

Abstract
Background and purpose
The aim of this study was to determine the frequency of over‐ and underdiagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and to identify related diagnostic pitfalls.
Methods
We conducted a retrospective study in Dutch patients referred to the Erasmus University Medical Centre Rotterdam between 2011 and 2017 with either a diagnosis of CIDP or another diagnosis that was revised to CIDP. We used the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) 2010 diagnostic criteria for CIDP to classify patients into three groups: overdiagnosis, underdiagnosis, or confirmed diagnosis of CIDP. Clinical and laboratory features and treatment history were compared between groups.
Results
A referral diagnosis of CIDP was revised in 32% of patients (31/96; overdiagnosis). Of 81 patients diagnosed with CIDP, 16 (20%) were referred with another diagnosis (underdiagnosis). In the overdiagnosed patients, 20% of muscle weakness was asymmetric, 48% lacked proximal muscle weakness, 29% only had distal muscle weakness, 65% did not fulfil the electrodiagnostic criteria for CIDP, 74% had an elevated cerebrospinal fluid (CSF) protein level, and 97% had another type of neuropathy. In the underdiagnosed patients, all had proximal muscle weakness, 50% had a clinically atypical CIDP, all fulfilled the electrodiagnostic criteria for CIDP, and 25% had an increased CSF protein level.
Conclusion
Over‐ and underdiagnosis of CIDP is common. Diagnostic pitfalls include lack of attention to proximal muscle weakness as a diagnostic hallmark of CIDP, insufficient recognition of clinical atypical phenotypes, overreliance on CSF protein levels, misinterpretation of nerve conduction studies and poor adherence to electrodiagnostic criteria, and failure to exclude other causes of polyneuropathy.
Keywords: chronic inflammatory demyelinating polyradiculoneuropathy, CIDP, peripheral nervous system diseases, cerebrospinal fluid, diagnostic errors, overdiagnosis, underdiagnosis
Over‐ and underdiagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is common. A referral diagnosis of CIDP was revised in 32% of patients (31/96; overdiagnosis). Of 81 patients diagnosed with CIDP, 16 (20%) were referred with another diagnosis (underdiagnosis). Almost all overdiagnosed patients had other forms of neuropathy, while underdiagnosed patients were referred with various neurological disorders. Diagnostic pitfalls included lack of attention to proximal muscle weakness as a diagnostic hallmark of CIDP, insufficient recognition of clinical atypical phenotypes, overreliance on cerebrospinal fluid protein levels, misinterpretation of nerve conduction studies and poor adherence to electrodiagnostic criteria, and failure to exclude other causes of polyneuropathy.
INTRODUCTION
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a treatable immune‐mediated neuropathy [1]. The diagnosis can be challenging due to the rarity of the disease, clinical and electrophysiological variation, and lack of reliable diagnostic biomarkers [2, 3, 4]. Although the differential diagnosis for typical CIDP is relatively narrow, the multitude of disorders that can mimic the atypical CIDP variants can be difficult to navigate [3, 4, 5, 6]. The observation that more than 15 sets of diagnostic criteria have been developed for CIDP highlights the challenge of defining a disorder without a diagnostic biomarker [7]. In the United States (US), at least half of patients diagnosed with CIDP have been found not to have that condition, but are nonetheless often exposed to expensive and potentially harmful long‐term immunotherapy [5, 8, 9, 10, 11]. The extent to which the US experience can be extrapolated to other parts of the world is unknown. Equally problematic is that underdiagnosis of CIDP also occurs [12, 13]. A recent study in the United Kingdom (UK) found that 68% of CIDP patients received an alternative prereferral diagnosis other than that of CIDP [13]. Recognition of CIDP is essential to initiate treatment at an early stage of disease and thereby prevent or reduce secondary and potentially irreversible axonal nerve damage and related disability [14].
The aim of the present study was to determine the frequency of both over‐ and underdiagnosis of CIDP and to identify diagnostic pitfalls. This insight is important to improve the diagnostic accuracy and treatment of patients with CIDP.
METHODS
Study design
We included adult patients who were referred by neurologists to the Erasmus University Medical Centre Rotterdam (Erasmus MC), a tertiary academic hospital and Centre of Excellence for Guillain–Barré syndrome (GBS) and CIDP, between April 2011 and March 2017. Eligible participants were required to have either a diagnosis of CIDP at the time of referral or another diagnosis that was revised to CIDP at the Erasmus MC. Patients were selected through screening of neurology outpatient clinic (discharge) letters by using disease‐specific search terms ([A‐]CIDP, chronic inflammatory demyelinating poly[radiculo]neuropathy, chronic idiopathic demyelinating poly[radiculo])neuropathy, DADS, MADSAM, Lewis[‐]Sumner, LSS). The Erasmus MC also keeps internal CIDP databases for clinical and research purposes. These databases were reviewed to ensure that all eligible patients were captured. This study was approved by the medical ethics committee of the Erasmus MC (MEC‐2018‐1569).
Data collection
We collected the available clinical, electrodiagnostic, laboratory and treatment data from both the referral centre and the Erasmus MC, derived from the medical records of the Erasmus MC. Electrodiagnostic data of nerve conduction studies (NCS) performed at the referring centre at the time of the initial CIDP diagnosis were inspected for quality and accuracy by an experienced clinical neurophysiologist of the Erasmus MC (J.D.). Erasmus MC interpretations were compared to those documented on the initial electrodiagnostic report. The same review process was performed for NCS completed at the Erasmus MC. We used reference values for nerve conduction parameters defined by Buschbacher [15]. If performed and available, cerebrospinal fluid (CSF), magnetic resonance imaging of spinal root and plexuses, nerve ultrasonography, nerve biopsy, paraprotein, and serum IgM antibodies to myelin‐associated glycoprotein (anti‐MAG antibodies) were recorded. Local hospital cut‐off values were used for an increased CSF protein level, which was 0.58 g/L for the Erasmus MC. We recorded the type and duration of initial and maintenance treatment regimens. Response to immunotherapy before referral, reported by both the patient and referring neurologist, was recorded.
All patients were discussed extensively in consensus meetings of four neuromuscular experts at the Erasmus MC (P.v.D., B.J., E.B., J.D.). For each patient all diagnostically relevant data were presented in a raw and systematic order. Patients were diagnosed as having “definite”, “probable” or “possible” CIDP according to the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) 2010 diagnostic criteria for CIDP [16, 17, 18]. Alternative diagnoses for patients that were determined not to have CIDP were documented if possible. Only patients where diagnostic consensus was reached were included in this analysis.
Definition of confirmed, over‐ and underdiagnosis of CIDP
All patients were classified in one of three groups: (1) overdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC; (2) underdiagnosis: patients referred with another diagnosis that was revised to CIDP at the Erasmus MC; and (3) confirmed CIDP diagnosis: patients referred with a diagnosis of CIDP that was confirmed at the Erasmus MC.
Statistical analyses
We used Statistical Package for Social Sciences (SPSS) version 25 for data analysis. Descriptive analyses were performed to study proportions and frequencies. We used the chi‐squared or Fisher's exact test to compare proportions (confirmed CIDP with overdiagnosis and underdiagnosis, respectively), and the Mann–Whitney U‐test to compare continuous variables. Statistical analyses were two‐tailed tested, and p values <0.05 were considered significant.
RESULTS
Diagnostic classification
A total of 122 patients were referred with a diagnosis of CIDP or with another diagnosis that was revised to CIDP at the Erasmus MC. We excluded 10 patients for the following reasons: no diagnostic consensus was reached (n = 6), final diagnosis (other than CIDP) was also part of the referral differential diagnosis list (n = 3), or the referral's differential diagnosis was too broad (n = 1), leaving 112 patients available for the analysis. In the group of patients referred with CIDP, the diagnosis was confirmed in 65/96 patients (68%; classified as ‘confirmed CIDP’), while the diagnosis was revised in the remaining 31 patients (32%; classified as ‘overdiagnosis’ [Figure 1]). Eighty‐one patients were diagnosed with CIDP at the Erasmus MC, of whom 16 (20%) were referred with a diagnosis other than CIDP (classified as ‘underdiagnosis’; Figure 1).
FIGURE 1.

Patients referred to or diagnosed with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) at Erasmus University Medical Centre (Erasmus MC). aOverdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC; bConfirmed CIDP: patients referred with a diagnosis of CIDP that was confirmed at the Erasmus MC
Characteristics
Clinical and laboratory data were compared between the confirmed CIDP group (n = 65) and both the overdiagnosis (n = 31) and the underdiagnosis group (n = 16; Table 1). Age and sex distribution were similar in all groups. Patients in the overdiagnosis group (p = 0.05) and the underdiagnosis group (p < 0.05) were more often referred by a non‐teaching hospital. No single neurologist or hospital provided a disproportionate number of referrals to either group.
TABLE 1.
Characteristics of patients with confirmed diagnosis, overdiagnosis and underdiagnosis of chronic inflammatory demyelinating polyradiculoneuropathy
| Confirmed diagnosis a (n = 65) | Overdiagnosis b (n = 31) | p c | Underdiagnosis d (n = 16) | p e | |
|---|---|---|---|---|---|
| Age at first visit Erasmus MC, years | 62 (17–82) | 62 (38–88) | 1.00 | 65 (52–81) | 0.37 |
| Male | 62 (40) | 68 (21) | 0.56 | 81 (13) | 0.14 |
| Symptom duration f , months | 9 (1–334) | 24 (1–146) | 0.05 | 24 (2–185) | 0.23 |
| Time since diagnosis g , months | 3 (0–294) | 3 (0–34) | 0.88 | 2 (0–33) | 0.43 |
| Time carrying “wrong” diagnosis, months | NA | 4 (0–34) h | NA | 3 (0–44)i | NA |
| Delay in CIDP diagnosis j , months | 9 (1–334) | NA | NA | 24 (2–190) | 0.53 |
| Referred with CIDP + ≥1 diagnoses | 37 (24) | 26 (8) | 0.28 | NA | NA |
| Referred by teaching hospital | 48 (31) | 27 (8/30) | 0.05 | 19 (3/16) | 0.04 |
Data presented as percentages (number) or median (range).
Abbreviations: CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; Erasmus MC, Erasmus University Medical Centre; NA, not applicable.
Confirmed diagnosis: patients referred with a diagnosis of CIDP that was confirmed at the Erasmus MC.
Overdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC.
Confirmed diagnosis vs. overdiagnosis.
Underdiagnosis: patients referred with another diagnosis that was revised to CIDP at the Erasmus MC.
Confirmed diagnosis vs. underdiagnosis.
Symptom duration: time between disease onset and date of referral diagnosis.
Time since diagnosis: time between date of referral diagnosis and first visit at Erasmus MC.
Time between date of referral diagnosis of CIDP and rejecting this diagnosis at Erasmus MC.
Time between date of referral diagnosis and date of CIDP diagnosis at Erasmus MC.
Time between disease onset and CIDP diagnosis
Overdiagnosis group
Clinical and diagnostic features
In almost all overdiagnosed patients (97%; 30/31), the diagnosis was revised to another form of neuropathy (Figure 2). Clinical and laboratory data are shown in Tables 2 and 3. Compared with the confirmed‐CIDP group, overdiagnosed patients were more likely to have only distal weakness (29% vs. 11%; p = 0.03) or asymmetric weakness (20% vs. 5%; p = 0.04), and were less likely to have proximal weakness (52% vs. 86%; p < 0.01). The majority of overdiagnosed patients (74%; 14/19) had an elevated CSF protein level with a normal cell count. Before referral, in 17% of overdiagnosed patients (4/20), paraprotein was not tested, and in one patient with a demonstrated IgM paraprotein, anti‐MAG antibodies were not determined.
FIGURE 2.

Alternative diagnosis for patients overdiagnoseda with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP; n = 31). Abbreviations: GBS, Guillain–Barré syndrome; IgM anti‐MAG, IgM antibodies to myelin‐associated glycoprotein; PN, polyneuropathy; ATTRv amyloidosis = hereditary transthyretin amyloidosis; Erasmus MC, Erasmus University Medical Centre. aOverdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC
TABLE 2.
Clinical and laboratory data in patients with confirmed diagnosis, overdiagnosis and underdiagnosis of chronic inflammatory demyelinating polyradiculoneuropathy
| Confirmed diagnosis a (n = 65) | Overdiagnosis b (n = 31) | p c | Underdiagnosis d (n = 16) | p e | |
|---|---|---|---|---|---|
| Clinical features f | |||||
| EFNS/PNS clinical criteria, typical | 78 (50/64) h | NA i | NA | 50 (8) j | 0.03 |
| Muscle weakness | 97 (63) | 81 (25) | 0.01 | 100 (16) | 1.00 |
| Asymmetric muscle weakness | 5 (3/63) | 20 (5/25) | 0.04 | 19 (3) | 0.09 |
| Proximal muscle weakness | 86 (56) | 52 (16) | <0.01 | 100 (16) | 0.19 |
| Distal muscle weakness | 95 (62) | 77 (24) | 0.01 | 100 (16) | 1.00 |
| Only distal muscle weakness | 11 (7) | 29 (9) | 0.03 | 0 (0) | 0.34 |
| Sensory dysfunction | 88 (57) | 94 (29) | 0.49 | 81 (13) | 0.45 |
| Reduced or absent reflexes | 95 (62) | 84 (26) | 0.11 | 100 (16) | 1.00 |
| Cranial nerve dysfunction | 15 (10) | 7 (2) | 0.33 | 6 (1) | 0.68 |
| Disease progression ≥2 months | 99 (64) g | 84 (26) | 0.01 | 100 (16) | 1.00 |
| Diagnostic features | |||||
| EFNS/PNS electrodiagnostic criteria | 100 (65) k | 35 (11) l | <0.01 | 100 (16) l | — |
| EFNS/PNS electrodiagnostic criteria, definite | 94 (60/64) k | 23 (7) l | <0.01 | 88 (14) l | 0.59 |
|
IgM paraprotein demonstrated With anti‐MAG |
10 (5/48) m 0 (0) |
10 (3/27) n 67 (2) |
— |
19 (3/15) o 0 (0) |
— |
| CSF protein level g/L | 0.83 (0.24–4.35) | 0.80 (0.21–4.48) | 1.00 | 0.56 (0.37–80) | 0.08 |
| Elevated CSF protein level with normal cell count | 67 (36/53) | 74 (14/19) | 0.51 | 50 (4/8) | 0.45 |
| > 0.58–1.00 g/L | 49 (17/35) | 40 (4/10) | — | 100 (4/4) | — |
| > 1.00 g/L | 51 (18/35) | 60 (6/10) | — | 0 (0/0) | — |
| MRI nerve root/plexuses enhancement and/or enlargement | 0 (0/5) | 0 (0/2) | — | 0 (0/1) | — |
| Nerve biopsy demyelination and/or remyelination | 25 (1/4) | 0 (0/3) | — | 0 (0/0) | — |
| EFNS/PNS diagnostic category | 100 (65) | NA | NA | 100 (16) | 1 |
Data presented as percentages (number) or median (range).
Abbreviations: CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CSF, cerebrospinal fluid; EFNS/PNS, European Federation of Neurological Societies/Peripheral Nerve Society; Erasmus MC, Erasmus University Medical Centre; IgM anti‐MAG antibodies, immunoglobulin isotype M antibodies to myelin‐associated glycoprotein; MRI, magnetic resonance imaging; NA, not applicable.
