

Abstract
Introduction
The association between cerebral amyloid‐β accumulation and downstream CSF biomarkers is not fully understood, particularly in asymptomatic stages.
Methods
In 318 cognitively unimpaired participants, we assessed the association between amyloid‐β PET (Centiloid), and cerebrospinal fluid (CSF) biomarkers of several pathophysiological pathways. Interactions by Alzheimer's disease risk factors (age, sex and APOE‐ε4), and the mediation effect of tau and neurodegeneration were also investigated.
Results
Centiloids were positively associated with CSF biomarkers of tau pathology (p‐tau), neurodegeneration (t‐tau, NfL), synaptic dysfunction (neurogranin) and neuroinflammation (YKL‐40, GFAP, sTREM2), presenting interactions with age (p‐tau, t‐tau, neurogranin) and sex (sTREM2, NfL). Most of these associations were mediated by p‐tau, except for NfL. The interaction between sex and amyloid‐β on sTREM2 and NfL was also tau‐independent.
Discussion
Early amyloid‐β accumulation has a tau‐independent effect on neurodegeneration and a tau‐dependent effect on neuroinflammation. Besides, sex has a modifier effect on these associations independent of tau.
Keywords: [18F]flutemetamol, Alzheimer, biomarkers, glial activation, inflammation, modulation, neuronal injury, preclinical
1. BACKGROUND
The pathological hallmarks of Alzheimer's disease (AD) are amyloid‐β (Aβ) plaques and neurofibrillary tau tangles. According to the amyloid cascade hypothesis, Aβ accumulation is the earliest pathological event, which can start decades before symptoms, and it is followed by tau accumulation and neurodegeneration. 1 In addition, recent studies have reported the involvement of many other downstream pathophysiological processes during the early stages of the Alzheimer's continuum, even in asymptomatic individuals, including neuroinflammation, synaptic dysfunction and neuronal injury. 2 , 3
Recent advances in the development of novel biomarkers have enabled us and others to track some of these processes through cerebrospinal fluid (CSF) or plasma biomarkers. 4 , 5 , 6 , 7 , 8 , 9 , 10 Studying the early changes in these biomarkers and their relationship with the main pathological hallmarks of Alzheimer's disease (AD) would allow us to better understand the role of these processes in the progression of AD. Understanding these mechanisms, especially in the earliest stages, can also be informative on novel possible drug targets for the prevention of AD.
In a previous study, we found that after CSF Aβ42/40 becomes positive, there is a steep increase in tau‐related (phosphorylated tau [p‐tau]) and synaptic dysfunction (neurogranin) CSF biomarkers and, to a lesser extent, in axonal injury (neurofilament light [NfL] and total tau [t‐tau]) and glial biomarkers (soluble triggering receptor on myeloid cells 2 [sTREM2], YKL40, glial fibrillary acidic protein [GFAP]). 11 Despite the novelty of these findings, there were still important questions to be addressed. First, to assess whether cerebral Aβ deposition, as measured by Aβ PET, would also be associated with CSF biomarkers of downstream pathophysiological mechanisms. To this regard, it is important to note that Aβ measured in CSF and in PET probe different pools of Aβ, 12 and that aggregated Aβ (or Aβ load, as measured by Aβ PET) may have a different effect than soluble Aβ (as measure by CSF Aβ42/40). Second, to investigate whether age, sex and APOE‐ε4 status, the main unmodifiable AD risk factors, had also a modulation effect over the association between Aβ PET and the rest of CSF biomarkers. Finally, since these CSF biomarkers show a high level of collinearity at early asymptomatic AD stages, it remained to be determined to what extent these associations represented unique downstream alterations associated to Aβ or were driven by other correlated biomarkers. In this respect, we hypothesized that CSF p‐tau would mediate several associations between cerebral Aβ load and CSF biomarkers.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg. PubMed) sources. Several studies have been recently published about the association between novel CSF biomarkers for Alzheimer's disease (AD) and core AD biomarkers (ie. amyloid‐β, t‐tau and p‐tau). Relevant citations are appropriately cited.
Interpretation: Unlike previous studies, we studied a wide range of biomarkers reflecting several pathophysiological mechanisms, we focused in the very early stages of the disease, and we assessed the modifier effects of AD risk factors. Moreover, we tested how tau pathology mediated the association between amyloid‐β pathology and downstream neurodegeneration and neuroinflammation.
Future directions: This is a cross‐sectional study. We will conduct a longitudinal study of these participants, which will allow us to have a better understanding of the evolution of these biomarkers in the early stages of AD.
With this in mind, we aimed to analyze the relationship between Aβ accumulation in the brain measured with [18F]flutemetamol Aβ PET and multiple biological pathways measured in CSF in the early Alzheimer's continuum. We also studied whether these associations were modified by the main risk factors for AD: age, sex and APOE‐ε4 status. We hypothesized that Aβ PET is associated with several downstream pathophysilogical processes, and these association are modified by age, sex and APOE‐ε4 status. Moreover, we assessed whether these associations were mediated by biomarkers of tau pathology and neurodegeneration. We hypothesized that CSF p‐tau would mediate several associations between cerebral Aβ load and CSF biomarkers. To achieve these aims, we analyzed CSF biomarkers reflecting multiple Alzheimer's pathophysiological, including: tau pathology (CSF p‐tau), neuronal injury (CSF t‐tau and NfL), synaptic dysfunction (CSF neurogranin), inflammation and glial activation (sTREM2, YKL‐40, GFAP, S100b and interleukin 6 [IL‐6]), and also total α‐synuclein. 2 , 4 All these analyses were performed in a cohort of 318 cognitively unimpaired participants. Remarkably, these participants had a relatively low mean [18F]flutemetamol Aβ PET uptake, which allowed us to study very early pathophysiological changes associated with a low load of cerebral Aβ deposition.
2. MATERIALS AND METHODS
2.1. Participants
Participants of this study were part of the ALFA+ cohort, nested in the ALFA (for ALzheimer's and FAmilies) parent cohort. 13 The ALFA cohort was established as a research platform to characterize preclinical AD in 2,743 cognitively unimpaired individuals, aged between 45 and 75 years old, and enriched for family history of sporadic AD. ALFA+ participants were selected for a more comprehensive evaluation including a lumbar puncture (LP) and a [18F]flutemetamol Aβ PET. All ALFA+ participants were cognitively unimpaired with a Mini‐Mental State Examination (MMSE) above 26 and a Clinical Dementia Rating (CDR) of zero and were enriched for APOE‐ ε4 allele carriership and family history of AD. We also measured delayed free recall from Free and Cued Selective Reminding Test (FCSRT, see Supplementary Material). 14 For this study, we included the first 318 consecutive participants that had usable CSF and Aβ PET data acquired in less than a year.
2.2. CSF sampling
CSF Aβ42 was measured using the Elecsys® β‐ amyloid(1‐42), 15 while t‐tau and p‐tau were measured using the electrochemiluminescence immunoassays Elecsys® Total‐Tau and Phospho‐Tau(181P) CSF on a fully automated cobas e 601 instrument (Roche Diagnostics International Ltd., Rotkreuz, Switzerland). The rest of the biomarkers (Aβ40, NfL, neurogranin, YKL‐40, GFAP, sTREM2, S100b, IL‐6 and α‐synuclein) were measured with robust prototype assays as part of the NeuroToolKit (Roche Diagnostics International Ltd., Rotkreuz, Switzerland) on a cobas e 411 and e 601 instruments). All available CSF biomarkers were used in this study. All measurements were performed at the Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden, by board‐certified laboratory technicians who were blinded to diagnostic and other clinical data.
