

Abstract
Biocatalysis for the synthesis of fine chemicals is highly attractive but usually requires organic (co‐)solvents (OSs). However, native enzymes often have low activity and resistance in OSs and at elevated temperatures. Herein, we report a smart salt bridge design strategy for simultaneously improving OS resistance and thermostability of the model enzyme, Bacillus subtilits Lipase A (BSLA). We combined comprehensive experimental studies of 3450 BSLA variants and molecular dynamics simulations of 36 systems. Iterative recombination of four beneficial substitutions yielded superior resistant variants with up to 7.6‐fold (D64K/D144K) improved resistance toward three OSs while exhibiting significant thermostability (thermal resistance up to 137‐fold, and half‐life up to 3.3‐fold). Molecular dynamics simulations revealed that locally refined flexibility and strengthened hydration jointly govern the highly increased resistance in OSs and at 50–100 °C. The salt bridge redesign provides protein engineers with a powerful and likely general approach to design OSs‐ and/or thermal‐resistant lipases and other α/β‐hydrolases.
Keywords: bacillus subtilits lipase A (BSLA), directed evolution, organic solvent resistance, rational design, salt bridge
By removing unfavorable surface salt bridges, the organic solvent and thermal resistance of enzymes can be significantly improved. This design strategy results in locally refined flexibility and strengthened hydration.
Introduction
The application of organic (co‐)solvents (OSs) as reaction media for biocatalysts is mandatory for a large number of applications in the chemical industries. OSs are needed to solubilize hydrophobic substrates and products, they allow for easy product recovery and shift the reaction equilibrium into the desired direction. [1] However, native enzymes often suffer from low activity and/or sensitivity in the presence of OSs, [2] constraining their extended application for biocatalysis. OSs can affect the catalytic activity of enzymes mainly for the following reasons: (a) they induce conformational changes, [3] (b) their presence results in a loss of essential water molecules on the protein surface, [4] (c) they cause competitive inhibition, [5] or (d) solubility changes of the substrate, [4d] and (e) they stabilize the charged transition state. [6]
Another important and attractive property of industrial biocatalysts is thermal resistance, which often correlates well with process and storage stability. Therefore, this property also needs to be present or improved for industrial chemical production. [7] Thermal‐resistant enzymes can be isolated from thermophilic and mesophilic microorganisms. [8] Further, protein engineering strategies, including directed evolution and (semi‐)rational design, have succeeded in improving the thermostability and other properties of enzymes (e.g., resistance in OSs). [9] Thermostability of enzymes is influenced by many factors, such as (a) the enzyme's solvent‐exposed surface area, [10] (b) the number and length of loops, [11] (c) metal‐binding capacity, [12] (d) oligomerization and intersubunit interactions.[ 12b , 13 ] (e) amino acid composition, [14] and their various interactions (e.g., hydrophobic interactions, [15] ion pairs, [11b] cation‐π interactions, [16] and salt bridge networks[ 14b , 17 ]).
Salt bridges play predominant roles for protein structure and function, e.g., protein recognition, [18] flexibility, [19] allosteric regulation,[ 19b , 20 ] and thermostability. [21] The formation of a salt bridge is determined by the protonation state of oppositely charged residues (<4 Å). [22] Basically, salt bridges in enzymes are considered to stabilize, [23] and expectedly, more salt bridges are found in thermophilic enzymes.[ 14b , 24 ] Generally, well‐designed salt bridge networks and geometries (e.g., fork‐fork, fork‐stick) play an essential role in producing thermostability[ 15c , 25 ] and protein folding/unfolding. [26] The loss of a conserved salt bridge can lead to higher flexibility in an enzymes’ active site and trade a decrease in thermal stability for improved catalytic efficiency. [27]
With respect to the influence of salt bridges on the OS resistance of enzymes, it is generally considered that nonpolar OSs often strengthen the salt bridges in the enzyme, which can impair the catalytic function by reducing the enzyme's structural flexibility. [28] In polar solvents like dimethyl sulfoxide (DMSO), the salt bridge with the zwitterionic form becomes much more favored. [29] But the role of salt bridges, especially those at the surface, on protein stability in the presence/absence of OSs is still controversial today.[ 23a , 30 ] For instance, Kawata et al. reported that the formation of a new salt bridge (D51‐R157) in Pseudomonas aeruginosa LST‐03 lipase increased the stability of the loop structure and thereby increased the stability of LST‐03 lipase in nonpolar and polar OSs (e.g., n‐octane, n‐hexane, and DMSO). [31] A similar result with the increase of thermal stability and tolerance toward methanol was obtained via building up a salt bridge in Proteus mirabilis lipase. [32] In contrast, the disruption of two salt bridges in a laccase from Bacillus sp. HR03 resulted in improved kinetic parameters in water and different OSs (e.g., methanol, ethanol, 1‐propanol). [33] And positive effects on OS resistance upon introduction of charged residues were not necessarily ascribed to the formation of new salt bridges. [34] Clearly, to obtain a fundamental understanding of how salt bridges cause these changes in enzyme stability and function in OSs and rationally tailor the enzyme's OS/thermal resistance, a systematic analysis concerning the salt bridge landscape of enzymes in OS environments is a prerequisite.
Indeed, very few reports have been conducted looking at the effect of naturally occurring substitutions on OS resistance of enzymes, but Bacillus subtilits Lipase A (BSLA) is a rare exception.[ 9b , 34 , 35 ] A site saturation library (BSLA‐SSM) was constructed in our previous study and screened for variants with improved resistance toward three water‐miscible OSs (1,4‐dioxane (DOX), DMSO, 2,2,2‐trifluoroethanol (TFE)).[ 9b , 34 , 35 , 36 ] This library covers the full natural diversity at each of 181 amino acid positions consisting in total of 3440 BSLA variants (19×181 + wild‐type(WT)=3440) and is therefore well‐suited to study a putative correlation between salt bridges and OS resistance, thus opening the way to guide salt bridge engineering for obtaining OS and thermal resistance. It is generally accepted that a good correlation exists between enzyme thermostability and robustness in OSs.[ 2c , 9e , 37 ] Integrating protein engineering and computational methods such as molecular dynamics (MD) simulation and thermodynamic stability analysis provides a way to understand the molecular mechanisms responsible for jointly changed thermostability and OS resistance and subsequently comprehend a sequence‐structure‐function relationship.