Confirmed diagnosis: patients referred with a diagnosis of CIDP that was confirmed at the Erasmus MC.
Overdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC.
Confirmed diagnosis vs. overdiagnosis.
Underdiagnosis: patients referred with another diagnosis that was revised to CIDP at the Erasmus MC.
Confirmed diagnosis vs. underdiagnosis.
Evaluation at Erasmus MC.
Progressing >4 weeks and IVIg‐dependent.
Clinical atypical CIDP phenotypes: predominantly distal (n = 3), asymmetric (n = 2), predominantly motor (n = 3), predominantly sensory (n = 5), chronic immune sensory polyradiculopathy (CISP) (n = 1).
Overdiagnosed patients were not classified according to the EFNS/PNS clinical criteria.
Clinical atypical CIDP phenotypes: predominantly distal (n = 2), asymmetric (n = 3), predominantly motor (n = 3).
All but 12 nerve conduction studies were performed at the Erasmus MC.
All nerve conduction studies were performed at the Erasmus MC.
Not tested (n = 6), test performed unknown/test results unknown (n = 11).
Not tested (n = 1), test performed unknown/test results unknown (n = 3).
Not tested (n = 1).
TABLE 3.
Patients overdiagnosed a with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) but fulfilling the European Federation of Neurological Societies/Peripheral Nerve Society electrodiagnostic criteria for CIDP
| Revised diagnosis | EFNS/PNS electrodiagnostic classification b | Clinical and diagnostic remarks | |
|---|---|---|---|
| 1. | Anti‐MAG PN | Definite | Proximal muscle weakness –, anti‐MAG antibodies +, elevated CSF protein level + |
| 2. | Anti‐MAG PN | Definite | Proximal muscle weakness –, anti‐MAG antibodies + |
| 3. | CMT1A | Definite | Muscle atrophy +, pes cavus +, proximal muscle weakness –, family history –, DNA confirmation CMT1A + (PMP2 mutation) |
| 4. | Guillain–Barré syndrome | Definite | Progression disease course <2 months + |
| 5. | CMT2A | Definite | Muscle atrophy +, pes cavus+, predominantly distal muscle weakness +, family history –, DNA confirmation CMT2A + (KIF1B mutation) |
| 6. | Waldenström macroglobulinemia PN | Definite | Proximal muscle weakness –, IgM paraprotein +, anti‐MAG antibodies –, elevated CSF protein level + |
| 7. | Axonal PN | Definite c | Slowly progressive disease course +, proximal muscle weakness –, NCS supportive for CIDP –, nerve ultrasound supportive for CIDP – |
| 8. | ATTRv amyloidosis | Possible c | Proximal muscle weakness +, family history +, DNA confirmation + (TTRMet30 mutation), elevated CSF protein level +, nerve biopsy amyloidosis + |
| 9. | Entrapment neuropathy | Possible c | NCS supportive for CIDP –, MRI plexus brachialis supportive for CIDP – |
| 10. | Guillain–Barré syndrome | Possible | Progression disease course <2 months +, proximal muscle weakness +, elevated CSF protein level – |
| 11. | Axonal PN combined with myopathy | Possible c | Proximal muscle weakness +, elevated CSF protein level +, NCS supportive for CIDP – |
Abbreviations: ATTRv amyloidosis, hereditary transthyretin amyloidosis, Erasmus MC, Erasmus University Medical Centre; CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CMT, Charcot‐Marie‐Tooth; CSF, cerebrospinal fluid; EFNS/PNS, European Federation of Neurological Societies/Peripheral Nerve Society; IgM, immunoglobulin M; KIF, kinesin family member 1B gene; MAG, myelin‐associated glycoprotein; MRI, magnetic resonance imaging; NCS, nerve conduction studies; PMP, peripheral myelin protein; PN, polyneuropathy.
Overdiagnosis: patients referred with a diagnosis of CIDP that was revised to another diagnosis at the Erasmus MC.
NCS performed at the Erasmus MC.
NCS showed abnormalities that according to the EFNS/PNS criteria are considered as a demyelinating feature, but in clinical practice are attributed to other factors. For instance reduced nerve conduction velocities in nerves with severely decreased compound muscle action potential (CMAP) amplitudes can be caused by a severe axonopathy and CMAP amplitude reductions of 50% in the tibial nerve are considered normal according to published reference values [15]. +Indicates the presence of feature; – Indicates the absence of feature.
Nerve conduction studies
In all overdiagnosed patients, NCS were performed both at the referral centre and at the Erasmus MC. Sixteen of the 31 NCS that were performed at the referring centre were interpreted as demyelinating or combined axonal and demyelinating by the referral centre (Table S1). Erasmus MC interpretations of referral study data were in agreement that 11 of the studies were demyelinating. The remaining five studies could not be assessed due to missing data (n = 4) or because insufficient nerves had been tested (n = 1). When repeating NCS at the Erasmus MC, only 8/31 were interpreted as demyelinating (Table S1). Eleven of 31 NCS performed at the Erasmus MC fulfilled the EFNS/PNS 2010 electrodiagnostic criteria for definite, probable or possible CIDP (Table 3). Of these, four formerly fulfilled the electrodiagnostic criteria but were not interpreted by the Erasmus MC as demyelinating. These four NCS showed abnormalities that, according to the EFNS/PNS criteria, are considered a demyelinating feature, but in clinical practice are attributed to other factors. For instance, reduced nerve conduction velocities in nerves with severely decreased compound muscle action potential (CMAP) amplitudes can be caused by a severe axonopathy, temporal dispersion across common compression sites can be caused by a compression neuropathy, and CMAP amplitude reductions of 50% in the tibial nerve are considered normal according to published reference values [15].
