

Abstract
Anomalous heat waves are causing a major decline of hard corals around the world and threatening the persistence of coral reefs. There are, however, reefs that have been exposed to recurrent thermal stress over the years and whose corals appear to have been tolerant against heat. One of the mechanisms that could explain this phenomenon is local adaptation, but the underlying molecular mechanisms are poorly known. In this work, we applied a seascape genomics approach to study heat stress adaptation in three coral species of New Caledonia (southwestern Pacific) and to uncover the molecular actors potentially involved. We used remote sensing data to characterize the environmental trends across the reef system, and sampled corals living at the most contrasted sites. These samples underwent next generation sequencing to reveal single nucleotide polymorphisms (SNPs), frequencies of which were associated with heat stress gradients. As these SNPs might underpin an adaptive role, we characterized the functional roles of the genes located in their genomic region. In each of the studied species, we found heat stress‐associated SNPs located in proximity of genes involved in pathways well known to contribute to the cellular responses against heat, such as protein folding, oxidative stress homeostasis, inflammatory and apoptotic pathways, and DNA damage‐repair. In some cases, the same candidate molecular targets of heat stress adaptation recurred among species. Together, these results underline the relevance and the power of the seascape genomics approach for the discovery of adaptive traits that could allow corals to persist across wider thermal ranges.
Keywords: coral bleaching, coral reef, heat stress, local adaptation, seascape genomics
1. INTRODUCTION
One of the most dramatic consequences of climate change is the worldwide decline of coral reefs, which are the most biodiverse ecosystems in the marine environment (Hughes et al., 2017). Among the main drivers of this decline is coral bleaching, a stress response to anomalous heat waves that eventually causes the death of hard corals (Bellwood et al., 2004; Hughes et al., 2017). In the most severe episodes, coral bleaching has provoked local coral cover loss of up to 50% (Hughes et al., 2017, 2018), with climate change projections expecting for bleaching conditions to be persistent worldwide by 2050 (Van Hooidonk et al., 2013).
Despite these alarming perspectives, a glimpse of hope is brought by coral reefs that show resistance after recurrent heat waves (Dance, 2019; Hughes et al., 2019; Krueger et al., 2017; Penin et al., 2013; Thompson & van Woesik, 2009). One of the mechanisms that might promote heat tolerance in corals is genetic adaptation (Sully et al., 2019). Indeed, genetic features potentially involved in thermal tolerance were recently identified in corals from reefs recurrently exposed to heat stress in Japan (Selmoni et al., 2020), on the Great Barrier Reef (Fuller et al., 2020) and along the western coast of Australia (Thomas et al., 2017). In recent years, there has been a growing body of literature investigating how coral thermal adaptation might alter the predictions of reef persistence, and how conservation policies could be modified accordingly (Logan et al., 2014; Matz et al., 2018; van Oppen et al., 2015).
Given the crucial role adaptation will play in long‐term reef persistence, there is an urgent need to characterize the adaptive potential of corals (Logan et al., 2014; van Oppen et al., 2015). For instance, there are still open questions concerning the spatial and temporal scales at which local adaptation operates (Matz et al., 2018; Roche et al., 2018). Changes in adaptive potential against heat stress have been observed along thermal gradients over hundreds of kilometres (e.g., Thomas et al., 2017), but also at reefs with distinct thermal variations located only a few hundred metres apart (e.g., Bay & Palumbi, 2014). Furthermore, different coral species are reported to show differential vulnerability against thermal stress, leading to the question of how different life‐history traits (e.g., reproductive strategies, morphology) drive the pace of adaptation (Darling et al., 2012; Hughes et al., 2018; Loya et al., 2001).
There are also open questions concerning the molecular mechanisms that might be targeted by heat stress adaptation in corals (Mydlarz et al., 2010; van Oppen & Lough, 2009; Palumbi et al., 2014). Some cellular responses to heat stress are now well characterized, such as DNA repair mechanisms, the activation of the protein folding machinery in the endoplasmic reticulum (ER) or the accumulation of reactive oxygen species (ROS, either endogenous or produced by the symbiont) that progressively elicits inflammatory and apoptotic responses (Maor‐Landaw & Levy, 2016; Mydlarz et al., 2010; Oakley et al., 2017; van Oppen & Lough, 2009; Patel et al., 2018). However, little is known about which of the many molecular actors participating in these cascades could be targeted by evolutionary processes to increase thermal tolerance.