Herein, we report a smart salt bridge design strategy to simultaneously improve OS resistance and thermostability of the model biocatalyst BSLA. The design principle utilizes computational MD simulations to identify salt bridges within enzymes, which are formed in OSs and are absent in water. The salt bridges formed in OSs are replaced by two residues with the same charge, which ensures local flexibility in OSs and a tight attraction and binding of water molecules. Surprisingly, these substitutions improve not only the activity of BSLA in OSs but also its thermal resistance in the aqueous environment.
Results and Discussion
MD simulation and salt bridge local landscape in the BSLA‐SSM library suggest a salt bridge redesign strategy
Before the MD simulation study, we analyzed the salt bridge landscape in the BSLA‐SSM library toward resistance against DOX, DMSO, and TFE (Table S1–S3 and Figure S1). See more details in Supporting Information (SI)). The results suggested that a balanced number of salt bridges is required within the BSLA structure to improve the OSs resistance of BSLA. And the native salt bridge positions might not be the best candidates for enzyme engineering. OS resistance of BSLA was evaluated as activity in the presence of OS divided by activity in the absence of OS [see Eq. (1) in SI].
100 ns MD simulations of BSLA WT in water and three OSs (DOX, DMSO, and TFE) were performed in our previous study (a summary is given in Figure S2 in SI). [9b] By comparing the salt bridge formation/disruption results of BSLA WT in water and three OSs, we found three “unfavorable salt bridges” on the BSLA WT surface formed between amino acids D34‐K35; K61‐D64; and K112‐D144 (see Figure 1 a and Table S2 in SI). We defined unfavorable salt bridges as salt bridges that are formed in the presence of OSs (but not in water) and which restrict the flexibility (and often also the activity) of the enzyme.
Figure 1.
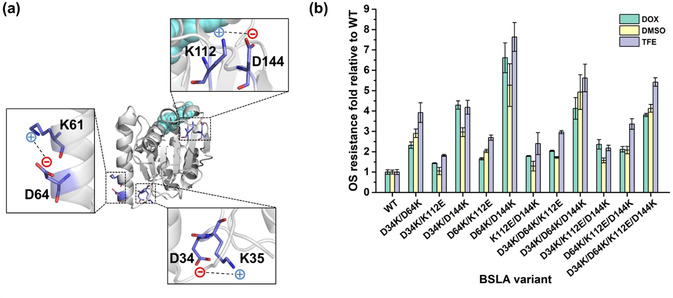
a) Location of three additional salt bridges in BSLA WT in the presence of OSs compared to water (“unfavorable salt bridges”). The BSLA structure is shown with a cartoon in grey. A close‐view of residues forming salt bridges is shown as sticks with carbon (marine), oxygen (red), and nitrogen (blue). The dashed line indicates the salt bridge. The catalytic triad residues (S77, D133, and H156) are shown as the cyan spheres. The structural model was generated using the structural model of BSLA WT (PDB ID: 1i6w, [42] Chain A) with Pymol. [43] b) OSs resistance of BSLA recombinants relative to BSLA WT. The OS resistance was measured in the absence or presence of 22 % (v/v) DOX, 60 % (v/v) DMSO, and 12 % (v/v) TFE as cosolvent after 2 h incubation with crude culture supernatant.
Salt bridges tended to be energetically stabilized in polar OSs and were mostly formed at the protein surface,[ 19b , 29 , 38 ] which has also been observed in other lipases, e.g., Geobacillus thermocatenulatus lipase in toluene, [28] Candida rugosa lipase in carbon tetrachloride, [39] and Bacillus thermocatenulatus lipase in ethanol and nonpolar OSs. [40] Thus, these OS‐stabilized salt bridges might be a clue to improve the OSs resistance also in BSLA. In combination with the three identified unfavorable salt bridges in BSLA, these findings led us to propose that breaking the unfavorable salt bridge(s) on the protein surface by well‐designed substitution(s) might result in a recovery of the interaction networks among surface residues and thereby enhance the stability of enzymes in OSs.
To validate this hypothesis, we examined the OS resistance pattern of all possible single amino acid substitutions at the three identified unfavorable salt bridges from the BSLA‐SSM library. The results of this analysis are depicted in Figure S3 and summarized in Table 1. Substitutions to charged amino acids predominantly improved OSs resistance (7–17 %) and resulted in the lowest rate of decreased (3–6 %) and inactive (0 %) variants when compared to polar, aromatic, and aliphatic substitutions (Table 1). Interestingly, the single amino acid substitutions in BSLA, including D34K, D34R, K112D, K112E, K112D, and D144K, which inverted the charge and thus disrupted the salt bridges at the respective positions, all showed elevated OSs resistance (Figure S3 and S4). Moreover, all the substitutions were located at a distance of at least 16 Å from the nearest active site residue and significantly exposed on the BSLA surface (Figure 1 a). Such residues are likely to increase the stability without compensating changes elsewhere. [41] In summary, introducing oppositely charged amino acids to break up unfavorable surface salt bridges could be a promising design principle for salt bridge engineering to improve the OSs resistance.
Table 1.
Classification of BSLA substitutions at six positions that form additional salt bridges in OSs.
|
OS resistance |
Classification of amino acid substitution % (variants)[a] |
|||
|---|---|---|---|---|
|
|
Charged |
Polar |
Aromatic |
Aliphatic |
|
Beneficial |
7–17 % (2–5) |
0–2 % (0–1) |
6–11 % (1–2) |
0–7 % (0–2) |
|
|
|
|
|
|
|
Unchanged |
77–87 % (23–26) |
71–81 % (30–34) |
72–78 % (13–16) |
73–93 % (22–26) |
|
|
|
|
|
|
|
Decreased |
3–6 % (1–2) |
7–17 % (3–7) |
6–22 % (1–4) |
3–23 % (1–7) |
|
|
|
|
|
|
|
Inactive |
0 % |
12 % (5) |
6 % (1) |
3 % (1) |
[a] The amino acid positions 34, 35, 61, 64, 112, 144 were selected for statistical analysis. OSs include 22 % (v/v) DOX, 60 % (v/v) DMSO, and 12 % (v/v) TFE. Charged substitutions were the best choice for improving the OS resistance comparing to polar, aromatic, and aliphatic. The results of all individual amino acid exchanges are depicted in Figure 2 b.