Treatment
Before referral, 52% of overdiagnosed patients (n = 16) were treated for CIDP with intravenous immunoglobulins (IVIg; n = 12), corticosteroids (n = 1), or IVIg and corticosteroids (n = 3). (Initial) improvement on therapy was reported by 44% of referring neurologists (n = 7/16) and by 56% of patients (n = 9/16). Of overdiagnosed patients that were treated with IVIg, three received IVIg maintenance therapy.
Underdiagnosis group
Clinical and diagnostic features
Underdiagnosed patients were referred with various other neurological diseases (Figure 3). Clinical and laboratory data are shown in Table 2. All 16 patients had symptoms for at least 2 months, with 25% having symptoms for 2–6 months, 13% having symptoms for 6–12 months and 63% having symptoms for >12 months. Underdiagnosed patients more often had a clinical atypical phenotype compared to the confirmed CIDP group (50% vs. 22%; p = 0.03), and included predominantly distal (n = 2), asymmetric (n = 3) and predominantly motor phenotypes (n = 3). All underdiagnosed patients had proximal muscle weakness when seen at Erasmus MC. Of these patients, in eight, no proximal muscle weakness was found at the referral centre and, in four, proximal muscle strength was not reported or not tested by the referral neurologist. Half of underdiagnosed patients (4/8) had elevated CSF protein levels with normal cell count.
FIGURE 3.
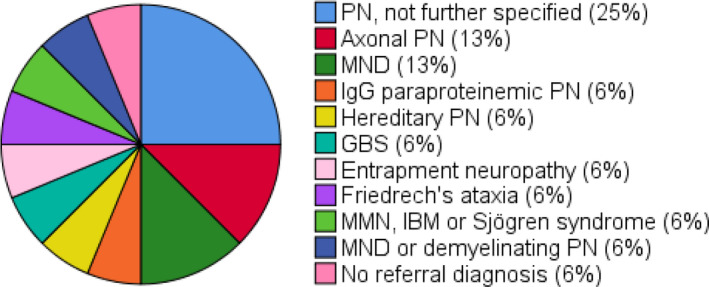
Referral (differential) diagnosis in underdiagnoseda chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) patients (n = 16). Erasmus MC, Erasmus University Medical Centre; GBS, Guillain–Barré syndrome; IBM, inclusion body myositis; MND, motor neuron disease; MMN, multifocal motor neuropathy; PN, polyneuropathy. aUnderdiagnosis: patients referred with another diagnosis that was revised to CIDP at the Erasmus MC
Nerve conduction studies
In all 16 underdiagnosed patients, NCS were performed both at the referral centre and at the Erasmus MC. When reviewing the raw data of NCS that were performed at the referring centre, 13 NCS were interpreted as demyelinating by the Erasmus MC and also fulfilled the EFNS/PNS electrodiagnostic criteria for CIDP (Table S1). Eight of those 13 NCS were also interpreted as demyelinating or combined axonal and demyelinating by the referral centre. Yet, although fulfilling the electrodiagnostic criteria, three of the 13 NCS were not interpreted as demyelinating by the referring centre. Based on the NCS performed at Erasmus MC, all 16 underdiagnosed patients fulfilled the EFNS/PNS electrodiagnostic criteria for either definite, probable or possible CIDP (Table 2).
DISCUSSION
This study showed that both over‐ and underdiagnosis of CIDP is common in the Netherlands. In almost one‐third of patients referred with CIDP, the diagnosis was revised and in approximately one‐fifth of patients with CIDP the patients were referred with another diagnosis. Almost all overdiagnosed patients had other forms of neuropathy, while underdiagnosed patients were referred with various neurological disorders. Several pitfalls related to over‐ and underdiagnosis were identified.
It has previously been indicated that a diagnosis of CIDP is relatively straightforward in patients with typical CIDP, but is more challenging in patients with clinically atypical features [4, 5]. Findings from our study support this claim: patients with clinical “atypical” phenotypes are both more likely to be falsely labelled as having CIDP and more likely to be falsely classified as having another disorder when a CIDP diagnosis is appropriate. Heightened diagnostic vigilance including referrals to GBS/CIDP centres of excellence is especially encouraged for these challenging “atypical” patients. Overdiagnosed patients often had only distal or asymmetric muscle weakness, and atypical CIDP variants were overrepresented in underdiagnosed patients. Proximal muscle weakness as a clinical hallmark of CIDP may be another factor in misdiagnosis. Approximately half of overdiagnosed patients had proximal muscle weakness, while in underdiagnosed patients a CIDP diagnosis was initially not suspected despite the presence of proximal muscle weakness in all these patients when seen at the Erasmus MC. Proximal muscle weakness should therefore be considered a sensitive sign of CIDP (apart from distal, sensory and perhaps asymmetric variants), but with limited specificity considering disorders in the differential diagnosis of CIDP. An (initial) GBS diagnosis was seen in both, over‐ and underdiagnosed patients. The differentiation between GBS and acute‐onset CIDP (A‐CIDP) within the first 8 weeks of presentation can be difficult and treatment may not be different. A‐CIDP with a very acute disease course or treatment‐related fluctuations, in particular, might initially suggest GBS.