Seascape genomics could contribute to filling these gaps. Seascape genomics is a budding field of population genomics that allows the study of local adaptation in wild populations (Riginos et al., 2016). This method combines the environmental characterization of the seascape with a genomic analysis of its population (Rellstab et al., 2015). The goal is to identify genetic variants that correlate with environmental gradients and that might underpin an adaptive role (Rellstab et al., 2015). Seascape genomics could enhance the characterization of coral adaptive potential because: (i) it requires an extensive sampling strategy that allows the study of adaptation at different geographical scales, and against different types of environmental constraints simultaneously (e.g., mean temperatures, standard deviations, accumulated heat stress; Leempoel et al., 2017; Selmoni et al., 2020); (ii) its experimental protocol is less laborious in comparison with traditional approaches used for studying coral adaptation (e.g., aquarium experiments, transplantations), and therefore facilitates scaling‐up to a multispecies analysis; and (iii) it is based on genomic data and thus reports candidate molecular targets of adaptation (Rellstab et al., 2015; Riginos et al., 2016). Moreover, recent work has described how the results of seascape genomics studies on corals can be directly transposed to a conservation perspective and support reef prioritization (Selmoni, Rochat, et al., 2020).
Here we applied the seascape genomics approach to uncover molecular actors potentially implicated in heat stress adaptation in three bleaching‐prone coral species of New Caledonia, in the southwestern Pacific (Figure 1). We first used publicly available satellite data to characterize the seascape conditions for over 1,000 km of the reef system. Coral samples were collected at 20 sites exposed to contrasted environmental conditions. The collected samples underwent a genotype‐by‐sequencing (DArT‐seq) genomic characterization, followed by a seascape genomics analysis accounting for the confounding role of demographic structure. This allowed us to uncover single nucleotide polymorphisms (SNPs) associated with heat stress. We then analysed the functional annotations of genes in proximity of these SNPs and found molecular targets that notably recurred among species and that referred to well‐established heat stress responses in coral cells. Our study highlights the relevance and power of seascape genomics to uncover candidate molecular targets of heat stress adaptation in corals.
FIGURE 1.
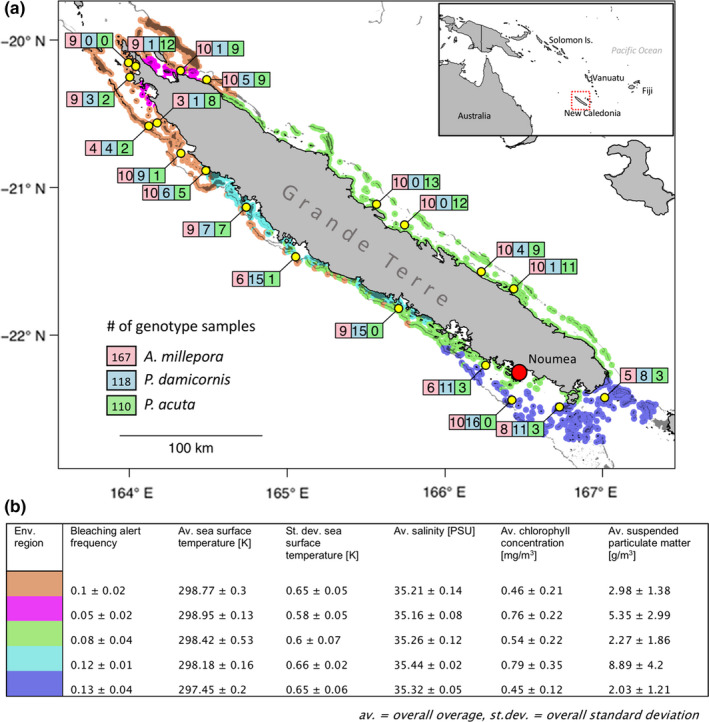
Study area, sampling sites and environmental regions. In (a), the 20 sampling sites around Grande Terre, the main island of New Caledonia (southwest Pacific), are shown in yellow. For every sampling site, the number of genotyped individuals per species (Acropora millepora: red, Pocillopora damicornis: blue, Pocillopora acuta: green) are given in the corresponding boxes. In the background, coral reefs surrounding Grande Terre are highlighted in five colours representing distinct environmental regions. In (b), environmental characteristics discriminating the five environmental regions are shown
2. MATERIALS AND METHODS
2.1. Environmental data
The seascape genomics approach requires an exhaustive description of the environmental conditions in order to prevent the misleading effect of collinear gradients (Leempoel et al., 2017; Riginos et al., 2016). For this reason, the seascape characterization we used encompassed seven environmental variables: sea water temperature (SST), chlorophyll concentration, sea surface salinity, sea current velocity, suspended particulate matter and bleaching alert frequencies (BAF; Table S1). The environmental characterization was performed in the R environment (R Core Team, 2009, 2016) using the raster package (Hijmans, 2016) and following the method described in previous work on coral seascape genomics (Selmoni, Rochat, et al., 2020) with some modifications outlined hereafter.
For the characterization of SST we used two different georeferenced data sets covering the extent of New Caledonia: (i) daily records of SST since 1981 at a spatial resolution of 5 km (SST5 km; EU Copernicus Marine Service, 2017); and (ii) daily records of SST since 2002 at resolution of 1 km (SST1 km; Group for High Resolution Sea Surface Temperature; Chao et al., 2009; Chin et al., 2017). The first data set covers a wider temporal range, therefore providing a more reliable characterization of historical trends. The second data set covers a smaller temporal window, but the higher geographical resolution allows the characterization of fine‐scale thermal patterns with a higher degree of confidence. Both data sets were used to compute, for each pixel of the study area, averages and standard deviations of the warmest month, the coldest month and the entire observational period. Furthermore, we computed the frequency of the bleaching warning conditions as defined by the Coral Reef Watch, corresponding to the accumulation of heat stress over a 3‐month rolling window (Liu et al., 2003). Two BAF variables were computed, one based on SST data at 5‐km resolution (hereafter referred to as BAF5) and one at 1‐km resolution (BAF1).
For the other data sets (chlorophyll concentration, sea surface salinity, sea current velocity and suspended particulate matter; EU Copernicus Marine Service, 2017), the spatial resolution ranged between 4 and 9 km (Table S1). All the data sets covered a temporal extent of at least 20 years before 2018 (the year of sampling) and were processed to compute: (i) highest monthly average, (ii) lowest monthly average and (iii) overall average. For all the data sets captured at daily resolution (i.e., all except suspended particulate matter), we also computed the standard deviation associated with the three averages.
In total, 35 environmental descriptors (Table S1) were computed. The polygons representing the reefs of New Caledonia (UNEP‐WCMC, WorldFish‐Center, WRI, & TNC, 2010) were reported in a regular grid (~3,000 reef cells of size: 2 × 2 km) using qgis (QGIS development team, 2009, 2016). Using the extract function of the raster package, we assigned a value of each of the 35 environmental descriptors to every reef cell.
2.2. Sampling
Twenty sampling sites were selected out of the ~3,000 reef cells surrounding Grande Terre, the main island of New Caledonia (Figure 1). Sampling sites were chosen following an approach that simultaneously maximized environmental contrasts and replicated them at distant sites (the “hybrid approach” described in Selmoni, Vajana, et al., 2020). The method consists of applying principal component analysis (PCA) and hierarchical clustering to the 35 environmental descriptors in order to separate the ~3,000 reef cells into distinct environmental regions. Next, the algorithm selects the same number of sampling sites within each region in order to maximize physical distance between sites. Increasing environmental variation is expected to raise the sensitivity of seascape genomics analysis, while the replication of environmental gradients is expected to reduce false discovery rates (Selmoni, Vajana, et al., 2020). Here the number of environmental clusters was five (Figure 1) and we established four sampling locations per cluster. When this was not possible (e.g., because of logistical constraints during the sampling campaign), additional sampling sites were added to the neighbouring clusters.
The sampling campaign was performed from February to May 2018 (under permit Nos. 609011‐/2018/DEPART/JJC and 783‐2018/ARR/DENV) and targeted three flagship species of the Indo‐Pacific: Acropora millepora, Pocillopora damicornis sensu Schmidt‐Roach et al. (2013)—corresponding to PSH04 sensu Gélin et al. (2017) ‐ and Pocillopora acuta sensu Schmidt‐Roach et al. (2013)—corresponding to PSH05 sensu Gélin et al. (2017). Of note, P. acuta and P. damicornis belong to the complex of species formerly named P. damicornis (Gélin et al., 2017; Johnston et al., 2017; Schmidt‐Roach et al., 2014). At every sampling site, we collected up to 20 samples of A. millepora and 20 of Pocillopora aff. damicornis (we did not discriminate between species while sampling as it can be difficult to distinguish them in the field, see section “Pocillopora species identification”). All the samples were collected within a radius of 1 km from the coordinate of the sampling site, and at a depth ranging between 2 and 4 m. Before sampling, each colony was imaged underwater, and then a piece of a branch was sampled with pliers. Each sample consisted of a 1‐ to 2‐cm branch that was immediately transferred to 80% ethanol and stored at −20°C. DNA from the 730 samples (370 A. millepora and 360 Pocillopora; Table S2) was extracted using the DNeasy Blood and Tissue 96 kit (Qiagen) following the manufacturer's instructions. Note that we did not apply mechanical shearing before DNA extraction, as this could have caused breaking of the cell wall of symbiotic algae and consequent contamination of the coral host DNA sample with high levels of symbiont DNA.
2.3. Pocillopora species identification
The 360 Pocillopora samples were identified molecularly a posteriori of sampling to be assigned to one species or the other (P. damicornis or P. acuta). Samples were genotyped using 13 microsatellite loci, as in Gélin et al. (2017). Then, colonies belonging to P. damicornis and to P. acuta were identified using assignment tests performed with structure (version 2.3.4; Pritchard et al., 2000; Figure S1, Table S2), as in Gélin et al. (2018). Colonies assigned to P. damicornis or P. acuta with a probability of at least.70 were retained in the final data set for this study. The Pocillopora sampling was composed of 148 P. damicornis (SSH04 sensu Gélin et al. 2017, more precisely SSH04a sensu Oury et al. 2020), 159 P. acuta colonies (a mix of SSH05a and SSH05b sensu Gélin, Fauvelot, et al., 2018) and 53 unassigned colonies (excluded from further analysis).
2.4. Acropora species identification
Acropora species are genetically and morphologically notoriously challenging in terms of identification and species boundaries detection. However, A. millepora can be recognized in the field based on its typical axial and radial corallite shape (Wallace, 1999). In situ images of each specimen were examined to look for the species diagnostic morphological characters and initial identifications were validated.
2.5. Screening and SNP genotyping
All DNA samples from A. millepora, P. damicornis and P. acuta were sent to the Diversity Arrays Technology (Canberra, Australia) for quality check screening and genotype‐by‐sequencing using the DArT‐sequencing method (DArT‐seq; Kilian et al., 2012). This approach, a variant of restriction enzyme‐associated DNA sequencing (RAD‐seq) techniques, employs restriction enzymes that are sensitive to DNA methylation. The result is a sequencing library that is enriched in hypomethylated regions of the genome. Such regions are more often functionally active regions (such as regulatory sequences or genes) and therefore of particular interest to study adaptation (Gawroński et al., 2016).
The restriction enzymes used for library preparation for A. millepora and Pocillopora samples were PstI and HpaII. Prior to sequencing, all the DNA samples underwent 1 hr of incubation with the digestion buffer, followed by quality check for integrity, purity and concentration running 1 µl per sample on a 0.8% agarose gel. Samples from each site were then ranked based on their quality (degree of smearing on the agarose gel). We then selected the samples with the best scores from each site and defined a list of 188 A. millepora, 128 P. damicornis and 150 P. acuta samples that proceeded to the sequencing step in four (A. millepora samples) and five lanes (Pocillopora samples) of an Illumina Hiseq2500. During each step of the workflow (DNA purification, library preparation and sequencing), A. millepora and Pocillopora samples were kept separated and randomly distributed across the respective batches (e.g., 96‐well plates, sequencing lanes) to minimize the risks of technical bias.
The processing of sequencing reads was performed by Diversity Arrays Technology using proprietary DArT analytical pipelines (Sansaloni et al., 2011). Raw sequencing reads were filtered based on PHRED quality scores (Q < 25) and demultiplexed on the barcodes. SNP calling was performed using the DArTSoft14 algorithm, which performs de novo alignment of reads with a procedure technically similar to stacks (Catchen et al., 2013).
2.6. SNP filtering
The DArT‐loci (i.e., the DNA sequences surrounding each SNP) initially underwent a sequence similarity search (blastn; version 2.7.1; Madden & Coulouris, 2008) against a reference genome to retain only those associated with the coral host (and discard possible contamination from the DNA of symbiotic algae or bacteria). For A. millepora, the reference genome was the A. millepora chromosome‐level assembly from Fuller and colleagues (version 2, Fuller et al., 2020), while for P. damicornis and P. acuta we used the P. damicornis sensu lato reference (version 1; Cunning et al., 2018). Only DArT‐loci scoring an E‐value below 10−6 were retained.