2.3. Image acquisition
Imaging acquisition and preprocessing protocols have been described previously. 16 Briefly, all participants had a [18F]flutemetamol PET scan and a T1‐weighted MRI acquired within one year. PET imaging was conducted in a Siemens Biograph mCT (Siemens, Munich, Germany), following a cranial CT scan for attenuation correction. Participants were injected with 185 MBq (range 166.5 to 203.5 MBq) of [18F]flutemetamol, and four frames of 5 min each were acquired 90 min post‐injection. Finally, the T1‐weighted 3D‐Turbo field echo (TFE) sequence was acquired in a Philips 3 T Ingenia CX scanner (Philips, Amsterdam, Netherlands).
2.4. Image processing
PET images were pre‐processed following the standard Centiloid pipeline using Statistical parametric mapping (SPM12). 17 In brief, PET frames were first realigned and summed to obtain a unique PET image, which was then co‐registered with the available T1‐weighted MRI scan of the same participant. Then, MRIs were normalized to the MNI space, and the same transformation was then applied to the PET image. All PET images were visually inspected as a quality control procedure. The intensity normalization was performed using the whole cerebellum as reference region, provided by the Centiloid working group on the GAAIN website (http://www.gaain.org/centiloid‐project). We quantified the global Aβ load using the standard Centiloid region of interest (ROI) that can also be found on the GAAIN website. The ratio of standardized uptake values (SUVr) were transformed to Centiloids using a previously validated linear regression. 16 From T1‐weighted images we derived normalized hippocampal volumes as described in the Supplementary Materials.
2.5. Statistical analyses
CSF biomarker determination were inspected before performing any correlation studies with [18F]flutemetamol PET uptake. Extreme values in CSF were excluded as specified in 11 (Supplementary Materials). We tested for normality of the distribution for each biomarker using the Kolmogorov‐Smirnov test and visual inspection of histograms. Those CSF biomarkers that did not follow a normal distribution log10‐transformed and Centiloid values were also log2‐transformed. Cross‐correlation between all CSF biomarkers were calculated using Pearson's correlation.
The first main analysis of this study aimed to assess the direct associations between Aβ load in the brain and all CSF biomarkers available. To this aim, we used each of the of the CSF biomarkers as variable of interest (dependent variable), and Aβ load (ie. Centiloids) as the independent variable in univariate general linear models (GLM). Age, sex, education and APOE‐ε4 status were added as covariates for all the models as shown in the following equation:
We also tested interaction effects between global cerebral Aβ load in the brain and main AD risk factors (ie, age, sex and APOE‐ε4 status) on each CSF biomarker. To test age interactions, we used a GLM model including a new variable that resulted from the product of age and Centiloids. To display the results of this interaction we divided the population in three age groups, using the tertiles of age, although this was not used for any statistical analysis. To test sex and APOE‐ε4 status interactions, we performed ANCOVAs using the same covariates as the previous models. The equations for the interaction are shown below:
As a complementary analysis we repeated the previous analyses replacing log(CL) by CSF Aβ42/40 ratio both only including the Aβ positive sub‐group, as the association between CSF Aβ42/40 ratio and the rest of CSF biomarkers showed a change of slope in the Aβ positivity threshold (Figure S3).
Finally, we performed a mediation analysis between cerebral Aβ load and each of the CSF biomarkers using p‐tau and/or NfL as mediators. The aim of this analysis was to understand whether the associations previously found between Aβ deposition and the other CSF biomarkers were partially, fully or not at all mediated by these biomarkers. We used the PROCESS version 3.4.1 toolbox from SPSS (www.processmacro.org) 18 to perform these analyses. In each model, Centiloid values were included as independent variable, CSF biomarkers as dependent variable, using the same covariates as in the previous analyses. Both p‐tau and NfL were included as mediators in all initial models (except the one studying NfL), but discarded if they did not show a significant mediation effect. As a complementary analysis, we also repeated mediation models using CSF Aβ42/40 ratio as independent variable in the Aβ positive sub‐group defined by CSF.
Finally, we repeated the main and interaction models including the significant mediators as covariates in each of the models to see whether these associations remained after adjusting by their mediators.
Other additional analyses regarding imaging biomarkers of neurodegeneration (hippocampal volumes) and cognition (MMSE and FCSRT) are presented in the Supplementary Materials, as well as Aβ PET analyses including only Aβ positive participants. Statistical significance was set at P < .05 without corrections for multiple comparisons for all analyses.
2.6. Ethical statement
The ALFA+ study (ALFA‐FPM‐0311) was approved by the Independent Ethics Committee “Parc de Salut Mar,” Barcelona, and registered at Clinicaltrials.gov (Identifier: NCT02485730). All participants signed the study's informed consent form that had also been approved by the Independent Ethics Committee “Parc de Salut Mar,” Barcelona.
3. RESULTS
Participants' demographics, CSF biomarker levels and PET measurements are summarized in Table 1. Their mean age was 61.4 years old, with a majority of women (62.6%) and more than half of the participants were APOE‐ε4 carriers (52.8%). The mean time difference between PET and LP was 97 days. Mean Aβ deposition for the whole cohort in the brain was low (mean CL: 2.7, range: −23.9 to 81.6). Importantly, those considered to be Aβ positive also have a low Aβ deposition compared to other cohorts (mean CL: 16.4, n = 109, Table S4), showing the early stage in the Alzheimer's continuum of this population. This can also be seen in the distribution of our participants in the A/T/(N) classification (Table S1). 19 All CSF biomarkers but sTREM2 were log10‐transformed for following analyses as were not normally distributed.
TABLE 1.
Participants' demographics and CSF and PET measures
| N = 318 | Mean (SD) |
|---|---|
| Demographics | |
| Age (years) [range] | 61.4 (4.6) [50.4 to 74.3] |
| Women, n(%) | 199 (62.6) |
| Education (years) | 13.4 (3.5) |
| APOE‐ε4 carriers, n(%) | 168 (52.8) |
| Time difference LP ‐ PET (days) | 96.7 (67.4) |
| MMSE | 29.2 (1.0) |
| FCSRT ‐ Delayed free recall | 11.6 (2.1) |
| CSF measures | |
| Aβ1‐42 (pg/mL) [range] | 1328 (569) [307 to 3595] |
| Aβ1‐40 (ng/mL) | 17.6 (5.0) |
| p‐tau (pg/mL) | 16.1 (6.3) |
| t‐tau (pg/mL) | 198 (68) |
| NfL (pg/mL) | 81.5 (26.8) |
| Neurogranin (pg/mL) | 805 (323) |
| GFAP (ng/mL) | 7.5 (2.3) |
| YKL‐40 (ng/mL) | 148 (53) |
| sTREM2 (ng/mL) | 7.9 (2.3) |
| S100b (ng/mL) | 1.01 (0.22) |
| IL‐6 (pg/mL) | 3.8 (1.4) |
| α‐synuclein (pg/mL) | 199 (81) |
| PET measures | |
| Centiloids [range] | 2.7 (16.6) [−23.9, 81.6] |
Mean (SD) values are shown unless otherwise stated.