The salt bridge redesign strategy showed additive effects and yielded a highly OSs tolerant BSLA variant
To examine putative additive effects of the design principle and further enhance the OSs resistance of BSLA, recombination among four BSLA substitutions (D34K, D64K, D144K, and K112E; Figure S5) was investigated. These four substitutions were selected since they exhibited improved resistance in at least two OSs. In total, eleven BSLA recombinants were constructed and examined for OS resistance with their crude culture supernatants (Figure 1 b). All recombinants exhibited elevated resistance toward three OSs compared to BSLA WT. Remarkably, the “best” variant (BSLA D64K/D144K) had a 6.6‐fold, 5.3‐fold, and 7.6‐fold improved resistance against DOX, DMSO, and TFE, respectively. In D34K/D144K, two salt bridges were broken, and ≈4.2‐fold higher OSs resistance was gained in the presence of DOX or TFE (3‐fold higher DMSO resistance). In variant BSLA D34K/D64K/D144K, three unfavorable salt bridges were broken, and the increased TFE resistance factor is 5.6‐fold (DMSO; 5‐fold). Moreover, the variant D34K/D64K/K112E/D144K, harboring four substitutions but with only two disrupted salt bridges, had a 3.8‐fold to 5.4‐fold improvement in OSs resistance. These results demonstrate that breaking unfavorable salt bridges results in increased OS resistance. The specific activity in buffer of the evolved variants showed comparable results with 43–96 % specific activity relative to BSLA WT (Figure S6a in SI). And the results of specific activity in OSs agree well with the observation within OS resistance (Figure S6b).
Investigation of OSs resistance profiles and kinetic characterization
The five most promising BSLA recombination variants and BSLA WT were purified (Figure S7 in SI), and their OSs resistance profiles (Figure 2) and catalytic kinetics were investigated (Table S4). Residual activity of purified BSLA WT and variants was measured at room temperature using pNPB as the substrate in the presence of various concentrations of three OSs (DOX, DMSO, and TFE) after 2 h incubation. Residual activity in buffer at room temperature (25 °C) was defined as 100 %. As shown in Figure 2 a–c, the profiles of the five purified recombination variants were shifted to higher residual activity over nearly the entire range of the investigated three OS concentrations in comparison with BSLA WT. This trend was most significant for DOX. For example, variants D64K/D144K and D34K/D64K/D144K still showed remarkable activity at high concentration of OSs: D34K/D64K/D144K had ≈50 % residual activity in >40 % (v/v) DOX, and ≈25 % residual activity in >80 % (v/v) DMSO. Moreover, almost all recombinants were activated in <15 % (v/v) DOX, <20 % (v/v) DMSO, and <6 % (v/v) TFE, as commonly observed for enzymes at low OSs concentration.[ 2c , 2d , 30b , 44 ] These results illustrated that BSLA variants of significantly higher OS resistance were obtained by applying the salt bridge design principle. Detailed kinetic characterization was shown in Table S4. Some variants showed a decreased K M with the substrate pNPB (e.g., D64K/D144K) or increased turnover numbers (k cat) (e.g., D34K/D64K) in the presence of OSs. Besides, the “best” resistant variant D64K/K144K showed slightly lower catalytic efficiency (k cat/K M) in both buffer and OSs than BSLA WT under the same conditions. However, a dramatically improved enzyme stability in OSs could enable higher total turnover numbers (TTNs) in a given biotransformation.
Figure 2.
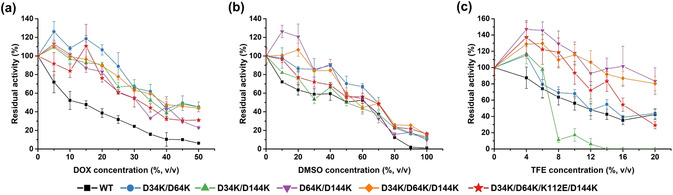
OSs resistance of the purified BSLA variants. Comparison of a) DOX, b) DMSO, and c) TFE resistance for WT and variants in different concentrations of OSs. Residual activities in OSs were measured with pNPB after 2 h incubation. All data shown were average values from measurements performed at least in triplicate.
Thermostability profiles of OS resistant BSLA variants
The global thermal profile of the five purified BSLA recombinants was investigated as well, including thermal resistance (Figure 3) and t 1/2 (t 1/2 being the enzyme's half‐life at 50 °C; Table 2 and Figure S8). BSLA WT lost >90 % activity after 1 h heat treatment at 50–100 °C (Figure 3). In comparison, all variants’ thermal resistance, except D34K/D64K/K112E/D144K, significantly increased over the entire temperature range of 40–100 °C. Notably, D34K/D144K showed an increase of up to 137‐fold in thermal resistance at 100 °C compared to BSLA WT. Interestingly, a re‐activation phenomenon of BSLA double and/or triple substitutions at high temperature (>70 °C) was observed, and the residual activities went up to ≈70 % residual activity. This phenomenon indicates that the denaturation mode might be different at <70 °C and >75 °C. The observed re‐activation effects of BSLA variants were also observed in a previous BSLA study. [45] Upon reaching the denaturation temperature (≈70 °C, Figure 3), the protein might unfold into the intermediate that was prone to irreversible aggregation and precipitation.[ 45 , 46 ]
Figure 3.
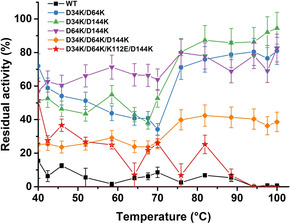
Comparison of thermal resistance of the purified BSLA WT and variants. The residual activities in buffer were measured after incubating at different temperatures from 25 °C to 100 °C for 60 min. Residual activity in buffer at room temperature (25 °C) was defined as 100 %. All data shown were average values from measurements performed at least in triplicate.
Table 2.
