S, et al. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e‐deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil‐dependent manner. PLoS One. 2013;8(2):e55784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kadoglou NPE, Moustardas P, Katsimpoulas M, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E‐deficient mice : dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther. 2012;26(5):367‐374. [DOI] [PubMed] [Google Scholar]
- 24. van Vlijmen BJ, Vant Hof HB, Mol MJ, et al. Modulation of very low density lipoprotein production and clearance contributes to age‐ and gender‐ dependent hyperlipoproteinemia in apolipoprotein E3‐Leiden transgenic mice. J Clin Invest. 1996;97(5):1184‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price PA, Kaneda Y. Vitamin K counteracts the effect of warfarin in liver but not in bone. Thromb Res. 1987;46(1):121‐131. [DOI] [PubMed] [Google Scholar]
- 26. Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle‐mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15(11):2857‐2867. [DOI] [PubMed] [Google Scholar]
- 27. Kapustin AN, Chatrou MLL, Drozdov I, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116(8):1312‐1323. [DOI] [PubMed] [Google Scholar]
- 28. Smyth GK. Limma: Linear Models for Microarray Data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York, NY: Springer New York; 2005. pp. 397‐420.(Bioinformatics and Computational Biology Solutions Using R and Bioconductor). [Google Scholar]
- 29. Raggi P, Callister TQ, Shaw LJ. Progression of coronary artery calcium and risk of first myocardial infarction in patients receiving cholesterol‐lowering therapy. Arterioscler Thromb Vasc Biol. 2004;24(7):1272‐1277. [DOI] [PubMed] [Google Scholar]
- 30. Boström KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108(4):446‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Byon CH, Javed A, Dai Q, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283(22):15319‐15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016;9:244‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Furmanik M, Chatrou M, van Gorp RH, et al. Reactive oxygen‐forming nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle‐mediated vascular calcification. Circ Res. 2020;22(1):185. [DOI] [PubMed] [Google Scholar]
- 34. Criqui MH, Knox JB, Denenberg JO, et al. Coronary artery calcium volume and density: potential interactions and overall predictive value: the multi‐ethnic study of atherosclerosis. JACC Cardiovasc Imaging. 2017;10(8):845‐854. [DOI] [PubMed] [Google Scholar]
- 35. van Gorp RH, Schurgers LJ. New insights into the pros and cons of the clinical use of vitamin K antagonists (VKAs) versus direct oral anticoagulants (DOACs). Nutrients. 2015;7(11):9538‐9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386(6620):78‐81. [DOI] [PubMed] [Google Scholar]
- 37. Proudfoot D, Shanahan CM. Molecular mechanisms mediating vascular calcification: role of matrix Gla protein. Nephrology (Carlton). John Wiley & Sons, Ltd (10.1111). 2006;11(5):455‐461. [DOI] [PubMed] [Google Scholar]
- 38. Schurgers LJ, Spronk HMH, Skepper JN, et al. Post‐translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;5(12):2503‐2511. [DOI] [PubMed] [Google Scholar]
- 39. Irkle A, Vesey AT, Lewis DY, et al. Identifying active vascular microcalcification by (18)F‐sodium fluoride positron emission tomography. Nat Comms. 2015;6:7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Florea A, Sigl JP, Morgenroth A, et al. Sodium 18[F]Fluoride PET can efficiently monitor in vivo atherosclerotic plaque calcification progression and treatment. Cells. 2021;10(2):275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petsophonsakul P, Furmanik M, Forsythe R, et al. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2019;39(7):1351‐1368. [DOI] [PubMed] [Google Scholar]
- 42. Yaghini FA, Song CY, Lavrentyev EN, et al. Angiotensin II–induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1–dependent superoxide generation. Hypertension. 2010;55(6):1461‐1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pingel S, Tiyerili V, Mueller J, Werner N, Nickenig G, Mueller C. Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch Med Sci. 2014;10(1):154‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee I‐O, Kratz MT, Schirmer SH, Baumhäkel M, Böhm M. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E‐deficient mice. J Pharmacol Exp Ther. 2012;343(2):253‐257. [DOI] [PubMed] [Google Scholar]
- 45. Yufu T, Hirano K, Bi D, et al. Rac1 regulation of surface expression of protease‐activated receptor‐1 and responsiveness to thrombin in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2005;25(7):1506‐1511. [DOI] [PubMed] [Google Scholar]
- 46. Ivey ME, Little PJ. Thrombin regulates vascular smooth muscle cell proteoglycan synthesis via PAR‐1 and multiple downstream signalling pathways. Thromb Res. 2008;123(2):288‐297. [DOI] [PubMed] [Google Scholar]
- 47. Libby P. Interleukin‐1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70(18):2278‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chatrou MLL, Cleutjens JP, van der Vusse GJ, Roijers RB, Mutsaers PHA, Schurgers LJ. Intra‐section analysis of human coronary arteries reveals a potential role for micro‐calcifications in macrophage recruitment in the early stage of atherosclerosis. PLoS One. 2015;10(11):e0142335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aikawa E, Nahrendorf M, Figueiredo J‐L, et al. Osteogenesis associates with inflammation in early‐stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116(24):2841‐2850. [DOI] [PubMed] [Google Scholar]
- 50. Mirkov I, Popov Aleksandrov A, Demenesku J, et al. Warfarin affects acute inflammatory response induced by subcutaneous polyvinyl sponge implantation in rats. Cutan Ocul Toxicol. 2017;36(3):283‐288. [DOI] [PubMed] [Google Scholar]
- 51. Belij S, Miljković D, Popov A, et al. Effects of subacute oral warfarin administration on peripheral blood granulocytes in rats. Food Chem Toxicol. 2012;50(5):1499‐1507. [DOI] [PubMed] [Google Scholar]
- 52. Roijers RB, Debernardi N, Cleutjens JPM, Schurgers LJ, Mutsaers PHA, van der Vusse GJ. Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am J Pathol. 2011;178(6):2879‐2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nadra I, Mason JC, Philippidis P, et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways. Circ Res. 2005;96(12):1248‐1256. [DOI] [PubMed] [Google Scholar]
- 54. Spronk HMH, de Jong AM, Verheule S, et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur Heart J. The Oxford University Press. 2016:ehw119. [DOI] [PubMed] [Google Scholar]
- 55. Kopec AK, Joshi N, Towery KL, et al. Thrombin inhibition with dabigatran protects against high‐fat diet‐induced fatty liver disease in mice. J Pharmacol Exp Ther. 2014;351(2):288‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fusaro M, Dalle Carbonare L, Dusso A, et al. Differential effects of dabigatran and warfarin on bone volume and structure in rats with normal renal function. PLoS One. 2015;10(8):e0133847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Millenaar D, Bachmann P, Böhm M, Custodis F, Schirmer SH. Effects of edoxaban and warfarin on vascular remodeling: Atherosclerotic plaque progression and collateral artery growth. Vascul Pharmacol. 2020;127:106661. [DOI] [PubMed] [Google Scholar]
- 58. Krüger T, Oelenberg S, Kaesler N, et al. Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol. 2013;33(11):2618‐2624. [DOI] [PubMed] [Google Scholar]
- 59. Libby P. How does lipid lowering prevent coronary events? New insights from human imaging trials. Eur Heart J. 2015;36(8):472‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1