Aβ, amyloid‐β; FCSRT, Free and Cued Selective Reminding Test; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; LP, lumbar puncture; MMSE, Mini‐Mental State Examination; NfL, neurofilament light; p‐tau, phosphorylated tau; SD, standard deviation; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2); t‐tau; total tau.
CSF levels for the different biomarkers were highly correlated. Except for CSF IL‐6, the rest of CSF biomarkers showed a significant correlation between them with high values (Figure S1).
3.1. Associations of CSF biomarkers with cerebral Aβ load
In our global main analysis, we calculated the association between a global measure of Aβ load in the brain and all CSF biomarkers (Table 2). CSF p‐tau, t‐tau, NfL, neurogranin, GFAP, YKL‐40, and sTREM2 showed a significant positive correlation with Centiloids. CSF S100b, IL‐6 and α‐synuclein did not show significant association. Similar associations were found when we took into account only the Aβ positive participants, except for CSF neurogranin and sTREM2 that became non‐significant (Table S5).
TABLE 2.
Main and interactions effects of Aβ on CSF biomarkers
| Main effect | Age interaction | Sex interaction (F > M) | APOE‐ε4 interaction (C > NC) | |||||
|---|---|---|---|---|---|---|---|---|
| Effect size [95% CI] | P | Effect size [95% CI] | P | Effect size [95% CI] | P | Effect size [95% CI] | P | |
| Aβ PET (all subjects) | ||||||||
| p‐tau | 0.30 [0.19 to 0.41] | <.001 | 0.17 [0.06 to 0.28] | .003 | 0.06 [−0.05 to 0.17] | .302 | −0.03 [−0.14 to 0.09] | .651 |
| t‐tau | 0.28 [0.17 to 0.39] | <.001 | 0.13 [0.02 to 0.24] | .017 | 0.03 [−0.08 to 0.14] | .584 | −0.04 [−0.15 to 0.07] | .483 |
| NfL | 0.25 [0.14 to 0.36] | <.001 | 0.07 [−0.04 to 0.18] | .201 | 0.11 [0.00 to 0.22] | .045 | 0.02 [−0.09 to 0.13] | .736 |
| Neurogranin | 0.15 [0.04 to 0.26] | .009 | 0.12 [0.01 to 0.23] | .031 | 0.04 [−0.07 to 0.15] | .451 | −0.03 [−0.14 to 0.08] | .584 |
| GFAP | 0.18 [0.07 to 0.29] | .001 | 0.04 [−0.07 to 0.15] | .461 | 0.04 [−0.07 to 0.15] | .527 | −0.04 [−0.15 to 0.07] | .452 |
| YKL‐40 | 0.21 [0.10 to 0.32] | <.001 | 0.06 [−0.05 to 0.17] | .261 | 0.10 [−0.01 to 0.21] | .085 | −0.02 [−0.13 to 0.09] | .681 |
| sTREM2 | 0.13 [0.02 to 0.24] | .026 | 0.11 [−0.00 to 0.22] | .051 | 0.13 [0.02 to 0.24] | .021 | −0.04 [−0.15 to 0.07] | .522 |
| S100b | 0.09 [−0.03 to 0.20] | .130 | −0.01 [−0.12 to 0.10] | .819 | 0.06 [−0.05 to 0.17] | .293 | −0.07 [−0.18 to 0.04] | .229 |
| IL‐6 | 0.06 [−0.06 to 0.17] | .326 | 0.09 [−0.02 to 0.20] | .099 | −0.01 [−0.12 to 0.10] | .857 | 0.01 [−0.10 to 0.12] | .856 |
| α‐synuclein | 0.07 [−0.04 to 0.18] | .216 | 0.09 [−0.02 to 0.20] | .112 | 0.04 [−0.07 to 0.15] | .479 | −0.11 [−0.21 to 0.01] | .065 |
| CSF Aβ42/40 ratio (Aβ positive subjects) | ||||||||
| p‐tau | −0.57 [−0.76, −0.38] | <.001 | −0.18 [0.37, 0.02] | .072 | 0.03 [−0.16, 0.22] | .758 | 0.19 [−0.01, 0.37] | .067 |
| t‐tau | −0.54 [−0.73, −0.35] | <.001 | −0.14 [0.32, 0.06] | .172 | 0.02 [−0.17, 0.21] | .851 | 0.15 [−0.05, 0.33] | .153 |
| NfL | −0.34 [−0.53, −0.14] | .001 | −0.05 [−0.23, 0.15] | .655 | −0.20 [−0.38, −0.00] | .047 | 0.15 [−0.05, 0.34] | .135 |
| Neurogranin | −0.48 [−0.67, −0.29] | <.001 | −0.08 [−0.27, 0.11] | .405 | 0.01 [−0.19, 0.20] | .951 | 0.19 [−0.00, 0.38] | .051 |
| GFAP | −0.27 [−0.46, −0.08] | .005 | 0.10 [−0.10, 0.29] | .323 | 0.08 [−0.12, 0.27] | .432 | −0.01 [−0.20, 0.18] | .935 |
| YKL‐40 | −0.46 [−0.65, −0.27] | <.001 | 0.02 [−0.17, 0.21] | .867 | 0.01 [−0.18, 0.21] | .884 | 0.02 [−0.17, 0.21] | .820 |
| sTREM2 | −0.19 [−0.38, −0.00] | .046 | −0.02 [−0.21, 0.17] | .815 | −0.06 [−0.25, 0.13] | .533 | −0.09 [−0.28, 0.11] | .382 |
| S100b | −0.08 [−0.27, 0.11] | .405 | 0.20 [−0.00, 0.38] | .051 | 0.07 [−0.13, 0.26] | .498 | −0.14 [−0.33, 0.05] | .260 |
| IL‐6 | −0.08 [−0.27, 0.11] | .413 | −0.11 [−0.29, 0.09] | .290 | −0.25 [−0.43, −0.04] | .017 | 0.12 [−0.07, 0.31] | .230 |
| α‐synuclein | −0.34 [−0.53, −0.15] | .001 | −0.01 [−0.20, 0.19] | .952 | −0.02 [−0.21, 0.17] | .829 | 0.20 [0.00, −0.38] | .047 |
Aβ was assessed both with PET and CSF. In the analysis using CSF Aβ42/40 ratio, only Aβ positive subjects were assessed due change of slopes between this Aβ marker and the rest of the CSF biomarkers in the Aβ cut‐off for positivity (see Figure S2). Aβ positive participants' were defined as having CSF Aβ42/40 ratio below 0.071. 11 Effect sizes are calculated as standardized betas. Significant effects (P < .05) are shown in bold. Models included age, sex, education and APOE‐ε4 status as covariates. Of note, the sign is reversed in the CSF analysis due to the inverse relationship between CSF Aβ and Aβ PET.
Aβ, amyloid‐β; C, carrier; CI, confidence interval; F, female; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; M, male; NC, non‐carrier; NfL, neurofilament light; p‐tau, phosphorylated tau; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2); t‐tau, total tau.
For comparison purposes, we also repeated this analysis using CSF Aβ42/40 ratio as biomarker Aβ pathology, instead of Aβ PET. For this analysis we only included Aβ positive subjects. Similar results were found with this approach with only a significant association with α‐synuclein as a difference (Table 2). Effect sizes found using CSF Aβ42/40 ratio were bigger than with those found using Aβ PET. However, this increase in effect sizes was also seen in PET analyses when we only considered Aβ positive subjects (Table S5). Therefore, we could say that analyses using CSF Aβ42/40 ratio were very similar to those using Aβ PET, although only considering Aβ positive participants.