Comparison of half‐life for BSLA WT and variants.
|
BSLA Variant |
t 1/2 [a] [min] |
|---|---|
|
WT |
18 |
|
D34K/D64K |
54 |
|
D34K/D144K |
53 |
|
D64K/D144K |
54 |
|
D34K/D64K/D144K |
60 |
|
D34K/D64K/K112E/D144K |
54 |
[a] t 1/2 is the half‐life at 50 °C.
Moreover, all variants exhibit an increase (2.9‐ to 3.3‐fold improvement) in t 1/2 compared to WT (Table 2). Notably, the t 1/2 value of D34K/D64K/D144K harboring three substitutions was shown to increase prominently from 18 min (BSLA WT) to 60 min (Table 2). It should be noted that these results with salt bridge disruption are different from most previous studies in which the formation of new salt bridges stabilized the enzyme at higher temperatures with increasing rigidity.[ 21 , 47 ]
Consequently, we have shown that BSLA recombinants evolved for enhanced OSs resistance by removing unfavorable salt bridges also show remarkably improved thermostability. Notably, the thermostability of BSLA can be gradually improved upon iteratively, disrupting the additional salt bridges by a recombination process. Screening for the enhanced particular property relating to thermostability (e.g., melting temperature, t 1/2) should also induce higher OS resistance, as it was previously shown.[ 37 , 48 ]
Computational analysis revealed the molecular mechanism of improved OS and thermal resistance of BSLA variants
MD simulation is a prominent technique to understand the physical basis of enzymes’ structure and function.[ 9b , 49 ] To reveal the predominant factor(s) that govern the OS resistance and thermostability simultaneously, we investigated in total 36 MD simulation runs of six BSLA variants (WT and the five most promising variants) under six conditions, including water at 25 °C (≈8363 water molecules in the system), 22 % (v/v) DOX at 25 °C (≈280 DOX molecules and ≈6950 water molecules), 60 % (v/v) DMSO at 25 °C (≈952 DMSO and ≈4200 water), 12 % (v/v) TFE at 25 °C (≈223 TFE and ≈7192 water), water at 50 °C (≈8363 water), water at 100 °C (≈8363 water) (Figure S2). In addition, the thermodynamic stability (ΔΔG fold) of six BSLA variants was investigated to reveal the intrinsic stability change introduced by amino acid substitutions (Table S5). Subsequently, a comprehensive computational analysis examined the overall/local structural stability, flexibility, and solvation phenomenon of the BSLA variants.
Changes in structure and flexibility were observed at local regions in OSs and at high temperatures. As shown in Figure 4 b and S9b, the internal H‐bond numbers within Loop 3 harboring D34K decreased from ≈4 to ≈2 compared to BSLA WT in all systems. Meanwhile, Loop 3 also represented higher flexibility with increased RMSF values (e.g., residue K34 in D34K/D64K/D144K increased from 1.20 Å to 1.94 Å in 50 °C system, Figure 4 a and S10). Similar results were obtained for Loop 7 & 9 harboring K112E and D144K, respectively (Figure S9c and S10). For instance, Loop 7 & 9 of D64K/D144K showed increased averaged‐RMSF value compared to BSLA WT in all systems, especially in TFE with increasing 1.24 Å from 1.73 Å (Table S6). Nevertheless, comparable flexibility was observed within α‐helix B (harboring D64K; Figure 4 a, S9a, and S10) in almost all BSLA variants compared to BSLA WT, e.g., the changes of averaged‐RMSF value were less than 0.15 Å in water, DOX, and 50 °C system (Table S6). Their corresponding RMSD values at local structure also confirmed these observations (Figure S11a‐c). This finding suggests that the substitutions to oppositely charged amino acids (e.g., D34K) not only resulted in the collapse of the salt bridges (e.g., D34‐K35) but also led to flexibility modifications, mostly to increased or comparable flexibility at locally defined regions under unnatural conditions (i.e., OSs and high temperatures). Apart from these findings, there is no significant and common overall structural change among variants in each system (Figure 4 c, S12–S15, and Table S5; see more details in SI).
Figure 4.
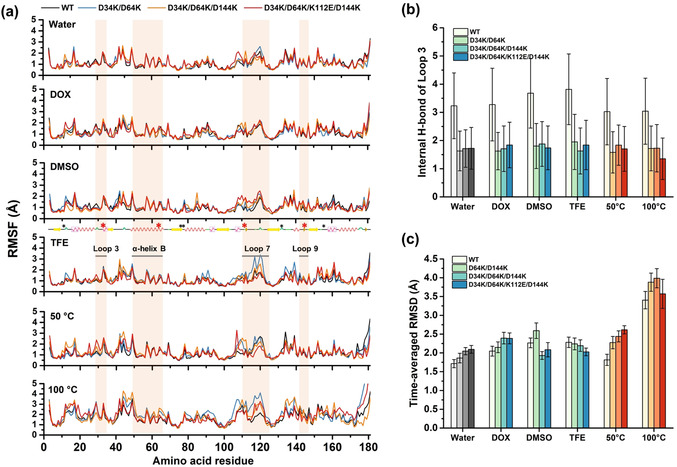
Overall and local structural and solvation change of BSLA variants in cosolvents and at different temperatures. a) RMSF of BSLA residues determined from the last 40 ns of MD simulations. The secondary structure elements were calculated using the DSSP program. [50] Secondary structure elements are shown by the following color Scheme: α‐helix (pink in box), 3/10‐helix (red), β‐sheet (yellow), and loop (green). Catalytic triad residues (Ser77, Asp133, and His156) and oxyanion hole residues (Ile12 and Met78) are labeled with black asterisks. The substituted sites (D34K, D64K, D144K, and K112E) and their located secondary structure (Loop 3 with residue 29–35, α‐helix B with residue 47–67, and Loop 7 & 9 with residue 109–124 and 142–147) are labeled with red asterisks and lines, respectively. b) Time‐averaged internal H‐bond of Loop 3 (residue 29–35) and c) time‐averaged RMSD of the heavy atoms determined from the last 40 ns of simulations under six conditions (water at 25 °C, DOX at 25 °C, DMSO at 25 °C, TFE at 25 °C, water at 50 °C, water at 100 °C). For better comparison, grey, blue, and red colors were used to indicate water, OSs, and high temperatures, respectively. Loop 3 includes residue 29–35. As the geometric cut‐off for the evaluation of hydrogen bond, distance 3.5 Å, and angle 30° were used.
