

Abstract
Objective
To establish clear priorities for the care of patients with acquired hemophilia A (AHA) by proposing 10 key principles of practical, holistic AHA management.
Method
These principles were developed by the Zürich Haemophilia Forum, an expert panel of European hemophilia specialists comprising physicians and nursing and laboratory specialists.
Results
The 10 proposed principles for AHA care are as follows: (a) Improving initial diagnosis of AHA; (b) Differential diagnosis of AHA: laboratory assessment of patients with unusual bleeding; (c) Effective communication between laboratories, physicians, and specialists; (d) Improving clinical care: networking between healthcare professionals in the treating hospital and specialist hemophilia centers; (e) Comprehensive assessment of bleeding; (f) Appropriate use of bypassing agents; (g) Long‐term follow‐up and monitoring for efficacy and safety of immunosuppressive treatment; (h) Inpatient/outpatient settings; (i) Access to innovative and disruptive treatments; (j) Promotion of international collaborative research.
Conclusion
The proposed principles for holistic AHA care aim to ensure swift diagnosis and optimal patient management. Key to achieving this goal is training for healthcare personnel in non‐specialist hospitals and collaboration between different specialists. We hope these principles will increase awareness of AHA in the wider medical community and catalyze efforts toward improving its practical, multidisciplinary management.
Keywords: acquired hemophilia, care, diagnosis, management, principles, treatment
1. What is the new aspect of your work?
We offer ten key principles for the practical and holistic management of individuals with acquired hemophilia A (AHA) care. The aim is to guide patient management from first presentation that often occurs outside specialist hematology settings, through to treatment and follow‐up.
2. What is the central finding of your work?
The timely diagnosis and effective management of AHA depend on a multidisciplinary approach founded on two key factors: educating healthcare personnel in smaller, non‐specialist settings about this rare disorder; and establishing good communication and collaboration between hemophilia specialists and the wide‐ranging healthcare professionals involved in the patient pathways.
3. What is (or could be) the specific clinical relevance of your work?
It is hoped that the 10 proposed principles will improve awareness of AHA in the wider medical community, help to ensure swifter diagnosis and more effective management of AHA, and act as a catalyst for further research and collaborative investment in the practical, multidisciplinary management of AHA.
1. INTRODUCTION
Acquired hemophilia A (AHA), a rare autoimmune bleeding disorder 1 , 2 characterized by the development of auto‐antibodies against endogenous factor VIII (FVIII), is characterized by spontaneous, mild to life‐threatening bleeds in (often elderly) individuals with no personal or family history of bleeding. 3 , 4 , 5 , 6 , 7 , 8 Rapid diagnosis and prompt treatment of bleeds are crucial to optimize outcomes. 9 However, effective care is constrained by the lack of awareness of the clinical features, the failure to appreciate the serious nature of the condition, and poor understanding of the laboratory assessment of acquired hemophilia.
Diagnosing and treating AHA are complex 5 and should always be managed directly by or jointly with specialist hemophilia care, even if patients do not present with significant bleeding. 10 While international recommendations and guidelines have been published, 1 , 6 , 11 , 12 , 13 they typically focus on medical and bleed treatment; furthermore, they are primarily targeted toward specialist audiences. Outside specialist settings, awareness of AHA remains low, resulting in delayed diagnosis and delayed and/or inappropriate treatment, which increases the risk of iatrogenic bleeds and ultimately affects the patient's prognosis. Accordingly, new and proactive strategies should be developed to improve awareness of AHA in the wider medical community, particularly in this era of a growing aging population, so that diagnosis and management of the disorder might be improved.
Therefore, we offer 10 key principles of practical, holistic AHA care focusing on critical steps from initial presentation through to diagnosis, treatment, and follow‐up (Table 1). Our overall goal is to establish clear priorities for patient care and to propose strategies for implementing such care through two key channels: first, by supporting the recognition and treatment of AHA when patients first present; and second, by encouraging the coordination and delivery of effective treatment by hemophilia specialists and other healthcare professionals (HCPs) involved in patient management.
TABLE 1.
Principles of acquired hemophilia A care
| Principle 1 | Improving initial diagnosis of AHA in all settings |
|---|---|
| Principle 2 | Differential diagnosis of AHA: Laboratory assessment of patients with unusual bleeding |
| Principle 3 | Effective communication between laboratories, physicians, and specialists |
| Principle 4 | Improving clinical care: networking between HCPs in the treating hospital and specialist hemophilia treatment centers |
| Principle 5 | Comprehensive assessment of bleeding is required |
| Principle 6 | Appropriate use of bypassing agents for prompt treatment |
| Principle 7 | Long‐term follow‐up and monitoring for efficacy and safety of immunosuppressive treatment |
| Principle 8 | Promotion of patient care and follow‐up in the home, inpatient and outpatient settings |
| Principle 9 | Access to innovative and disruptive treatments will be important |
| Principle 10 | Promotion of collaborative research to improve patient outcomes |
Abbreviations: AHA, acquired hemophilia A; HCP, healthcare professionals.
2. METHODS
The principles of AHA care were developed by members of the Zürich Haemophilia Forum, an expert panel of European hemophilia specialists, sponsored by a grant from Novo Nordisk. When the Forum convened for their 21st meeting in May 2019, their aim was to consider and formulate consensus on optimal management strategies for AHA, focusing on the needs of patients and the various HCPs who provide care. The principles outlined in this document were derived from the panel's discussions and consensus. For the purpose of providing a comprehensive, holistic perspective, the panel for this meeting included physicians, and nursing and laboratory specialists.
