

Abstract
The CEP83 protein is an essential part in the first steps of ciliogenesis, causing a ciliopathy if deficient. As a core component of the distal appendages of the centriole, CEP83 is located in almost all cell types and is involved in the primary cilium assembly. Previously reported CEP83 deficient patients all presented with nephronophthisis and kidney dysfunction. Despite retinal degeneration being a common feature in ciliopathies, only one patient also had retinitis. Here, we present two unrelated patients, who both presented with retinitis pigmentosa, without nephronophthisis or any form of kidney dysfunction. Both patients harbor bi‐allelic variants in CEP83. This report expands the current clinical spectrum of CEP83 deficiency. For timely diagnosis of CEP83 deficiency, we advocate that CEP83 should be included in gene panels for inherited retinal diseases.
Keywords: CEP83, ciliopathy, retinal dystrophy, retinitis pigmentosa
1. INTRODUCTION
Ciliopathies form a group of monogenetic diseases, characterized by abnormal formation or function of cilia (Braun & Hildebrandt, 2017; Wheway & Mitchison, 2019). Cilia are microtubule‐based, filamentous cytoplasmic protrusions on the outside of the cell surface. Due to either their motile function in motile cilia or their osmosensory, chemosensory, or phototransduction function in nonmotile cilia, they are essential for many developmental and physiological processes in almost every tissue type (Waters & Beales, 2011). Ciliopathies manifest particularly during early childhood or adolescence and can affect almost every organ system (Braun & Hildebrandt, 2017; Waters & Beales, 2011), although some tissues are more frequently affected, causing characteristic ciliopathy‐associated symptoms. These predominantly affected tissues are the retina, kidney, liver, and brain, resulting in retinal degeneration, cystic kidney disease, fibrocystic liver disease, and a variation of abnormalities of the central nervous system (Thomas et al., 2019; Waters & Beales, 2011). Other frequent symptoms in ciliopathies are diabetes mellitus, obesity, and skeletal dysplasia. The function of the defective protein and the severity of the mutation determines the intensity of the phenotype (Waters & Beales, 2011).
CEP83 is one of 190 proteins that are known to cause a ciliopathy if deficient (Failler et al., 2014; Wheway & Mitchison, 2019). CEP83 is a centrosomal protein, located in almost all cell types as component of the distal appendages (DAP) of the centriole (Failler et al., 2014; Yang et al., 2018). Early in the centriole‐maturation, CEP83 is recruited by the centrioles. It anchors vesicles originating from the Golgi‐complex to the DAP of the mother centriole. This vesicle‐centriole linkage subsequently initiates primary cilium assembly by anchoring the DAP of the mother centriole also to the plasma membrane (Joo et al., 2013). Besides this main function, CEP83 has two additional functions. First, it ensures removal of CP110, which is a protein that inhibits ciliogenesis. Second, CEP83 ensures recruitment of other DAPs, in particular CEP89, CEP164, and LLRC45 (Failler et al., 2014; Lo et al., 2019). Thus, due to its involvement in primary cilium assembly, CEP83 is an essential part of the first steps of ciliogenesis (Failler et al., 2014; Joo et al., 2013).
To date, bi‐allelic mutations in CEP83 (OMIM# 615847, previous HGNC symbol CCDC41) have been reported in eight individuals who all presented with early‐onset nephronophthisis (Failler et al., 2014), and in one individual who presented with nonsyndromic retinitis pigmentosa (Haer‐Wigman et al., 2017). In the patients reported by Failler et al., next to kidney dysfunction these patients also showed intellectual disability, cholestasis, portal fibrosis, hydrocephalus, and strabismus. Despite retinal degeneration being a common feature in ciliopathies, only one patient was reported to have an ophthalmologic defect, described as retinitis (Failler et al., 2014). In this report, we present two unrelated patients with bi‐allelic variants in CEP83 (NM_016122.2), who both presented with retinitis pigmentosa, without any kidney dysfunction. These patients expand the current clinical spectrum of CEP83 deficiency, which is important for both a timely diagnosis and for diligent patient monitoring.
1.1. Clinical report
P1 is an eight‐year‐old girl. She is the second child of nonconsanguineous Dutch parents, who both originate from the same region in the Netherlands, but who are not knowingly related. The girl was born after an uneventful pregnancy and delivery. There is no relevant family history and her older sister is healthy. Early development was normal, with the exception of repeatedly subnormal eye tests routinely performed at the children's healthcare center. At the age of four, she was referred to an ophthalmologist, who noted a suboptimal to normal visual acuity and hypermetropia, for which glasses were prescribed. In the following years, her distance vision remained suboptimal and she stumbled frequently. In addition, she experienced nyctalopia. To investigate cerebral visual impairment as cause for the persisting and varying suboptimal vision in combination with the stumbling, she was referred to the national diagnostic center for complex visual disorders at the age of eight
There, diagnostic analyses confirmed a mild vision loss with a best‐corrected visual acuity of 0.5 (20/40, OD) and 0.4 (20/50, OS). Color vision test results were not reliable. Visual field testing (Goldmann) showed intact peripheral fields, but reduced central sensitivity in the posterior fields. Imaging analyses revealed both central and peripheral retinal abnormalities (Figure 1). To assess retinal function, an electroretinogram (ERG) was performed revealing low—within normal limits—light adapted amplitudes, indicative of slightly impaired cone function. Dark‐adapted responses were both reduced and delayed, indicative of impaired rod function. The ERG was typical for rod‐cone dystrophy (Figure S1), resulting in a clinical diagnosis of retinitis pigmentosa.
FIGURE 1.
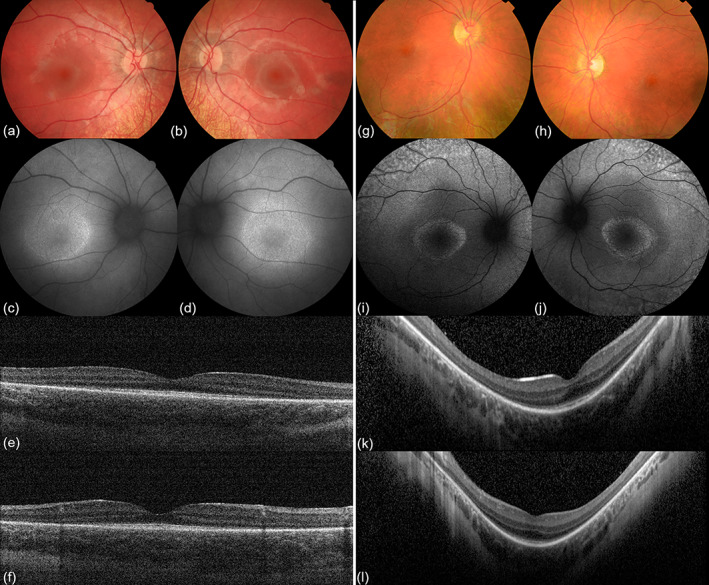
Images of P1 (left) and P2 (right). P1's right (a) and left (b) eye showed dull reflexes of the macula and normal optic discs. Peripheral retinal atrophy is visible, without typical bone‐spicule pigmentation. Fundus autofluorescence (FAF) images of the right (c) and left (d) eye demonstrated a hypofluorescent ring on the foveal area. Optical coherence tomography (OCT) in right (e) and left (f) eye revealed peripheral retinal thinning with photoreceptor loss. These paracentral abnormalities are characteristic for retinal dystrophy in P1, specifically rod‐cone dystrophy. In P2's right (g) and left (h) eye, the foveal reflex was absent and the macula appeared granularly with a patchy peripheral retina. Optic discs appeared normal. On FAF, a bull's eye maculopathy was seen in the right (i) and left (j) eye, consisting of an increased autofluorescent ring in the posterior pole. Both increased and decreased autofluorescent patches were found. OCT of the right (k) and left (l) eye revealed absence of the IS/OS junction in the extra‐foveal region. These findings in P2 were suggestive for the diagnosis retinitis pigmentosa [Color figure can be viewed at wileyonlinelibrary.com]
In addition, during the first grades of primary school, the patient's development seemed somewhat delayed in comparison to her classmates. The delay mainly comprised her gross motor skills, memory, and attention span, and to a lesser extent her fine motor skills. The Dutch Wechsler Intelligence Scale for Children V revealed an IQ of 87 [95% confidence interval: 83–96] at the age of eight, classified as low average up to average (Ruiter et al., 2017). The just below average IQ score and the recent diagnosis of retinitis pigmentosa, in combination with a slightly corpulent appearance (weight‐to‐age and weight‐to‐length both +2SD), led to the suspicion of an underlying metabolic condition or mitochondrial disease, or a ciliopathy, for which she was referred to the department for pediatric metabolic diseases of the University Medical Center in Utrecht.
Here, physical examination, screening laboratory tests and basic metabolic screening provided no explanation for her problems. In particular, no increased LAMP‐1 expression or C vacuolated lymphocytes—as seen in Batten disease (CLN3 disease)—were found (Anderson et al., 2005; Kuper et al., 2020). Trio whole exome sequencing (WES) revealed a homozygous missense variant in CEP83 (c.581 T > C; p.Leu194Pro), associated with a ciliopathy involving kidney disease. Hence, ultrasound of the liver and kidneys, and repeated laboratory tests evaluating liver and especially kidney function in blood and urine were normal and showed no renal concentration defect (Table 1).
TABLE 1.
Laboratory tests for kidney function evaluation
| Plasma | Urine | ||||
|---|---|---|---|---|---|
| Creatinine (μmol/L) | Urea (mmol/L) | eGFR (mL/min/1.73 m2) | Creatinine (mmol/L) | Protein/creatinine ratio (mg/mmol) | |
| P1, first visit | 38 | 5.0 | NA | 11.0 | 48.9 |
| P1, after 9 months | 37 | 5.3 | NA | 5.7 | NA |
| P2, first visit | 46 | 6.2 | >90 | 2.5 | 20.0 |
| P2, after 7 months | 45 | 6.0 | >90 | 10.2 | 12.0 |
| Reference range | 21–65 | 3.0–7.5 | ≥90 | Not available | Not available |
P2 is a five‐year‐old boy. He has a half‐sister and a half‐brother, sharing the same father. His half‐brother passed away at birth due to prematurity. His parents are nonconsanguineous, from Dutch origin, and living in the same region in the Netherlands as the parents of P1 originate from, but there is no known relatedness. Mother has congenital hearing loss, a common affliction in her family, which the boy also suffers from. Next to the hearing loss, his parents noticed esotropia since he was 18 months old. An alternating esotropia and hypermetropia was found. During ophthalmic examination a visual acuity of 0.4 (20/50, OD) and 0.32 (20/63, OS) was measured. Fundoscopy revealed an absent foveal reflex and a granular appearance of the macula, as well as a patchy peripheral retina. The optic disc appeared normal. A bull's eye maculopathy was seen on fundus autofluorescence, consisting of an increased autofluorescent ring in the posterior pole. Also, increased and decreased autofluorescent patches were found. Optical coherence tomography revealed absence of the inner segment/outer segment junction in the extra‐foveal region (Figure 1). The ERG yielded pathological scotopic and photopic responses (Figure S1). These findings resulted in the clinical diagnosis of retinitis pigmentosa under the suspicion of an underlying Usher syndrome, the boy was referred to the Radboud University Medical Center in Nijmegen for evaluation and genetic testing.
Extraocular features manifested as congenital hearing loss, low birth weight (2660 g, 39 + 5 weeks of gestation), frequent upper airway infections, hypermobility, balance problems, and exostosis on his right fibula. He attends a special needs school, because of his visual and hearing impairment. His IQ will be evaluated since regular education required slightly more effort than his classmates, although this might also be due to his hearing and visual impairment. Gene panel analysis (panels blindness and deafness, https://order.radboudumc.nl/producten/wes-visusstoornissen?s=31; https://order.radboudumc.nl/producten/wes-doofheid?s=31) of WES data was performed and revealed two heterozygous variants in CEP83: the same missense variant (c.581 T > C; p.Leu194Pro) as P1 and a nonsense variant (c.1278 T > A; p.Tyr426*). Genetic analysis could not explain the hearing loss in the family of P2, as no potentially disease‐causing variants (including deletions) were detected in genes associated with hearing impairment, including GJB2 and GJB6 (Santos et al., 2005).
Evaluation of kidney function by abdominal ultrasound revealed slightly swollen kidneys and an echogenic aspect of the renal parenchyma. However, repeated tests evaluating his kidney function were normal (Table 1).
1.2. Genetic studies
Informed consent was obtained from the patients' parents. WES was performed in the Wilhelmina Children's Hospital Utrecht and the Radboud University Medical Center Nijmegen, the Netherlands, on genomic DNA isolated from peripheral blood. Genomic DNA was hybridized by an Agilent SureSelectXT system, sequenced with the Illumina system and compared to the human reference genome (NCBI build37/hg19 version), according to the best practice guidelines for next‐generation sequencing (Weiss et al., 2013). Detected variants were annotated, filtered and prioritized using the Bench NGS Lab platform. Variants dominantly inherited from one of the parents were excluded, and variants that fitted a de novo or recessive inheritance model were analyzed. Reporting of de novo variants in genes not yet associated with genetic disease was restricted to putative protein changing variants in genes that are intolerant to missense and loss‐of‐function variants, according to gnomAD (Karczewski et al., 2019). For the recessive inheritance hypothesis, homozygous and compound heterozygous putative protein changing variants were filtered using a population allele frequency cutoff of 0.5% (Karczewski et al., 2019). Variants in candidate recessive genes were only reported if at least one allele carried a putative loss‐of‐function variant. Variant pathogenicity was predicted by SIFT, Polyphen‐2, CADD, and MutationTaster software. Nucleotide conservation prediction was performed using GERP. Larger deletions/duplications, missense, synonymous, and intronic variants affecting protein function of other genes cannot be excluded.
WES revealed in both patients a missense variant in CEP83 (NM_016122.2, g.94772787A > G; c.518 T > C), resulting in a substitution of proline for leucine (p.Leu194Pro). Both parents of P1 and the mother of P2 are heterozygous carriers of this variant. The p.Leu194Pro variant was found only once in heterozygous fashion in >120,000 controls in gnomAD, and it was not found in homozygous fashion (Karczewski et al., 2019). The variant has not been reported in ClinVar (Landrum et al., 2018). However, one patient with nonsyndromic retinitis pigmentosa has been reported to harbor the p.Leu194Pro variant in homozygous fashion as well. In this report, the variant was labeled as likely causative (Haer‐Wigman et al., 2017). The in silico algorithms MutationTaster and Polyphen2 predicted the variant to be disease‐causing and probably damaging, respectively (Adzhubei et al., 2010; Schwarz et al., 2014). Both PROVEAN and SIFT predicted the substitution to be deleterious, with an PROVEAN‐score of −3.48 (Choi & Chan, 2015; Sim et al., 2012). According to the American College of Medical Genetics and Genomics (ACMG), it is a variant of unknown significance (Richards et al., 2015).
In P2, WES revealed a nonsense variant in CEP83 (NM_016122.2, g.94761635A > T; c.1278 T > A), resulting in truncation of the protein (p.Tyr426*). The father of P2 is a heterozygous carrier of this variant. The p.Tyr426* variant has not been identified in gnomAD, and has not been reported in ClinVar (Karczewski et al., 2019; Landrum et al., 2018). The in silico algorithms MutationTaster and Polyphen2 predicted the variant to be pathogenic, and according to the ACMG guidelines, the variant should indeed be classified as pathogenic (Adzhubei et al., 2010; Richards et al., 2015; Schwarz et al., 2014).
Next to the identification of these variants in CEP83, five other variants of unknown significance (three heterozygous, one homozygous, and one de novo) were identified in P1 and six heterozygous variants of unknown significance were detected in P2. These variants were excluded from the final report as they were variants in genes not (yet) associated with a genetic disease, or variants considered unlikely to be disease‐causing based on the ACMG criteria (Richards et al., 2015). More details on the identified genetic variants are available upon request.
2. DISCUSSION
Here, we report two children presenting with retinitis pigmentosa without kidney dysfunction. Both patients harbor bi‐allelic variants in CEP83. To the best of our knowledge, these patients are the first with a CEP83 deficiency without kidney disease.
Although pathogenicity is not yet been proven, we consider the p.Leu194Pro variant very likely to be disease‐causing in our reported patients. First, based on the ACMG guidelines, pathogenicity of this variant is supported by lines of evidence supporting evolutionary conservation (pathogenic supporting argument 3) and almost complete absence of the variant in population databases (pathogenic moderate argument 2) (Richards et al., 2015). Pathogenicity is also supported by the predicted loss of function effect of the variant and the fact that in P1, WES revealed no other variants that could explain the phenotype. Moreover, the phenotype suggests pathogenicity (pathogenic supporting argument 4) of the variant, as retinal dystrophy has also been reported in two other patients with CEP83 deficiency: one patient with retinitis pigmentosa who is homozygous for the same p.Leu194Pro variant (Haer‐Wigman et al., 2017), and a second patient with retinitis and a truncating p.Arg209* mutation, located in the same coiled‐coil domain as the p.Leu194Pro variant (Failler et al., 2014). Interestingly, as the parents of both patients originate from the same region in the Netherlands, and the patient reported by Haer‐Wigman et al. is also from Dutch origin, it might be considered that p.Leu194Pro is a founder mutation. It would be interesting to analyze the exome data of these three families to see if a common haplotype could be delineated, but unfortunately the families did not give permission for such an analysis.
It is likely that a CEP83 deficiency could result in retinal dystrophy, as degeneration of retinal photoreceptors is common in many ciliopathies (Waters & Beales, 2011). CEP83 is localized in the cilia of the photoreceptors that are located in the light‐sensitive outer layer of the retina. The outer segments of these photoreceptors are formed by modified sensory cilia. They function as cellular antenna and are essential for proper functioning of the photoreceptors in the retina (Yildiz & Khanna, 2012). The central strand of the cilium—the axoneme—connects the outer and inner segment of the photoreceptor and acts as a backbone for membranic discs, loaded with proteins essential for phototransduction. These peripheral and integral membrane proteins create a platform from the cilia that is essential for signaling cascades by acting as an entrance for extrinsic signals to the internal part of the photoreceptor (Bujakowska et al., 2017; Yildiz & Khanna, 2012). In CEP83 knockdown human retinal epithelial cells, ciliogenesis is inhibited, evidencing the essential role of CEP83 in ciliogenesis (Joo et al., 2013). Moreover, both reduced protein expression and partial impairment of the protein already affect assembly of the cilium to the mother centriole, as well as affected recruitment of CEP164 in the DAP of the mother centriole (Failler et al., 2014). Thus, in summary, CEP83 deficiency results in defective primary cilium assembly, leading to cilia dysfunction in photoreceptors of the retina, and can thereby cause retinal dystrophy (Bujakowska et al., 2017; Waters & Beales, 2011).
Next to retinal dystrophy, the other features that the patients present with could theoretically all be linked to the ciliopathy spectrum. Though, both patients lack the most remarkable and more serious features, such as kidney dysfunction, liver abnormalities and neurological abnormalities. Absence of these features could reflect a milder phenotype, but it is possible that other symptoms will manifest at a later age. The patient reported by Haer‐Wigman et al., that is also homozygous for the p.Leu194Pro variant, was diagnosed with retinitis pigmentosa at the age of 56, and mild renal insufficiency was noted at the age of 47 (Haer‐Wigman et al., 2017). For this reason, we will perform a meticulous follow‐up of our reported patients to screen for signs of kidney dysfunction, liver abnormalities, and observation of potential neurological symptoms. Likewise, for patients with CEP83 deficiency without retinal dystrophy, we urge awareness of the potential vision problems, and we recommend regular consultations with an ophthalmologist.
To date, gene panels for retinal dystrophy do not include CEP83 (RetNet, http://sph.uth.edu/RetNet/), except for the vision gene panel of the Radboud UMC Nijmegen (Radboud UMC Nijmegen, https://order.radboudumc.nl/producten/wes-visusstoornissen?s=31). As we here present patients with CEP83 deficiency that presented with retinitis pigmentosa without any form of kidney dysfunction, we advocate inclusion of CEP83 in all gene panels for inherited retinal diseases. In addition, we speculate that in the future, these gene panels might also require further inclusion of other genes associated with ciliopathies.
In conclusion, we here present the first two patients that presented with retinitis pigmentosa in the absence of kidney dysfunction in childhood, as a result of bi‐allelic variants in CEP83. Hereby, we expand the phenotypic spectrum of CEP83 deficiency. We urge awareness of potential vision problems in CEP83 deficient patients, and we advocate that CEP83 should be included in gene panels for inherited retinal diseases.
CONFLICT OF INTEREST
The authors have no potential conflict of interest.
AUTHORS' CONTRIBUTIONS
Bram C. F. Veldman, Willemijn F. E. Kuper, Peter M. van Hasselt, and Hanneke A. Haijes conceptualized and designed the study. Marc Lilien, Janneke H. M. Schuurs‐Hoeijmakers, Ymkje Hettinga, and Peter M. van Hasselt were involved in the acquisition of clinical data. Janneke H. M. Schuurs‐Hoeijmakers, Carlo Marcelis, and Peter M. van Hasselt contributed with the acquisition of exome data and genomic sequencing annotation. Ymkje Hettinga and Herman E. Talsma interpreted the ophthalmological examination of P1, Milan Phan of P2. Bram C. F. Veldman and Herman E. Talsma were involved in the visualization of the ophthalmological examination. Bram C. F. Veldman wrote the manuscript, Willemijn F. E. Kuper, and Hanneke A. Haijes revised and edited the manuscript. All authors reviewed the manuscript critically for important intellectual content and approved the final version of the manuscript for publication. Hanneke A. Haijes supervised the complete project.
Supporting information
Figure S1 ISCEV Standard electroretinogram (ERG) showing normal responses (top) and the responses of P1 (middle) and P2 (bottom). Particularly the dark adapted responses are impaired with reduced amplitudes and delayed a‐ and b‐ waves, confirmative of rod‐cone dystrophies.
ACKNOWLEDGMENTS
The authors would like to thank the patients and their families for participating in this study. Special thanks to K.L.I. van Gassen from the University Medical Center Utrecht and L. Haer‐Wigman from the Radboud University Medical Center Nijmegen for their contribution and full support.
Veldman BCF, Kuper WFE, Lilien M, et al. Beyond nephronophthisis: Retinal dystrophy in the absence of kidney dysfunction in childhood expands the clinical spectrum of CEP83 deficiency. Am J Med Genet Part A. 2021;185A:2204–2210. 10.1002/ajmg.a.62225
Funding information Radboud University Medical Center
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, G. , Smith, V. V. , Malone, M. , & Sebire, N. J. (2005). Blood film examination for vacuolated lymphocytes in the diagnosis of metabolic disorders; retrospective experience of more than 2500 cases from a single centre. Journal of Clinical Pathology, 58(12), 1305–1310. 10.1136/jcp.2005.027045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, D. A. , & Hildebrandt, F. (2017). Ciliopathies. Cold Spring Harbor Perspectives in Biology, 9(3), a028191. 10.1101/cshperspect.a028191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujakowska, K. M. , Liu, Q. , & Pierce, E. A. (2017). Photoreceptor cilia and retinal ciliopathies. Cold Spring Harbor Perspectives in Biology, 9(10). 10.1101/cshperspect.a028274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. , & Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and Indels. Bioinformatics, 31(16), 2745–2747. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failler, M. , Gee, H. Y. , Krug, P. , Joo, K. , Halbritter, J. , Belkacem, L. , Filhol, E. , Porath, J. D. , Braun, D. A. , Schueler, M. , Frigo, A. , Alibeu, O. , Masson, C. , Brochard, K. , de Ligny, B. H. , Novo, R. , Pietrement, C. , Kayserili, H. , Salomon, R. , … Saunier, S. (2014). Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. American Journal of Human Genetics, 94(6), 905–914. 10.1016/j.ajhg.2014.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haer‐Wigman, L. , Van Zelst‐Stams, W. A. G. , Pfundt, R. , Van Den Born, L. I. , Klaver, C. C. W. , Verheij, J. B. G. M. , Haer‐Wigman, L. , van Zelst‐Stams, W. A. , Pfundt, R. , van den Born, L. I. , Klaver, C. C. , Verheij, J. B. , Hoyng, C. B. , Breuning, M. H. , Boon, C. J. , Kievit, A. J. , Verhoeven, V. J. , Pott, J. W. , Sallevelt, S. C. , … Yntema, H. G. (2017). Diagnostic exome sequencing in 266 Dutch patients with visual impairment. European Journal of Human Genetics, 25(5), 591–599. 10.1038/ejhg.2017.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo, K. , Kim, C. G. , Lee, M. S. , Moon, H. Y. , Lee, S. H. , Kim, M. J. , Kweon, H. S. , Park, W. Y. , Kim, C. H. , Gleeson, J. G. , & Kim, J. (2013). CCDC41 is required for ciliary vesicle docking to the mother centriole. Proceedings of the National Academy of Sciences of the United States of America, 110(15), 5987–5992. 10.1073/pnas.1220927110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , Seaby, E. G. , Kosmicki, J. A. , Walters, R. K. , … MacArthur, D. G. (2019). Variation across 141,456 human exomes and genomes reveals the Spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv, 531210. 10.1101/531210 [DOI] [Google Scholar]
- Kuper, W. F. E. , Oostendorp, M. , Van den Broek, B. T. A. , Van Veghel, K. , Nonkes, L. J. P. , Nieuwenhuis, E. E. S. , Fuchs, S. A. , Veenendaal, T. , Klumperman, J. , Huisman, A. , Nierkens, S. , & Van Hasselt, P. M. (2020). Quantifying lymphocyte vacuolization serves as a measure of CLN3 disease severity. JIMD Reports, 54(1), 87–97. 10.1002/jmd2.12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Benson, M. , Brown, G. R. , Chao, C. , Chitipiralla, S. , Gu, B. , Hart, J. , Hoffman, D. , Jang, W. , Karapetyan, K. , Katz, K. , Liu, C. , Maddipatla, Z. , Malheiro, A. , McDaniel, K. , Ovetsky, M. , Riley, G. , Zhou, G. , … Maglott, D. R. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Research, 46(D1), D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, C. H. , Lin, I. H. , Yang, T. T. , Huang, Y. C. , Tanos, B. E. , Chou, P. C. , Chang, C. W. , Tsay, Y. G. , Liao, J. C. , & Wang, W. J. (2019). Phosphorylation of CEP83 by TTBK2 is necessary for cilia initiation. The Journal of Cell Biology, 218(10), 3489–3505. 10.1083/jcb.201811142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Sue. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter, S. A. J. , Hurks, P. P. M. , & Timmerman, M. E. (2017). IQ‐score is Dringend Aan Modernisering toe: Naar Een Nieuwe Interpretatie En Classificatie van Geschatte Intelligentie [The IQ score is in dire need of modernisation: Towards a new interpretation and classification for estimating intelligence]. Kind En Adolescent Praktijk, 16, 16–23. 10.1007/s12454-017-0005-y [DOI] [Google Scholar]
- Santos, R. L. P. , Aulchenko, Y. S. , Huygen, P. L. M. , Van der Donk, K. P. , De Wijs, I. J. , Kemperman, M. H. , Admiraal, R. J. C. , Kremer, H. , Hoefsloot, L. H. , & Cremers, C. W. R. J. (2005). Hearing impairment in Dutch patients with Connexin 26 (GJB2) and Connexin 30 (GJB6) mutations. International Journal of Pediatric Otorhinolaryngology, 69(2), 165–174. 10.1016/j.ijporl.2004.08.015 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). Mutationtaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Sim, N. L. , Kumar, P. , Hu, J. , Henikoff, S. , Schneider, G. , & Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research, 40(W1), 452–457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S. , Boutaud, L. , Reilly, M. L. , & Benmerah, A. (2019). Cilia in hereditary cerebral anomalies. Biology of the Cell, 211, 217–231. 10.1111/boc.201900012 [DOI] [PubMed] [Google Scholar]
- Waters, A. M. , & Beales, P. L. (2011). Ciliopathies: An expanding disease spectrum. Pediatric Nephrology, 26(7), 1039–1056. 10.1007/s00467-010-1731-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, M. M. , Van der Zwaag, B. , Jongbloed, J. D. H. , Vogel, M. J. , Brüggenwirth, H. T. , Lekanne Deprez, R. H. , Mook, O. , Ruivenkamp, C. A. L. , van Slegtenhorst, M. A. , van den Wijngaard, A. , Waisfisz, Q. , Nelen, M. R. , & Van der Stoep, N. (2013). Best practice guidelines for the use of next‐generation sequencing applications in genome diagnostics: A national collaborative study of Dutch genome diagnostic laboratories. Human Mutation, 34(10), 1313–1321. 10.1002/humu.22368 [DOI] [PubMed] [Google Scholar]
- Wheway, G. , & Mitchison, H. M. (2019). Opportunities and challenges for molecular understanding of ciliopathies—The 100,000 genomes project. Frontiers in Genetics, 10, 1–21. 10.3389/fgene.2019.00127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. T. , Chong, W. M. , Wang, W. J. , Mazo, G. , Tanos, B. , Chen, Z. , Tran, T. M. N. , Chen, Y.‐D. , Weng, R. R. , Huang, C.‐E. , Jane, W.‐N. , Tsou, M.‐F. B. , & Liao, J. C. (2018). Super‐resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nature Communications, 9(1), 1–11. 10.1038/s41467-018-04469-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz, O. , & Khanna, H. (2012). Ciliary signaling cascades in photoreceptors. Vision Research, 75, 112–116. 10.1016/j.visres.2012.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ISCEV Standard electroretinogram (ERG) showing normal responses (top) and the responses of P1 (middle) and P2 (bottom). Particularly the dark adapted responses are impaired with reduced amplitudes and delayed a‐ and b‐ waves, confirmative of rod‐cone dystrophies.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.