Solvation of the enzyme plays a critical role in OS systems. [52] Therefore, local and overall solvation phenomena were investigated by analyzing the spatial distribution function (SDF), the hydration shell, the OS solvation layer, the water/OS molecules around the substituted sites and in the substrate‐binding cleft (Figure 5 and S16,S17). Notably, observations from each system show that all recombinants (especially, D34K/D64K/D144K; Figure 5 a) gain great hydration around the substituted sites and comparable even slightly intensive overall hydration shells compared to WT (Figure 5 b,c), indicating that the hydration is a predominant factor to affect the multiple OSs and thermal resistance of BSLA. [2d] In terms of the other solvation phenomena (i.e., solvation in the substrate binding cleft and overall/local OS solvation), BSLA substitutions did not lead to a noticeable difference (Figure S16a,b and S17a–c). These results indicate that the broken salt bridges within the recombinants “uncage” the charged amino acids that were previously trapped in salt bridges, ultimately leading to the increased attraction of water molecules. On the other hand, the removal of detrimental salt bridges did not result in changes in the orientation, distribution, and assembly of OS molecules. This finding agrees well with the generally accepted concepts that water molecules bonded to a protein surface are universally essential to maintain the catalytically active enzyme conformation, [53] and enzyme activity is often dependent on bound water and/or water content.[ 4a , 5b , 33b ]
Figure 5.
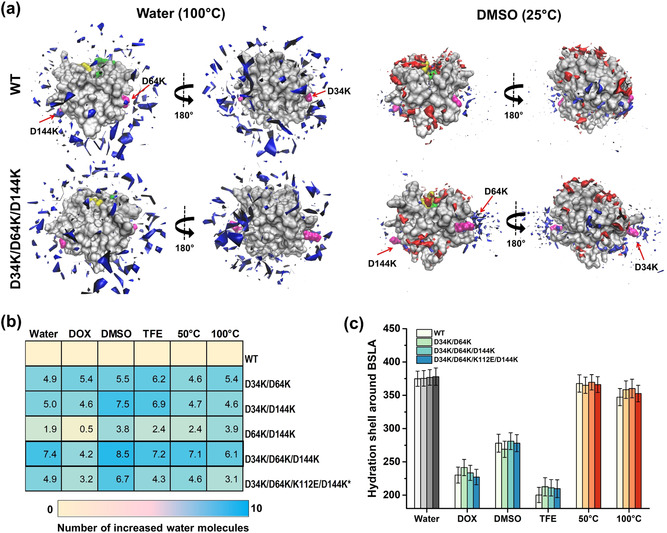
Solvation phenomenon of BSLA variants in cosolvents and at different temperatures. a) Spatial distribution of water and/or OS molecules at the molecular surface of the BSLA WT and D34K/D64K/D144K in water (100 °C) and DMSO (25 °C). The BSLA surface is shown in grey, Ser77, Asp133, His156 (the catalytic triad) in green, and Ile12, Met78 (oxyanion hole) in yellow; the OS molecules in red, the water molecules in blue. The substitutions are shown in magenta and indicated by the red arrow. The 180‐degree rotation is offered to give a complete view of BSLA. The contours are shown with isovalue 10 for water and isovalue 14 for DMSO molecules. b) Heatmap indicating the number of water molecules around the substituted sites averaged over the last 40 ns of MD trajectories. The substituted sites include positions 34, 64, 112, and 144. Asterisks: only two unfavorable salt bridges were removed in D34K/D64K/K112E/D144K. c) Hydration shell around BSLA variants averaged over the last 40 ns of MD trajectories. The hydration shell is defined as water molecules whose oxygen atom is localized at a distance ≤3.5 Å from any non‐hydrogen atom of the protein.[ 9b , 51 ] The number of water molecules is defined as hydration level. [51] For better comparison, grey, blue, and red colors were used to indicate water, OSs, and high temperatures, respectively.
In summary, we found the enhanced OSs and thermal resistant variants mainly benefit from two factors: (i) refined flexibility at a local region, and (ii) improved hydration around the substituted sites and the entire BSLA enzyme.
Conclusion
A salt bridge design strategy was reported that enabled us to improve the OSs and thermal resistance of the model lipase, BSLA. Lipases are of high industrial relevance for the synthesis of chemicals, and their stability under the process conditions is a crucial parameter. By introducing oppositely charged amino acids, three salt bridges on the BSLA surface that were specifically formed in the presence of OSs (termed “unfavorable salt bridges”) were disrupted. By recombination, five OS‐resistant BSLA variants were obtained with an up to 7.6‐fold (D64K/D144K) improvement, an up to 137‐fold higher thermal resistance, and an up to 3.3‐fold enhanced half‐life at 50 °C (t 1/2). MD simulations revealed that the refined local flexibility and the strengthened hydration near the substitution sites were the main factors leading to the improvements. In summary, we present here a rational approach to improve OS resistance by protein engineering. We anticipate that the given strategy can be transferred to other α/β‐hydrolases and different enzyme classes and empower researchers to expand the industrial application range of biocatalysis in OSs and at elevated temperatures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Haiyang Cui is supported by a Ph.D. scholarship from the China Scholarship Council (CSC No. 201604910840). Simulations were performed with computing resources granted by JARA‐HPC from RWTH Aachen University under projects JARA0169 and JARA0189. Part of the work of Karl‐Erich Jaeger and Ulrich Schwaneberg is funded by the German Federal Ministry of Education and Research in the framework of the project LipoBiocat (project‐no. 031B0837A). The authors thanks Dr. Daniel Friedrich Sauer, Isabell Hofmann, Francisca Contreras Leiva, and Dr. Kevin Herrmann for the discussion and help on thermostability experiments. Open access funding enabled and organized by Projekt DEAL.
H. Cui, L. Eltoukhy, L. Zhang, U. Markel, K.-E. Jaeger, M. D. Davari, U. Schwaneberg, Angew. Chem. Int. Ed. 2021, 60, 11448.
Contributor Information
Dr. Mehdi D. Davari, Email: m.davari@biotec.rwth-aachen.de.