3.2. Age, sex, and APOE‐ε4 interactions with Aβ load on CSF biomarkers
The first interaction effect tested in the association between Centiloids and CSF biomarkers was age. We found that this interaction was significant for CSF p‐tau, t‐tau and neurogranin (Table 2 and Figure 1). This was also true when we only included Aβ positive subjects (Table S5). Age‐interaction also shows a trend to significance on sTREM2 and IL‐6. All CSF biomarkers showed a more pronounced positive association with Centiloids with older age. No significant interactions with age were observed using CSF Aβ42/40 as a marker of Aβ pathology when only studying Aβ positive subjects (Table 2).
FIGURE 1.
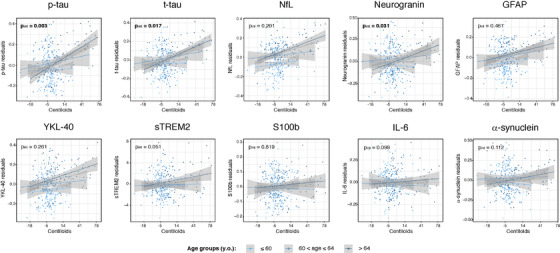
Age and Aβ interaction effects on CSF biomarkers. CSF biomarkers residuals, after adjusting by covariates (sex, education, and APOE‐ε4 status), are compared to global Aβ load measured as Centiloids. Light, medium and dark blue colors depict the three age groups (tertiles): below 60, between 60 and 64 and, above 64 years old, respectively. These groups were used for visualization purposes only. P‐value of each interaction effect is shown in the upper left corner. Statistically significant effects (P < .05) are shown in bold. x axis is depicted in logarithmic scale. Aβ, amyloid‐β; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; NfL, neurofilament light; p‐tau, phosphorylated tau; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2); t‐tau, total tau
Regarding the interaction between sex and Centiloids, it was only significant for CSF NfL and sTREM2 (Table 2 and Figure 2). In both cases, women presented a higher correlation between CSF levels and Centiloids. Together with these two biomarkers, YKL‐40 also showed a sex interaction when only Aβ positive participants were included (Table S5). In contrast, the sex interaction with CSF Aβ42/40 was also significant for CSF NfL and IL‐6 for Aβ positive subjects (Table 2).
FIGURE 2.
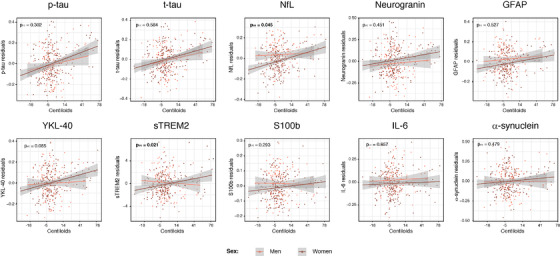
Sex and Aβ interaction effects on CSF biomarkers. CSF biomarkers residuals, after adjusting by covariates (age, education, and APOE‐ε4 status), are compared to global Aβ load measured as Centiloids. Colors represent the two sex groups, with women depicted in dark coral. P‐value of each interaction effect is shown in the upper left corner. Statistically significant effects (P < .05) are shown in bold. x axis is depicted in logarithmic scale. Aβ, amyloid‐β; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; NfL, neurofilament light; p‐tau, phosphorylated tau; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2); t‐tau, total tau
On the contrary, none of the CSF biomarkers showed a significant interaction between APOE‐ε4 status and Centiloids (both with the whole sample and Aβ positive participants). However, the interaction between CSF Aβ42/40 and APOE‐ε4 status was significant for α‐synuclein in Aβ positive subjects (Table 2).
3.3. Mediation effects of tau and neuronal injury biomarkers
Here, we tested whether the association between Centiloids and CSF biomarkers were mediated by tau pathology and/or neuronal injury, as measured by CSF p‐tau and CSF NfL, respectively. We performed the mediation on those CSF biomarkers that showed a significant or a trend to a significant effect in the main association analyses (ie. CSF NfL, GFAP, YKL‐40, and sTREM2). We did not include CSF t‐tau or neurogranin in this analysis due to its high colinearity with CSF p‐tau, (r>0.9, Figure S1). Figure 3 shows the final models whose mediators (CSF p‐tau and/or NfL) showed at least a trend to significance in the mediation path. First, we observed that the Centiloids effect on CSF NfL was only partially mediated by CSF p‐tau. This is shown by the fact that the Centiloids direct effect on CSF NfL still showed a trend effect after adjusting by CSF p‐tau (40.5% of total effect). In contrast, we found that the effect of Centiloids on CSF sTREM2 was fully mediated by CSF p‐tau, but not by CSF NfL. Regarding GFAP and YKL‐40, both had a significant two‐step indirect effect of p‐tau and NfL (ie. Centiloids → p‐tau → GFAP/YKL‐40 and Centiloids → NfL → GFAP/YKL‐40); and also a three‐step indirect effect of p‐tau and NfL (ie. Centiloids → p‐tau → NfL → GFAP/YKL‐40). However, the biggest effect in both cases was seen in the indirect path involving p‐tau (ie, Centiloids → p‐tau → GFAP/YKL‐40, GFAP: 56.6% and YKL‐40: 95.3% of the total effect).
FIGURE 3.
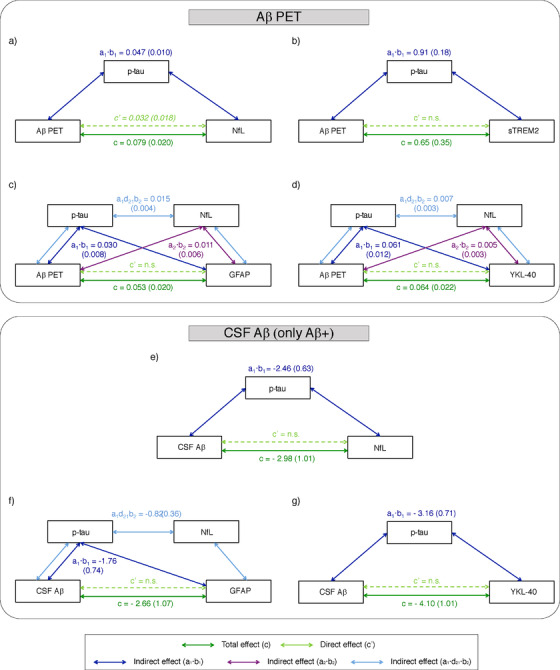
Aβ mediated effects on CSF biomarkers. Models of mediation for CSF NfL (A,E), sTREM2 (B), GFAP (C,F) and YKL‐40 (D,G) with Aβ PET (upper part) and CSF Aβ (lower part). Models with CSF Aβ as independent variable only included Aβ positive participants (n = 109). The values of each path show the effect (SE). Total effect between Aβ load and each CSF biomarker is shown in dark green (path c); direct effect, after adjusting by mediators, is shown in light green (path c'); and indirect effects are shown in dark blue (path a1b1, mediation effect of p‐tau), in purple (path a2b2, mediation effect of NfL) and in light blue (path a1·d21·b2, mediation effect of p‐tau and NfL). All paths depicted are significant (P < .05), except for direct effect between Aβ and NfL that showed a trend to significance (P < .10, in italics). All paths are adjusted by covariates (age, sex, education, and APOE‐ε4 status). Aβ, amyloid‐β; GFAP, glial fibrillary acidic protein; IL‐6, interleukin 6; NfL, neurofilament light; p‐tau, phosphorylated tau; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2).
