

Abstract
We report herein a facile and generalized approach to the modification of solid surfaces with polymer brushes under ambient conditions: filter paper‐assisted surface‐initiated Cu0‐mediated controlled radical polymerization (PSI‐CuCRP). The polymerization solution wetted filter paper is sandwiched between a copper plate and an initiator‐modified substrate, which allows the creation of a surface‐initiated polymerization (SIP) “band‐aid” so that everyone can perform the surface grafting selectively with good control over the quality of the polymer brushes employing low concentration and microliter amounts of the monomer solution. The versatility of this method is demonstrated by grafting different homo‐, block‐, and multicomponent polymer brushes by using the same activation system and reaction conditions, the polymerization process can be precisely controlled to yield uniform polymers and show high chain‐end functionality which is exemplified by in situ tetra‐copolymerization. The combination of photolithography and paper cutting enables to prepare arbitrary three‐dimensional patterned polymer brushes on the surface.
Keywords: ambient condition, copper, polymer brush, surface grafting, surface polymerization
A facile and generalized approach is reported for the modification of solid surfaces with polymer brushes under ambient conditions: filter paper‐assisted surface‐initiated Cu0‐mediated controlled radical polymerization. A surface‐initiated “band‐aid” of the monomer solution wetted filter paper covered with a thin copper plate allows to perform the surface grafting selectively with polymer brushes.
Polymer brushes showing many interesting physicochemical properties have been widely explored in a myriad of applications, such as antifouling, [1] smart materials, [2] biomaterials, [3] actuators, [4] and surface adhesives. [5] Surface‐initiated reversible deactivation radical polymerizations have emerged as a viable strategy for the preparation of functional polymeric surfaces. [6] Surface‐initiated atom transfer radical polymerization (SI‐ATRP) is one of the most popular methods, which allows polymers to be synthesized with unrivaled chain uniformity, this method is also tolerant to impurities and even allows the polymerization to proceed at physiological conditions. [7] Though promising, SI‐ATRP is always based on complicated, high‐cost, multi‐stage processes. Moreover, to conduct a successful SI‐ATRP, parameters such as the type of initiators, added salts, reducing agents and oxygen removal, need to be optimized to achieve well‐defined polymer brushes on surfaces. [8] This is challenging for the “experts”, and particularly for nonexperts, as choosing the appropriate conditions for a successful surface polymerization can be difficult and time‐consuming, which limits its application in many fields.
To overcome these limitations and gain better control over the polymerization process, various external stimuli‐mediated ATRP approaches have been developed recently for surface grafting, including activator regenerated by electron transfer radical polymerization (ARGET ATRP), [9] electrochemical‐(eATRP), [10] and light stimulus (light ATRP), [11] as well as Cu0‐mediated processes, [12] which enable the synthesis of polymer brushes with a remarkable degree of control under less rigorous standards. However, the choice of an appropriate method depends highly on the intrinsic nature of the monomers (e.g., structure, chain propagation rate, solubility), which can also be challenging. Hence, a universal activation system is highly desirable where identical components (e.g., catalyst, ligand, solvent, and temperature) can be used for the surface‐initiated polymerization (SIP) of a wide range of monomer families under environmentally friendly conditions.
Herein, we report a universal method for the surface‐initiated polymerization of various types of monomers based on filter paper‐assisted surface‐initiated CuCRP strategy (PSI‐CuCRP) for the fast fabrication of well‐defined polymer brushes under ambient conditions. Filter paper wetted by the polymerization solution (monomer, ligand and solvent) is sandwiched between a copper plate and an initiator‐modified substrate. We demonstrate a proof of concept design, SIP “band‐aid”, which makes polymer brush growth on arbitrary substrates easily achievable. The filter paper is used as a monomer reservoir for the polymerization while using the same catalyst, ligand, and solvent. Cu0 acts as the source of CuI activator and CuΙΙ deactivator and the reducing agent for the CuΙΙ complex to generate active CuΙ species through the comproportionation (Scheme 1, inset). A [CuΙ/CuΙI]/ligand complex diffused to the vicinity of the initiator layer, reacts with the initiator as a consequence of producing free radicals to initiate the polymerization reaction. A wide range of hydrophobic and hydrophilic monomers including acrylates, acrylamides, methacrylates, methacrylamides, styrene, and 4‐vinylpyridine were successfully polymerized. The easy setup, as simple as a medical band‐aid that “everyone” can perform the surface polymerization accurately and reproducibly.
Scheme 1.
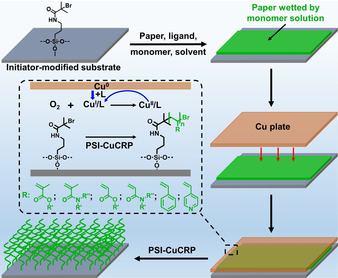
Schematic view of polymer brush grafting through PSI‐CuCRP under ambient conditions. The filter paper acts as a polymerization solution reservoir and a spacer to separate the copper plate and the initiator‐modified substrate.
Scheme 1 shows the procedure for the preparation of the surface grafted polymer brushes via PSI‐CuCRP. A piece of filter paper wetted by the polymerization solution which contains monomer (e.g., methacrylates, methacrylamides, acrylates, acrylamides, styrene (St) or 4‐vinyl pyridine (4VP)), ligand N,N,N′,N′,N′‐pentamethyldiethylenetriamine (PMDETA), and solvent (water/methanol, v/v=2:1) was placed onto the ATRP initiator‐modified substrate (Scheme S1), a thin and flat copper plate (200 nm‐thick) was covered onto the filter paper to form a sandwiched structure with the initiator‐modified substrate. The copper plate acts as the source of CuI and CuΙΙ complexes in the presence of ligand. CuΙΙ complex was reduced by Cu0 to form the catalytic CuΙ species through the comproportionation, spatiotemporal distribution of [CuI]/[CuII] as a function of the distance between the paper and the copper plate mediated the polymerization kinetics. As a proof‐of‐concept, we demonstrated the polymerization of the thermo‐responsive N‐isopropylacrylamide (NIPAAm) on the initiator‐modified silicon substrate (see the details of the initiator immobilization in SI) by using our method, a filter paper (15 mm×8 mm, 75 μm‐thick with the average pore size around 0.45 μm) was wetted by 15 μL of the monomer solution (2.36 M, 4 mg/15 μL) and was placed on the top of the initiator‐modified substrate, the copper plate was covered onto the filter paper, the polymerization occurred in the small compartment between the “band‐aid” and the substrate. The growth of PNIPAAm brushes on the substrate was confirmed by FT‐IR (Figure S1a). The amide stretch and bend of PNIPAAm at 1640 and 1540 cm−1 can be assigned to the amide groups of the PNIPAAm, indicating the successful grafting of PNIPAAm brushes. The growth kinetics of PNIPAAm on the substrate was tracked by ellipsometry. Even in the presence of air, no induction period was observed due to the fast consumption of limited amounts of oxygen in the system. One can see in Figure 1 a that the polymerization is very fast and PNIPAAm reaches the maximum growth at around half an hour with a thickness around 440 nm (average growth rate: 14.7 nm min−1). The grafting density is about 0.29 chain/nm2 (estimated from the swelling ratio of the dry and swollen thickness of PNIPAAm, Table S1). [13] We also compared the PNIPAAm brush growth via other SIP methods such as SI‐CuCRP (4.7 or 3.2 nm min−1 in the presence of air or oxygen‐free), [14] SI‐photo ATRP (1.6 nm min−1), [15] SI‐sa‐ATRP (1.7 nm min−1), [16] and eATRP (0.33–2.1 nm min−1) [17] (Table S2) and our method gave the fastest growth rate. The fast polymerization rate was observed due to the fast reduction reaction of CuΙΙ to CuΙ so as to a high concentration of [CuI]/[CuII], while fast termination reactions occurring at the same time limit the chain propagations. [8a] Additionally, when a polymerization mixture is placed between a flat copper plate and an initiator‐modified substrate, the vertical distance between these two overlaying surfaces determines the tolerance of the grafting process toward the oxygen, at a very small distance between the copper plate and the initiator‐modified surface, the oxygen dissolved in the solution is rapidly scavenged by CuI or through the oxidation of the metallic substrate, while CuΙ was (re)generated by the continuous reduction of CuΙΙ and made the whole process cycle. In the presence of ligand, CuI species diffuse to the initiator surface and trigger the polymerization. Hence, the CuI (re)generation rate, together with the intrinsic property of the monomer which is related to the activation process (k act) of ATRP determines the growth rate of polymer brushes. [18]
Figure 1.
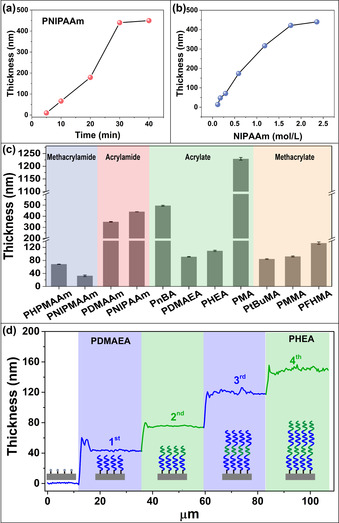
Thickness evolution of PNIPAAm brushes on silicon substrate against the polymerization time, [NIPAAm]=2.36 M (a) and, the thickness vs. the concentration of NIPAAm, polymerization time, 30 min (b). c) Thickness characterization of 11 different polymer‐coated surfaces (polymerization time, 30 min). d) Copolymerization of two different monomers, [DMAEA]=1.65 M, [HEA]=2.18 M, PDMAEA‐b‐PHEA‐b‐PDMAEA‐b‐PHEA via PSI‐CuCRP.
Since the initiator forms a monolayer on the surface, the monomer consumed in the polymer growth can be ignored in comparison to that in the bulk solution. [19] However, a high concentration of monomer solution is still required to get the maximum growth of the polymer brushes, [17] but it is hard to recycle, in particular for ionic monomers and very expensive ones. Moreover, the recycling process can be time‐consuming. The new method reports here allows the polymer brushes to be prepared at low monomer concentration and microliter volumes of the polymerization solution. PSI‐CuCRP of NIPAAm at various concentrations was conducted and the relationship between the thickness and monomer concentration was shown in Figure 1 b. We observed that the PNIPAAm brush growth showed a “quasi‐linear” growth with the monomer concentration below 2 M. Maximum growth of the PNIPAAm growth (440 nm) was observed at the concentration of 2.36 M, and 13 nm of PNIPAAm was observed when the concentration was as low as milligram per milliliter (mg mL−1), which offers an alternate way to tune the polymer growth on the surface. The versatility of this universal method was demonstrated by the controlled polymerization of different monomer families, such as methacrylamides, acrylamides, acrylates, and methacrylates, these monomers were successfully polymerized and the thicknesses of the corresponding polymer brushes were shown in Figure 1 c. However, structural differences of these monomers yielded different polymerization kinetics even if the same catalytic system was chosen. [20]
The “living” nature and the end‐group fidelity was exemplified by in situ sequential re‐initiation of 2‐(Dimethylamino)ethyl acrylate (DMAEA) and 2‐hydroxyethyl acrylate (HEA), after 4 times of polymerization, the polymers are still showing the “living” end functionality. The thickness of each block was characterized by AFM and showed 40, 35, 45, and 30 nm in thickness for each block (Figure 1 d, Figure S2). We compared the polymer growth with other SIP methods such as SI‐CuCRP (in the presence of air or oxygen‐free condition) and SI‐ATRP, by testing the polymerization of different types of monomers, zwitterionic [2‐(methacryloyloxy)ethyl] dimethyl‐(3‐sulfopropyl) ammonium hydroxide (SBMA), hydrophobic St and 4VP by using the same monomer concentration. The results are shown in Figure S1b–d and Table S2 in supporting information, and the thicknesses of PSBMA, PS, and P4VP brushes via PSI‐CuCRP are 80, 31, and 61 nm, respectively. PSI‐CuCRP method yielded the highest polymer thickness, while there are almost no polymer brushes observed by SI‐CuCRP or SI‐ATRP. The results demonstrate our method reported here is more universal.
To elucidate the role of the filter paper in PSI‐CuCRP, we designed three control experiments (Figure S3). The arrangement of the filter paper in between the copper plate and the initiator surface controls the polymerization kinetics, the filter paper that is close to the initiator surface showed the highest polymer brush growth. Thus, we believe the filter paper provides a microenvironment for the polymerization with a relatively high amount of active cuprous species but with less oxygen in the polymerization medium. In addition, the filter paper was characterized by the scanning electron microscope (SEM) before and after the polymerization, no obvious morphology or pore size change was observed (Figure S4).
Besides, PSI‐CuCRP also provide a facile and economic way for fabrication of functional and patterned surfaces. As Figure 2 a shows, a dual‐functional surface with hydrophobic and hydrophilic brushes on the same substrate by using the same initiating conditions. Half of the substrate was grafted with the hydrophobic poly(1H,1H,2H,2H‐nonafluorohexyl methacrylate) (PFHMA) brushes, and another half of the substrate was grafted with the hydrophilic poly(3‐sulfopropyl methacrylate potassium salt) (PSPMA) brushes. SEM image (Figure 2 b) and EDS of fluorine distribution map (Figure 2 c) clearly show the boundary between the PFHMA and PSPMA brush regions. The static water‐contact angles were 116±2° for PFHMA surface and <10° for PSPMA surface (Figure 2 d). Following this strategy, multiple polymer brush patterns can be easily created on the same substrate by the simple paper cutting without using other lithography methods (Figure 2 e). In addition, different patterns can be fabricated by using different‐shaped SIP “band‐aid” (Figure 2 f). Moreover, various patterns (negative, positive, and binary patterns) with smaller sizes can be also obtained by taking the advantage of the photolithographic technology (Figure S5). Thus, PSI‐CuCRP offers an efficient and facile way to prepare various patterned polymer brushes with different functionalities.
Figure 2.
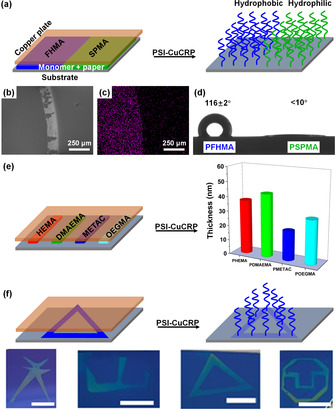
a) Illustration of the synthesis of dual‐functional binary polymer brushes. b) SEM image and c) EDS fluorine (belongs to the functional group of FHMA) distribution map and d) contact angle analysis of binary PFHMA and PSPMA brushes. e) Fabrication of polymer brush arrays via PSI‐CuCRP without using PDMS mask and the thickness of the as‐prepared polymer brushes. f) Preparation of patterned polymer brushes via a combination of PSI‐CuCRP and paper cutting (Scale bar: 5 mm).
Usually, to conduct the surface modification on a solid substrate, the substrate needs to be completely immersed into the monomer solution to achieve homogeneous polymer growth, and the polymerization solution requires careful degassing prior to polymerization, which makes the SIP methods not ideal to modify large and irregular shaped substrate, in some cases, is practically impossible. Furthermore, it is challenging to modify a certain area of interest instead of the whole surface. While, with the PSI‐CuCRP method, surface‐attached polymers can be prepared on substrate of irregular shape selectively yet using microliter amounts of polymerization solution as long as the “band‐aid” covers the surface of interest. In Figure 3 a,b, we made a conceptional “band‐aid” to conduct surface grafting on large and irregular‐shaped surfaces, one can observe the wetting property change of the glass vial surface after grafting with polystyrene. As shown in Figure 3 c,d, surfaces grafted with hydrophilic PSPMA brushes show good antifog and anti‐icing properties, the modified areas of the glass surface exhibit very good fidelity under high humidity and low temperature (−20 °C), which have wide applications such as producing antifog and anti‐icing coatings for the glass windows of ships that sail in foggy or icy weather conditions.
Figure 3.
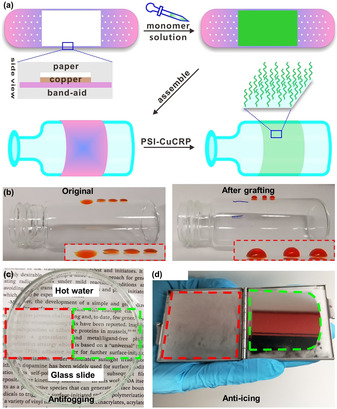
a) schematic illustration of SIP “band‐aid” for polymer brush growth. (b) Photographs of glass bottle before and after grafting polymer brushes via SIP “band‐aid”. A glass slide (c) and mirror (d) with half side modified with hydrophilic PSPMA for antifogging and anti‐icing purposes.
In conclusion, PSI‐CuCRP was successfully explored to fabricate different types of polymer brushes by using the same reaction conditions (same catalyst/ligand, solvent) with low concentration and microliter volumes of monomers under the ambient atmosphere, enables the synthesis of multifunctional polymer brushes on arbitrary substrates with a remarkable degree of control over modification areas on‐demand without inert atmosphere protection nor deoxygenation process. The polymer brushes on the surface show high living nature, which has been demonstrated by stepwise block copolymerization. The versatile nature of this method, together with readily commercially available reagents, will greatly expand the access and availability of polymer brushes to “everyone” but not limited to researchers only. We envision that polymer brushes will be widely used both in scientific research fields and in our daily life.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
WL acknowledges the financial support from the China Scholarship Council (CSC) of the People′s Republic of China (Ph.D. grant, No. 201609505009). WS acknowledges PhD research funding from the Initiative and Networking Fund of the Helmholtz Association of German Research Centers through the International Helmholtz Research School for Nanoelectronic Networks, IHRS NANONET (VH‐KO‐606). The authors also acknowledge the use of the facilities in the Dresden Center for Nanoanalysis (DCN) of Technische Universität Dresden. Open access funding enabled and organized by Projekt DEAL.
W. Li, W. Sheng, B. Li, R. Jordan, Angew. Chem. Int. Ed. 2021, 60, 13621.
Contributor Information
Dr. Wenbo Sheng, Email: wenbo.sheng@tu-dresden.de.
Dr. Bin Li, Email: bin1.li@tum.de.
Prof. Rainer Jordan, Email: rainer.jordan@tu-dresden.de.
References
- 1.
- 1a. Jiang S., Cao Z., Adv. Mater. 2010, 22, 920–932; [DOI] [PubMed] [Google Scholar]
- 1b. Ma H., Hyun J., Stiller P., Chilkoti A., Adv. Mater. 2004, 16, 338–341. [Google Scholar]
- 2.
- 2a. Stuart M. A., Huck W. T., Genzer J., Muller M., Ober C., Stamm M., Sukhorukov G. B., Szleifer I., Tsukruk V. V., Urban M., Winnik F., Zauscher S., Luzinov I., Minko S., Nat. Mater. 2010, 9, 101–113; [DOI] [PubMed] [Google Scholar]
- 2b. Zhou F., Huck W. T. S., Phys. Chem. Chem. Phys. 2006, 8, 3815–3823; [DOI] [PubMed] [Google Scholar]
- 2c. Sheng W., Li W., Li B., Li C., Xu Y., Guo X., Zhou F., Jia X., Macromol. Rapid Commun. 2015, 36, 1640–1645; [DOI] [PubMed] [Google Scholar]
- 2d. Sheng W., Li B., Wang X., Dai B., Yu B., Jia X., Zhou F., Chem. Sci. 2015, 6, 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Hafner D., Ziegler L., Ichwan M., Zhang T., Schneider M., Schiffmann M., Thomas C., Hinrichs K., Jordan R., Amin I., Adv. Mater. 2016, 28, 1489–1494; [DOI] [PubMed] [Google Scholar]
- 3b. Senaratne W., Andruzzi L., Ober C. K., Biomacromolecules 2005, 6, 2427–2448. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Li B., Du T., Yu B., van der Gucht J., Zhou F., Small 2015, 11, 3494–3501; [DOI] [PubMed] [Google Scholar]
- 4b. Hosono N., Kajitani T., Fukushima T., Ito K., Sasaki S., Takata M., Aida T., Science 2010, 330, 808–811. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Lamping S., Otremba T., Ravoo B. J., Angew. Chem. Int. Ed. 2018, 57, 2474–2478; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2499–2503; [Google Scholar]
- 5b. Ma S., Zhang X., Yu B., Zhou F., NPG Asia Mater. 2019, 11, 24. [Google Scholar]
- 6.
- 6a. Tria M. C. R., Grande C. D. T., Ponnapati R. R., Advincula R. C., Biomacromolecules 2010, 11, 3422–3431; [DOI] [PubMed] [Google Scholar]
- 6b. Gonzato C., Courty M., Pasetto P., Haupt K., Adv. Funct. Mater. 2011, 21, 3947–3953; [Google Scholar]
- 6c. Andruzzi L., Hexemer A., Li X., Ober C. K., Kramer E. J., Galli G., Chiellini E., Fischer D. A., Langmuir 2004, 20, 10498–10506; [DOI] [PubMed] [Google Scholar]
- 6d. Zoppe J. O., Ataman N. C., Mocny P., Wang J., Moraes J., Klok H. A., Chem. Rev. 2017, 117, 1105–1318; [DOI] [PubMed] [Google Scholar]
- 6e. Gupta S., Agrawal M., Conrad M., Hutter N. A., Olk P., Simon F., Eng L. M., Stamm M., Jordan R., Adv. Funct. Mater. 2010, 20, 1756–1761; [Google Scholar]
- 6f. Steenackers M., Gigler A. M., Zhang N., Deubel F., Seifert M., Hess L. H., Lim C. H., Loh K. P., Garrido J. A., Jordan R., Stutzmann M., Sharp I. D., J. Am. Chem. Soc. 2011, 133, 10490–10498. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Li B., Yu B., Ye Q., Zhou F., Acc. Chem. Res. 2015, 48, 229–237; [DOI] [PubMed] [Google Scholar]
- 7b. Narupai B., Page Z. A., Treat N. J., McGrath A. J., Pester C. W., Discekici E. H., Dolinski N. D., Meyers G. F., Read de Alaniz J., Hawker C. J., Angew. Chem. Int. Ed. 2018, 57, 13433–13438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13621–13626; [Google Scholar]
- 7c. Layadi A., Kessel B., Yan W., Romio M., Spencer N. D., Zenobi-Wong M., Matyjaszewski K., Benetti E. M., J. Am. Chem. Soc. 2020, 142, 3158–3164; [DOI] [PubMed] [Google Scholar]
- 7d. Navarro L. A., Enciso A. E., Matyjaszewski K., Zauscher S., J. Am. Chem. Soc. 2019, 141, 3100–3109. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Li B., Yu B., Zhou F., Macromol. Rapid Commun. 2013, 34, 246–250; [DOI] [PubMed] [Google Scholar]
- 8b. Matyjaszewski K., Tsarevsky N. V., J. Am. Chem. Soc. 2014, 136, 6513–6533; [DOI] [PubMed] [Google Scholar]
- 8c. Yan W., Dadashi-Silab S., Matyjaszewski K., Spencer N. D., Benetti E. M., Macromolecules 2020, 53, 2801–2810. [Google Scholar]
- 9.
- 9a. Byard S. J., Williams M., McKenzie B. E., Blanazs A., Armes S. P., Macromolecules 2017, 50, 1482–1493; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Min K., Jakubowski W., Matyjaszewski K., Macromol. Rapid Commun. 2006, 27, 594–598. [Google Scholar]
- 10.
- 10a. Magenau A. J. D., Strandwitz N. C., Gennaro A., Matyjaszewski K., Science 2011, 332, 81–84; [DOI] [PubMed] [Google Scholar]
- 10b. Li B., Yu B., Huck W. T. S., Zhou F., Liu W., Angew. Chem. Int. Ed. 2012, 51, 5092–5095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5182–5185. [Google Scholar]
- 11. Fors B. P., Poelma J. E., Menyo M. S., Robb M. J., Spokoyny D. M., Kramer J. W., Waite J. H., Hawker C. J., J. Am. Chem. Soc. 2013, 135, 14106–14109. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Yan W., Fantin M., Ramakrishna S., Spencer N. D., Matyjaszewski K., Benetti E. M., ACS Appl. Mater. Interfaces 2019, 11, 27470–27477; [DOI] [PubMed] [Google Scholar]
- 12b. Yan W., Fantin M., Spencer N. D., Matyjaszewski K., Benetti E. M., ACS Macro Lett. 2019, 8, 865–870; [DOI] [PubMed] [Google Scholar]
- 12c. Li W., Sheng W., Jordan R., Zhang T., Polym. Chem. 2020, 11, 6971–6977; [Google Scholar]
- 12d. Fantin M., Ramakrishna S. N., Yan J., Yan W., Divandari M., Spencer N. D., Matyjaszewski K., Benetti E. M., Macromolecules 2018, 51, 6825–6835; [Google Scholar]
- 12e. Li W., Sheng W., Wegener E., Du Y., Li B., Zhang T., Jordan R., ACS Macro Lett. 2020, 9, 328–333. [DOI] [PubMed] [Google Scholar]
- 13. Jordan R., Ulman A., Kang J. F., Rafailovich M. H., Sokolov J., J. Am. Chem. Soc. 1999, 121, 1016–1022. [Google Scholar]
- 14.
- 14a. Zhang T., Du Y., Kalbacova J., Schubel R., Rodriguez R. D., Chen T., Zahn D. R. T., Jordan R., Polym. Chem. 2015, 6, 8176–8183; [Google Scholar]
- 14b. Zhang T., Du Y., Müller F., Amin I., Jordan R., Polym. Chem. 2015, 6, 2726–2733. [Google Scholar]
- 15. Yan J., Li B., Zhou F., Liu W., ACS Macro Lett. 2013, 2, 592–596. [DOI] [PubMed] [Google Scholar]
- 16. Yan J., Li B., Yu B., Huck W. T., Liu W., Zhou F., Angew. Chem. Int. Ed. 2013, 52, 9125–9129; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9295–9299. [Google Scholar]
- 17. Shida N., Koizumi Y., Nishiyama H., Tomita I., Inagi S., Angew. Chem. Int. Ed. 2015, 54, 3922–3926; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3994–3998. [Google Scholar]
- 18.
- 18a. Zhang Q., Wilson P., Li Z., McHale R., Godfrey J., Anastasaki A., Waldron C., Haddleton D. M., J. Am. Chem. Soc. 2013, 135, 7355–7363; [DOI] [PubMed] [Google Scholar]
- 18b. Turgman-Cohen S., Genzer J., J. Am. Chem. Soc. 2011, 133, 17567–17569. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Harris B. P., Metters A. T., Macromolecules 2006, 39, 2764–2772; [Google Scholar]
- 19b. Dunderdale G. J., Urata C., Miranda D. F., Hozumi A., ACS Appl. Mater. Interfaces 2014, 6, 11864–11868. [DOI] [PubMed] [Google Scholar]
- 20. Jones D. M., Huck W. T., Adv. Mater. 2001, 13, 1256–1259. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary