

Abstract
Objective
To evaluate the efficacy and safety of lasmiditan in Japanese adults with migraine.
Background
Global clinical studies have demonstrated the efficacy and safety of lasmiditan in the acute treatment of migraine.
Methods
This was a multicenter, randomized, double‐blind, placebo‐controlled, phase 2 study in Japan (NCT03962738), which enrolled adults with migraine with or without aura. Participants were randomized 7:3:7:6 to placebo, lasmiditan 50 mg, 100 mg, or 200 mg to be self‐administered orally within 4 h of onset of a single moderate‐to‐severe migraine attack. Participants recorded their response to treatment prior to dosing and for 48 h postdose. The primary endpoint was headache pain freedom at 2 h postdose.
Results
Participants (N = 846) were randomized and treated (N = 691, safety; N = 682, modified intent‐to‐treat). At 2 h postdose, a significantly higher proportion of participants were headache pain‐free in the lasmiditan 200 mg (40.8%, 73/179; odds ratio 3.46 [95% confidence interval 2.17 to 5.54]; p < 0.001; primary objective) and 100 mg groups (32.4%, 67/207; odds ratio 2.41 [1.51 to 3.83]; p < 0.001) compared with the placebo group (16.6%, 35/211), whereas the lasmiditan 50 mg group had a numerically higher proportion of participants headache pain‐free (23.5%, 20/85; odds ratio 1.55 [0.83 to 2.87]; p = 0.167) compared with placebo. A statistically significant linear dose–response relationship for pain freedom was achieved at 2 h by a Cochran–Armitage trend test (p < 0.001). Lasmiditan treatment was also associated with headache pain relief, most bothersome symptom freedom, and improvement on disability and Patient Global Impression of Change outcomes. The majority of treatment‐emergent adverse events were mild and of short duration, the most common of which were dizziness (39.4%; 188/477), somnolence (19.3%; 92/477), and malaise (10.5%; 50/477) in all lasmiditan groups, with no serious adverse events reported.
Conclusions
Lasmiditan was well tolerated and effective for the acute treatment of Japanese patients with migraine, consistent with global phase 3 studies.
Keywords: 5‐HT1F‐receptor, acute treatment, headache, lasmiditan, migraine, phase 2
Abbreviations
- 5‐HT
serotonin
- AE
adverse event
- BMI
body mass index
- CI
confidence interval
- e‐diary
electronic diary
- h
hours
- IHS
International Headache Society
- ITT
intent‐to‐treat
- MBS
most bothersome symptom
- MedDRA
Medical Dictionary for Regulatory Activities
- MIDAS
Migraine Disability Assessment Score
- mITT
modified intent‐to‐treat
- N
number of participants in analysis population
- n
number of participants in indicated category
- Nr
number of subjects for responder analysis with severity >0 at baseline and nonmissing response at each time point
- Nt
number of participants in the analysis population at a given time point
- PGIC
Patient Global Impression of Change
- SD
standard deviation
- TEAE
treatment‐emergent adverse event
INTRODUCTION
Migraine is a disabling neurologic disease with an estimated global prevalence of nearly 12%. 1 Neuropeptide‐mediated pain pathways within the trigeminovascular system play key roles in the pathophysiology of migraine. 2 Lasmiditan, the first of a new class of antimigraine therapeutics known as ditans, is a centrally penetrant serotonin (5‐HT) receptor agonist that decreases neuropeptide release in the trigeminal nerve. 3 , 4 , 5 Lasmiditan has a >440‐fold selectivity for the 5‐HT1F receptor relative to the 5‐HT1B receptor, the activation of which is vasoconstrictive. 3 , 5 Lasmiditan did not cause vasoconstriction in the human arteries tested to date, including the proximal coronary, distal coronary, intermammary, and middle meningeal arteries. 6
Lasmiditan was approved in October 2019 in the United States for the acute treatment of migraine with or without aura in adults. 7 Two randomized, double‐blind, placebo‐controlled phase 3 studies, SAMURAI and SPARTAN, demonstrated the efficacy of lasmiditan in treating a single migraine attack. In these studies, compared with the placebo, 50, 100, and 200 mg lasmiditan showed statistically significant superiority on pain freedom, freedom from the most bothersome symptom (MBS), and pain relief, with a manageable safety profile. 8 , 9 Lasmiditan was well tolerated by patients with cardiovascular risk factors in global phase 3 studies, with no significant cardiovascular adverse events (AEs) reported in this subgroup of patients. 8 , 9
Consistent with the United States and Europe, available studies indicate a high prevalence of migraine and significant migraine burden in East Asia and suggest that there are unmet needs for improvements in the treatment of migraine in Asian populations. 10 Lasmiditan is currently being developed for patients with migraine in Japan. The objective of the current study, MONONOFU, was to examine the efficacy and safety of oral lasmiditan (50, 100, and 200 mg) versus placebo for the acute treatment of migraine in Japanese patients. We hypothesized that lasmiditan would show efficacy and safety in Japanese patients and that the outcomes in Japanese patients would be consistent with those of the global populations in prior clinical studies.
METHODS
Study design
This was a prospective, multicenter, randomized, double‐blind, placebo‐controlled phase 2 study (NCT03962738) conducted across 34 sites in Japan. The primary objective was to evaluate the efficacy of lasmiditan 200 mg on migraine headache pain freedom compared with placebo in Japanese adult patients suffering from migraine with or without aura. The key secondary objectives were to evaluate the dose response of lasmiditan 50, 100, and 200 mg on migraine headache pain freedom compared with placebo and the efficacy of lasmiditan 100 mg on migraine headache pain relief compared with placebo. The study design included a screening visit, randomization visit, treatment period of ≤8 weeks, and an end of study visit between 3 and 28 days after treating the migraine attack (Figure S1).
Participants
Eligible participants were aged ≥18 years who had a history of migraine with or without aura for ≥1 year (International Headache Society [IHS] diagnostic criteria 1.1 and/or 1.2.1 11 ); onset at <50 years; three to eight migraine attacks per month (<15 headache days/month for the past 3 months); and disabling migraine defined as a Migraine Disability Assessment (MIDAS 12 , 13 ) score ≥ 11. Key exclusion criteria included participants with known lasmiditan sensitivity; history of chronic migraine or other chronic headache disorders with ≥15 headache days/month within the past 12 months; hemorrhagic stroke, epilepsy, or any other condition placing the participant at increased risk of seizures, recurrent dizziness, and/or vertigo; diabetes mellitus with complications; orthostatic hypotension with syncope; significant renal or hepatic impairment; and participants who, in the investigator’s judgment, were a significant suicide risk.
The study was conducted in accordance with the Declaration of Helsinki, the relevant laws and regulations in Japan, and the Guidance on Ethnic Factors in the Acceptability of Foreign Clinical Data (ICH‐E5). 14 The protocol was reviewed by a central ethical review board and approved by the appropriate institutional review board for each site. All participants provided written, informed consent.
Treatments and procedures
Participants were randomly allocated 7:3:7:6 to receive oral placebo or lasmiditan 50 mg, 100 mg, or 200 mg; this ratio was chosen as it provided the highest statistical power for all primary and key secondary endpoints through statistical simulation. The sponsor randomized participants via a central randomization process using a computer‐generated random sequence and an interactive web‐response system. Participants and investigators/sponsor staff involved in the treatment or clinical evaluation of participants were masked to treatment allocation. Randomization was stratified for current use of concomitant preventive migraine medication(s) (yes/no). Participants were asked to treat a single migraine attack of moderate‐to‐severe intensity with the study drug within 4 h of headache pain onset on an outpatient basis, provided that the migraine was not improving and no other acute treatment for migraine had been taken within the previous 24 h. Rescue medication (nonsteroidal anti‐inflammatory drugs, acetaminophen, and/or antiemetics) for persistent or recurrent migraine was allowed after completion of 2 h postdose assessments. Triptans, ergots, opioids, and barbiturates were not permitted within 24 h postdose.
Efficacy data were collected in an electronic diary (e‐diary), which the participants completed. Participants recorded the date and time of migraine onset and the time at which the study drug was taken. Migraine severity was recorded in the e‐diary using a 4‐point IHS headache severity rating scale (0 = no pain; 1 = mild pain; 2 = moderate pain; and 3 = severe pain), and the definitions of pain‐free (defined as moderate or severe headache pain becoming none) and pain relief (defined as moderate or severe headache pain becoming mild or none) were based on this rating scale. 15 Migraine severity was rated both prior to taking the study drug (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 24, and 48 h postdose. At these time points, participants also recorded the presence or absence of accompanying symptoms (yes/no) of photophobia, phonophobia, nausea, and vomiting; time to meaningful relief and to becoming headache pain‐free; disability level; and Patient Global Impression of Change (PGIC). 16 From the migraine‐associated symptoms of photophobia, phonophobia, and nausea, participants selected the symptom that they considered their MBS at baseline. At each time point, the e‐diary asked “Do you feel anything unusual since you took the study medication that you have not felt with a migraine before?” For affirmative responses, the e‐diary instructed the patient to record the symptom in a paper journal. Patients were also instructed to record any AEs, concomitant medication use, rescue and recurrence medication use, and menstrual cycle status in a paper diary. Patients were instructed to refrain from driving for 8 h after dosing and to complete a driving questionnaire on motor vehicle accidents and moving violations/citations at baseline and at end of study/early discontinuation.
Participants underwent physical examination and had an electrocardiogram, vital signs, and clinical laboratory tests conducted at screening and follow‐up and additionally at the discretion of the investigator. Safety was assessed in the safety population as treatment‐emergent adverse events (TEAEs), defined as onset or worsening of an AE within 48 h postdose. AEs were classified according to the Medical Dictionary for Drug Regulatory Activities (version 23) and assessed for seriousness. Investigators determined whether a TEAE was related to study treatment.
Endpoints
The primary endpoint was the proportion of participants in each group who were pain‐free at 2 h postdose. The key secondary endpoint was the proportion of patients with pain relief in each group at 2 h postdose. Additional secondary endpoints included pain‐free or pain relief at all time points; MBS free at 2 h postdose (MBS identified by the participant at baseline); sustained pain freedom (defined as headache pain‐free at both 2 h and 24 or 48 h, without rescue/recurrence medications); freedom from symptoms associated with migraine; total migraine freedom (defined as migraine pain‐free and not experiencing any other migraine‐associated symptoms) at 2 h; migraine‐related disability level at 2 h (based on response to the question “How much is your migraine interfering with your normal activities?”, rated on a 4‐point scale from “0 = not at all” to “3 = completely, needs bed rest,” where those self‐rating as “0” were considered responders); and PGIC at 2 h (based on response to the question “How do you feel after taking study medication?”, rated on a 7‐point scale from “very much better” to “very much worse” where participants in the top two categories were considered responders). 16
Statistical analyses
Power calculations centered around multiplicity‐adjusted endpoints, which included the placebo versus lasmiditan 200 mg comparison for pain‐free at 2 h in the primary analysis as well as two key secondary endpoints, the linear dose–response trend test for pain‐free at 2 h (Cochran–Armitage trend test of lasmiditan 0 mg [placebo], 50, 100, 200 mg) and the placebo versus lasmiditan 100 mg comparison for pain relief at 2 h. Sample size was based on the results of the global phase 3 study, SPARTAN, 8 which reported that 21.3%, 28.6%, 31.4%, and 38.8% of participants were headache pain‐free at 2 h postdose and 47.7%, 59.0%, 64.8%, and 65.0% of participants had headache pain relief at 2 h postdose in the placebo, lasmiditan 50 mg, lasmiditan 100 mg, and lasmiditan 200 mg treatment groups, respectively. Assuming 29% of participants would be untreated during the 8‐week study, 880 randomized patients (i.e., 624 patients in the efficacy analysis population) were needed to provide 85.6% power for achieving statistical significance for the multiplicity‐adjusted primary and two key secondary analyses, simultaneously, with a two‐sided alpha of 0.05. Sample size and optimal allocation ratio were calculated through statistical simulations using Monte Carlo sampling of random numbers from binomial distributions. Enrollment was ended when the target sample size (i.e., 624) was expected to be met, based on blinded monitoring.
The safety population included all randomized participants who used a dose of study drug, regardless of whether they completed any assessments. The intent‐to‐treat (ITT) population included all randomized participants who used a dose of study drug and provided postdose headache severity or symptom assessment data. The modified ITT (mITT) population included all participants in the ITT population who treated a migraine attack within 4 h of onset. Primary efficacy and key secondary analyses were conducted on the mITT population. Additional efficacy analyses were conducted on the ITT population, as indicated. Descriptive statistics were used for demographic and safety data, with continuous measures summarized by sample size, mean plus standard deviation (SD), and median plus interquartile range and categorical variables summarized by sample size, frequency counts, and percentages. The estimated odds ratio of achieving a response, 95% confidence intervals (CIs), and p values for comparisons between the proportions of participants across treatment groups, including the analyses for the primary and key secondary endpoints, were derived using Wald’s test by logistic regression (with treatment [placebo, lasmiditan 50 mg, lasmiditan 100 mg, and lasmiditan 200 mg] and baseline usage of preventive medications to reduce the frequency of migraine [Yes/No] as factors). The dose response of lasmiditan 0 mg (placebo), 50, 100, and 200 mg for “pain‐free at 2 hours” was based on the Cochran–Armitage trend test.
The primary and two key secondary analyses were conducted sequentially by a gatekeeping method to prevent inflation of the overall type 1 error. Specifically, the analyses occurred in the following order: (1) a primary analysis comparing the proportion of participants headache pain‐free at 2 h postdose in the lasmiditan 200 mg versus the placebo groups, (2) a key secondary analysis examining the proportion of participants who were headache pain‐free at 2 h in each treatment group, and (3) a key secondary analysis comparing the proportion of participants with pain relief at 2 h postdose in the lasmiditan 100 mg versus the placebo groups. Tests of significance were conducted at the two‐sided significance level of 5%. Multiplicity adjustments were not made for other analyses. For analyses without multiplicity adjustment, nominal p values are provided for reference.
A participant with missing data at a particular time point was considered a “nonresponder” for pain‐free, pain relief, and MBS analyses. If a participant took a rescue/recurrent medication post‐baseline, then all evaluations thereafter were considered “nonresponder,” even if the evaluation was missing. For other analyses, participants who failed to record information at an analysis time point had that value considered missing (no imputation). Participants with disability level >0 at baseline and nonmissing response at 2 h postdose were included in the responder analysis. Statistical analyses were performed using SAS version 9.4 (SAS Institute).
RESULTS
Participant disposition
The study was conducted between May 30, 2019 and June 8, 2020. A total of 863 participants were screened, and 846 (98.0%) participants were randomized (Figure 1). A total of 691 (81.7%) participants were treated with study intervention (safety population), including 687 (81.2%) who provided postdose headache severity and/or symptom assessment data (ITT population) and 682 (80.6%) who provided postdose assessment data and treated their migraine within 4 h (mITT population). Across all treatment groups, 836 (98.8%) of the randomized participants completed the study. Of those participants who received a dose of the study drug, 691 (100%) completed the study; in addition, 145 participants (17.1% of those randomized) who did not treat with the study drug completed the study. Ten participants (1.2%) discontinued the study after randomization and prior to taking a dose of the study drug, most frequently due to protocol deviations (n = 5; Figure 1).
FIGURE 1.
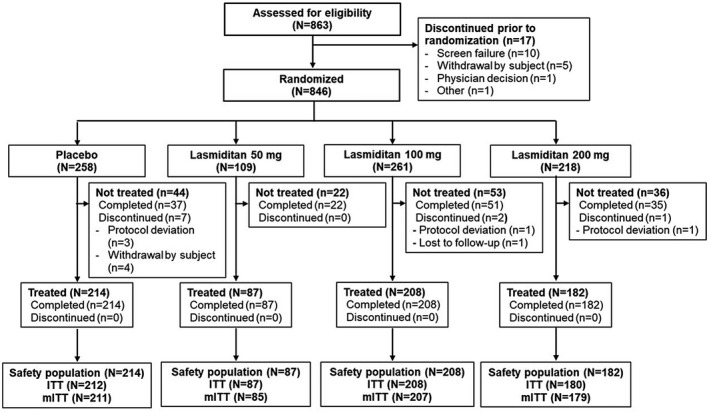
Participant disposition. Safety population included all subjects who dosed with study drugs. The ITT population included all participants who dosed with the study drug and provided assessment data after administration, whereas the mITT population included ITT participants who treated their migraine attacks within 4 h of pain onset. ITT, intent‐to‐treat; mITT, modified intent‐to‐treat; N, number of participants in indicated category or population; n, number of participants in subgroup of category or population
Demographic and baseline characteristics
Demographic and baseline clinical characteristics of participants were similar across treatment groups (Table 1). In the safety population, the majority of participants were female (83.1%), with a mean age (SD) of 45.2 (9.7) years, a mean weight (SD) of 58.3 (11.5) kg, and a mean body mass index (BMI) (SD) of 22.6 (3.8) kg/m2. Participants had a history of migraine for a mean (SD) 24.2 (11.9) years, 14.8% with and 85.2% without aura. The mean (SD) MIDAS total score was 22.3 (11.4). Preventive medication for migraine was used by 37.5% of the participants at randomization, and the majority had experience in using triptans (95.7%). Overall, 45.7% of the participants had ≥2 cardiovascular risk factors, with no major differences among treatment groups. Common cardiovascular risk factors included having a family history of cardiovascular disease (43.4%), being male or postmenopausal female (40.8%), or having obesity (21.7%) or hypertension (17.8%) (Table S1). Overall, a medical history of cardiovascular diseases was noted in 8.0% of participants in the lasmiditan groups compared with 10.7% in the placebo group, the majority of whom had a history of hypertension (lasmiditan, 4.8%; placebo, 7.9%) or arrhythmias (lasmiditan, 2.1%; placebo, 1.4%).
TABLE 1.
Demographic and baseline clinical characteristics
| Characteristic a | Placebo | Lasmiditan | ||
|---|---|---|---|---|
| 50 mg | 100 mg | 200 mg | ||
| Safety population | N = 214 | N = 87 | N = 208 | N = 182 |
| Demographic characteristics | ||||
| Age, years, mean (SD) | 45.2 (9.0) | 44.9 (10.2) | 45.7 (9.7) | 44.7 (10.4) |
| Female, n (%) | 178 (83.2) | 75 (86.2) | 176 (84.6) | 145 (79.7) |
| Body weight (kg), mean (SD) | 58.1 (12.2) | 57.8 (10.8) | 58.4 (11.7) | 58.8 (10.9) |
| BMI (kg/m2), mean (SD) | 22.6 (4.1) | 22.4 (3.7) | 22.6 (3.7) | 22.7 (3.4) |
| Clinical characteristics | ||||
| Duration of migraine history, years, mean (SD) | 24.4 (11.5) | 23.8 (12.4) | 24.7 (12.0) | 23.7 (12.2) |
| MIDAS total score b , mean (SD) | 22.3 (11.0) | 25.0 (14.1) | 22.5 (11.6) | 20.8 (9.8) |
| Migraine attacks/month in past 3 months c , mean (SD) | 5.7 (1.6) | 5.6 (1.6) | 5.6 (1.6) | 5.6 (1.6) |
| Days with headache pain in past 3 months c , mean (SD) | 21.8 (7.7) | 21.4 (7.6) | 21.2 (7.4) | 21.8 (7.7) |
| History of migraine with and without aura, n (%) | ||||
| With aura | 34 (15.9) | 11 (12.6) | 33 (15.9) | 24 (13.2) |
| Without aura | 180 (84.1) | 76 (87.4) | 175 (84.1) | 158 (86.8) |
| Use of preventive migraine medication, n (%) | 83 (38.8) | 34 (39.1) | 75 (36.1) | 67 (36.8) |
| Prior triptan use (ever), n (%) | 208 (97.2) | 84 (96.6) | 198 (95.2) | 171 (94.0) |
| Triptan use within 3 months of informed consent, n (%) | 196 (91.6) | 82 (94.3) | 186 (89.4) | 162 (89.0) |
| Family history of migraine | 133 (62.1) | 52 (59.8) | 134 (64.4) | 111 (61.0) |
| Presence of cardiovascular risk factors d , n (%) | ||||
| ≥2 risk factors at baseline | 95 (44.4) | 38 (43.7) | 101 (48.6) | 82 (45.1) |
| mITT population | N = 211 | N = 85 | N = 207 | N = 179 |
| Characteristics of treated migraine attacks | ||||
| Baseline migraine severity, n (%) | ||||
| Severe | 15 (7.1) | 10 (11.8) | 16 (7.7) | 10 (5.6) |
| Moderate | 196 (92.9) | 75 (88.2) | 191 (92.3) | 169 (94.4) |
| Baseline symptoms, n (%) | ||||
| Nausea | 66 (31.3) | 26 (30.6) | 55 (26.6) | 45 (25.1) |
| Phonophobia | 51 (24.2) | 21 (24.7) | 60 (29.0) | 55 (30.7) |
| Photophobia | 117 (55.5) | 34 (40.0) | 103 (49.8) | 81 (45.3) |
| None | 54 (25.6) | 26 (30.6) | 57 (27.5) | 58 (32.4) |
| Baseline MBS, n (%) e | ||||
| Nausea | 48 (22.7) | 24 (28.2) | 36 (17.4) | 37 (20.7) |
| Phonophobia | 18 (8.5) | 10 (11.8) | 34 (16.4) | 25 (14.0) |
| Photophobia | 91 (43.1) | 25 (29.4) | 80 (39.6) | 59 (33.0) |
| Time to dosing from migraine attack (h), median (interquartile range) | 1.2 (0.6, 2.3) | 1.5 (0.6, 2.4) | 1.6 (0.8, 2.5) | 1.3 (0.5, 2.2) |
| Dosed between 4 a.m. and 8 a.m., n (%) | 29 (13.7) | 9 (10.6) | 29 (14.0) | 20 (11.2) |
| Dosed during menstrual period, n (%) f | ||||
| Yes | 19 (10.9) | 5 (6.8) | 27 (15.4) | 27 (18.9) |
Abbreviations: BMI, body mass index; h, hours; MBS, most bothersome symptom; MIDAS, Migraine Disability Assessment Score; mITT, modified intent‐to‐treat; N, number of participants in population; n, number of participants meeting criteria; Nt, number of participants in the analysis population at a given time point; SD, standard deviation.
Demographic characteristics, clinical characteristics, and medical history are shown for the safety population; characteristics of treated migraine attacks are shown for the mITT population.
MIDAS total score was calculated as the sum of the answers to the five questions on the MIDAS questionnaire.
Duration of migraine history and frequency of migraine attacks in the last 3 months are measured from the date of Visit 1.
Cardiovascular risk factors were defined by the Japanese Council on cerebro‐cardiovascular disease, comprehensive risk management chart for cerebro‐cardiovascular disease 21 and are shown in Table S1.
Proportion baseline MBS was calculated based on Nt as follows: placebo: Nt = 157; lasmiditan 50 mg: Nt = 59; lasmiditan 100 mg: Nt = 150; lasmiditan 200 mg: Nt = 121.
“Dosed during menstrual period” included participants in the mITT population who dosed during a 5‐day window beginning 2 days prior to the onset of menses. The denominator has been adjusted due to sex‐specific event for females.
The characteristics of the treated migraine attacks were similar across treatment groups (Table 1). Overall, participants reported moderate (92.5%) or severe (7.5%) migraine, with 71.4% reporting the presence of nausea, phonophobia, and/or photophobia at baseline. Photophobia was the most commonly reported migraine‐associated symptom in all groups (49.1%) and the most commonly chosen MBS at baseline (37.4%). Headaches were treated within a median (interquartile range) 1.4 (0.6, 2.4) h of onset, with 12.8% of the participants treating migraine attacks between 4 a.m. and 8 a.m., which could be considered early morning migraine. Attacks were treated during menstruation (which included participants who dosed during a 5‐day window beginning 2 days prior to the onset of menses) in 13.8% of female participants.
Efficacy
As the primary endpoint, the lasmiditan 200 mg group had a statistically significantly higher proportion of participants (40.8%) who were headache pain‐free at 2 h compared with placebo (16.6%; Table 2). Compared with placebo at 2 h, the lasmiditan 100 mg group also had a significantly higher proportion of participants (32.4%) headache pain‐free, and the lasmiditan 50 mg group had a numerically higher proportion of participants (23.5%) headache pain‐free. There was a statistically significant linear dose–response relationship (placebo < lasmiditan 50 mg < 100 mg < 200 mg) regarding headache pain freedom at 2 h (p < 0.001). Compared with the placebo group, higher proportions of pain freedom were achieved starting at 30 and 60 min in the lasmiditan 200 mg and 100 mg groups, respectively (Figure 2).
TABLE 2.
Summary of efficacy endpoints by treatment group
| Endpoint a | Placebo | Lasmiditan | ||
|---|---|---|---|---|
| 50 mg | 100 mg | 200 mg | ||
| mITT population | N = 211 | N = 85 | N = 207 | N = 179 |
| Pain‐free at 2 h, n (%) | 35 (16.6) | 20 (23.5) | 67 (32.4) | 73 (40.8) |
| Odds ratio (95% CI) | 1.55 (0.83, 2.87) | 2.41 (1.51, 3.83) | 3.46 (2.17, 5.54) | |
| p value | 0.167 | <0.001 | <0.001 | |
| Trend test b | p < 0.001 | |||
| Pain relief at 2 h, n (%) | 116 (55.0) | 58 (68.2) | 166 (80.2) | 140 (78.2) |
| Odds ratio (95% CI) | 1.76 (1.03, 2.99) | 3.34 (2.16, 5.17) | 2.95 (1.89, 4.62) | |
| p value | 0.037 | <0.001 | <0.001 | |
| MBS free at 2 h, n (%) c | 73 (46.5) | 33 (55.9) | 87 (58.0) | 73 (60.3) |
| Odds ratio (95% CI) | 1.49 (0.81, 2.72) | 1.59 (1.01, 2.50) | 1.75 (1.08, 2.83) | |
| p value | 0.179 | 0.044 | 0.023 | |
| ITT population | N = 212 | N = 87 | N = 208 | N = 180 |
| Sustained pain‐free d at 24 h, n (%) | 22 (10.4) | 13 (14.9) | 42 (20.2) | 42 (23.3) |
| Odds ratio (95% CI) | 1.52 (0.73, 3.17) | 2.19 (1.26, 3.82) | 2.63 (1.50, 4.61) | |
| p value | 0.268 | 0.006 | <0.001 | |
| Sustained pain‐free d at 48 h, n (%) | 26 (12.3) | 13 (14.9) | 41 (19.7) | 38 (21.1) |
| Odds ratio (95% CI) | 1.28 (0.61, 2.58) | 1.77 (1.04, 3.03) | 1.93 (1.12, 3.33) | |
| p value | 0.535 | 0.036 | 0.018 | |
| Total migraine freedom e at 2 h, n (%) | 21 (14.6) | 18 (20.7) | 58 (27.9) | 58 (32.2) |
| Odds ratio (95% CI) | 1.52 (0.80, 2.90) | 2.26 (1.39, 3.68) | 2.78 (1.70, 4.55) | |
| p value | 0.200 | 0.001 | <0.001 | |
| Disability level responder (no disability) f , n (%) | 38 (18.4) | 21 (24.7) | 51 (25.4) | 49 (28.7) |
| Odds ratio (95% CI) | 1.44 (0.79, 2.64) | 1.50 (0.94, 2.41) | 1.80 (1.11, 2.92) | |
| p value | 0.233 | 0.092 | 0.017 | |
| PGIC responder (much or very much better) f , n (%) | 50 (23.6) | 31 (35.6) | 93 (44.7) | 87 (48.3) |
| Odds ratio (95% CI) | 1.78 (1.04, 3.07) | 2.67 (1.75, 4.06) | 3.27 (2.11, 5.06) | |
| p value | 0.037 | <0.001 | <0.001 | |
Abbreviations: CI, confidence interval; ITT, intent‐to‐treat; MBS, most bothersome symptom; mITT, modified intent‐to‐treat; N, number of participants in population; n, number of participants meeting criteria; Nr, number of subjects for responder analysis with severity >0 at baseline and nonmissing response at each time point; Nt, number of participants in the analysis population at a given time point; PGIC, Patient Global Impression of Change.
Odds ratio, CIs, and p values for comparisons of lasmiditan treatment groups versus placebo were derived using logistic regression analysis with treatment and baseline usage of preventive medications to reduce the frequency of migraine frequency (Yes/No) as factors.
The dose response of placebo, lasmiditan 50, 100, and 200 mg (pain‐free at 2 h) was based on the Cochran–Armitage trend test.
Proportion MBS free was calculated based on Nt as follows: placebo: Nt = 157; lasmiditan 50 mg: Nt = 59; lasmiditan 100 mg: Nt = 150; lasmiditan 200 mg: Nt = 121.
Sustained pain freedom was defined as headache pain‐free at both 2 h and 24 or 48 h, without rescue or recurrence medications.
Total migraine freedom was defined as free from migraine pain and not experiencing any other migraine symptoms.
Disability responders are defined as having a score of 0 (no disability) at 2 h postdose. PGIC responders are defined as having a rating in the top two categories of “very much better” and “much better” at 2 h postdose. Responder analyses conducted on participants with severity >0 at baseline and nonmissing response at 2 h postdose. Percentage of response for disability is calculated by n/Nr * 100%. (placebo: Nr = 206; lasmiditan 50 mg: Nr = 85; lasmiditan 100 mg: Nr = 201; lasmiditan 200 mg: Nr = 171). Comparisons are of lasmiditan treatment groups versus placebo, performed using logistic regression analysis with treatment and baseline usage of preventive migraine medications (Yes/No) as factors.
FIGURE 2.
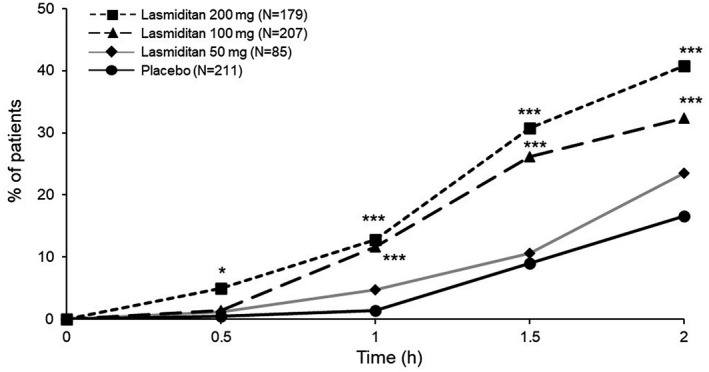
Headache pain‐free postdose. Modified intent‐to‐treat population. *p < 0.05, ***p < 0.001 versus placebo, Wald’s test by logistic regression. h, hours; N, number of participants in analysis population
Headache pain relief was a key secondary endpoint (Table 2). Compared with placebo, a statistically significantly higher proportion of participants reported headache pain relief at 2 h in each of the lasmiditan groups (placebo, 55.0% vs. lasmiditan 200 mg, 78.2%; lasmiditan 100 mg, 80.2%; lasmiditan 50 mg, 68.2%). Compared with placebo, higher rates of pain relief were achieved at 30 min, 1 h, and 2 h in the lasmiditan 200, 100, and 50 mg groups, respectively (Figure 3).
FIGURE 3.
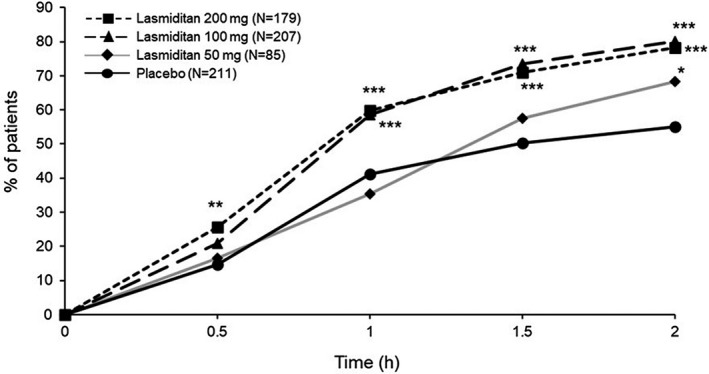
Headache pain relief postdose. Modified intent‐to‐treat population. *p < 0.05; **p < 0.01; ***p < 0.001 versus placebo, Wald’s test by logistic regression. h, hours; N, number of participants in analysis population
Of the other secondary endpoints, a significantly higher proportion of participants in the lasmiditan 100 mg (27.9%) and 200 mg (32.2%) groups reported total migraine freedom at 2 h compared with those in the placebo group (14.6%) and sustained headache pain freedom at both 24 and 48 h (Table 2). The proportions of participants MBS free at 2 h were greater at higher doses of lasmiditan than for placebo (Table 2; Figure S2). For pain‐free, pain relief, and MBS analyses, sensitivity analyses excluding the nonresponders due to missing data showed similar results to the original analyses (Table S2). The proportion of participants with individual migraine‐associated symptoms was numerically lower in the lasmiditan groups compared with the placebo group for phonophobia and photophobia (Table S3). At 2 h, the proportion of participants reporting no disability was statistically significantly higher in the lasmiditan 200 mg group than in the placebo group (Tables 2 and S4). The proportion of participants who were responders at 2 h on the PGIC assessment was statistically significantly higher in all lasmiditan groups than in the placebo group (Tables 2 and S4).
Safety
No serious AEs, deaths, or discontinuations due to AEs were reported (Table 3). TEAE incidence was higher in each lasmiditan group than in the placebo group and increased with increasing lasmiditan dose (placebo: 23.4%; lasmiditan 50 mg: 50.6%; 100 mg: 70.7%; 200 mg: 80.8%). Most TEAEs were mild in severity (placebo, 96.0%; lasmiditan 50 mg, 90.9%; 100 mg, 89.1%; 200 mg, 84.4%). Only one participant (0.2%) in the lasmiditan 200 mg group reported severe TEAEs of dizziness, somnolence, malaise, and headache. The most common TEAEs reported by lasmiditan‐treated participants were dizziness (39.4%), somnolence (19.3%), malaise (10.5%), asthenia (7.8%), hypoesthesia (7.8%), and nausea (6.3%) (Table 3). The median time to onset for these TEAEs was 0.5–1.0 h postdose, and the median duration was 1.1–3.6 h, with 75% of the participants reporting these events ending within 3.0–8.0 h. No cases of injury or accidents temporally associated with a neurologic TEAE were reported in any lasmiditan group. No patients reported motor vehicle accidents or moving violations/citations after taking the study drug. Cardiovascular TEAEs were relatively uncommon. Events that were identified in the lasmiditan groups as likely cardiovascular TEAEs included 17 (3.6%) TEAEs of palpitations (placebo, n = 3, 1.4%). All likely cardiovascular TEAEs were nonserious and recovered with no action, and all were of mild severity except one TEAE of palpitations of moderate severity in the lasmiditan 200 mg group.
TABLE 3.
Overview of AEs and most commonly reported treatment‐emergent adverse events
| AEs | Placebo (N = 214) | Lasmiditan | ||
|---|---|---|---|---|
| 50 mg (N = 87) | 100 mg (N = 208) | 200 mg (N = 182) | ||
| Overview of events, n (%) | ||||
| Deaths | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Serious AEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Total AEs | 101 (47.2) | 57 (65.5) | 168 (80.8) | 160 (87.9) |
| Discontinuations due to AE | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| TEAEs a | 50 (23.4) | 44 (50.6) | 147 (70.7) | 147 (80.8) |
| TEAEs related to study treatment | 31 (14.5) | 41 (47.1) | 134 (64.4) | 146 (80.2) |
| TEAEs occurring in ≥5% in any lasmiditan group (preferred term b ), n (%) c | ||||
| Dizziness | 7 (3.3) | 18 (20.7) | 79 (38.0) | 91 (50.0) |
| Somnolence | 11 (5.1) | 7 (8.0) | 44 (21.2) | 41 (22.5) |
| Malaise | 3 (1.4) | 6 (6.9) | 23 (11.1) | 21 (11.5) |
| Asthenia | 1 (0.5) | 5 (5.7) | 14 (6.7) | 18 (9.9) |
| Hypoesthesia | 0 (0) | 1 (1.1) | 12 (5.8) | 24 (13.2) |
| Nausea | 5 (2.3) | 3 (3.4) | 11 (5.3) | 16 (8.8) |
| Muscular weakness | 0 (0) | 1 (1.1) | 11 (5.3) | 7 (3.8) |
Abbreviations: AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities; N, number of participants in analysis population; n, number of participants reporting event; TEAE, treatment‐emergent adverse event.
TEAEs are any AEs with onset or worsening within 48 h after a dose of study drug.
MedDRA Version 23.0.
Proportions were calculated based on the safety population.
The majority of TEAEs were considered treatment‐related by the investigator in the lasmiditan groups, and the incidence in lasmiditan groups was higher than in the placebo group (placebo, 14.5%; lasmiditan 50 mg, 47.1%; 100 mg, 64.4%; 200 mg, 80.2%; Table S5). No clinically meaningful differences were observed between treatment groups in the mean change from baseline in any vital sign or laboratory test values (hematology, blood chemistry, and urinalysis parameters).
DISCUSSION
This is a report on the primary findings from the phase 2 MONONOFU study of the efficacy and safety of lasmiditan in Japanese adults experiencing migraine with or without aura. Notably, on the primary endpoint, this study demonstrated that a statistically significantly greater proportion of participants were pain‐free at 2 h after receiving a single dose of lasmiditan 200 mg or 100 mg compared with placebo, with a linear dose–response trend for lasmiditan efficacy. Similarly, on the key secondary endpoint, lasmiditan demonstrated a significantly greater proportion of participants with pain relief at 2 h after receiving a single dose of lasmiditan 200 mg, 100 mg, or 50 mg. Compared with placebo, lasmiditan 100 and 200 mg resulted in both an early onset of efficacy in terms of pain freedom and sustained pain freedom up to 48 h. In addition, lasmiditan showed benefits over placebo at 2 h in terms of the proportions of participants free from their MBS and in the patient‐reported outcomes of PGIC and disability. These results are similar to those from the prior global phase 3 studies, SAMURAI and SPARTAN, which showed that lasmiditan 50, 100, and 200 mg were significantly superior to placebo for pain freedom, pain relief, and MBS freedom, 8 , 9 , 17 , 18 collectively indicating that a single dose of lasmiditan is efficacious in the acute treatment of migraine.
Lasmiditan was well tolerated in Japanese patients, with no severe AEs or deaths reported in this study. The incidence of TEAEs was higher in all lasmiditan groups compared with placebo, with the proportion of participants reporting at least one TEAE increasing with higher lasmiditan doses, and the majority of TEAEs were deemed related to lasmiditan treatment. However, most TEAEs were mild in severity and of short duration, with durations similar to those observed in previous studies. 19 In addition, there were no reported events of accident or injury associated with lasmiditan. Although this study included participants with comorbid cardiovascular disease and/or cardiovascular risk factors (e.g., 45.7% with ≥2 cardiovascular risk factors), the incidence of cardiovascular TEAEs was low. Importantly, no major cardiovascular TEAEs consistent with vasoconstriction were reported. The types of common TEAEs in the current study (e.g., dizziness, somnolence, malaise) were similar to those reported in the global phase 3 studies, SPARTAN and SAMURAI, 8 , 9 broadly attributed to the actions of lasmiditan on the central nervous system. 8 , 9 However, TEAEs were more frequently reported in the current study (e.g., incidence of ≥1 TEAE for lasmiditan 200 mg was 80.8% in MONONOFU compared with 39.0% in SPARTAN and 42.7% in SAMURAI). Variation in the frequency of reported TEAEs among lasmiditan phase 2 and phase 3 studies has been previously observed, which was attributed to differences in AE data collection methods, informed consent documents, and other factors. 20
This study is the first to investigate lasmiditan in a Japanese population and enrolled a broad population of patients, including those with comorbid cardiovascular disease and/or cardiovascular risk factors, on preventive medications, and prior triptan experience. A unique feature of this study is that the majority of participants were relatively small (mean BMI of 22.6 kg/m2 as compared to the mean BMI of 30–31 kg/m2 of prior global phase 3 studies 8 , 9 ), and this study is the first to demonstrate the safety and efficacy of lasmiditan in a population of smaller and leaner patients.
However, there are limitations to this study. Notably, the safety and efficacy of a single dose of lasmiditan were assessed over a 48 h period, and long‐term data and data on multiple attacks are not available in this study population. In addition, postdose efficacy assessments were obtained starting at 30 min, and some efficacy outcomes may have had an earlier onset. For lasmiditan 50 mg, the sample size was not calculated to ensure certain statistical power compared with placebo; thus, interpretation of these results is limited. Furthermore, as this study only enrolled patients ≥18 years old, the findings may not be generalizable to pediatric patients; this is the subject of an ongoing study.
CONCLUSIONS
This study demonstrated that a single dose of lasmiditan was effective in improving or eliminating moderate‐to‐severe migraine pain and migraine‐associated symptoms in Japanese patients experiencing migraine with or without aura. Compared with controls, participants in this study who treated their migraine with lasmiditan reported less or no disability and a higher proportion improved in overall clinical status (as measured by PGIC). Lasmiditan was well tolerated in Japanese patients, with dizziness, somnolence, and malaise the most frequently reported TEAEs, the majority of which were mild in severity and of short duration. The findings of this study were consistent with those from global phase 3 studies and support the use of lasmiditan in treating migraine pain and migraine‐associated symptoms in Japanese patients. A plain language summary is available for this manuscript as Figure S3.
INSTITUTIONAL REVIEW BOARD APPROVAL
IRB approval was obtained at each of the 34 study sites.
CONFLICT OF INTEREST
Fumihiko Sakai reports consulting for Eli Lilly Japan K.K., Amgen, and Otsuka Pharmaceutical Co. Ltd. Takao Takeshima reports personal fees from Pfizer Japan Inc., Eisai Co. Ltd., Otsuka Pharmaceutical Co. Ltd., Kyowa Kirin Co. Ltd., Sawai Pharmaceutical Co. Ltd., Amgen Astellas BioPharma, Eli Lilly Japan K.K., Daiichi Sankyo Co. Ltd., AbbVie GK, Ono Pharmaceutical Co. Ltd., FUJIFILM Toyama Chemical Co. Ltd., Bayer Yakuhin Ltd., Nippon Boehringer Ingelheim Co. Ltd., Alexion Pharma Godo Kaisha, Sumitomo Dainippon Pharma Co. Ltd., Teijin Pharma Ltd., Novartis Pharma K.K., FP Pharmaceutical Corporation, Takeda Pharmaceutical Company Limited, Mitsubishi Tanabe Pharma Corporation, and Nippon Zoki Pharmaceutical Co. Ltd. Gosuke Homma, Yuka Tanji, Hideaki Katagiri, and Mika Komori are employees and minor shareholders of Eli Lilly Japan K.K.
AUTHOR CONTRIBUTIONS
Study concept and design: Fumihiko Sakai, Mika Komori, Hideaki Katagiri. Acquisition of data: Takao Takeshima, Yuka Tanji, Mika Komori. Analysis and interpretation of data: Fumihiko Sakai, Takao Takeshima, Gosuke Homma, Yuka Tanji, Hideaki Katagiri, Mika Komori. Drafting of the manuscript: Gosuke Homma, Yuka Tanji, Hideaki Katagiri, Mika Komori. Revising it for intellectual content: Fumihiko Sakai, Takao Takeshima, Gosuke Homma, Yuka Tanji, Hideaki Katagiri, Mika Komori. Final approval of the completed manuscript: Fumihiko Sakai, Takao Takeshima, Gosuke Homma, Yuka Tanji, Hideaki Katagiri, Mika Komori.
CLINICAL TRIALS REGISTRATION NUMBER
NCT03962738; U.S. National Library of Medicine: https://clinicaltrials.gov/ct2/show/NCT03962738.
Supporting information
Fig S1
{kind=link}
Fig S2
{kind=link}
Fig S3
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the patients, investigators, and site staff involved in this study. The authors would also like to thank Kraig Kinchen and John Krege (Eli Lilly and Company) for supporting clinical trial implementation and Aki Yoshikawa (Eli Lilly Japan K.K.) for project management during the production of this manuscript. Medical writing assistance (Kaye L. Stenvers, PhD) and editorial assistance (Cynthia Abbott and Antonia Baldo) were provided by Syneos Health and supported by Eli Lilly Japan K.K., Kobe, Japan.
REFERENCES
- 1. Woldeamanuel YW, Cowan RP. Migraine affects 1 in 10 people worldwide featuring recent rise: a systematic review and meta‐analysis of community‐based studies involving 6 million participants. J Neurol Sci. 2017;372:307‐315. [DOI] [PubMed] [Google Scholar]
- 2. Clemow DB, Johnson KW, Hochstetler HM, Ossipov MH, Hake AM, Blumenfeld AM. Lasmiditan mechanism of action—review of a selective 5‐HT1F agonist. J Headache Pain. 2020;21:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nelson DL, Phebus LA, Johnson KW, et al. Preclinical pharmacological profile of the selective 5‐HT1F receptor agonist lasmiditan. Cephalalgia. 2010;30:1159‐1169. [DOI] [PubMed] [Google Scholar]
- 4. Labastida‐Ramirez A, Rubio‐Beltran E, Garrelds IM, et al. Lasmiditan inhibits CGRP release in the mouse trigeminovascular system. 18th International Headache Congress; September 7‐10, 2017; Vancouver, Canada. [Google Scholar]
- 5. Rubio‐Beltran E, Labastida‐Ramirez A, Villalon CM, MaassenVanDenBrink A. Is selective 5‐HT1F receptor agonism an entity apart from that of the triptans in antimigraine therapy? Pharmacol Ther. 2018;186:88‐97. [DOI] [PubMed] [Google Scholar]
- 6. Rubio‐Beltran E, Labastida‐Ramirez A, Haanes KA, et al. Characterization of binding, functional activity, and contractile responses of the selective 5‐HT1F receptor agonist lasmiditan. Br J Pharmacol. 2019;176:4681‐4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lamb YN. Lasmiditan: first approval. Drugs. 2019;79:1989‐1996. [DOI] [PubMed] [Google Scholar]
- 8. Goadsby PJ, Wietecha LA, Dennehy EB, et al. Phase 3 randomized, placebo‐controlled, double‐blind study of lasmiditan for acute treatment of migraine. Brain. 2019;142:1894‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuca B, Silberstein SD, Wietecha L, et al. Lasmiditan is an effective acute treatment for migraine: a phase 3 randomized study. Neurology. 2018;91:e2222‐e2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takeshima T, Wan QI, Zhang Y, et al. Prevalence, burden, and clinical management of migraine in China, Japan, and South Korea: a comprehensive review of the literature. J Headache Pain. 2019;20:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Headache Classification Subcommittee of the International Headache Society . The International Classification of Headache Disorders: 2nd edition. Cephalalgia. 2004;24(suppl. 1):9‐160. [DOI] [PubMed] [Google Scholar]
- 12. Lipton RB, Stewart WF, Sawyer J, Edmeads JG. Clinical utility of an instrument assessing migraine disability: the Migraine Disability Assessment (MIDAS) questionnaire. Headache. 2001;41:854‐861. [PubMed] [Google Scholar]
- 13. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache‐related disability. Neurology. 2001;56:S20‐S28. [DOI] [PubMed] [Google Scholar]
- 14. European Medicines Agency . ICH‐E5: Guidance on Ethnic Factors in the Acceptability of Foreign Clinical Data; 1998. https://www.fda.gov/science‐research/guidance‐documents‐including‐information‐sheets‐and‐notices/ich‐guidance‐documents. Accessed October 18, 2020. [Google Scholar]
- 15. Tfelt‐Hansen P, Pascual J, Ramadan N, et al. Guidelines for controlled trials of drugs in migraine: third edition. A guide for investigators. Cephalalgia. 2012;32:6‐38. [DOI] [PubMed] [Google Scholar]
- 16. Hurst H, Bolton J. Assessing the clinical significance of change scores recorded on subjective outcome measures. J Manipulative Physiol Ther. 2004;27:26‐35. [DOI] [PubMed] [Google Scholar]
- 17. Ashina M, Vasudeva R, Jin L, et al. Onset of efficacy following oral treatment with lasmiditan for the acute treatment of migraine: integrated results from 2 randomized double‐blind placebo‐controlled phase 3 clinical studies. Headache. 2019;59:1788‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doty EG, Krege JH, Jin L, Raskin J, Halker Singh RB, Kalidas K. Sustained responses to lasmiditan: results from post‐hoc analyses of two phase 3 randomized clinical trials for acute treatment of migraine. Cephalalgia. 2019;39:1569‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krege JH, Rizzoli PB, Liffick E, et al. Safety findings from phase 3 lasmiditan studies for acute treatment of migraine: results from SAMURAI and SPARTAN. Cephalalgia. 2019;39:957‐966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kudrow D, Krege JH, Hundemer HP, et al. Issues impacting adverse event frequency and severity: differences between randomized phase 2 and phase 3 clinical trials for lasmiditan. Headache. 2020;60:576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. The Japanese Council on Cerebro‐Cardiovascular Disease . Comprehensive risk management chart for cerebro‐cardiovascular disease 2019. J‐STAGE. 2019;108:1024‐1074. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material