Similar results were observed when using CSF Aβ42/40 as independent variable in Aβ positive participants (Figure 3). CSF p‐tau also mediated the association between CSF Aβ42/40 and NfL, but in this case, CSF Aβ42/40 direct effect on NfL was no longer significant. CSF GFAP was also mediated by both p‐tau (66.2% of the total effect) and NfL (30.8% of the total effect). On the other hand, CSF YKL‐40 was only mediated by p‐tau. Finally, we did not perform the mediation analysis with CSF sTREM2 in this case, as it did not show a significant total effect.
3.4. Associations between CSF biomarkers and Aβ load after adjusting for mediators
Finally, we repeated the main association models as well as the ones including interaction effects but adjusting by the significant mediators found in the previous analyses. These analyses were only repeated for those CSF biomarkers and associations that were significant in the main analyses using Aβ PET. These analyses had the objective of disentangling whether these associations or interactions were independent of p‐tau and/or NfL indirect effects. As shown in Table 3, none of the main associations between Aβ load and the CSF biomarkers studied remained significant after adjusting by their specific mediators. Similarly, CSF NfL and age interaction became non‐significant after including mediators as covariates. Finally, we found that the interaction effect between sex and Aβ load on CSF sTREM2 was still significant after adjusting by CSF p‐tau.
TABLE 3.
Main and interactions effects of Aβ load on CSF biomarkers after adjusting by specific mediators
| Main effect | Age interaction | Sex interaction (F > M) | ||||
|---|---|---|---|---|---|---|
| CSF biomarker | Effect size [95% CI] | P | Effect size [95% CI] | P | Effect size [95% CI] | P |
| NfL (adj p‐tau) | 0.10 [−0.01 to 0.21] | .082 | 0.00 [−0.11 to 0.11] | .958 | 0.10 [−0.01 to 0.21] | .077 |
| GFAP (adj p‐tau & NfL) | −0.01 [−0.12 to 0.10] | .853 | ‐ | ‐ | ‐ | ‐ |
| YKL‐40 (adj p‐tau & NfL) | −0.03 [−0.14 to 0.08] | .632 | ‐ | ‐ | ‐ | ‐ |
| sTREM2 (adj p‐tau) | −0.05 [−0.16 to 0.06] | .410 | ‐ | ‐ | 0.13 [0.02 to 0.24] | .023 |
Association parameters between CSF biomarkers and global Aβ deposition for the main and interaction effects after adjusting by significant mediators. Only those associations that were significant without mediators (shown in Table 2) were tested. Effect sizes are calculated as standardized betas. Significant effects (P < .05) are shown in bold.
Aβ, amyloid‐β; CI, confidence interval; F, female; GFAP, glial fibrillary acidic protein; M, male; NfL, neurofilament light; p‐tau, phosphorylated tau; sTREM2, soluble triggering receptor on myeloid cells 2 (TREM2).
4. DISCUSSION
In this study, we investigated the relationship between Aβ load in the brain, as measured by PET, and CSF biomarkers of pathophysiological processes downstream to Aβ in a cohort of cognitively unimpaired participants. Remarkably, some of them were in a very early stage of the Alzheimer's continuum, as shown by a very low load of Aβ load. Our first finding was that markers of tau pathology (CSF p‐tau), neuronal injury (CSF t‐tau and NfL), synaptic dysfunction (CSF neurogranin), and neuroinflammation (CSF GFAP, YKL‐40, and sTREM2) showed a positive association with Aβ load, even at this early stage. Interestingly, we found that age and sex, but not APOE‐ε4 status, had a modulatory influence on some of these relationships. There was a stronger association between Aβ PET and CSF biomarkers in older individuals and in women. Finally, as some of these CSF biomarkers were highly correlated, we performed mediation analyses to disentangle the ones driving the observed associations. We found that the association of Aβ PET with CSF NfL, a marker of neuronal injury, was only partially mediated by tau pathology (CSF p‐tau) suggesting a direct effect of Aβ deposition on neurodegeneration. In contrast, almost all the associations of Aβ PET with inflammatory markers (ie. CSF GFAP, YKL‐40, and sTREM2), as well as their respective interactions with AD risk factors, were mediated by CSF biomarkers of tau pathology (CSF p‐tau), alone or in combination with neuronal injury (CSF NfL). The only exception was the interaction between sex and Aβ load on CSF sTREM2 (and between sex and CSF NfL, at a trend level) that remained significant after correction for CSF p‐tau.
Previously, it was reported in older subjects with cognitive impairment associations between Aβ PET and several CSF biomarkers, including CSF p‐tau, t‐tau, neurogranin, NfL and YKL‐40. 20 , 21 However, less is known in preclinical Alzheimer. Herein, we extend these findings to younger individuals without objective cognitive impairment associations. In line with our findings, a recent study by Palmqvist et al. showed changes in CSF p‐tau, neurogranin, NfL, and YKL‐40 associated with Aβ load measured with PET. 20 Similar results were also found in another recent study by Bos et al., in which the authors reported an association between CSF Aβ and NfL, neurogranin and YKL‐40 in cognitively unimpaired participants. 21 With respect to these reports, our sample is younger (mean age of 61.4 vs 72.1 years old) and does not include mild cognitive impairment (MCI) patients. Importantly, we included a relatively high percentage of individuals (25%, see Table S1) that were Aβ positive (as defined by CSF Aβ42/40) but still tau negative (as defined by CSF p‐tau). It can thus be considered that our study focuses on an earlier stage in the Alzheimer's continuum and in late‐/middle‐aged individuals, when Alzheimer's pathology most likely starts. In addition, we also detected associations between Aβ load and additional neuroinflammatory markers (CSF GFAP and sTREM2) that, to the best of our knowledge, have not been previously described. It is important to note that the vast majority of these associations remained significant even when we only analyzed Aβ positive participants, both measuring CSF Aβ42/40 and Aβ PET load (see Table 2 and Table S5).
Mounting evidence suggests that women are at increased risk of AD, 22 but the biological explanation of this difference is still unknown. Neither Aβ load nor its accumulation over time seem to be the cause as multiple studies found no difference in Aβ by sex, 23 , 24 , 25 , 26 which is replicated in our study (data not shown). In our previous study, we found that CSF NfL was significantly lower than in women. 11 But in the present study we discovered that this difference was related to Aβ load; as we revealed that CSF NfL levels had a steeper increase in women with higher Aβ load than men, with women having lower CSF NfL with low Aβ load but higher CSF NfL with high Aβ load. This effect was also seen even after correcting for CSF p‐tau (trend level) or when measuring Aβ pathology in the CSF. Previous studies already reported higher levels of neurodegeneration in women, 27 , 28 particularly in APOE‐ε4 carriers, and have been related to their worse clinical output, although this was not replicated in all cases. 26 Our result confirmed this in a younger and earlier in the Alzheimer's continuum population. What is new in our study is that we find that these differences are related to Aβ pathology load and irrespective of tau pathology. This result suggests that women may show a tighter coupling between Aβ pathology and neurodegeneration, which may contribute to explain their higher risk of AD, independently of their tau levels.
Another novelty of our study was finding an interaction between sex and Aβ load on sTREM2. Women presented more elevated levels of CSF sTREM2 with increasing Aβ load than men, although their global measures did not differ. 11 Of note, this difference in slope with increasing Aβ was independent of tau pathology. Whether this is a protective or detrimental effect for women against Aβ cannot be stated. The role of microglial activation, and in particular of TREM2‐mediated microglia function, in AD is still not fully understood, and it has been suggested that could have different effects depending on the stage of the disease, being beneficial at early stages but detrimental at later ones. 3 , 29 , 30 , 31 Sex differences in microglial activation have been reported before in mice. 32 Also, recently early sex differential patterns of atrophy were related to glial biomarkers with a greater impact in areas of the Papez circuit in women and greater impact in men in lateral parietal and paracentral areas. 33
We also found an interaction between age and Aβ load on p‐tau and t‐tau, suggesting that for the same levels of Aβ PET‐measured pathology, older individuals have increased tau pathology. To the best of our knowledge, this has not been reported before and, therefore, needs replication. However, multiple hypotheses may explain this behavior. On one hand, older subjects have an increased probability to have other comorbidities which can decrease their resistance to tau pathology and neurodegeneration. 34 On another hand, it is possible that biological pathways that may help to slow‐down the spreading of tau pathology once Aβ is present (ie. glial activation), become less effective in older ages. 35 CSF neurogranin, sTREM2 and IL‐6 (the two last at a trend level) also had a significant Aβ*age interaction, however, these interactions became non‐significant when we adjusted by tau pathology. This suggests that their initial relationship was mainly driven by the actual interaction of age and Aβ on p‐tau.
The association between CSF NfL, a marker of neuronal injury, and Aβ PET or CSF Aβ has been reported before. 11 , 20 , 21 However, whether this association is mediated by tau pathology is unknown and, therefore, we conducted a mediation analysis. Interestingly, we found a tau‐dependent but also a relevant tau‐independent effect of Aβ PET on CSF NfL (40.5% of the total effect, Figure 3). This is in line with a recent publication that shows a direct effect of Aβ accumulation on CSF NfL levels and neurodegeneration in AD‐regions in a rat model with minimal tau pathology. 36 In the same study, these results were replicated in humans even when accounting for tau pathology. Altogether, this result suggests that Aβ load has, at least, a partial direct effect on neurodegeneration in early stages of Alzheimer's continuum. It is important to note, that this Aβ‐direct effect over CSF NfL did not remain significant when we studied this model using CSF Aβ42/40 in positive participants. We speculate that there may be two reasons underlying these findings, which are not mutually exclusive. First, that soluble Aβ changes first triggers tau metabolism changes and, after that, neurodegeneration occurs. This is supported by the fact that there is an active secretion of p‐tau in response of Aβ early changes. 37 , 38 , 39 , 40 Second, there is the possibility that Aβ‐direct effect on neurodegeneration may be most important in later stages of the Alzheimer's continuum, when the progression of AD pathology can still be tracked by PET and no longer by CSF Aβ and due to other mechanisms, such as inflammation or actual physical damage by the plaques. Then, after both Aβ and tau pathology increase, this association may be fully driven by tau pathology, which agrees with previous studies finding a direct association between tau ‐but not Aβ‐ and neurodegeneration. 41 , 42 However, longitudinal investigations in participants in the very early stages of the Alzheimer's continuum are needed to confirm this hypothesis.
Overall, three markers of neuroinflammation showed associations with Aβ pathology in this study: one related to microglial activation (sTREM2), and two related to astroglial reactivity (YKL‐40 and GFAP). sTREM2 is the soluble ectodomain of the TREM2 receptor, mainly expressed in microglia in the central nervous system. Previous studies have shown that CSF sTREM2 levels are elevated in late asymptomatic (once participants have changes in tau‐related biomarkers) and early symptomatic stages of familial and sporadic AD. 43 , 44 , 45 , 46 , 47 Recently, we found that CSF sTREM2 was increased in Aβ and tau positive cognitively unimpaired individuals but not in those that were Aβ positive but tau negative. 11 Considering that Aβ PET changes later than CSF Aβ, 48 , 49 , 50 the associations that we observe here between Aβ PET and CSF sTREM2 reinforce the idea that CSF sTREM2 increases are not as soon as the first changes in CSF Aβ42/40. Also supporting this hypothesis, we found that CSF sTREM2 only showed an association with CSF Aβ42/40 in our cohort when Aβ‐positive participants were considered, suggesting that this association may relate to later stages of preclinical AD pathophysiology. Our mediation analyses allowed us to determine, whether this association was possibly due to collinearity with other CSF biomarkers or a singular association with Aβ PET load. The fact that this association was fully mediated by CSF p‐tau is in line with the hypothesis that CSF sTREM2 increases with the early changes in tau biomarkers.
We also found associations between cerebral Aβ load ‐and CSF Aβ‐ and astroglial markers (YKL‐40 and GFAP). To our knowledge, this is the first study to show an association between Aβ load and CSF GFAP, although we previously showed this association with CSF Aβ42/40. 11 On the other hand, previous studies only reported associations between CSF YKL‐40 with tau levels but not with Aβ in CSF, 51 , 52 except for our previous study only in Aβ positive participants. 11 To this regard, and like CSF sTREM2, we found the association between astroglial markers and Aβ to be fully mediated by p‐tau levels. This pinpoints the importance of this analysis to understand the underlying associations between a set of CSF biomarkers that were highly correlated. The fact that p‐tau fully mediated the association between Aβ and astroglial markers is in line with the hypothesis that levels of glial markers parallel those of tau biomarkers. 43 , 44 , 45 , 46 , 47 , 51 , 52 Some of these previous publications also found an association of glial markers, not only with tau, but also with markers of neuronal injury (ie, t‐tau and NfL) and imaging markers of neurodegeneration. 43 , 45 , 46 , 52 , 53 In line with these, we also found CSF NfL to mediate the association between Aβ load and astroglial markers, but not CSF sTREM2. However, both for CSF GFAP and YKL‐40, the effect size of this mediation was considerably lower than that of CSF p‐tau (GFAP: p‐tau effect = 56.6%; YKL‐40: p‐tau effect = 95.3%). Moreover, NfL mediation effect did not remain significant when only Aβ positive subjects were studied. Such a weak association may be explained by the almost complete lack of neurodegeneration positive individuals in our sample: only three participants (0.9%) were A+T+N+, see Table S1). 19 The follow‐up of these participants will enable us to study whether this association is replicated when they will be more advanced in the Alzheimer's continuum. Altogether, we hypothesize that the neuroinflammatory response in this early phase of AD probably follows the early changes of tau pathology and, to a lower extent, of neurodegeneration. Of note, in the mediation analysis, we used CSF NfL, and not CSF t‐tau, as a marker of neurodegeneration due to the colinearity observed between the latter and p‐tau (r>0.98, Figure S1). Moreover, recent publications suggest that CSF NfL may be a better neurodegeneration biomarker than CSF t‐tau for AD. 54 , 55 , 56
In this study, we considered the associations, and interactions, found between Aβ‐downstream CSF biomarkers and Aβ load as the main results. However, we also performed complementary analyses using CSF Aβ42/40 ratio as a proxy of Aβ pathology. In these last analyses, we focused on Aβ positive participants only, as we found that associations between CSF Aβ and the rest of CSF biomarkers were highly modified by Aβ status (see Figure S3). However, even only focusing in a subset of participants, we found that the results of Aβ main effects on the other CSF biomarkers to be very similar. On the contrary, many of the interactions found between AD risk factors and Aβ load were not significant with CSF Aβ and, also other became significant such as with APOE‐ε4 status on α‐synuclein when using CSF Aβ. This result may be related to the fact that, although both measures are usually used as Aβ biomarkers, they are measuring different pools of Aβ, 12 and although they are usually used as indistinguishable clinical biomarkers, differences between them has shown to have consequences on future tau deposition. 57 Therefore, it is possible that AD risk factors affect differently the Aβ production/clearance imbalance (as measured in CSF) and Aβ plaque production (as measured in PET), as we have seen in a previous study of our group with APOE‐ε4 status. 58
The main strengths of this study are: (1) the availability of many different CSF biomarkers that allowed us to study, in the same individuals, the relationship between Aβ and many other pathophysiological processes, which their role in the AD development is still unknown; (2) the inclusion of many cognitively unimpaired participants with low or very low levels of Aβ deposited in the brain resulting in an increased statistical power to detect the earliest changes in the Alzheimer's continuum; and (3) the comparison between associations with Aβ both measured in PET and in CSF. Nonetheless, there are also some limitations to note. The cross‐sectional design of this study gives us a picture of all the processes occurring but cannot tell us its implications on future developments. In particular, it is worth noting that mediation models in cross‐sectional data do not allow testing for causality. This will be analyzed in the follow‐up of these participants that is already on‐going. Another limitation was the high correlation between almost all CSF biomarkers that makes it difficult to disentangle each specific association with Aβ. Finally, due to the exploratory nature of this study, we did not correct for multiple comparisons. Worth to note, all main associations survived FDR‐correction, but none of the interaction effects did, which may be due to limited statistical power. Further investigations are needed to validate these results in independent cohorts.
As a conclusion, our results suggest that Aβ deposition in the brain is associated with many biological pathways in very early stages of the Alzheimer's continuum, notably including neurodegeneration, even after accounting for the effect of tau physiopathology. On the other hand, tau, alone or in combination with neurodegeneration, fully accounts for the observed association between Aβ deposition and glial response. From a clinical point of view, a better understanding of the pathophysiological mechanisms triggered by early AD pathology may provide novel insights to develop therapeutical strategies to interfere with the course of the disease in preclinical AD stages. Our results may help to better understand the sequence of events that occur in preclinical Alzheimer, which can be very valuable in trials design. More specifically, our results suggest that removing deposited Aβ in the brain may impact on future neurodegeneration. And also that targeting tau pathology, may have consequences on other pathophysiological mechanisms in Alzheimer's, such as inflammation, even at the earliest stages of the Alzheimer's continuum. Finally, in the context of precision medicine for future AD treatments, as suggested by our results, it may be important to also take into account subject's characteristics that may modify the course of these mechanisms, such as age or sex.
CONFLICT OF INTERESTS
JLM is currently a full time employee of H. Lundbeck A/S and priory has served as a consultant or at advisory boards for the following for‐profit companies, or has given lectures in symposia sponsored by the following for‐profit companies: Roche Diagnostics, Genentech, Novartis, Lundbeck, Oryzon, Biogen, Lilly, Janssen, Green Valley, MSD, Eisai, Alector, BioCross, GE Healthcare, ProMIS Neurosciences. HZ has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics and CogRx, has given lectures in symposia sponsored by Fujirebio, Alzecure and Biogen, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). KB has served as a consultant, at advisory boards, or at data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu. Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers, and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. GK is a full time employee of Roche Diagnostics GmbH. The remaining authors declare that they have no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This publication is part of the ALFA study (ALzheimer and FAmilies). The authors would like to express their most sincere gratitude to the ALFA project participants and relatives without whom this research would have not been possible. Collaborators of the ALFA study are: Annabella Beteta, Raffaele Cacciaglia, Alba Cañas, Carme Deulofeu, Ruth Dominguez, Maria Emilio, Carles Falcon, Sherezade Fuentes, Oriol Grau‐Rivera, Laura Hernandez, Gema Huesa, Jordi Huguet, Eider M. Arenaza‐Urquijo, Paula Marne, Tania Menchón, Grégory Operto, Albina Polo, Sandra Pradas, Aleix Sala‐Vila, Gonzalo Sánchez‐Benavides, Anna Soteras, Marc Vilanova and Natalia Vilor‐Tejedor. The authors thank Roche Diagnostics International Ltd. for providing the kits to measure CSF biomarkers and GE Healthcare for providing the doses for [18F]flutemetamol PET. ELECSYS, COBAS, and COBAS E are trademarks of Roche.
Salvadó G, Milà‐Alomà M, Shekari M, et al. Cerebral amyloid‐β load is associated with neurodegeneration and gliosis: Mediation by p‐tau and interactions with risk factors early in the Alzheimer's continuum . Alzheimer's Dement. 2021;17:788–800. 10.1002/alz.12245
Funding information
The project leading to these results has received funding from “la Caixa” Foundation (ID 100010434), under agreement LCF/PR/GN17/50300004 and the Alzheimer's Association and an international anonymous charity foundation through the TriBEKa Imaging Platform project (TriBEKa‐17‐519007). Additional support has been received from the Universities and Research Secretariat, Ministry of Business and Knowledge of the Catalan Government under the grant no. 2017‐SGR‐892. MSC received funding from the European Union's Horizon 2020 Research and Innovation Program under the Marie Sklodowska‐Curie action grant agreement No 752310, and currently receives funding from Instituto de Salud Carlos III (PI19/00155) and from the Spanish Ministry of Science, Innovation and Universities (Juan de la Cierva Programme grant IJC2018‐037478‐I). JDG is supported by the Spanish Ministry of Science and Innovation (RYC‐2013‐13054). HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), and the UK Dementia Research Institute at UCL. KB holds the Torsten Söderberg Professorship in Medicine at the Royal Swedish Academy of Sciences, and is supported by the Swedish Research Council (#2017‐00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), a grant (#ALFGBG‐715986) from the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement, and European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236).