Prof. Dr. Ulrich Schwaneberg, Email: u.schwaneberg@biotec.rwth-aachen.de.
References
- 1.
- 1a. Gupta M. N., Eur. J. Biochem. 1992, 203, 25–32; [DOI] [PubMed] [Google Scholar]
- 1b. Klibanov A. M., Nature 2001, 409, 241; [DOI] [PubMed] [Google Scholar]
- 1c. Carrea G., Riva S., Angew. Chem. Int. Ed. 2000, 39, 2226–2254; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2312–2341. [Google Scholar]
- 2.
- 2a. Wang S., Meng X., Zhou H., Liu Y., Secundo F., Liu Y., Catalysts 2016, 6, 32; [Google Scholar]
- 2b. Lombard C., Saulnier J., Wallach J., Protein Pept. Lett. 2005, 12, 621–629; [DOI] [PubMed] [Google Scholar]
- 2c. Cui H., Jaeger K.-E., Davari M. D., Schwaneberg U., Chem. Eur. J. 2021, 27, 2789; [DOI] [PubMed] [Google Scholar]
- 2d. Cui H., Zhang L., Eltoukhy L., Jiang Q., Korkunç S. K., Jaeger K.-E., Schwaneberg U., Davari M. D., ACS Catal. 2020, 10, 14847–14856. [Google Scholar]
- 3.
- 3a. Yaacob N., Ahmad Kamarudin N. H., Leow A. T. C., Salleh A. B., Raja Abd Rahman R. N. Z., Mohamad Ali M. S., Molecules 2017, 22, 1312; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Dutta Banik S., Nordblad M., Woodley J. M., Peters G. n. H., ACS Catal. 2016, 6, 6350–6361; [Google Scholar]
- 3c. Watanabe K., Yoshida T., Ueji S., Bioorg. Chem. 2004, 32, 504–515. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Valivety R. H., Halling P. J., Peilow A. D., Macrae A. R., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1992, 1122, 143–146; [DOI] [PubMed] [Google Scholar]
- 4b. Wangikar P. P., Michels P. C., Clark D. S., Dordick J. S., J. Am. Chem. Soc. 1997, 119, 70–76; [Google Scholar]
- 4c. Serdakowski A. L., Dordick J. S., Trends Biotechnol. 2008, 26, 48–54; [DOI] [PubMed] [Google Scholar]
- 4d. Klibanov A. M., Trends Biotechnol. 1997, 15, 97–101; [DOI] [PubMed] [Google Scholar]
- 4e. Chaudhary A. K., Kamat S. V., Beckman E. J., Nurok D., Kleyle R. M., Hajdu P., Russell A. J., J. Am. Chem. Soc. 1996, 118, 12891–12901. [Google Scholar]
- 5.
- 5a. Graber M., Irague R., Rosenfeld E., Lamare S., Franson L., Hult K., Biochim. Biophys. Acta Proteins Proteomics 2007, 1774, 1052–1057; [DOI] [PubMed] [Google Scholar]
- 5b. Valivety R. H., Halling P. J., Macrae A. R., Biotechnol. Lett. 1993, 15, 1133–1138; [Google Scholar]
- 5c. Bovara R., Carrea G., Ottolina G., Riva S., Biotechnol. Lett. 1993, 15, 937–942; [Google Scholar]
- 5d. Foresti M. L., Galle M., Ferreira M. L., Briand L. E., J. Chem. Technol. Biotechnol. 2009, 84, 1461–1473. [Google Scholar]
- 6.
- 6a. Kim A. S., Kakalis L. T., Abdul-Manan N., Liu G. A., Rosen M. K., Nature 2000, 404, 151–158; [DOI] [PubMed] [Google Scholar]
- 6b. Xu Z., Affleck R., Wangikar P., Suzawa V., Dordick J. S., Clark D. S., Biotechnol. Bioeng. 1994, 43, 515–520. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Haki G., Rakshit S., Bioresour. Technol. 2003, 89, 17–34; [DOI] [PubMed] [Google Scholar]
- 7b. Rigoldi F., Donini S., Redaelli A., Parisini E., Gautieri A., APL Bioeng. 2018, 2, 011501; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Li Y., Drummond D. A., Sawayama A. M., Snow C. D., Bloom J. D., Arnold F. H., Nat. Biotechnol. 2007, 25, 1051–1056. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Liao H., McKenzie T., Hageman R., Proc. Natl. Acad. Sci. USA 1986, 83, 576–580; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Vieille C., Zeikus G. J., Microbiol. Mol. Biol. Rev. 2001, 65, 1–43; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Chadha B., Kaur B., Basotra N., Tsang A., Pandey A., Bioresour. Technol. 2019, 277, 195–203; [DOI] [PubMed] [Google Scholar]
- 8d. Atalah J., Cáceres-Moreno P., Espina G., Blamey J. M., Bioresour. Technol. 2019, 280, 478–488. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Contreras F., Thiele M. J., Pramanik S., Rozhkova A. M., Dotsenko A. S., Zorov I. N., Sinitsyn A. P., Davari M. D., Schwaneberg U., ACS Sustainable Chem. Eng. 2020, 8, 12388–12399; [Google Scholar]
- 9b. Cui H., Stadtmüller T. H., Jiang Q., Jaeger K. E., Schwaneberg U., Davari M. D., ChemCatChem 2020, 12, 4073; [Google Scholar]
- 9c. Zhang L., Cui H., Zou Z., Garakani T. M., Novoa-Henriquez C., Jooyeh B., Schwaneberg U., Angew. Chem. Int. Ed. 2019, 58, 4562–4565; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4610–4613; [Google Scholar]
- 9d. Bornscheuer U. T., Hauer B., Jaeger K. E., Schwaneberg U., Angew. Chem. Int. Ed. 2019, 58, 36–40; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 36–41; [Google Scholar]
- 9e. Cui H., Cao H., Cai H., Jaeger K. E., Davari M. D., Schwaneberg U., Chem. Eur. J. 2020, 26, 643–649; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Cui H., Jaeger K.-E., Davari M. D., Schwaneberg U., Chem. Eur. J. 2021, 27, 2789–2797; [DOI] [PubMed] [Google Scholar]
- 9g. Cui H., Zhang L., Söder D., Tang X., Davari M. D., Schwaneberg U., ACS Catal. 2021, 11, 2445–2453. [Google Scholar]
- 10.