3. PRINCIPLES OF ACQUIRED HEMOPHILIA CARE
3.1. Principle 1: Improving initial diagnosis of AHA in all settings
3.1.1. Rationale
The rarity of AHA, combined with its heterogeneous bleeding phenotype and association with advanced age and multiple comorbidities, 5 , 6 means that diagnosis can be challenging.
This difficulty is compounded by the low awareness of AHA in the wider medical community. This is problematic, as patients with AHA typically present to a wide range of clinical teams (eg, internal medicine, emergency, or intensive care physicians; geriatricians; obstetricians; rheumatologists; oncologists; or surgeons). 1 , 3
Lack of familiarity with AHA may mean that patients are not referred to the correct specialist care, or that they remain undiagnosed. Thus, it is likely that AHA will be frequently under‐ or misdiagnosed and inadequately treated in real‐world clinical practice. 1 , 3 , 6
3.1.2. Improving recognition of AHA
AHA is extremely rare, affecting approximately 1.5 individuals per million population per year. 11 Even specialists within hemophilia treatment centers (HTCs), such as those in this panel, only see a few cases per year, and suspected cases of AHA that present outside of these specialist centers are not systematically referred to experts in HTCs. Further, the bleeding pattern in AHA is heterogeneous: It is typically characterized by subcutaneous bleeds, bleeding into other soft tissues or mucous membranes, and muscle hematomas, 3 , 5 , 8 , 11 and bleeds may range from mild enough that they do not require treatment to so severe that they threaten life, limbs, or organs 5 , 6 , 8 , 11 , 14 (Figure 1). Additionally, although 50% of AHA cases are associated with underlying medical conditions, including autoimmune disease, malignancy, infections, postpartum, and drug‐related conditions, 5 , 8 , 11 , 15 the remaining 50% are idiopathic.
FIGURE 1.
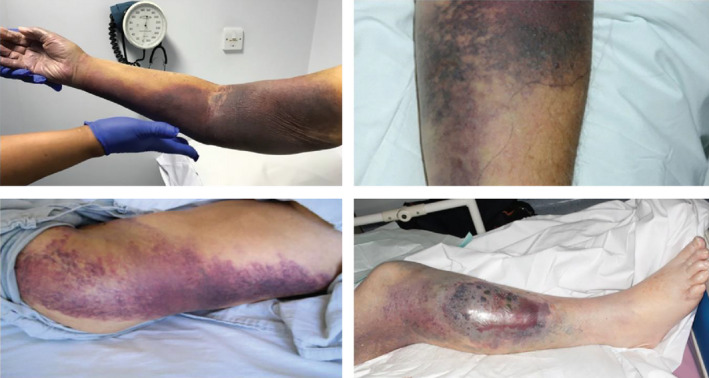
Bleeding manifestations in AHA. Subcutaneous ecchymoses associated with AHA. For example, the top left image is a typical presentation of bleeding post venipuncture in a Caucasian lady; note the tracking of blood all the way into the hand/fingers. Patient consent and full permission for reproduction and publication were obtained for these images. AHA, acquired hemophilia A
Recognizing AHA can therefore be extremely difficult. Nonetheless, some diagnostic indicators can be proposed. When faced with unexpected bleeding in older males and females or in postpartum women in the absence of a personal or family history of bleeding, physicians should first evaluate the patient's medical history, including current medication use. If the effects of anticoagulant therapy can be ruled out as the cause of bleeding, physicians should suspect AHA and investigate accordingly. It should be noted, however, that AHA may develop in individuals already on anticoagulant therapy. Although AHA predominantly affects elderly individuals and postpartum women, it can also occur in patients of any age with malignancy or autoimmune disease; therefore, unexpected bleeding in these patients should also be investigated. The first step to confirming a diagnosis of AHA is a thorough laboratory investigation (see Section 3.2: Principle 2).
3.1.3. Improving awareness in the wider medical community
Increasing awareness of AHA among the wider medical community will improve recognition of AHA and ultimately improve patient outcomes. Multiple and varied initiatives should be considered in the attempt to improve awareness, including awareness campaigns, webinars, scientific meetings, leaflets, brochures, training programs, networking events, dedicated sessions on AHA at national and international conferences, and the publication of recommendations and other relevant articles in general medicine journals with open access.
Efforts to improve awareness of AHA among specialists in other, non‐hemophilia fields should position AHA as a medical emergency, as well as highlight its clinical presentation and bleeding phenotype and differentiate it from congenital hemophilia (Table 2).
TABLE 2.
Advice for non‐specialists: improving awareness, diagnosis, and treatment of AHA outside specialist centers
| Diagnosing AHA |
Awareness of the following features of AHA will aid recognition of the disease and facilitate referral of patients to specialist centers:
|
| Laboratory investigation |
|
| Patient management: advice for nurses |
|
Abbreviations: AHA, acquired hemophilia A; aPTT, activated partial thromboplastin time; FVIII, factor VIII; PT, prothrombin time.
Key takeaway
Increasing awareness of AHA in the wider medical community will aid recognition of the disease, facilitate patient referral to specialist care, and ultimately improve outcomes.
3.2. Principle 2: Differential diagnosis of AHA: Laboratory assessment of patients with unusual bleeding
3.2.1. Rationale
An isolated, prolonged activated partial thromboplastin time (aPTT) is often the first clue and recognition of this feature is key to diagnosing AHA.
Assessing the differential diagnoses may be challenging in smaller, non‐specialist centers that do not have access to laboratories with the relevant expertise.