REFERENCES
- 1. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Milà‐Alomà M, Suárez‐Calvet M, Molinuevo JL. Latest advances in cerebrospinal fluid and blood biomarkers of Alzheimer's disease. Ther Adv Neurol Disord 2019;12:1‐23. 10.1177/https. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, Fagan AM, Hampel H, Mielke MM, Mikulskis A, O’Bryant S, Scheltens P, Sevigny J, Shaw LM, Soares HD, Tong G, Trojanowski JQ, Zetterberg H, Blennow K. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathologica 2018;136(6):821‐853. 10.1007/s00401-018-1932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med 2019;11:e11170. 10.15252/emmm.201911170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal‐dominant Alzheimer's disease. Sci Transl Med 2014;6:226ra30. 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schindler SE, Li Y, Todd KW, et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer's disease. Alzheimers Dement 2019;15:655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 2018;91:E1295‐E306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleisher AS, Chen K, Quiroz YT, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross‐sectional study. JAMA Neurol 2015;72:316‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lleó A, Alcolea D, Martínez‐Lage P, et al. Longitudinal cerebrospinal fluid biomarker trajectories along the Alzheimer's disease continuum in the BIOMARKAPD study. Alzheimers Dement 2019;15:742‐753. [DOI] [PubMed] [Google Scholar]
- 11. Milà‐Alomà M, Salvadó G, Gispert JD, et al. Amyloid‐β, tau, synaptic, neurodegeneration and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimers Dement 2020;16:1358‐1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roberts BR, Lind M, Wagen AZ, et al. Biochemically‐defined pools of amyloid‐β in sporadic Alzheimer's disease: correlation with amyloid PET. Brain 2017;140:1486‐1498. [DOI] [PubMed] [Google Scholar]
- 13. Molinuevo JL, Gramunt N, Gispert JD, et al. The ALFA project: a research platform to identify early pathophysiological features of Alzheimer's disease. Alzheimers Dement 2016;2:82‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buschke H, Kuslansky G, Katz M, et al. Screening for dementia with the Memory Impairment Screen. Neurology 1999;52:231‐238. [DOI] [PubMed] [Google Scholar]
- 15. Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β‐amyloid (1‐42) in human cerebrospinal fluid. Alzheimers Dement 2016;12:517‐526. [DOI] [PubMed] [Google Scholar]
- 16. Salvadó G, Molinuevo JL, Brugulat‐Serrat A, et al. Centiloid cut‐off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther 2019;11:27. 10.1186/s13195-019-0478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 2015;11:1‐15.e4. 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayes AF. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression‐Based Approach. 2nd ed. New York, NY: Guilford Press; 2018. [Google Scholar]
- 19. Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmqvist S, Insel PS, Stomrud E, et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol Med 2019;11:e11170. 10.15252/emmm.201911170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bos I, Vos S, Verhey F, Scheltens P, et al. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer's disease spectrum. Alzheimers Dement 2019;15:644‐654. [DOI] [PubMed] [Google Scholar]
- 22. Fisher DW, Bennett DA, Dong H. Sexual dimorphism in predisposition to Alzheimer's disease. Neurobiol Aging 2018;70:308‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buckley RF, Mormino EC, Amariglio RE, et al. Sex, amyloid, and APOE ε4 and risk of cognitive decline in preclinical Alzheimer's disease: Findings from three well‐characterized cohorts. Alzheimers Dement 2018;14:1193‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE‐related risk of developing Alzheimer disease. Ann Neurol 2014;75:563‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiner MW, Veitch DP, Aisen PS, et al. 2014 Update of the Alzheimer's Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement 2015;11:e1‐120. 10.1016/j.jalz.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jack CR, Wiste HJ, Weigand SD, et al. Age, sex, and APOE ϵ4 effects on memory, brain structure, and β‐Amyloid across the adult life Span. JAMA Neurol 2015;72:511‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sampedro F, Vilaplana E, de Leon MJ, et al. APOE‐by‐sex interactions on brain structure and metabolism in healthy elderly controls. Oncotarget 2015;6:26663‐26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holland D, Desikan RS, Dale AM, McEvoy LK. Higher rates of decline for women and apolipoprotein e ε4 carriers. Am J Neuroradiol 2013;34:2287‐2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamelin L, Lagarde J, Dorothée G, et al. Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain 2018;141:1855‐1870. [DOI] [PubMed] [Google Scholar]
- 30. Parhizkar S, Arzberger T, Brendel M, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque‐associated ApoE. Nat Neurosci 2019;22:191‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer's disease. Mol Neurodegener 2018;13:66. 10.1186/s13024-018-0298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang JT, Wang ZJ, Cai HY, et al. Sex differences in neuropathology and cognitive behavior in APP/PS1/tau Triple‐transgenic mouse model of Alzheimer's disease. Neurosci Bull 2018;34:736‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Falcon C, Grau‐Rivera O, Suárez‐Calvet M, et al. Sex differences of longitudinal brain changes in cognitively unimpaired adults. J Alzheimers Dis 2020;1‐9. 10.3233/JAD-160256. [DOI] [PubMed] [Google Scholar]
- 34. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease. Neurology 2018;90:695‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iram T, Trudler D, Kain D, et al. Astrocytes from old Alzheimer's disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol Dis 2016;96:84‐94. [DOI] [PubMed] [Google Scholar]
- 36. Kang MS, Aliaga AA, Shin M, et al. Amyloid‐beta modulates the association between neurofilament light chain and brain atrophy in Alzheimer's disease. Mol Psychiatry 2020. 10.1038/s41380-020-0818-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chai X, Dage JL, Citron M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis 2012;48:356‐366. [DOI] [PubMed] [Google Scholar]
- 38. Karch CM, Jeng AT, Goate AM. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J Biol Chem 2012;287:42751‐42762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 2013;14:389‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018;97:1284‐1298.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iaccarino L, Tammewar G, Ayakta N, et al. Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer's disease. NeuroImage Clin 2018;17:452‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 2016;139:1551‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suárez‐Calvet M, Caballero MÁA, Kleinberger G, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 2016;8:34‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Suárez‐Calvet M, Morenas‐Rodríguez E, Kleinberger G, et al. Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related‐neurodegeneration but not with amyloid‐β pathology. Mol Neurodegener 2019;14:1‐14. 10.1186/s13024-018-0301-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rauchmann BS, Schneider‐Axmann T, Alexopoulos P, Perneczky R. CSF soluble TREM2 as a measure of immune response along the Alzheimer's disease continuum. Neurobiol Aging 2019;74:182‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suárez‐Calvet M, Kleinberger G, Araque Caballero MÁ, et al. sTREM 2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early‐stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 2016;8:466‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heslegrave A, Heywood W, Paterson R, et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener 2016;11:3. 10.1186/s13024-016-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement 2018;14:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaw LM, Waligorska T, Fields L, et al. Derivation of cutoffs for the Elecsys ® amyloid β (1‐42) assay in Alzheimer's disease. Alzheimers Dement 2018;10:698‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gispert JD, Monté GC, Suárez‐Calvet M, et al. The APOE ε4 genotype modulates CSF YKL‐40 levels and their structural brain correlates in the continuum of Alzheimer's disease but not those of sTREM2. Alzheimers Dement 2017;6:50‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alcolea D, Martínez‐Lage P, Sánchez‐Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology 2015;85:626‐633. [DOI] [PubMed] [Google Scholar]
- 53. Alcolea D, Vilaplana E, Pegueroles J, et al. Relationship between cortical thickness and cerebrospinal fluid YKL‐40 in predementia stages of Alzheimer's disease. Neurobiol Aging 2015;36:2018‐2023. [DOI] [PubMed] [Google Scholar]
- 54. Kern S, Syrjanen JA, Blennow K, et al. Association of cerebrospinal fluid neurofilament light protein with risk of mild cognitive impairment among individuals without cognitive impairment. JAMA Neurol 2019;76:1413‐1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mattsson‐Carlgren N, Leuzy A, Janelidze S, et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology 2020;94:e2233‐e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sugarman MA, Zetterberg H, Blennow K, et al. A longitudinal examination of plasma neurofilament light and total tau for the clinical detection and monitoring of Alzheimer's disease. Neurobiol Aging 2020;94:60‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reimand J, Collij L, Scheltens P, Bouwman F, Ossenkoppele R. Association of amyloid‐β CSF/PET discordance and tau load five years later. Neurology 2020;95:e2648‐e2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cacciaglia R, Molinuevo JL, Salvadó G, et al. Impact of the APOE gene on amyloid deposition in participants with abnormal soluble amyloid levels. AAIC, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.