- 10a. Veltman O. R., Vriend G., Hardy F., Mansfeld J., Van Den Burg B., Venema G., Eijsink V. G. H., Eur. J. Biochem. 1997, 248, 433–440; [DOI] [PubMed] [Google Scholar]
- 10b. Suvd D., Fujimoto Z., Takase K., Matsumura M., Mizuno H., J. Biochem. 2001, 129, 461–468. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Thompson M. J., Eisenberg D., J. Mol. Biol. 1999, 290, 595–604; [DOI] [PubMed] [Google Scholar]
- 11b. Yano J. K., Poulos T. L., Curr. Opin. Biotechnol. 2003, 14, 360–365; [DOI] [PubMed] [Google Scholar]
- 11c. Yu H., Yan Y., Zhang C., Dalby P. A., Sci. Rep. 2017, 7, 41212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Sharma S., Vaid S., Bhat B., Singh S., Bajaj B. K. in Advances in Enzyme Technology (Eds.: Singh R. S., Singhania R. R., Pandey A., Larroche C.), Elsevier, Amsterdam, 2019, pp. 469–495; [Google Scholar]
- 12b. Choi J. M., Lee Y.-J., Cao T.-P., Shin S.-M., Park M.-K., Lee H.-S., di Luccio E., Kim S.-B., Lee S.-J., Lee S. J., Lee S. H., Lee D.-W., Arch. Biochem. Biophys. 2016, 596, 51–62. [DOI] [PubMed] [Google Scholar]
- 13. Lokanath N. K., Shiromizu I., Ohshima N., Nodake Y., Sugahara M., Yokoyama S., Kuramitsu S., Miyano M., Kunishima N., Acta Crystallogr. Sect. D 2004, 60, 1816–1823. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Fukuchi S., Nishikawa K., J. Mol. Biol. 2001, 309, 835–843; [DOI] [PubMed] [Google Scholar]
- 14b. Kumar S., Tsai C.-J., Nussinov R., Protein Eng. Des. Sel. 2000, 13, 179–191; [DOI] [PubMed] [Google Scholar]
- 14c. Van Boxstael S., Cunin R., Khan S., Maes D., J. Mol. Biol. 2003, 326, 203–216. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Rathi P. C., Jaeger K. E., Gohlke H., PLoS One 2015, 10, e0130289; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Spassov V. Z., Karshikoff A. D., Ladenstein R., Protein Sci. 1995, 4, 1516–1527; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Folch B., Rooman M., Dehouck Y., J. Chem. Inf. Model. 2008, 48, 119–127; [DOI] [PubMed] [Google Scholar]
- 15d. Kim T., Joo J. C., Yoo Y. J., J. Biotechnol. 2012, 161, 49–59. [DOI] [PubMed] [Google Scholar]
- 16. Gromiha M. M., Thomas S., Santhosh C., Prep. Biochem. Biotechnol. 2002, 32, 355–362. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Wintrode P. L., Miyazaki K., Arnold F. H., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2001, 1549, 1–8; [DOI] [PubMed] [Google Scholar]
- 17b. Paiardini A., Gianese G., Bossa F., Pascarella S., Proteins: Struct. Funct. Genet. 2003, 50, 122–134. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Xu D., Tsai C., Nussinov R., Protein Eng. 1997, 10, 999–1012; [DOI] [PubMed] [Google Scholar]
- 18b. Donald J. E., Kulp D. W., DeGrado W. F., Proteins Struct. Funct. Bioinf. 2011, 79, 898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Basu S., Biswas P., Biochim. Biophys. Acta Proteins Proteomics 2018, 1866, 624–641; [DOI] [PubMed] [Google Scholar]
- 19b. Costantini S., Colonna G., Facchiano A. M., Bioinformation 2008, 3, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Lounnas V., Wade R. C., Biochemistry 1997, 36, 5402–5417; [DOI] [PubMed] [Google Scholar]
- 20b. Valentini G., Chiarelli L., Fortin R., Speranza M. L., Galizzi A., Mattevi A., J. Biol. Chem. 2000, 275, 18145–18152; [DOI] [PubMed] [Google Scholar]
- 20c. Ferrari D., Niks D., Yang L. H., Miles E. W., Dunn M. F., Biochemistry 2003, 42, 7807–7818. [DOI] [PubMed] [Google Scholar]
- 21. Wu J.-P., Li M., Zhou Y., Yang L.-R., Xu G., Biotechnol. Lett. 2015, 37, 403–407. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Meuzelaar H., Vreede J., Woutersen S., Biophys. J. 2016, 110, 2328–2341; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Jelesarov I., Karshikoff A. in Protein Structure, Stability, and Interactions, Springer, Heidelberg, 2009, pp. 227–260. [Google Scholar]
- 23.
- 23a. Makhatadze G. I., Loladze V. V., Ermolenko D. N., Chen X., Thomas S. T., J. Mol. Biol. 2003, 327, 1135–1148; [DOI] [PubMed] [Google Scholar]
- 23b. Ge M., Xia X.-Y., Pan X.-M., J. Biol. Chem. 2008, 283, 31690–31696. [DOI] [PubMed] [Google Scholar]
- 24. Behera R. K., Mazumdar S., Int. J. Biol. Macromol. 2010, 46, 412–418. [DOI] [PubMed] [Google Scholar]
- 25. Missimer J. H., Steinmetz M. O., Baron R., Winkler F. K., Kammerer R. A., Daura X., van Gunsteren W. F., Protein Sci. 2007, 16, 1349–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Meuzelaar H., Tros M., Huerta-Viga A., van Dijk C. N., Vreede J., Woutersen S., J. Phys. Chem. Lett. 2014, 5, 900–904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Basu S., Mukharjee D., J. Mol. Model. 2017, 23, 206. [DOI] [PubMed] [Google Scholar]
- 27. Mhaindarkar D., Gasper R., Lupilov N., Hofmann E., Leichert L. I., Commun. Biol. 2018, 1, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yenenler A., Venturini A., Burduroglu H. C., Sezerman O. U., J. Mol. Model. 2018, 24, 229. [DOI] [PubMed] [Google Scholar]
- 29. Zheng Y.-J., Ornstein R. L., J. Am. Chem. Soc. 1996, 118, 11237–11243. [Google Scholar]
- 30.
- 30a. Takano K., Tsuchimori K., Yamagata Y., Yutani K., Biochemistry 2000, 39, 12375–12381; [DOI] [PubMed] [Google Scholar]
- 30b. Zou Z., Alibiglou H., Mate D. M., Davari M. D., Jakob F., Schwaneberg U., Chem. Commun. 2018, 54, 11467–11470. [DOI] [PubMed] [Google Scholar]
- 31. Kawata T., Ogino H., Biochem. Biophys. Res. Commun. 2010, 400, 384–388. [DOI] [PubMed] [Google Scholar]
- 32. Korman T. P., Sahachartsiri B., Charbonneau D. M., Huang G. L., Beauregard M., Bowie J. U., Biotechnol. Biofuels 2013, 6, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Mollania N., Khajeh K., Ranjbar B., Hosseinkhani S., Enzyme Microb. Technol. 2011, 49, 446–452; [DOI] [PubMed] [Google Scholar]
- 33b. Rasekh B., Khajeh K., Ranjbar B., Mollania N., Almasinia B., Tirandaz H., Eng. Life Sci. 2014, 14, 442–448. [Google Scholar]
- 34. Markel U., Zhu L., Frauenkron-Machedjou V. J., Zhao J., Bocola M., Davari M. D., Jaeger K. E., Schwaneberg U., Catalysts 2017, 7, 142. [Google Scholar]
- 35.
- 35a. Frauenkron-Machedjou V. J., Fulton A., Zhao J., Weber L., Jaeger K. E., Schwaneberg U., Zhu L., Bioresour. Bioprocess. 2018, 5, 2; [Google Scholar]
- 35b. Rathi P. C., Fulton A., Jaeger K.-E., Gohlke H., PLoS Comput. Biol. 2016, 12, e1004754; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35c. Nutschel C., Fulton A., Zimmermann O., Schwaneberg U., Jaeger K.-E., Gohlke H., J. Chem. Inf. Model. 2020, 60, 1568–1584. [DOI] [PubMed] [Google Scholar]
- 36. Fulton A., Frauenkron-Machedjou V. J., Skoczinski P., Wilhelm S., Zhu L., Schwaneberg U., Jaeger K. E., ChemBioChem 2015, 16, 930–936. [DOI] [PubMed] [Google Scholar]
- 37. Reetz M. T., Soni P., Fernández L., Gumulya Y., Carballeira J. D., Chem. Commun. 2010, 46, 8657–8658. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Sarakatsannis J. N., Duan Y., Proteins Struct. Funct. Bioinf. 2005, 60, 732–739; [DOI] [PubMed] [Google Scholar]
- 38b. Arnold F. H., Trends Biotechnol. 1990, 8, 244–249. [DOI] [PubMed] [Google Scholar]
- 39. Tejo B. A., Salleh A. B., Pleiss J., J. Mol. Model. 2004, 10, 358–366. [DOI] [PubMed] [Google Scholar]
- 40. Shehata M., Timucin E., Venturini A., Sezerman O. U., J. Mol. Model. 2020, 26, 122. [DOI] [PubMed] [Google Scholar]
- 41. Zhao H., Arnold F. H., Protein Eng. 1999, 12, 47–53. [DOI] [PubMed] [Google Scholar]
- 42. van Pouderoyen G., Eggert T., Jaeger K. E., Dijkstra B. W., J. Mol. Biol. 2001, 309, 215–226. [DOI] [PubMed] [Google Scholar]
- 43.W. L. DeLano, http://www.pymol.org 2002.
- 44. Wong T. S., Arnold F. H., Schwaneberg U., Biotechnol. Bioeng. 2004, 85, 351–358. [DOI] [PubMed] [Google Scholar]
- 45. Ahmad S., Rao N. M., Protein Sci. 2009, 18, 1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou Y., Pérez B., Hao W., Lv J., Gao R., Guo Z., Biochem. Eng. J. 2019, 148, 195–204. [Google Scholar]
- 47.
- 47a. Elcock A. H., J. Mol. Biol. 1998, 284, 489–502; [DOI] [PubMed] [Google Scholar]
- 47b. Dominy B. N., Minoux H., C. L. Brooks III , Proteins Struct. Funct. Bioinf. 2004, 57, 128–141; [DOI] [PubMed] [Google Scholar]
- 47c. Lam S. Y., Yeung R. C., Yu T., Sze K., Wong K., PLoS Biol. 2011, 9, e1001027; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47d. Chan C.-H., Yu T.-H., Wong K.-B., PLoS One 2011, 6, e21624; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47e. Wang G., Wu J., Lin J., Ye X., Yao B., Biochem. Biophys. Res. Commun. 2016, 481, 139–145. [DOI] [PubMed] [Google Scholar]
- 48. Sun Z., Liu Q., Qu G., Feng Y., Reetz M. T., Chem. Rev. 2019, 119, 1626–1665. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Karplus M., McCammon J. A., Nat. Struct. Biol. 2002, 9, 646–652; [DOI] [PubMed] [Google Scholar]
- 49b. Zhang L., Cui H., Dhoke G. V., Zou Z., Sauer D. F., Davari M. D., Schwaneberg U., Chem. Eur. J. 2020, 26, 4974–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kabsch W., Sander C., Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 51. Wedberg R., Abildskov J., Peters G. H., J. Phys. Chem. B 2012, 116, 2575–2585. [DOI] [PubMed] [Google Scholar]
- 52. Stepankova V., Bidmanova S., Koudelakova T., Prokop Z., Chaloupkova R., Damborsky J., ACS Catal. 2013, 3, 2823–2836. [Google Scholar]
- 53.
- 53a. Bellissent-Funel M., Hassanali A., Havenith M., Henchman R., Pohl P., Sterpone F., van der Spoel D., Xu Y., Garcia A. E., Chem. Rev. 2016, 116, 7673–7697; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53b. Serdakowski A. L., Munir I. Z., Dordick J. S., J. Am. Chem. Soc. 2006, 128, 14272–14273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary