

Abstract
TRIM28 was recently identified as a Wilms' tumour (WT) predisposition gene, with germline pathogenic variants identified in around 1% of isolated and 8% of familial WT cases. TRIM28 variants are associated with epithelial WT, but the presence of other tumour components or anaplasia does not exclude the presence of a germline or somatic TRIM28 variant. In children with WT, TRIM28 acts as a classical tumour suppressor gene, with both alleles generally disrupted in the tumour. Therefore, loss of TRIM28 (KAP1/TIF1beta) protein expression in tumour tissue by immunohistochemistry is an effective strategy to identify patients carrying pathogenic TRIM28 variants. TRIM28 is a ubiquitously expressed corepressor that binds transcription factors in a context‐, species‐, and cell‐type‐specific manner to control the expression of genes and transposable elements during embryogenesis and cellular differentiation. In this review, we describe the inheritance patterns, histopathological and clinical features of TRIM28‐associated WT, as well as potential underlying mechanisms of tumourigenesis during embryonic kidney development. Recognizing germline TRIM28 variants in patients with WT can enable counselling, genetic testing, and potential early detection of WT in other children in the family. A further exploration of TRIM28‐associated WT will help to unravel the diverse and complex mechanisms underlying WT development. © 2021 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: TRIM28, KAP1, TIF1beta, Wilms' tumour, nephroblastoma, cancer predisposition, embryonic kidney development
Background
Wilms' tumour (WT) is the most common renal malignancy of childhood, with a median age at diagnosis of 3 years, the majority of patients being diagnosed before the age of 7 years. Morphologically, WTs present with a triphasic histology composed of stromal, epithelial, and blastemal cells in variable proportions, but often two, or even only one, of these components predominate [1].
WTs originate from a developmental arrest during nephrogenesis [1, 2, 3]. Manifestations of this developmental arrest include nephrogenic rests, which are embryonic remnants found in the surrounding kidney tissue of ~40% of WTs (~100% in bilateral cases) and are considered to be WT precursor lesions. Whereas intralobar rests are centrally located in the kidney and thought to arise in early nephrons, perilobar rests are located towards the periphery and thought to arise in a later stage of gestation [1]. Apart from nephrogenic rests, it was recently reported that WT precursor clones that genetically resemble the tumour can also exist within morphologically normal‐appearing kidney tissue, a phenomenon referred to as clonal nephrogenesis [4]. For malignant transformation of these precursor clones or for nephrogenic rests to develop into WT, additional events are necessary.
Currently, approximately 40 different genes have been identified as possible drivers of WT development, with the most commonly mutated and established drivers being WT1, WTX/AMER1, CTNNB1, SIX1, SIX2, DROSHA, DICER1, DCGR8, and TP53 [5, 6, 7]. However, given that a considerable proportion of WTs do not harbour mutations in any of these genes, the spectrum of driver mutations will likely be larger and also epigenetic mechanisms are thought to play an important role in WT development [2, 8].
A subset of WT patients has an underlying tumour predisposition syndrome. Whereas 1–2% of all WT cases are familial, most WT predisposition syndromes are caused by de novo (epi)mutations [9, 10]. The most well‐known examples include Beckwith–Wiedemann syndrome (BWS) and syndromes caused by germline WT1 variants or deletions [5, 11]. In recent years, novel WT predisposition genes (such as TRIM28, CTR9, and REST) have been identified, each in itself accounting for ≤1% of WT cases [12]. For many of these genes, the mechanisms by which they predispose to WT development are incompletely understood. Unravelling these mechanisms and the associated clinical and histopathological features will help to advance our understanding of WT pathogenesis. In this literature review, we will focus on one of the recently discovered WT predisposition genes, TRIM28. We describe the histopathological and clinical features of TRIM28‐associated WT, as well as potential underlying mechanisms.
TRIM28 variants in patients with WT
Pathogenic TRIM28 variants have currently been reported in 46 patients with WT (Table 1), including 27 cases where the variant was detected in lymphocyte DNA, eight cases where the variant was detected in DNA derived from resected normal kidney tissue (lymphocyte DNA not available), and 11 cases where the variant was shown to be only present in the tumour [12, 13, 14, 15, 16]. Nineteen familial cases were reported in nine families [12, 14, 15, 16]. TRIM28 variants were considered to be germline events in 30 patients, based on their confirmation in heterozygosity in lymphocyte DNA (N = 27) or in kidney tissue in the case of familial WT (N = 3). In five patients, TRIM28 variants were originally reported as germline variants [13, 15] but may represent clonal nephrogenesis [4], since lymphocyte DNA for confirmation of germline status was not available and no other relatives were (known to be) affected. With one exception, the reported variants are truncating or splice site variants located throughout all protein coding domains of the TRIM28 gene (Figure 1).
Table 1.
Reported Wilms' tumour patients with TRIM28 variants in blood, kidney, and/or tumour (N = 46).
| ID in original report [reference] | Mutation identified in: | Familial WT? | M/F | Age | Inheritance | Mutation | Histology | NR | LOH/IHC, other findings in tumour | FU | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0477_01 [12] | Blood | Familial | F | 24 | Mat | p.Gly310Asp | Epithelial predominant† | NA | NA | NA | |
| 0477_02 [12] | Blood | Familial | M | 84 | Mat | p.Gly310Asp | Epithelial† | NA | NA | NA | |
| 0477_03 [12] | Blood | Familial | F | 93 | Mat | p.Gly310Asp | NA | NA | NA | NA | |
| Blood & tumour | Familial | M | 8 | Mat | p.Glu583Argfs*93 | Monomorphic epithelial† | NA | LOH | 30 | ||
| Blood & tumour | Familial | F | 5 | Mat | p.Glu583Argfs*93 | Monomorphic epithelial | No | LOH | 29 | ||
| 0498_03 [12] | Blood | Familial | F | 6 | NA | p.Glu583Argfs*93 | Epithelial† | NA | NA | NA | |
| 0487_01 [12] | Blood | Familial | M | 15 | Mat | p.Thr144Hisfs*12 | Epithelial predominant † | NA | NA | 18 | |
| 0487_02 [12] | Blood | Familial | M | 18 | NA | p.Thr144Hisfs*12 | NA | NA | NA | NA | |
| 0506_01 [12] / 37 [14] | Blood & tumour | Familial | M | 39 | Mat | p.Thr176Profs*32 | Monomorphic epithelial† | No | CN‐LOH, TRIM28 IHC loss | 20 | |
| 0506_02 [12] / 39 [14] | Blood & tumour | Familial | F | 8 | Mat | p.Thr176Profs*32 | L | Monomorphic epithelial† | No | CN‐LOH, TRIM28 IHC loss | 20 |
| R | Monomorphic epithelial† | ||||||||||
| 7487_01 [12] | Blood | Isolated | F | 118 | Mat | p.Leu80Profs*11 | Epithelial predominant with diffuse anaplasia† | NA | NA | 3†† | |
| 1982 [12] | Blood | Isolated | M | 11 | DN | p.Leu653Cysfs*23 | L | Epithelial predominant† | NA | NA | 15 |
| R | Epithelial predominant† | ||||||||||
| 6530 [12] | Blood | Isolated | M | 15 | DN | p.Glu70Alafs*19 | Epithelial + blastemal† | NA | NA | 5 | |
| 1969 [12] | Blood | Isolated | M | 118 | DN | Splice, c.840–2A>G | Epithelial + blastemal† | NA | NA | 10 | |
| 7574 [12] | Blood | Isolated | M | 13 | DN | p.*836Trpext*? | Epithelial predominant† | NA | NA | NA | |
| 0902 [12] | Blood | Isolated | F | 12 | Mat | p.Ser417* | Epithelial predominant† | NA | NA | NA | |
| 0692 [12] | Blood | Isolated | F | 13 | NA | p.Arg487* | L | NA | NA | NA | 36 |
| R | NA | ||||||||||
| 6671 [12] | Blood | Isolated | F | 10 | NA | p.Arg230* | L | Epithelial predominant† | NA | NA | 5 |
| R | Epithelial predominant† | ||||||||||
| 0796 [12] | Blood | Isolated | F | 61 | NA | p.Leu362* | NA | NA | NA | 28 | |
| 0866 [12] | Blood | Isolated | F | 90 | NA | p.Gln435Serfs*35 | Epithelial predominant† | NA | NA | 22 | |
| 0936 [12] | Blood | Isolated | M | 8 | NA | p.Glu384* | NA | NA | NA | NA | |
| 1 [15] | Blood & tumour | Familial | F | 5 | Mat | p.Cys83Phefs*6 | L | Epithelial type † | PLNR | CN‐LOH, TRIM28 IHC loss | |
| R | Epithelial type † | ||||||||||
| 2 [15] | Blood & tumour | Familial | F | 18 | Mat | p.Cys83Phefs*6 | Epithelial type† | PLNR | CN‐LOH, TRIM28 IHC loss | NA | |
| 3 [15] | Blood & tumour | Familial | M | 69 | Mat | p.Arg524Leufs*155 | Mixed type† | PLNR | No LOH, TRIM28 IHC loss, mutations in DICER1 & AMER1 | NA | |
| 4 [15] | Blood & tumour | Familial | M | 7 | Mat | p.Arg524Leufs*155 | L | Epithelial type† | PLNR | CN‐LOH, TRIM28 IHC loss, NF1 mutation | NA |
| R | Blastemal type† | ||||||||||
| 5 [15] | Healthy kidney & tumour | Familial | F | 6 | NA | p.Gln283* | Epithelial type† | PLNR | NA | NA | |
| 6 [15] | Healthy kidney & tumour | Familial | F | 7 | NA |
p.Gln283* |
L | Epithelial type† | PLNR | NA | NA |
| R | Nephroblastomatosis† | ||||||||||
| 7 [15] | Both kidneys & tumour | Familial | M | 6 | Mat‡ | p.Gln339* | L | Epithelial type† | PLNR | CN‐LOH, TRIM28 IHC loss | NA |
| R | Epithelial type† | ||||||||||
| 1 [16] | Blood & tumour | Familial | F | 12 | NA | p.Gln701* | L | Epithelial type† | NA | CN‐LOH | NA |
| R | Epithelial type† | NA | CN‐LOH | NA | |||||||
| 2 [16] | Blood & tumour | Familial | F | 14 | NA | p.Gln701* | L | Epithelial type† | NA | CN‐LOH | 8 |
| R | Epithelial type† | NA | CN‐LOH | 4 | |||||||
| 8 [15] | Both kidneys & tumour | Isolated | M | 17 | NA | Splice, c.586+2T>C | L | Nephroblastomatosis† | PLNR | CN‐LOH, TRIM28 IHC loss | NA |
| R | Epithelial type† | ||||||||||
| 9 [15] | Healthy kidney & tumour | Isolated | F | 7 | NA | p.Leu59Trpfs*34 | Epithelial type† | PLNR | NA | NA | |
| 11 [15] | Healthy kidney & tumour | Isolated | F | 75 | NA | p.Cys174Argfs*4 | L | Nephroblastomatosis† | PLNR | NA | |
| R | Epithelial type† | ||||||||||
| PAKVET [13] | Healthy kidney & tumour | NA | NA | 13 | NA | Splice, c.839+1G>A | Monomorphic epithelial | No | CN‐LOH | NA | |
| 10 [15] | Healthy kidney & tumour | Isolated | F | 40 | Mosaic§ | p.Ala544Profs*132 | Epithelial type with diffuse anaplasia† | No | NA | NA | |
| 12 [15] | Tumour|| | Isolated | F | 8 | Somatic | p.Met389Argfs*2 | Epithelial type† | NA | NA | NA | |
| PADWNP [13] | Tumour|| | Isolated | NA | 18 | Somatic | p.Gln233* | Monomorphic epithelial | No | CN‐LOH | NA | |
| PAJMKN [13] | Tumour|| | Isolated | NA | 17 | Somatic |
p.Gly107Argfs*75 |
Monomorphic epithelial | No | CN‐LOH | NA | |
| PAJMZF [13] | Tumour|| | Isolated | NA | 8 | Somatic | p.Arg487* | Monomorphic epithelial | No | No LOH, promoter hypermethylation¶ | NA | |
| PADDLL [13] | Tumour|| | Isolated | NA | 6 | Somatic | p.Phe645Leufs*29 | Monomorphic epithelial | No | NA | NA | |
| PAJPER [13] | Tumour|| | Isolated | NA | 15 | Somatic | Splice, c.839+1G>A and p.Arg487* | Monomorphic epithelial | No | NA | NA | |
| PAKSJN [13] | Tumour|| | Isolated | NA | 91 | Somatic | p.Arg230* | Monomorphic epithelial | No | NA | NA | |
| PAJNYM [13] | Tumour|| | Isolated | NA | 10 | Somatic | Splice, c.340+2T>G | Monomorphic epithelial | No | CN‐LOH | NA | |
| PAKYLT [6, 13] | Tumour|| | Isolated | NA | NA | Somatic | Splice, c.839+1G>A | Anaplastic, epithelial | NA | CN‐LOH, TP53 mutation | NA | |
| W117 [14] | Tumour|| | Isolated | M | 7 | Somatic | p.Phe645Leufs*30 | Monomorphic epithelial | No | No LOH, TRIM28 IHC loss, exon 1 hypermethylation | NA | |
| WESK150 (this report) | Tumour|| | Isolated | M | 7 | Somatic | p.Thr154Tyrfs*2 | Epithelial type† | PLNR | CN‐LOH, TRIM28 IHC loss | NA | |
M, male; F, female; Age, age at Wilms' tumour diagnosis (months); DN, de novo; Mat, maternal; NR, nephrogenic rests; PLNR, perilobar nephrogenic rests; LOH, loss of heterozygosity; IHC, immunohistochemistry; CN‐LOH, copy‐neutral loss of heterozygosity; FU, duration of follow‐up (years); NA, Not available.
†(Presumably) after preoperative chemotherapy.
‡Assumed that mutation was inherited from mother, who was not tested but had bilateral Wilms' tumour at age 8 years.
§Based on variant allele frequency.
||Absent in adjacent kidney tissue.
¶Not presumed to be responsible for silencing the wild‐type allele.
Variants are described on transcript NM_005762.2 according to the Human Genome Variation Society (HGVS) recommendations. ††Patient deceased. The protein annotation of the original publication has been changed according to HGVS recommendations.
Figure 1.
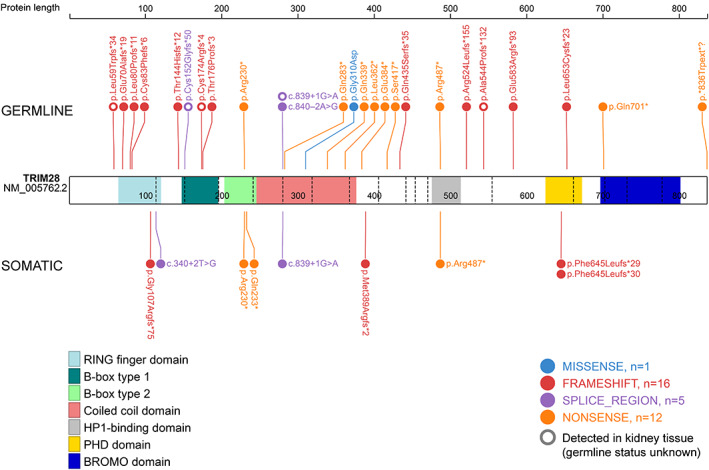
Schematic representation of the TRIM28 protein and reported germline and somatic variants in patients with Wilms' tumour. Variants identified in adjacent normal kidney tissue in non‐familial cases (N = 5) are included in this figure as potential germline variants, marked as open circles. Protein annotations follow the recommendations of the Human Genome Variation Society (HGVS).
Histological features of TRIM28‐mutated tumours
The comparison of WT histology in TRIM28‐mutated WTs is complicated by the use of two distinct histological classification systems: the Children's Oncology Group (COG) classification and the SIOP classification of renal tumours. The two classification systems apply to WTs treated with primary surgery and preoperatively treated WT, respectively [17]. Generally, preoperative chemotherapy is recommended in SIOP Renal Tumour Study Group (RTSG) protocols for all children aged ≥6 months at diagnosis [18], while in North American COG protocols it is only recommended for children with a known genetic predisposition and/or bilateral WT [19]. In most cases after preoperative chemotherapy, part of the tumour has become necrotic and because the undifferentiated, blastemal cells are more sensitive to chemotherapy, the initial composition of epithelium, stroma, and blastema may have shifted [20]. In the reviewed studies on TRIM28, it was frequently not specified whether tumours had been pretreated and/or which histological classification system had been used. Therefore, in this review, we will describe histology according to the terminology in the original reports.
Histological characterization was reported for 51 tumours from 46 patients [12, 13, 14, 15, 16]. Out of the 51 tumours, 44 (86%) were described as (monomorphic) epithelial (type or predominant) WTs, three (6%) as epithelial (type or predominant) with (diffuse) anaplasia, one as blastemal‐type WT (2%), and two (4%) as ‘epithelial and blastemal’ WTs. Thus, although epithelial tumours appear to be the predominant subtype among TRIM28‐mutated tumours, the presence of other tumour components (particularly blastema) or anaplasia does not exclude the presence of (germline or somatic) TRIM28 variants.
The presence or absence of nephrogenic rests was specified for 24 patients with TRIM28 variants. Nephrogenic rests were reported in 11 patients, including 7/10 (70%) with germline TRIM28 variants, 3/5 (60%) patients with TRIM28 variants that were confirmed in kidney tissue, and 1/9 (11%) patients with somatic TRIM28 mutations in their tumours. All reported nephrogenic rests were perilobar rests.
TRIM28 acts a tumour suppressor in patients with WT
TRIM28 acts as a classical tumour suppressor gene in WT patients, where disruption of both alleles appears to be required to initiate tumour development. In ten TRIM28‐mutated tumours in which immunohistochemistry (IHC) was performed (Figure 2), including seven with a germline variant, tumour cells had lost expression of TRIM28, in contrast to the surrounding non‐malignant cells, that showed retained nuclear expression (Table 1) [14, 15]. Loss of heterozygosity (LOH) was found to be the most common mechanism for this second hit, which was confirmed in 17 out of 20 cases. In 13 of these 17 tumours, B‐allele frequency and/or SNP array data were available, revealing that in all these cases LOH was caused by a somatic recombination event on the q‐arm of chromosome 19, resulting in (copy‐neutral) homozygosity of the mutated allele. The size of the LOH region (if reported) varied from regions encompassing almost the entire chromosome arm (19q13.11–19q13.43) [13] to regions less than 0.5 Mb [15].
Figure 2.

Loss of TRIM28 protein expression in TRIM28‐mutated Wilms' tumour. Top: immunohistochemical staining with anti‐KAP1 antibody (ab10484) in an epithelial Wilms' tumour (WT) of a 7‐month‐old boy with a somatic TRIM28 mutation showing absent nuclear staining in tumour cells, with retained expression of KAP1 in non‐tumoural cells. Bottom: retained expression of KAP1 in adjacent normal kidney tissue. The counterstaining with Mayer's haematoxylin (blue) appears more intense in the tumour, due to the fact that the tumour slice is slightly thicker and lacks KAP1 (brown) staining.
Mutations in other known WT driver genes were assessed in whole exome sequencing (WES) data of 11 TRIM28‐mutated tumours. Eight tumours (72%) did not reveal any driver gene mutation [14, 15]. One tumour revealed a TP53 mutation, which was likely related to its diffuse anaplastic histology [13, 21]. In the study by Diets et al, two tumours revealed somatic mutations in DICER1, AMER1 (individual 3), and NF1 (individual 4) [15].
Recently, Brzezinski et al observed that TRIM28‐mutated tumours belong to a subgroup of WT with genome‐wide dysregulation of DNA methylation [22] and display a very distinct and recognizable DNA methylation pattern (Brzezinski, personal communication).
Biological functions of TRIM28
TRIM28 (also known as KAP1 or TIF1beta) is a multi‐domain protein that is part of the tripartite motif (TRIM)‐containing protein family. Proteins in this family are associated with a wide variety of physiological processes [23]. Although TRIM28 is ubiquitously expressed, its functions are context‐, species‐, and/or cell type‐dependent [24, 25].
TRIM28 is a central regulator of transcription that can either promote or repress chromatin accessibility. TRIM28 does not have a DNA‐binding domain, but is indirectly recruited to genomic loci through its interaction with a variety of transcription factors that determine target specificity [26]. An important group of transcription factors is the large family of Krüppel‐associated box‐containing zinc‐finger proteins (KRAB‐ZFPs, also known as KRAB‐ZNF proteins) that control transcriptional repression during embryogenesis and tissue differentiation [27, 28, 29]. These KRAB‐ZFP–TRIM28 complexes subsequently recruit multiple chromatin‐modifying proteins, including the histone deacetylase complex NuRD, heterochromatin protein 1 (HP1), and the histone H3 lysine 9 (H3K9me3)‐specific methyltransferase SETDB1 [30]. This transcriptional effect of TRIM28 appears to depend on the post‐translational modifications of TRIM28 [24, 31]. Specifically, SUMOylated TRIM28 acts as a scaffold for heterochromatin inducing factors, whereas phosphorylated TRIM28 promotes chromatin accessibility and enables transcriptional elongation by releasing paused RNA polymerase II [32]. Targets of TRIM28‐mediated transcriptional regulation include protein‐coding as well as promoter regions, imprinting control regions, long non‐coding RNAs (lncRNAs), and transposable elements [25, 33, 34].
Through this extensive protein–protein interaction network, TRIM28 is involved in a wide variety of cellular processes, including cell differentiation [24], stem cell maintenance [34], DNA damage repair [35], establishment of genomic imprints [36, 37], apoptosis [38], and autophagy [39]. Therefore, it is perhaps not surprising that loss of TRIM28 is lethal in mouse embryos [37] and overexpression of TRIM28 is observed in many cancer types [31].
TRIM28 and WT development
As is true for many of the recently discovered WT predisposition genes, much needs to be unravelled about how pathogenic TRIM28 variants lead to WT development (Figure 3). WTs result from maldevelopment of the embryonic kidney and many WT predisposition genes are involved in the transcriptional regulation of nephrogenesis, WT1 being the most extensively studied. As yet, however, the exact mechanisms of WT development in the context of these germline variants are still not fully elucidated [1, 40].
Figure 3.
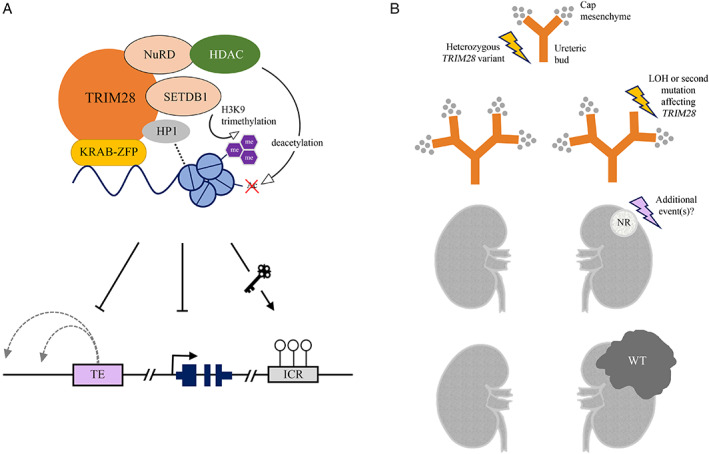
Model for TRIM28‐mutated Wilms' tumour development. TRIM28 is thought to act as a transcriptional corepressor during the early stages of kidney development, through its interaction with one of the Krüppel‐associated box‐containing zinc‐finger proteins (KRAB‐ZFPs). H3K9, histone H3 lysine 9; me, methyl group; Ac, acetyl group; TE, transposable element; ICR, imprinting control region; LOH, loss of heterozygosity; WT, Wilms' tumour. (A) The TRIM28–KRAB‐ZFP complex acts as a scaffold for chromatin‐modifying proteins that regulate local chromatin accessibility and gene expression, including SET domain bifurcated histone lysine methyltransferase 1 (SETDB1), the nucleosome remodelling and deacetylase complex (NuRD), histone deacetylases (HDACs), and heterochromatin protein 1 (HP1). Targeted transposable elements (TEs) and genes are repressed, whereas imprinting control regions (ICRs) are maintained. (B) Loss of TRIM28 in the embryonic kidney leads to a branching arrest which may cause nephrogenic rests (NRs) to persist in the postnatal kidney. Additional events are necessary for NRs to develop into WT.
When compared with germline WT1 variants which are associated with intralobar nephrogenic rests, the identification of perilobar nephrogenic rests in patients with germline TRIM28 variants suggests a relatively late disturbance of nephrogenesis, which is normally completed by 34–37 weeks of gestation [41, 42]. The predominance of epithelial WT suggests that the arrested renal mesenchyme is somehow directed towards epithelial differentiation.
In embryonic rat kidneys, Dihazi et al demonstrated that knockdown of TRIM28 indeed resulted in reduced ureteric bud branching or even branching arrest, which provides a potential model of how TRIM28 mutations could lead to the formation of nephrogenic rests and WT (Figure 3B). In their study, TRIM28 protein was expressed in the ureteric bud, cap mesenchyme, and renal vesicle, but downregulated in comma‐ and S‐shaped bodies, the subsequent stages that develop into the mature nephron [43]. Based on bioinformatics analysis of chromatin immunoprecipitation (ChIP) data previously generated by O'Geen et al [44], Dihazi et al identified 22 genes involved in kidney development among the ~7000 potential binding sites of TRIM28 [43]. These included WT1, BMP4, BMP7, GDNF, and RET, which are known to play important roles in ureteric bud branching [45]. Of these genes, BMP4 [25], BMP7 [26], and RET [25, 26] were also among the significantly upregulated genes in TRIM28 knockdown HEK293 cell lines [26] and/or TRIM28 knockout human ESCs [25].
In WTs studied by Armstrong et al [13] and Halliday et al [14], pathogenic TRIM28 variants were correlated to a specific gene expression pattern that had previously been labelled the S1 subtype, described as a post‐induction gene expression pattern [6]. Compared with other WTs, TRIM28‐mutated and S1‐subtype WTs had 18 differentially expressed genes in common, including lower expression of SIX2 [13]. SIX2 is a homeobox protein, normally expressed in the cap mesenchyme, which is responsible for maintaining the undifferentiated state of blastemal cells [46]. Additionally, TRIM28‐mutated WTs revealed an increased expression of four KRAB‐ZFP genes, namely ZNF728, ZNF676, ZNF208, and ZNF780A. Presumably, these four KRAB‐ZFPs play crucial roles in TRIM28‐mediated silencing of specific genomic loci in the developing kidney. The overexpression of these genes may be explained by the fact that the expression of KRAB‐ZFP genes appears to be controlled by a TRIM28‐dependent auto‐regulatory mechanism [44]. Finally, a large number of transposable elements across the genome were found to show differential expression, the majority of which were overexpressed [13].
Transposable elements
TRIM28 is known to be involved in the silencing of a wide range of transposable elements (TEs), including LINE‐1, LTRs, HERVs, and SVAs (Figure 3A) [25, 34, 47]. TEs are repetitive DNA sequences that comprise about half of the human genome, most of them remnants of ancient proviral infections [48]. In recent years, it has been shown that specific TEs can be expressed and (retro)transpose themselves into new genomic locations, in germ cells, embryonic stem cells, and cancer cells [49, 50, 51, 52].
In cancer cells, TEs can disrupt protein coding or regulatory sequences of specific tumour suppressor genes [52]. Additionally, global hypomethylation of TEs has been associated with genomic instability in various adult cancer types [51]. Although WTs generally harbour few mutations or copy number changes compared with adult cancer, TRIM28‐mutated WTs were recently shown to be part of a subgroup of WTs which are less stable genomically [22].
In embryonic stem cells (ESCs), the expression of TEs was shown to correlate with changes in chromatin accessibility and DNA methylation, and it is thought that TRIM28‐mediated TE silencing may have evolved to regulate germline competency and somatic lineage differentiation [25, 36, 53]. As in HEK293 cells [26], human ESCs with TRIM28 knockout showed an extensive number of differentially expressed TEs and KRAB‐ZNF genes [25]. In contrast to TRIM28‐deficient mouse ESCs [53], human ESCs with TRIM28 knockout retained self‐renewal capacity and even displayed a growth advantage [25]. Yet TRIM28 knockout ESCs seemed less capable of producing primordial germ cells and cardiomyocytes, and it was suggested that specific cell lineages with a very narrow developmental window are affected by TRIM28 loss [25]. We hypothesize that this balance between differentiation and proliferation is also disturbed in nephron progenitor cells that lack TRIM28, probably resulting in an extensively deregulated transcriptional landscape that blocks normal differentiation and favours tumourigenesis.
Maternal inheritance
A remarkable observation in the families identified thus far was that in all 15 patients with WT for whom parental inheritance could be established, the pathogenic TRIM28 variant was inherited from the mother (three of whom were also diagnosed with WT) [12, 15]. The underlying cause of this maternal inheritance pattern is currently unknown.
A recently proposed explanation is related to the PEG3 imprinting control region (ICR), which is a paternally expressed ICR located in close vicinity to TRIM28 on the tip of chromosome arm 19q [12]. PEG3 was suggested to function as a tumour suppressor gene, which is inactivated by the somatic loss of the paternal 19q arm in the case of a germline TRIM28 mutation on the maternal allele. Although this scenario requires further analysis, the LOH region in at least two published TRIM28‐mutated tumours did not include PEG3 [12, 15].
Another explanation for the maternal inheritance pattern could be that pathogenic TRIM28 variants impair spermatogenesis and result in male subfertility or infertility, as was suggested by a recent study in mice with heterozygous loss of TRIM28 [54]. This would prevent male carriers from passing the variant on to offspring. In published pedigrees of families with carriers of pathogenic TRIM28 variants, all male carriers were affected with WT and none were reported to have children carrying the variant [12, 15], although case 37 [14] fathered a wildtype daughter (unpublished data, February 2021). Fertility assessment in male carriers, as well as determining the parental origin of de novo TRIM28 mutations, will help to clarify whether genomic imprinting or male infertility, or a combination of both, explains the maternal inheritance pattern.
TRIM28 interacts with other WT genes
Two WT‐associated genes, REST and AMER1, have been reported to interact with TRIM28. The REST gene which, like TRIM28, was recently identified as a WT predisposition gene, encodes a KRAB‐ZFP which binds to DNA targets and recruits TRIM28 as a corepressor in the regulation of genes involved in neuronal development [55]. The AMER1 gene, somatically mutated in ~18% of WTs, encodes the WTX protein which was demonstrated to be a binding partner of TRIM28 [56]. Further research is needed to characterize the networks in which these genes, including TRIM28, are involved.
Clinical implications
WT risk and age at diagnosis
Among the 30 patients with germline TRIM28 variants (17 female, 13 male), ten (33%) had bilateral disease. Median age at WT diagnosis was 13 months (range 5–118 months), which is younger compared with general WT cohorts [57]. However, compared with WT patients with germline WT1 variants, where >95% of tumours are diagnosed before the age of 5 years [58], a relatively large proportion of patients with TRIM28 variants presented at older ages. We found that 25/30 patients (83%) were diagnosed before the age of 7 years and 28/30 (93%) before the age of 8 years, which may encourage continuing surveillance until the age of 8 years (Figure 4). Additionally, based on two families in which all affected individuals were diagnosed before the age of 8 months, it is conceivable that other unidentified genetic factors play a role in the age of onset [12, 15].
Figure 4.

Age at Wilms' tumour diagnosis (in years) of patients with germline TRIM28 variants (N = 30) versus an unselected reference cohort of patients with WT (N = 126). The reference cohort includes all patients diagnosed with WT in The Netherlands in a 5‐year period.
Pedigrees from families with germline pathogenic TRIM28 variants suggest a disease penetrance of ~67%, with 18 affected individuals out of a combined total of 27 (obligate) carriers [12, 15]. Only one pedigree showed the presence of TRIM28 variants in more than two generations. In this pedigree (ID_0477 in Mahamdallie et al [12]), four unaffected obligate carriers and six affected individuals were identified. Since reported families were identified based on the presence of multiple affected individuals, this estimated penetrance is likely biased, but certainly supports offering surveillance to children with germline TRIM28 variants.
Prognosis
In the reviewed studies, metastatic disease was not reported. Follow‐up data were available for 13 patients with germline pathogenic variants in TRIM28, none of whom relapsed. The duration of follow‐up ranged from 3 to 36 years, with a median of 20 years for patients with follow‐up data. One patient with diffuse anaplastic WT died of an unspecified cause, 3 years after WT diagnosis [12]. It has been previously suggested that TRIM28‐mutated WTs represent a subgroup of WTs with a low risk of metastases or relapse. This may be attributed to the fact that the majority are epithelial WTs, which are known to have a good outcome [59, 60]. This information can be reassuring for families with young carriers of pathogenic TRIM28 variants.
Additional phenotypes
Despite the involvement of TRIM28 in a wide variety of cellular processes, there is no strong evidence suggesting that germline pathogenic TRIM28 variants cause a phenotype other than WT predisposition in humans. Additional clinical findings were only documented in 4/33 patients, although phenotypic data may have been incompletely reported. For example, Mahamdallie et al only reported that patients had no other cancers [12] and no phenotypic data were available for the patient reported by Armstrong et al [13]. Patients with additional clinical findings included two unrelated patients with autism and speech delay/intellectual disability [12] and two siblings with congenital heart defects, in one of them accompanied by oesophageal atresia and retinopathy [15]. For the two siblings, a different (genetic) cause of their congenital heart defect cannot be excluded, even though this was not identified with WES [15]. As mentioned previously, the male infertility observed in haploinsufficient mice [54] has not been documented in humans, but may warrant attention during the clinical follow‐up of TRIM28 mutation carriers.
Recommendations for the genetic analysis of TRIM28 in patients with WT
To enable counselling, genetic testing, and early detection of WTs in young family members, it is important to recognize germline pathogenic TRIM28 variants in patients with WT. Depending on local infrastructure and resources, some paediatric oncology centres may offer routine genetic testing to all patients, while others select those who are clinically suspected of having a genetic predisposition syndrome [61].
To identify patients with germline variants in TRIM28, we would recommend routine assessment of WTs for TRIM28 loss by IHC with the anti‐KAP1 antibody (ab10484) [15], which is a relatively simple and inexpensive test. Even though the majority of TRIM28‐mutated tumours are epithelial (predominant) WT, we would recommend including all WT subtypes in this assessment, as other histological subtypes have also been reported and an accurate distribution of TRIM28 mutations among the different histological subtypes has not yet been determined. Subsequently, genetic analysis of TRIM28 in blood‐derived DNA can be performed in all patients who display loss of TRIM28 in the tumour.
Directions for future research
A further exploration of TRIM28‐associated WT will help to unravel the diverse mechanisms that can lead to WT development. In vitro models suggest that loss of TRIM28 leads to a loss of (epigenetic) transcriptional regulation. This may upregulate specific signalling pathways in the ureteric bud and metanephric mesenchyme, resulting in a disturbed balance between proliferation and differentiation, and in a branching arrest in the embryonic kidney. Further studies in embryonic kidney models are needed to determine exactly which signalling pathways are deregulated upon loss of TRIM28. This also includes the direct epigenetic impact of TRIM28 deficiency, i.e. changes in DNA methylation and chromatin organization, in the developing kidney. Although we have gained many insights from mouse studies, additional studies are preferably conducted in human kidney models, given the recently described differences between human and mouse developmental programs during nephrogenesis [42, 62].
For this purpose, organoid models may provide valuable opportunities. Organoid models can be established directly from tumour‐ and adjacent‐kidney tissue of patients with germline pathogenic TRIM28 variants [63]. Since such a model may not recapitulate the crucial effects of TRIM28 loss during the earliest stages of nephrogenesis, TRIM28‐deficient human pluripotent stem cells (hPSCs) could be an interesting alternative. We speculate that differentiation of these hPSCs into kidney organoids will enable us to study the consequences of TRIM28 loss during the earliest stages of nephrogenesis, which is not possible in patient‐derived organoids [62]. By additionally knocking out REST and AMER1, more insight into potential TRIM28–REST and TRIM28–AMER1 regulatory effects may also be provided.
The role of TEs in human embryonic kidney and WT development warrants further investigation. In addition to the TRIM28‐mediated transcriptional repression of TEs, recent evidence suggests that post‐transcriptional repression of TEs is mediated by miRNAs [64], which is intriguing because miRNA processing genes (DROSHA, DICER1, DIS3L2, DGCR8) represent an important group of WT driver genes [65].
Similar to some other WT predisposition genes [12], such as WT1, IGF2, and DICER1, TRIM28 seems to promote WT development in both a germline and a somatic context. Given its role in early nephrogenesis and the high rate of germline variants, TRIM28 mutations are considered early events. We speculate that the identified somatic mutations may have been present in a mosaic state in adjacent normal kidney tissue, as was demonstrated in one patient by Diets et al [15]. This could be further investigated by assessing multiple samples from adjacent normal kidney tissue of somatically TRIM28‐mutated WT.
Finally, from a clinical perspective, it is relevant to collect more data on both healthy and affected carriers of pathogenic TRIM28 variants. This will require international collaboration, and will help to improve the counselling of patients and their families.
Author contributions statement
RPK was responsible for conceptualization. JAH wrote the original draft. All the authors wrote, reviewed and edited the final paper.
Acknowledgement
JH is funded by Stichting Kinderen Kankervrij (KiKa), Award Number 278.
No conflicts of interest were declared.
References
- 1. Hohenstein P, Pritchard‐Jones K, Charlton J. The yin and yang of kidney development and Wilms' tumors. Genes Dev 2015; 29: 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Charlton J, Williams RD, Sebire NJ, et al. Comparative methylome analysis identifies new tumour subtypes and biomarkers for transformation of nephrogenic rests into Wilms tumour. Genome Med 2015; 7: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bharathavikru R, Hastie ND. Overgrowth syndromes and pediatric cancers: how many roads lead to IGF2? Genes Dev 2018; 32: 993–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coorens THH, Treger TD, Al‐Saadi R, et al. Embryonal precursors of Wilms tumor. Science 2019; 366: 1247–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Treger TD, Chowdhury T, Pritchard‐Jones K, et al. The genetic changes of Wilms tumour. Nat Rev Nephrol 2019; 15: 240–251. [DOI] [PubMed] [Google Scholar]
- 6. Gadd S, Huff V, Walz AL, et al. A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 2017; 49: 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wegert J, Ishaque N, Vardapour R, et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high‐risk blastemal type Wilms tumors. Cancer Cell 2015; 27: 298–311. [DOI] [PubMed] [Google Scholar]
- 8. Anvar Z, Acurzio B, Roma J, et al. Origins of DNA methylation defects in Wilms tumors. Cancer Lett 2019; 457: 119–128. [DOI] [PubMed] [Google Scholar]
- 9. Ruteshouser EC, Huff V. Familial Wilms tumor. Am J Med Genet C Semin Med Genet 2004; 129C: 29–34. [DOI] [PubMed] [Google Scholar]
- 10. Scott RH, Stiller CA, Walker L, et al. Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 2006; 43: 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Segers H, Kersseboom R, Alders M, et al. Frequency of WT1 and 11p15 constitutional aberrations and phenotypic correlation in childhood Wilms tumour patients. Eur J Cancer 2012; 48: 3249–3256. [DOI] [PubMed] [Google Scholar]
- 12. Mahamdallie S, Yost S, Poyastro‐Pearson E, et al. Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 2019; 3: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Armstrong AE, Gadd S, Huff V, et al. A unique subset of low‐risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: a Children's Oncology Group study. PLoS One 2018; 13: e0208936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halliday BJ, Fukuzawa R, Markie DM, et al. Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet 2018; 14: e1007399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diets IJ, Hoyer J, Ekici AB, et al. TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer 2019; 145: 941–951. [DOI] [PubMed] [Google Scholar]
- 16. Moore C, Monforte H, Teer JK, et al. TRIM28 congenital predisposition to Wilms' tumor: novel mutations and presentation in a sibling pair. Cold Spring Harb Mol Case Stud 2020; 6: a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dome JS, Graf N, Geller JI, et al. Advances in Wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol Off J Am Soc Clin Oncol 2015; 33: 2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van den Heuvel‐Eibrink MM, Hol JA, Pritchard‐Jones K, et al. Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP–RTSG 2016 protocol. Nat Rev Urol 2017; 14: 743–752. [DOI] [PubMed] [Google Scholar]
- 19. Ehrlich PF, Chi YY, Chintagumpala MM, et al. Results of treatment for patients with multicentric or bilaterally predisposed unilateral Wilms tumor (AREN0534): a report from the Children's Oncology Group. Cancer 2020; 126: 3516–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vujanić GM, Sandstedt B, Harms D, et al. Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Med Pediatr Oncol 2002; 38: 79–82. [DOI] [PubMed] [Google Scholar]
- 21. Gadd S, Huff V, Huang CC, et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 2012; 14: 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brzezinski J, Choufani S, Romao R, et al. Clinically and biologically relevant subgroups of Wilms tumour defined by genomic and epigenomic analyses. Br J Cancer 2021; 124: 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watanabe M, Hatakeyama S. TRIM proteins and diseases. J Biochem 2017; 161: 135–144. [DOI] [PubMed] [Google Scholar]
- 24. Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem 2011; 286: 26267–26276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tao Y, Yen MR, Chitiashvili T, et al. TRIM28‐regulated transposon repression is required for human germline competency and not primed or naive human pluripotency. Stem Cell Reports 2018; 10: 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iyengar S, Ivanov AV, Jin VX, et al. Functional analysis of KAP1 genomic recruitment. Mol Cell Biol 2011; 31: 1833–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsui T, Leung D, Miyashita H, et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010; 464: 927–931. [DOI] [PubMed] [Google Scholar]
- 28. Rowe HM, Friedli M, Offner S, et al. De novo DNA methylation of endogenous retroviruses is shaped by KRAB‐ZFPs/KAP1 and ESET. Development 2013; 140: 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turelli P, Castro‐Diaz N, Marzetta F, et al. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res 2014; 24: 1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schultz DC, Ayyanathan K, Negorev D, et al. SETDB1: a novel KAP‐1‐associated histone H3, lysine 9‐specific methyltransferase that contributes to HP1‐mediated silencing of euchromatic genes by KRAB zinc‐finger proteins. Genes Dev 2002; 16: 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Czerwińska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. J Biomed Sci 2017; 24: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bunch H, Zheng X, Burkholder A, et al. TRIM28 regulates RNA polymerase II promoter‐proximal pausing and pause release. Nat Struct Mol Biol 2014; 21: 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bunch H, Lawney BP, Burkholder A, et al. RNA polymerase II promoter‐proximal pausing in mammalian long non‐coding genes. Genomics 2016; 108: 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rowe HM, Jakobsson J, Mesnard D, et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010; 463: 237–240. [DOI] [PubMed] [Google Scholar]
- 35. White D, Rafalska‐Metcalf IU, Ivanov AV, et al. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res 2012; 10: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cammas F, Mark M, Dollé P, et al. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 2000; 127: 2955–2963. [DOI] [PubMed] [Google Scholar]
- 37. Messerschmidt DM, de Vries W, Ito M, et al. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012; 335: 1499–1502. [DOI] [PubMed] [Google Scholar]
- 38. Yang B, O'Herrin SM, Wu J, et al. MAGE‐A, mMage‐b, and MAGE‐C proteins form complexes with KAP1 and suppress p53‐dependent apoptosis in MAGE‐positive cell lines. Cancer Res 2007; 67: 9954–9962. [DOI] [PubMed] [Google Scholar]
- 39. Yang Y, Fiskus W, Yong B, et al. Acetylated hsp70 and KAP1‐mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A 2013; 110: 6841–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hastie ND. Wilms' tumour 1 (WT1) in development, homeostasis and disease. Development 2017; 144: 2862–2872. [DOI] [PubMed] [Google Scholar]
- 41. Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet 1998; 79: 268–273. [DOI] [PubMed] [Google Scholar]
- 42. Lindström NO, McMahon JA, Guo J, et al. Conserved and divergent features of human and mouse kidney organogenesis. J Am Soc Nephrol 2018; 29: 785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dihazi GH, Jahn O, Tampe B, et al. Proteomic analysis of embryonic kidney development: heterochromatin proteins as epigenetic regulators of nephrogenesis. Sci Rep 2015; 5: 13951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Geen H, Squazzo SL, Iyengar S, et al. Genome‐wide analysis of KAP1 binding suggests autoregulation of KRAB‐ZNFs. PLoS Genet 2007; 3: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishinakamura R, Sakaguchi M. BMP signaling and its modifiers in kidney development. Pediatr Nephrol 2014; 29: 681–686. [DOI] [PubMed] [Google Scholar]
- 46. Self M, Lagutin OV, Bowling B, et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 2006; 25: 5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Castro‐Diaz N, Ecco G, Coluccio A, et al. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev 2014; 28: 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol 2012; 10: 395–406. [DOI] [PubMed] [Google Scholar]
- 49. Hancks DC, Kazazian HH Jr. Roles for retrotransposon insertions in human disease. Mob DNA 2016; 7: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grow EJ, Flynn RA, Chavez SL, et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015; 522: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burns KH. Transposable elements in cancer. Nat Rev Cancer 2017; 17: 415–424. [DOI] [PubMed] [Google Scholar]
- 52. Kaer K, Speek M. Retroelements in human disease. Gene 2013; 518: 231–241. [DOI] [PubMed] [Google Scholar]
- 53. Rowe HM, Kapopoulou A, Corsinotti A, et al. TRIM28 repression of retrotransposon‐based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res 2013; 23: 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tan JHL, Wollmann H, van Pelt AMM, et al. Infertility‐causing haploinsufficiency reveals TRIM28/KAP1 requirement in spermatogonia. Stem Cell Reports 2020; 14: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee N, Park SJ, Haddad G, et al. Interactomic analysis of REST/NRSF and implications of its functional links with the transcription suppressor TRIM28 during neuronal differentiation. Sci Rep 2016; 6: 39049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim WJ, Wittner BS, Amzallag A, et al. The WTX tumor suppressor interacts with the transcriptional corepressor TRIM28. J Biol Chem 2015; 290: 14381–14390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nakata K, Colombet M, Stiller CA, et al. Incidence of childhood renal tumours: an international population‐based study. Int J Cancer 2020; 147: 3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scott RH, Walker L, Olsen ØE, et al. Surveillance for Wilms tumour in at‐risk children: pragmatic recommendations for best practice. Arch Dis Child 2006; 91: 995–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Verschuur AC, Vujanic GM, Van Tinteren H, et al. Stromal and epithelial predominant Wilms tumours have an excellent outcome: the SIOP 93 01 experience. Pediatr Blood Cancer 2010; 55: 233–238. [DOI] [PubMed] [Google Scholar]
- 60. Parsons LN, Mullen EA, Geller JI, et al. Outcome analysis of stage I epithelial‐predominant favorable‐histology Wilms tumors: a report from Children's Oncology Group study AREN03B2. Cancer 2020; 126: 2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cullinan N, Villani A, Mourad S, et al. An eHealth decision‐support tool to prioritize referral practices for genetic evaluation of patients with Wilms tumor. Int J Cancer 2020; 146: 1010–1017. [DOI] [PubMed] [Google Scholar]
- 62. Gupta N, Dilmen E, Morizane R. 3D kidney organoids for bench‐to‐bedside translation. J Mol Med (Berl) 2021; 99: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Calandrini C, Schutgens F, Oka R, et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat Commun 2020; 11: 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pedersen IM, Zisoulis DG. Transposable elements and miRNA: regulation of genomic stability and plasticity. Mob Genet Elem 2016; 6: e1175537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wegert J, Bausenwein S, Roth S, et al. Characterization of primary Wilms tumor cultures as an in vitro model. Genes Chromosomes Cancer 2012; 51: 92–104. [DOI] [PubMed] [Google Scholar]