Nerve conduction studies were found to play an important role in misdiagnosis. Overdiagnosed patients often did not meet the EFNS/PNS electrodiagnostic criteria, and were frequently referred with a diagnosis of CIDP despite confirmed axonal polyneuropathy on NCS. Importantly, 35% of overdiagnosed patients actually did fulfil the EFNS/PNS electrodiagnostic criteria for CIDP. In these patients, demyelination was explained by another condition mimicking CIDP, or could not be reliably assessed due to severely reduced CMAP amplitudes. In patients with clinical atypical CIDP features, it is especially important to consider other causes of a demyelinating neuropathy, in particular IgM paraproteinemia with anti‐MAG antibodies and genetically determined causes. The interpretation of NCS is particularly difficult in studies when the CMAP amplitudes are very low (<1 mV). Latency onsets can be difficult to determine and moderate amplitude‐dependent slowing can be misinterpreted as demyelinating when it is better explained by fast conducting fibre loss or by slowly regenerating immature nerve fibres [10]. NCS also played an important role in underdiagnosis. In underdiagnosed patients, demyelinating features were sometimes missed. On the other hand, the majority of underdiagnosed patients were often not referred with a diagnosis of CIDP despite a confirmed demyelinating polyneuropathy on NCS by the referral centre. Careful performance and interpretation of NCS is therefore of great importance.
We found that CSF protein level was not an accurate diagnostic marker for CIDP. Although an elevated CSF level is considered supportive of the diagnosis of CIDP, in the present study, CSF protein level was often elevated in overdiagnosed patients and normal in patients with CIDP. A range of cut‐off values for elevated CSF protein is used in current clinical practice, which influences the predictive value of CSF examination [19]. CSF protein levels of any level should not be considered diagnostic of CIDP without supporting clinical and electrodiagnostic features of CIDP. CSF examination in the diagnostic evaluation of CIDP can be relevant to exclude disorders characterized by pleiocytosis.
Our findings are partly in line with previous studies on overdiagnosis of CIDP in the United States [5, 9, 10]. Although 47% of patients that carried a diagnosis of CIDP in the United States were found to be misdiagnosed at a tertiary academic center, the frequency of misdiagnosis in patients outside of that setting may be higher [5, 11]. In the present series, we observed that 32% of patients were overdiagnosed as having CIDP. Differences in healthcare systems, such as the accessibility of a neuromuscular specialist visit or consultation, might explain the difference between the Dutch and US experience. In line with the present study, common diagnostic pitfalls in CIDP included overreliance on an elevated CSF protein level, dismissal of NCS that showed only axonal features or were normal, and misinterpretation of axonal NCS findings as demyelinating [5, 10]. In addition, perception of treatment benefit was described as a diagnostic pitfall of overdiagnosis of CIDP [5, 10]. In the present study, we did not conduct an analysis of the treatment response because of the retrospective study design, insufficient data and absence of predefined criteria for subjective and objective treatment response. Treatment may also be considered in patients with a relatively low suspicion of CIDP just to prevent withholding a chance of recovery. In contrast to the present study, overdiagnosed patients in the United States were treated more often before referral (86% vs. 52%) and for a longer period of time [5]. Furthermore, revised diagnosis in overdiagnosed patients in the United States frequently uncovered non‐neuropathy conditions, while in almost all patients (97%) in our study CIDP diagnosis was revised to another form of polyneuropathy [5]. In one patient, a CIDP diagnosis was revised in hereditary transthyretin (ATTRv) amyloidosis. Several studies on misdiagnosis of ATTRv amyloidosis reported on initial CIDP diagnosis [20, 21, 22, 23]. Furthermore, studies on misdiagnosis of other diseases showed that CIDP also could be a mimic for POEMS syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal protein, Skin changes), CANOMAD (chronic ataxic neuropathy, ophthalmoplegia, immunoglobulin M [IgM] paraprotein, cold agglutinins, and disialosyl antibodies), lymphoma and progressive spinal muscular atrophy [22, 24, 25].
Comparable studies on underdiagnosis of CIDP are limited [12, 13]. One study on diagnostic delay in Dutch patients with CIDP found that 15% were initially diagnosed with various other diseases, including other forms of polyneuropathy (n = 10), myasthenia gravis (n = 1), bulging disc/tendomyalgia (n = 4), and motor neuron disease (n = 2) [12]. A recent UK study found that 68% of CIDP patients received an alternative prereferral diagnosis other than that of CIDP [13], which is considerably higher than found in the present study (20%). Differences in healthcare systems, such as more centralized care with specialized (neuromuscular) centres in the United Kingdom, and thereby possibly more referrals without an extensive diagnostic evaluation, might explain the difference between the Dutch and UK experience. As in the present study, common diagnostic pitfalls leading to underdiagnosis were a lack of attention to proximal muscle weakness and misinterpretation of NCS [13]. Furthermore, an atypical clinical presentation seemed to be more common in the underdiagnosed group than in the correctly diagnosed group, but this did not reach statistical significance [13]. In contrast to the present study, initial diagnosis in underdiagnosed patients in the United Kingdom were frequently other forms of polyneuropathy, especially GBS, [13] while in the present study patients were initially diagnosed with various neurological disorders.
A limitation of the present study is its retrospective nature. Based on referral records, no reliable classification of objective improvement could be made. Therefore, we could not include an objective treatment response as a supportive criterion in the EFNS/PNS diagnostic criteria. We also realize that the diagnosis in individual patients may have become more evident over time, which was a clear advantage when assessing patients later in the disease course.
The present study shows that CIDP overdiagnosis is common not just in the United States, but in the Netherlands as well. Furthermore, we have provided insight into the frequency of CIDP underdiagnosis. Both over‐ and underdiagnosis have detrimental consequences that impact individual patients and society. As a means to improve CIDP diagnostic accuracy, we highlight the importance of: (i) identifying proximal muscle weakness; (ii) recognition of atypical CIDP variants; (iii) avoiding overreliance on elevated CSF protein level; (iv) accurate interpretation of NCS with adherence to electrodiagnostic criteria; and (v) exclusion of other causes of demyelinating polyneuropathy. Although EFNS/PNS electrodiagnostic criteria may not capture all patients with CIDP, we encouraged diagnostic re‐exploration for patients not meeting electrodiagnostic standards. Conversely, patients who fulfil electrodiagnostic criteria but have atypical phenotypes also need consideration of diagnostic alternatives. Updates to the EFNS/PNS 2010 CIDP diagnostic criteria that provide improved guidance to these challenging patients are anticipated. Furthermore, we believe it would be helpful if the revised diagnostic criteria for CIDP were further improved to increase their use in current clinical practice. Given the complexity of the disease, with distinct diagnostic pitfalls, we encourage referrals to CIDP expertise centres, especially in cases of diagnostic ambiguity or if there is an unexpectedly poor treatment response.
CONFLICT OF INTEREST
Merel C. Broers and Hester F. Lingsma report grants from the Dutch Prinses Beatrix Spierfonds, during the conduct of the study. Carina Bunschoten, Judith Drenthen, Tiago A. O. Beck and Esther Brusse report no disclosures. Jeffrey A. Allen reports consultations with Akcea, Alexion, Argenx, Momenta, CSL Behring, Grifols and Biotest, outside the submitted work. Richard A. Lewis reports Consulting fees from Argenx, CSL Behring, Biotest, Annexon, Pharnext, Momenta, Pfizer, Sanofi and Takeda, and speaker honoraria for Akcea, Alnylam, CSL Behring and Medschape, outside the submitted work. Pieter A. van Doorn reports fees from Octapharma, Kedrion, CSL Behring, Grifols and Hansa (all fees to departmental research fund), grants from Dutch Prinses Beatrix Spierfonds, Sanquin, Takeda, Baxalta, Shire and Grifols, outside the submitted work. Bart C. Jacobs reports grants from the Dutch Prinses Beatrix Spierfond, during the conduct of the study, and grants from the Dutch Prinses Beatrix Spierfonds, Horizon 2020, GBS‐CIDP Foundation International, Baxalta, Grifols, CSL Behring, Annexon and Hansa Biopharma, outside the submitted work.
AUTHOR CONTRIBUTIONS
Merel Broers: Conceptualization (equal); Data curation (lead); Formal analysis (lead); Methodology (equal); Writing – original draft (lead); Writing – review and editing (equal). Carina Bunschoten: Data curation (supporting); Formal analysis (supporting); Writing – original draft (supporting); Writing – review and editing (equal). Judith Drenthen: Data curation (supporting); Supervision (supporting); Writing – original draft (supporting); Writing – review and editing (equal). Tiago Beck: Data curation (supporting); Writing – original draft (supporting). Esther Brusse: Data curation (supporting); Writing – review and editing (equal). Hester F. Lingsma: Supervision (supporting); Writing – review and editing (equal). Jeffrey A. Allen: Conceptualization (equal); Methodology (supporting); Writing – review and editing (equal). Richard A. Lewis: Conceptualization (equal); Methodology (supporting); Writing – review and editing (equal). Pieter A Van Doorn: Conceptualization (equal); Data curation (supporting); Methodology (equal); Writing – review and editing (equal). Bart C Jacobs: Conceptualization (equal); Methodology (equal); Supervision (lead); Writing – original draft (supporting); Writing – review and editing (equal).
Supporting information
Table S1
Funding information
This study was funded by the Dutch Prinses Beatrix Spierfonds (grant number: W.OR16‐18).
DATA AVAILABILITY STATEMENT
Anonymized (raw) data supporting the findings of this study are available on reasonable request within the current regulations.
REFERENCES
- 1. Oaklander AL, Lunn MP, Hughes RA, van Schaik IN, Frost C, Chalk CH. Treatments for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): an overview of systematic reviews. Cochrane Database Syst Rev. 2017;1:CD010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broers MC, Bunschoten C, Nieboer D, Lingsma HF, Jacobs BC. Incidence and prevalence of chronic inflammatory demyelinating polyradiculoneuropathy: a systematic review and meta‐analysis. Neuroepidemiology. 2019;52(3–4):161‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bunschoten C, Jacobs BC, Van den Bergh PYK, Cornblath DR, van Doorn PA. Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Lancet Neurol. 2019;18(8):784‐794. [DOI] [PubMed] [Google Scholar]
- 4. Neligan A, Reilly MM, Lunn MP. CIDP: mimics and chameleons. Pract Neurol. 2014;14(6):399‐408. [DOI] [PubMed] [Google Scholar]
- 5. Allen JA, Lewis RA. CIDP diagnostic pitfalls and perception of treatment benefit. Neurology. 2015;85(6):498‐504. [DOI] [PubMed] [Google Scholar]
- 6. Van den Bergh PYK, van Doorn PA, Jacobs BC, Querol L, Bunschoten C, Cornblath DR. Boundaries of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2020;25(1):4‐8. [DOI] [PubMed] [Google Scholar]
- 7. Bromberg MB. Review of the evolution of electrodiagnostic criteria for chronic inflammatory demyelinating polyradicoloneuropathy. Muscle Nerve. 2011;43(6):780‐794. [DOI] [PubMed] [Google Scholar]
- 8. Kaplan A, Brannagan TH 3rd. Evaluation of patients with refractory chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2017;55(4):476‐482. [DOI] [PubMed] [Google Scholar]
- 9. Allen JA, Gorson KC, Gelinas D. Challenges in the diagnosis of chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2018;8(3):e00932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allen JA, Ney J, Lewis RA. Electrodiagnostic errors contribute to chronic inflammatory demyelinating polyneuropathy misdiagnosis. Muscle Nerve. 2018;57(4):542‐549. [DOI] [PubMed] [Google Scholar]
- 11. Levine TD, Katz JS, Barohn R, et al. Review process for IVIg treatment: Lessons learned from INSIGHTS neuropathy study. Neurol Clin Pract. 2018;8(5):429‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bunschoten C, Blomkwist‐Markens PH, Horemans A, van Doorn PA, Jacobs BC. Clinical factors, diagnostic delay, and residual deficits in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2019;24(3):253‐259. [DOI] [PubMed] [Google Scholar]
- 13. Chaudhary UJ, Rajabally YA. Underdiagnosis and diagnostic delay in chronic inflammatory demyelinating polyneuropathy. J Neurol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bouchard C, Lacroix C, Plante V, et al. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52(3):498‐503. [DOI] [PubMed] [Google Scholar]
- 15. Buschbacher RM, Prahlow ND. Manual of nerve conduction studies. 2nd ed. New York, NY: Demos Medical Publishing. 2006. (ISBN 1‐888799‐94‐3). [Google Scholar]
- 16. Van den Bergh PYK, Hadden RD, Bouche P, et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society‐First Revision. Eur J Neurol. 2010;17:356‐363. [DOI] [PubMed] [Google Scholar]
- 17. Corrigendum. Eur J Neurol. 2011(18):796. 10.1111/j.1468-1331.2011.03403.x. [DOI] [Google Scholar]
- 18. Mitsuma S, Van den Bergh P, Rajabally YA, et al. Effects of low frequency filtering on distal compound muscle action potential duration for diagnosis of CIDP: a Japanese‐European multicenter prospective study. Clin Neurophysiol. 2015;126(9):1805‐1810. [DOI] [PubMed] [Google Scholar]
- 19. Breiner A, Bourque PR, Allen JA. Updated cerebrospinal fluid total protein reference values improve chronic inflammatory demyelinating polyneuropathy diagnosis. Muscle Nerve. 2019;60(2):180‐183. [DOI] [PubMed] [Google Scholar]
- 20. Mathis S, Magy L, Diallo L, Boukhris S, Vallat JM. Amyloid neuropathy mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2012;45(1):26‐31. [DOI] [PubMed] [Google Scholar]
- 21. Luigetti M, Papacci M, Bartoletti S, Marcaccio A, Romano A, Sabatelli M. AL amyloid neuropathy mimicking a chronic inflammatory demyelinating polyneuropathy. Amyloid. 2012;19(1):53‐55. [DOI] [PubMed] [Google Scholar]
- 22. Karam C, Moshe‐Lilie O, Chahin N, Ragole T, Medvedova E, Silbermann R. Diagnosis of paraproteinemic neuropathy: room for improvement. J Neurol Sci. 2020;415:116902. [DOI] [PubMed] [Google Scholar]
- 23. Koike H, Hashimoto R, Tomita M, et al. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid. 2011;18(2):53‐62. [DOI] [PubMed] [Google Scholar]
- 24. Tomita M, Koike H, Kawagashira Y, et al. Clinicopathological features of neuropathy associated with lymphoma. Brain. 2013;136(Pt 8):2563‐2578. [DOI] [PubMed] [Google Scholar]
- 25. Visser J, van den Berg‐Vos RM, Franssen H, et al. Mimic syndromes in sporadic cases of progressive spinal muscular atrophy. Neurology. 2002;58(11):1593‐1596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
Anonymized (raw) data supporting the findings of this study are available on reasonable request within the current regulations.