3.2.2. Laboratory investigation
The finding of a low FVIII coagulant activity (FVIII:C) is crucial in the diagnosis of AHA. Thereafter confirmatory tests to demonstrate the antibody‐mediated or inhibitory activity against FVIII will establish the diagnosis. Several diagnostic algorithms have been proposed to confirm AHA (see, for example, Collins et al, 3 Kruse‐Jarres et al, 1 Tiede et al 16 ); an overview of key diagnostic steps is provided in Figure 2.
FIGURE 2.
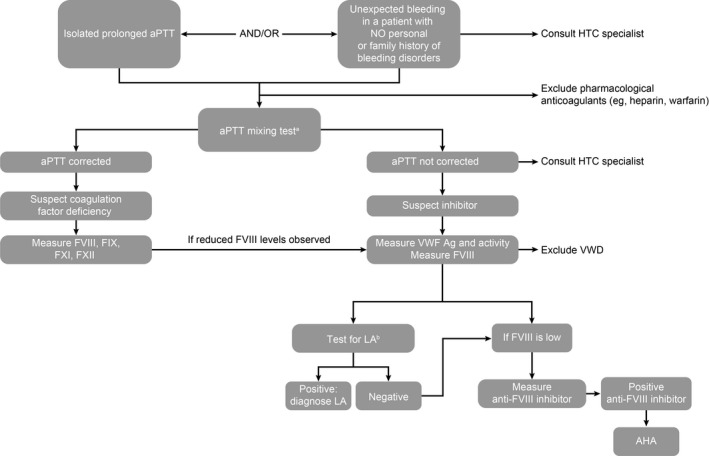
Key steps in diagnosing AHA. AHA, acquired hemophilia A; aPTT, activated partial thromboplastin time; FVIII, factor VIII; FIX, factor IX; FXI, factor XI; FXII, factor XII; HTC, hemophilia treatment center; LA, lupus anticoagulant; VWD, von Willebrand disease; VWF, von Willebrand factor
It is important for smaller hospitals and laboratories in non‐hematology specialist centers to establish a strategy for dealing with a prolonged aPTT, especially in cases where discrimination between AHA and other potential causes of a prolonged aPTT is difficult. 16 This strategy should involve referring unexpected prolonged aPTT results and details of any bleeding to a specialist (see Section 3.3: Principle 3). To raise awareness of AHA and the associated diagnostic work‐up, more attention should also be given to presenting case studies at laboratory educational initiatives.
Key takeaway
AHA should be suspected in patients with unexplained bleeding and a prolonged aPTT.
3.3. Principle 3: Effective communication between laboratories, physicians, and specialists
3.3.1. Rationale
Laboratory test results can be confusing and difficult to interpret, especially for non‐specialists (see Section 3.2: Principle 2).
It is crucial to support clinicians in smaller, non‐HTC hospitals, and to improve organization, collaboration, and information‐sharing between physicians and laboratory scientists.
3.3.2. How to improve communication
The key to accurate interpretation of laboratory test results and prompt AHA diagnosis is effective communication and collaboration between the laboratory, hemophilia/coagulation specialists, and the physician requesting the tests.
When requesting laboratory tests, physicians should tell the laboratory staff whether or not the patient is actively bleeding and provide comprehensive information regarding the patient's history, symptoms, and clinical scenario. Physicians should also indicate to whom test results should be returned—time is often wasted in trying to find the clinician who requested the test and the laboratory personnel must be aware that this is an urgent clinical situation and that the processing of tests should be prioritized. In turn, laboratories could better support physicians by providing a written explanation and interpretation of the results, rather than returning the results without comment.
Further, it is important to establish a system that flags changes in aPTT results over time. In the event of abnormal test results, such as a prolonged aPTT, the laboratory could suggest next steps to take, such as contacting the hematology team immediately if further information is needed. It may also be useful for hospitals to define which other clinical and laboratory scenarios trigger a request for the hematologist to see the patient and/or consult with the patient's physician.
Key takeaway
The laboratory diagnosis of AHA requires: (a) collaboration and information‐sharing between the specialist, the treating physician, and laboratory personnel; and (b) a system by which important changes in laboratory results and/or clinical conditions are flagged up to specialists.
3.4. Principle 4: Improving clinical care—networking between HCPs in the treating hospital and specialist HTCs
3.4.1. Rationale
When a patient with suspected AHA presents in a non‐specialist setting, collaboration with hemostasis experts is vital for ensuring accurate diagnosis and optimal management. Therefore, every non‐HTC hospital and laboratory should know whom to contact when treating patients with unexplained bleeding.
Effective networking between HCPs in specialist HTCs and non‐specialist hospitals will facilitate the recognition of AHA, swift referral to specialist care, and delivery of effective treatment.
Patients with AHA are prone to subcutaneous, mucosal, and soft tissue bleeding 3 , 5 , 8 , 11 that can occur after minimal trauma 3 , 15 , 17 and that can quickly become life threatening. 11 Further, invasive procedures should be avoided unless they are absolutely necessary, in which case they should only be performed under bypassing agent coverage. Therefore, collaboration can also help ensure that patients are handled and managed in a way that minimizes the risk of iatrogenic injury.
3.4.2. Networking to improve patient care
A key principle of care must include collaboration between HCPs in the treating hospital and those in a specialist HTC. This collaboration needs to begin as soon as a patient is diagnosed and should involve a multidisciplinary approach to ensure effective assessment of bleeding, timely treatment, and appropriate care. The multidisciplinary team responsible for patient care should include hemophilia specialists, physiotherapists, laboratory staff, and other relevant specialists (such as physicians specializing in internal medicine, emergency medicine, geriatrics, obstetrics, rheumatology, and oncology).
Collaboration should include education of relevant staff. For example, specialist nurses in HTCs should provide training and education to nurses and key staff in non‐specialist centers about the early recognition of signs and symptoms of bleeds and the need for prompt treatment. Education about appropriate patient handling in order to minimize the risk of inducing bleeds is also important. (Table 2). Individuals with AHA should, where possible, be treated in a specialist environment, however, the treating physician and HTC staff may decide if the patient should remain in the non‐specialist center (for example, if the patient is too ill to be transferred). In such circumstances, collaboration between the treating staff and their HTC counterparts ensures effective delivery of comprehensive care. Networks of hospitals who collaborate in managing patients with bleeding disorders should also establish counseling programs for such patients, including those with AHA.
Key takeaway
Collaboration with hematology experts and the development of multidisciplinary care teams are vital for the optimal management of patients with AHA.
3.5. Principle 5: Comprehensive assessment of bleeding is required
3.5.1. Rationale
While treatment of acute bleeds is a key priority in AHA care, 6 , 9 , 15 not all patients bleed, and not all bleeds require intervention. 1 , 6 , 11
Elderly patients with multiple comorbidities—often the case with AHA patients—may face an increased risk of thrombosis during treatment with bypassing agents 1 , 3 , 14 , 18 , 19 ; therefore, the benefits and disadvantages of initiating hemostatic treatment must be carefully weighed.
3.5.2. Bleed assessment and rationale for initiating hemostatic treatments
Recent recommendations on hemostatic therapy and treatment evaluation have been published, 12 , 13 so these aspects are not covered in detail here. Instead, our focus concerns the decision about whether to treat a bleed.
The type of bleed is important when determining whether hemostatic treatment is needed. Generally, retroperitoneal or retropharyngeal hematomas, muscle bleeds, intracranial hemorrhage, severe hematuria, bleeds in multiple sites, and gastrointestinal, pulmonary, or postoperative bleeding all require hemostatic treatment. However, ecchymosis and mild to moderate subcutaneous bleeds may not require hemostatic treatment, and close observation may be sufficient. 6 Bleeds for which treatment has been initiated should also be assessed regularly to evaluate treatment efficacy. 12 , 13 The use of a body map can be a useful tool in serial assessment (Figure 3).
FIGURE 3.
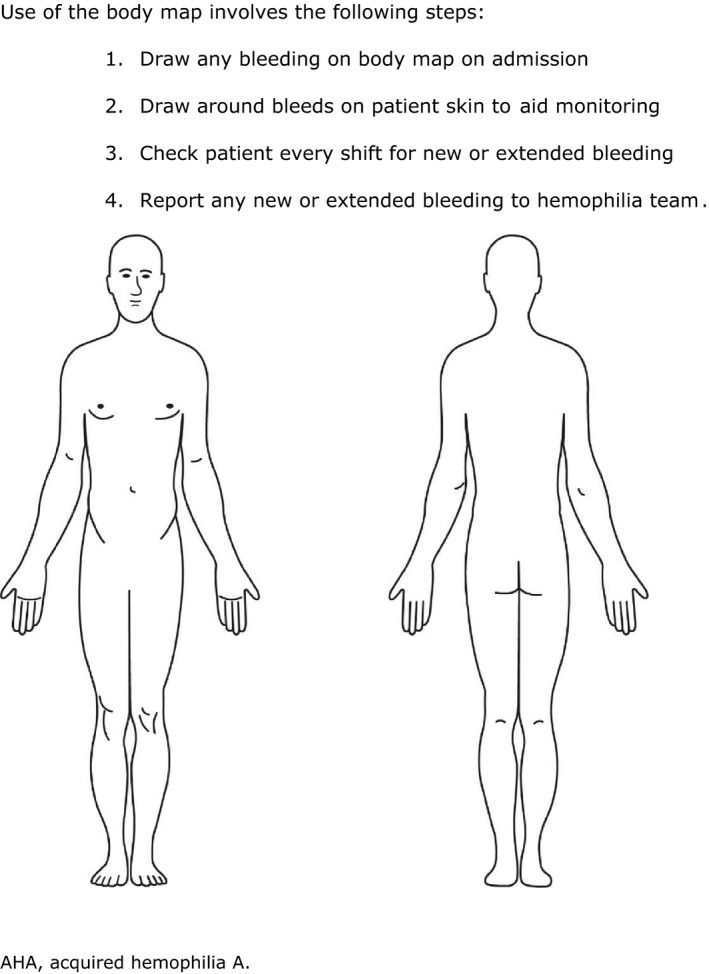
Example of a body map for monitoring bleeds in AHA. Use of the body map involves the following steps: (1) Draw any bleeding on body map on admission. (2) Draw around bleeds on patient skin to aid monitoring. (3) Check patient every shift for new or extended bleeding (4) Report any new or extended bleeding to hemophilia team. AHA, acquired hemophilia A
Treatment is recommended in patients with severe bleeds and/or a significant decrease in hemoglobin levels, irrespective of the inhibitor titer. 1 , 6 Additional considerations are also important. First, treatment benefits must be weighed against the risks: for example, the use of bypassing agents may put patients at risk for arterial and venous thromboembolism, particularly in elderly patients and those with predisposing conditions (eg, underlying malignancy or history of thrombosis). 1 Second, severe bleeds may require aggressive treatment; however, surgical removal of hematomas should be avoided.
Key takeaway
It is crucial to know which bleeds and which clinical scenarios require hemostatic intervention, and the benefits of initiating hemostatic treatment must be weighed against the potential risks.
3.6. Principle 6: Appropriate use of bypassing agents for prompt treatment
3.6.1. Rationale
When it has been ascertained that a bleed requires treatment, a key priority in AHA care is to start hemostatic treatment promptly to control bleeding, especially when severe. 6 , 9 , 15
Bypassing agents (recombinant activated factor VII [rFVIIa; NovoSeven®, Novo Nordisk, Bagsvaerd, Denmark] and plasma‐derived activated prothrombin complex concentrate [pd‐aPCC; FEIBA, Baxter, Deerfield, IL, USA]) are used as first‐line treatment for bleed control. 3 , 6 , 9 Recombinant porcine FVIII (rpFVIII; Obizur®, Baxalta/Shire) is also available; rpFVIII is similar enough to human FVIII to provide clotting activity in humans, but its lack of complete homology with human FVIII means that it may not be neutralized by inhibitory anti‐human FVIII antibodies. 20 , 21 However, experience with rpFVIII in the acute clinical setting is more limited. 20 , 21 , 22 , 23
Three key studies support the use of rFVIIa and pd‐aPCC in AHA, 14 , 24 , 25 and similar efficacy between the two agents has been reported. 14 However, the feasibility of administering prompt treatment with bypassing agents may be influenced by various practical considerations.
3.6.2. Managing bypassing agent use
First‐line treatment for bleed control in patients with AHA may be provided by either rFVIIa, pd‐aPCC, or rpFVIII. Recombinant FVIIa is administered at an initial dose of 90 µg/kg, with repeat doses administered every 2‐3 hours, if necessary, to control bleeding. A higher initial dose may be required, for example, 270 µg/kg, depending on the clinical situation. 26 FEIBA is generally administered at doses of 50‐100 U/kg repeated every 6‐12 hours, depending on the location and severity of bleeding. 27 The maximum recommended dose is 200 U/kg/day. The dosing of pFVIII is usually 200 U/kg initially with further dosing depending on plasma levels of pFVIII and the clinical situation. Efficacy may be influenced by anti‐porcine FVIII titer.
Administering these bypassing agents may be impacted by several considerations including availability of the therapeutic agent since these are not widely stocked. First, ideally, the patient should be transferred to a facility where these drugs are available. This treating facility should have sufficient availability of trained staff experienced using these drugs and in reconstituting and administering bypassing agents. An additional consideration may be the time required to administer these agents: pd‐aPCC, for example, has longer mixing and administration time (30‐60 minutes per dose); rFVIIa requires dosing every 2‐3 hours until the bleed is controlled. 26 Also, venous access must be assessed, managed, and preserved in patients receiving bypassing agents. A peripherally inserted central catheter (PICC) line can be beneficial: some clinicians prefer to use a PICC line in all patients with AHA who require hospital admission, frequent venous sampling, and intravenous therapy, while others reserve its use for patients with difficult venous access and/or high risk of cutaneous bleeding with further venepuncture (Table 2).
Where rFVIIa is concerned, treatment feasibility might be enhanced by using an infusion pump method designed to deliver timed, accurate bolus doses. 28 However, this novel delivery method is not currently widely available for routine use. For rpFVIII, a validated laboratory assay for monitoring treatment is important.
Regular clinical monitoring of vital signs and site of bleeding is essential to confirm efficacy and switching to an alternative therapy should be considered with suboptimal response. 29 The duration of treatment will depend on the clinical circumstance and as a minimum, treatment should continue until there are no signs of bleeding.
Key takeaway:
Bypassing agents (rFVIIa and pd‐aPCC) and rpFVIII are first‐line treatments for bleeds that require intervention; where possible, patients should be transferred to a facility where bypassing agents (and staff trained in their use) are available.
3.7. Principle 7: Long‐term follow‐up and monitoring for efficacy and safety of immunosuppressive treatment
3.7.1. Rationale
Although the anti‐FVIII inhibitor that causes AHA may spontaneously disappear without treatment in ~ 30% of cases, 30 remission occurrence is unpredictable 6 , 31 and a patient remains at risk of serious bleeding while the inhibitor persists. 1 , 11 , 32 , 33
Therefore, it is currently recommended that all patients with AHA receive immunosuppressive therapy (IST) to eradicate the inhibitor. 1 , 3 , 6 First‐line options for IST include corticosteroids alone or in combination with cyclophosphamide; if initial treatment fails, subsequent attempts with IST might be initiated with other immunosuppressive agents such as azathioprine, vincristine, mycophenolate, cyclosporine, or rituximab. 1 , 3 , 6
However, up to 20% of patients receiving IST experience a relapse, 4 , 11 and most IST drugs carry potential side effects. 6 Prolonged follow‐up for efficacy and safety is therefore required.
3.7.2. Long‐term follow‐up for efficacy
Monitoring IST may depend on individual clinical situations and practicability. 1 Current recommendations suggest that patients on IST can be monitored weekly on an outpatient basis during the first 6 weeks of therapy unless there is active bleeding or comorbidities involving hospitalization, surgery, invasive procedures, or delivery. If patients are hospitalized, they should be monitored twice weekly. 6 Routine monitoring should include physical examination, full blood count, aPTT, FVIII activity, and anti‐FVIII inhibitor titer 3 , 6 ; FVIII levels and anti‐FVIII inhibitor titers should be monitored until FVIII levels normalize and the inhibitor becomes undetectable although anti‐FVIII inhibitor titers may not to be tested as frequently as FVIII:C levels. 1
After achieving a sustained response, IST can be tapered. Once it has been discontinued completely, monitoring for aPTT and plasma FVIII levels should continue so that any inhibitor recurrence can be detected. 1 , 6 It has been suggested that aPTT and FVIII levels should be monitored monthly during the first 6 months after achieving remission, every 2‐3 months up to 12 months, and every 6 months during the second year and beyond to assess for relapse. 6 Patients who experience bleeding after successful IST should be carefully evaluated, as such bleeding could indicate inhibitor recurrence. 1 , 3
3.7.3. Long‐term follow‐up for safety
IST is associated with high morbidity rates, as most immunosuppressive drugs are associated with frequent complications that can be fatal in this (typically) frail, elderly population. 1 , 6 , 15 Infection (including sepsis) is a common cause of IST‐related morbidity and can lead to higher mortality rates than bleeding. 1 , 6 , 10 , 24 , 34 Other adverse effects commonly associated with IST include raised blood sugar/worsened diabetes mellitus, psychiatric disorders, and mucous membrane ulcers. 10 , 24 Patients should be monitored closely 1 , 6 and any signs of infection, sepsis, or high fever of unknown origin should be considered as either contraindications for IST or as grounds for adjusting ongoing treatment. The patient's age and presence of severe, life‐limiting comorbidities should also be considered when making decisions about starting, continuing, or adjusting IST. 6 Physicians should aim to balance the need for swift inhibitor eradication with the need to minimize exposure to potential side effects. 6
Key takeaway
Immunosuppressive therapy (IST) to eradicate the inhibitor is recommended for all AHA patients, but regular monitoring is crucial to maintain a balance between rapid inhibitor eradication and minimizing complications, which can be frequent and severe.
3.8. Principle 8: Promotion of patient care and follow‐up in the home, inpatient and outpatient settings
3.8.1. Rationale
Patients with AHA must be monitored carefully, as the rates of adverse events, morbidity, and mortality are high. 32 Further, as FVIII:C in AHA is not predictive of bleeding risk, patients can have significant bleeding despite a modest reduction in FVIII:C. 1 , 33 It is also crucial to monitor efficacy, response, and side effects associated with hemostatic treatments and IST. 32
However, maintaining patient follow‐up for long periods of time can be difficult, especially when frail, elderly patients live at home or in nursing homes and are unable to travel to clinic appointments at the hospital. Strategies to encourage and facilitate such regular monitoring should be devised.
3.8.2. Patient care and follow‐up in the home, inpatient and outpatient settings
Numerous factors affect not only the feasibility but also the location (home or hospital) of long‐term patient monitoring and care. Logistics—such as the patient's geographical location—play a key role. For example, it can be challenging to obtain follow‐up samples for measurement of FVIII:C from patients in the community who live at home or in a nursing home and who are unable to travel to clinic appointments at the hospital. For some (but not all) of these patients, it is possible for community nurses or nursing home nurses to facilitate long‐term follow‐up by obtaining regular blood samples in a timely manner. Other significant factors affecting how and where treatment is monitored and delivered include the availability of facilities and resources, along with the organization and infrastructure of healthcare systems. Thus, whether treatment and monitoring take place in the hospital (in the inpatient/outpatient setting) or in the patient's home depends on a variety of factors.
Priority should also be given to educating patients about how to recognize bleed symptoms, report them, and urgently access care so that hemostatic treatment can be initiated promptly (see Section 3.6: Principle 6). When a patient has a significant, active bleed that requires treatment, they should be monitored closely in the hospital, and the patient may need to be admitted as an inpatient. It has been recommended that assessment of bleed location and severity should be performed every 6‐12 hours 32 ; while it may be unrealistic to see significant changes with such frequent monitoring, we consider 12‐hourly monitoring to be helpful for new bleeds in inpatients, some of whom may not advocate well for themselves. As soon as complete remission with normal FVIII:C activity has been achieved, either by AHA treatment or by spontaneous remission, it should be evaluated whether prophylaxis is indicated to prevent venous thromboembolism.
Key takeaway
Long‐term patient care and monitoring are affected by numerous factors and challenges, including logistics, resource availability, and organization of healthcare systems. Appropriate proactive strategies are required to ensure and facilitate continued care and follow‐up.
3.9. Principle 9: Access to innovative and disruptive treatments will be important
3.9.1. Rationale
While bypassing agents are the current treatment of choice for managing bleeding in patients with AHA, they are not without clinical shortcomings. For instance, neither is predictably effective 35 ; patients may exhibit differential responses to treatment 35 , 36 ; treatment cannot be monitored with conventional laboratory assays 32 ; and bypassing agents increase the risk for thromboembolism in elderly patients with comorbidities. 32 , 35
3.9.2. Emerging and innovative therapies
In the future various new and emerging non‐factor therapies may offer treatment options for patients with AHA. To date, no emerging non‐factor therapies have been approved for AHA.
Hemlibra® (Emicizumab, Roche, Basel, Switzerland) is a bispecific antibody that mimics FVIII. 37 It is currently approved for use only in congenital hemophilia A with or without inhibitors, 32 , 38 but some clinical experience in patients with AHA has recently been published. 39 , 40 , 41 , 42 , 43
Other potential new non‐factor treatments in development for use in congenital hemophilia include anti‐tissue factor pathway inhibitor (TFPI) antibodies 32 ; inhibition of anti‐thrombin (fitusiran), and inhibition of proteins C and S. 32 , 44 However, clinical studies of all these therapies in AHA would be required before recommending use. To date, no such clinical studies have been planned.
Key takeaway
Several new and emerging non‐factor treatments (including emicizumab, anti‐TFPI antibodies, anti‐thrombin inhibitors, and inhibitors of protein C and protein S) may offer new treatment options for AHA in the future.
3.10. Principle 10: Promotion of collaborative research to improve patient outcomes
3.10.1. Rationale
As AHA is rare, complex, and most commonly seen in elderly, multimorbid patients, it is one of the most challenging hematologic diseases to treat 32 and many unmet needs and knowledge gaps remain.
Therefore, every effort should be made to promote international collaborative research to improve our knowledge base and ultimately improve patient outcomes.
3.10.2. Areas for international collaborative research
Key AHA areas for development include the need for less toxic immunosuppressive regimens and new therapies to treat bleeds. 32 , 35 Highly individualized therapy would benefit from collaborative research, where treatment (eg, choice of hemostatic agent, response criteria, immunosuppressive regimen, monitoring schedule, and adjunct medications) are tailored according to the patient's individual clinical condition. 32
Currently, the only large‐scale data available for AHA are from observational registers and it is vital that further cooperative efforts are made to continue and optimize registry data collection. 7 Outcome data should be regularly and systematically included in registries and should cover long‐term follow‐up data and detailed information regarding dosing regimens and local clinical practice. 7 Such data would also allow detailed analyses of certain patient subgroups and analyses to identify risk factors for bleeding and/or poor outcomes. 7
Finally, additional work to further refine the principles outlined in this article is warranted. For instance, these principles were developed by an expert panel of European hemophilia specialists; although principles of care arguably remain the same all over the world, regional or national adaptation of the principles may help to facilitate their implementation in differing healthcare systems and to accommodate cultural and socio‐economic diversity, as seen for principles of care developed for congenital hemophilia. 45 , 46 Further, input from additional experts in other specialties, such as geriatricians and endocrinologists, would allow greater insights into a more holistic approach for treating AHA. A Delphi process might be useful in this regard.
Key takeaway
International collaborative research is essential to increase knowledge of AHA, enhance data collection in registries, and improve both treatment regimens and management strategies.
4. CONCLUSIONS
This document sets out key principles for comprehensive, holistic AHA care to ensure swift diagnosis and optimal management, regardless of whether patients present to an HTC or a non‐specialist hospital. Key factors in achieving effective holistic care are education and training for healthcare personnel in non‐specialist hospitals and collaboration between hemophilia specialists and physicians in other specialties. It is hoped that the proposed principles will increase awareness of AHA in the wider medical community and catalyze renewed efforts and resource contribution toward improving the practical and multidisciplinary treatment of AHA.
ACKNOWLEDGEMENTS
Medical writing and editorial assistance were provided by Julie Smith and Jo Fetterman, Parexel, and funded by a grant from Novo Nordisk. All authors wrote, and directed the development of, the manuscript and take full responsibility for the content and conclusions stated in this manuscript. Novo Nordisk neither influenced the content of this publication nor was it involved in the interpretation.
Dolan G, Benson G, Bowyer A, et al. Principles of care for acquired hemophilia. Eur J Haematol. 2021;106:762–773. 10.1111/ejh.13592
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created in this study.
REFERENCES
- 1. Kruse‐Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017;927:695‐705. [DOI] [PubMed] [Google Scholar]
- 2. Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia. 2010;16(Suppl 3):41‐45. [DOI] [PubMed] [Google Scholar]
- 3. Collins P, Baudo F, Huth‐Kuhne A, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010;3:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collins P, Baudo F, Knoebl P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;1201:47‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: a concise review. Am J Hematol. 2005;801:55‐63. [DOI] [PubMed] [Google Scholar]
- 6. Huth‐Kuhne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;944:566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kessler CM, Knobl P. Acquired haemophilia: an overview for clinical practice. Eur J Haematol. 2015;95(Suppl 81):36‐44. [DOI] [PubMed] [Google Scholar]
- 8. Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;104:622‐631. [DOI] [PubMed] [Google Scholar]
- 9. Collins PW, Percy CL. Advances in the understanding of acquired haemophilia A: implications for clinical practice. Br J Haematol. 2010;1482:183‐194. [DOI] [PubMed] [Google Scholar]
- 10. Collins PW. Management of acquired haemophilia A. J Thromb Haemost. 2011;9(Suppl 1):226‐235. [DOI] [PubMed] [Google Scholar]
- 11. Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2‐year national surveillance study by the United Kingdom Haemophilia Centre Doctors' Organisation. Blood. 2007;1095:1870‐1877. [DOI] [PubMed] [Google Scholar]
- 12. Tiede A, Giangrande P, Teitel J, et al. Clinical evaluation of bleeds and response to haemostatic treatment in patients with acquired haemophilia: A global expert consensus statement. Haemophilia. 2019;256:969‐978. [DOI] [PubMed] [Google Scholar]
- 13. Tiede A, Collins P, Knoebl P, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. 2020;1057:1791‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baudo F, Collins P, Huth‐Kuhne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;1201:39‐46. [DOI] [PubMed] [Google Scholar]
- 15. Delgado J, Jimenez‐Yuste V, Hernandez‐Navarro F, Villar A. Acquired haemophilia: review and meta‐analysis focused on therapy and prognostic factors. Br J Haematol. 2003;1211:21‐35. [DOI] [PubMed] [Google Scholar]
- 16. Tiede A, Werwitzke S, Scharf RE. Laboratory diagnosis of acquired hemophilia A: limitations, consequences, and challenges. Semin Thromb Hemost. 2014;407:803‐811. [DOI] [PubMed] [Google Scholar]
- 17. Araf S, Aleem S, Liu B, Balikai G. An unusual cause of a haemorrhagic stroke: acquired haemophilia A. BMJ Case Rep. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sumner MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of acquired haemophilia with recombinant activated FVII: a critical appraisal. Haemophilia. 2007;135:451‐461. [DOI] [PubMed] [Google Scholar]
- 19. Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH‐AH 01/2010 study. Blood. 2015;1257:1091‐1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tarantino MD, Cuker A, Hardesty B, Roberts JC, Sholzberg M. Recombinant porcine sequence factor VIII (rpFVIII) for acquired haemophilia A: practical clinical experience of its use in seven patients. Haemophilia. 2017;231:25‐32. [DOI] [PubMed] [Google Scholar]
- 21. Khan D, Raza‐Burton S, Baker P, et al. Acquired haemophilia A treated with recombinant porcine factor VIII: a single centre UK experience. Br J Haematol. 2020;189:e119‐e187. [DOI] [PubMed] [Google Scholar]
- 22. Kruse‐Jarres R, St‐Louis J, Greist A, et al. Efficacy and safety of OBI‐1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015;212:162‐170. [DOI] [PubMed] [Google Scholar]
- 23. Turkantoz H, Konigs C, Knobl P, et al. Cross‐reacting inhibitors against recombinant porcine factor VIII in acquired hemophilia A: data from the GTH‐AH 01/2010 Study. J Thromb Haemost. 2020;181:36‐43. [DOI] [PubMed] [Google Scholar]
- 24. Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost. 1997;786:1463‐1467. [PubMed] [Google Scholar]
- 25. Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia. 2004;102:169‐173. [DOI] [PubMed] [Google Scholar]
- 26. Novo Nordisk A/S . NovoSeven® Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product‐information/novoseven‐epar‐product‐information_en.pdf. Accessed May 6, 2020.
- 27. Shire Pharmaceuticals . FEIBA Summary of Product Characteristics. https://www.medicines.org.uk/emc/product/9192/smpc#gref. Accessed October 1, 2020.
- 28. Pollard DSB, Chowdary P, Gomez K. Use of an innovative syringe pump to deliver bolus rFVIIa for patients with haemophilia and inhibitors undergoing surgery. J Haemophilia Pract. 2018;41:35‐39. [Google Scholar]
- 29. Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;1602:153‐170. [DOI] [PubMed] [Google Scholar]
- 30. Lottenberg R, Kentro TB, Kitchens CS. Acquired hemophilia. A natural history study of 16 patients with factor VIII inhibitors receiving little or no therapy. Arch Intern Med. 1987;1476:1077‐1081. [DOI] [PubMed] [Google Scholar]
- 31. Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost. 1993;705:753‐757. [PubMed] [Google Scholar]
- 32. Knobl P. Prevention and Management of Bleeding Episodes in Patients with Acquired Hemophilia A. Drugs. 2018;7818:1861‐1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holstein K, Liu X, Smith A, et al. Bleeding and response to hemostatic therapy in acquired hemophilia A (AHA): results from the GTH‐AH 01/2010 study. Blood. 2020;136:279‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Borg JY, Guillet B, Le Cam‐Duchez V, et al. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l'Hemophilie Acquise) registry. Haemophilia. 2013;194:564‐570. [DOI] [PubMed] [Google Scholar]
- 35. Janbain M, Leissinger CA, Kruse‐Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med. 2015;6:143‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Astermark J, Donfield SM, DiMichele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) Study. Blood. 2007;1092:546‐551. [DOI] [PubMed] [Google Scholar]
- 37. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;13023:2463‐2468. [DOI] [PubMed] [Google Scholar]
- 38. European Medicines Agency . Hemlibra Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product‐information/hemlibra‐epar‐product‐information_en.pdf. Accessed June 4, 2020.
- 39. Knoebl P, Thaler J, Jilma P, et al. Emicizumab for the treatment of acquired hemophilia A. Blood. 2021;137:410‐419. [DOI] [PubMed] [Google Scholar]
- 40. Al‐Banaa K, Alhillan A, Hawa F, et al. Emicizumab Use in Treatment of Acquired Hemophilia A: A Case Report. Am J Case Rep. 2019;20:1046‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dane KE, Lindsley JP, Streiff MB, et al. Successful use of emicizumab in a patient with refractory acquired hemophilia A and acute coronary syndrome requiring percutaneous coronary intervention. Res Pract Thromb Haemost. 2019;33:420‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hess KJ, Patel P, Joshi AM, Kotkiewicz A. Utilization of Emicizumab in Acquired Factor VIII Deficiency. Am J Case Rep. 2020;21:e922326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mohnle P, Pekrul I, Spannagl M, et al. Emicizumab in the Treatment of Acquired Haemophilia: A Case Report. Transfus Med Hemother. 2019;462:121‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hartmann J, Croteau SE. 2017 Clinical trials update: Innovations in hemophilia therapy. Am J Hematol. 2016;9112:1252‐1260. [DOI] [PubMed] [Google Scholar]
- 45. Dunkley S, Lam JCM, John MJ, et al. Principles of haemophilia care: the Asia‐Pacific perspective. Haemophilia. 2018;243:366‐375. [DOI] [PubMed] [Google Scholar]
- 46. Makris M, Kasper C. The World Federation of Hemophilia guideline on management of haemophilia. Haemophilia. 2013;191:1. [DOI] [PubMed] [Google Scholar]
- 47. Giangrande P.Acquired Hemophilia: revised version; (2012). World Federation of Hemophilia. http://www1.wfh.org/publications/files/pdf‐1186.pdf. Accessed May 06, 2020.
- 48. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;191:e1‐47. [DOI] [PubMed] [Google Scholar]
- 49. Reding MT, Cooper DL. Barriers to effective diagnosis and management of a bleeding patient with undiagnosed bleeding disorder across multiple specialties: results of a quantitative case‐based survey. J Multidiscip Healthc. 2012;5:277‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created in this study.