The processing of the SNP data followed a pipeline from previous work on coral seascape genomics (Selmoni, Rochat, et al., 2020). For each species’ data set, we removed SNPs and individuals with high missing rates (> 50%) by using custom functions (available with the study data, see “Data Accessibility” section) in the R environment. Next, we proceeded with imputation of missing genotypes using the linkimpute algorithm (based on k‐nearest‐neighbours imputation; Money et al., 2015) implemented in tassel 5 (Bradbury et al., 2007) using the default settings. We then repeated the filtering of SNPs and individuals for missing rates, but this time using a more stringent threshold (5%). We also applied a filter to exclude rare alleles (minor allele frequency <5%) and highly frequent genotypes (major genotype frequency >95%). The goal of these filtering steps was to remove either highly rare or highly frequent genetic variants, as these have been shown to create bias in population structure analyses (Roesti et al., 2012). SNPs were then filtered for linkage disequilibrium using the R package snprelate (function snpsgdsLDpruning, LDthreshold =0.3, version 1.16; X. Zheng et al., 2012). This filtering step anticipated the issue of spurious genotype–environment associations due to SNPs that were physically close (genetic hitchhiking, Rellstab et al., 2015). Finally, we applied a filter for clonality: when groups of colonies shared highly correlated genotypes (Pearson correlation >.9) only one colony per group was kept. As fragmentation is a common reproductive strategy in corals (Highsmith, 1982), this filter prevented us from retaining clonal individuals in the data set.
2.7. Neutral genetic structure analysis
Prior to the genotype–environment association analysis, we assessed the neutral genetic structure of the studied populations. For each of the studied species, the analysis was conducted in two steps: (i) identification of neutral SNPs and (ii) investigation of population structure using discriminant analysis of principal components (DAPC).
We first identified the principal components (PCs) underlying the neutral genetic structure (with p <.01: 11 in A. millepora, 19 in P. damicornis and 13 in P. acuta) using Tracy–Widom test as implemented in the R package assoctests (version 0.4; Wang et al., 2017). We then used the R package pcadapt (version 4.3.1; Luu et al., 2017) to perform a genome scan and detect outlier SNPs (i.e., SNPs with diverging allele frequencies, compared to the natural genetic structure). Outlier SNPs were discarded from the neutral genotype matrix employed in the second step of the analysis (genotype–environment association analysis).
The population structure was analysed using the DAPC method (Jombart et al., 2010) as implemented in the adegenet R package (version 2.1.2, Jombart, 2008). For a given genotype matrix, this method investigates the axes of variation (discriminant functions) maximizing the variance between groups of individuals. Groups of individuals can be either specified with a priori information (e.g., sampling sites) or inferred without a priori information using the find.cluster function. We tested both approaches, following the recommended practices described in DAPC documentation as outlined hereafter.
The find.cluster method combines k‐means clustering with a Bayesian inference criterion to select the optimal number of groups. We investigated this optimal number of groups using all the PCs of the genotype matrix. For the DAPC computation using sampling locations as a priori, we used the optim.a.score function to determine the optimal number of PCs to retain for the discriminant analysis (19 for A. millepora, 12 for P. damicornis and 19 for P. acuta). We assessed the population structure by comparing the distribution of samples across the first two discriminant functions with their spatial distribution across the archipelago.
In addition, we estimated the levels of genetic diversity at every sampling location by computing the minor allele frequency of every SNP. This calculation was applied to the three species and concerned sampling locations having at least five samples.
2.8. Genotype–environment association analysis
The genotype–environment association analyses were performed separately on the three species using the LFMM method implemented in the lea R package (version 2.4.0; Frichot & François, 2015; Frichot et al., 2013). This method associates single environmental gradients and individual SNP variations in mixed models, where the confounding effect of neutral genetic variation is accounted for through latent factors (Frichot et al., 2013).
Briefly, the first step of the LFMM pipeline is to estimate the number of latent factors (K; Frichot & François, 2015). This parameter corresponds to the number of ancestral populations and can be estimated by using the snmf function of the lea package. The method processes a genotype matrix to estimate individual admixture coefficients under different values of K, and then evaluates the quality of fit for each K via cross‐validation (Frichot & François, 2015). We ran 10 replicates of this analysis for all the studied species, and found that the optimal number of K (according to the lowest entropy criterion) ranged from 2 to 4 for A. millepora, 6 to 8 for P. damicornis, and 10 to 12 for P. acuta (Figure S2).
We then proceeded to the genotype–environment association analyses with LFMM. To avoid redundant signals of genotype–environment associations due to collinearity between environmental variables, we preliminarily grouped highly collinear environmental variables (absolute value of Pearson correlation >.9). This resulted in 16 groups of collinear variables in A. millepora (Table S3a), 15 in P. damicornis (Table S3b) and 19 in P. acuta (Table S3c). For every group, we randomly selected one environmental variable to be employed in the association analyses. The association analysis was performed using the lfmm function, setting K to the ranges previously estimated for each species and running five replicates of each analysis.
LFMM returns p‐values describing the statistical significance of every genotype–environment association under different values of K. For each association model related to the same environmental variable, p‐values were corrected for multiple testing using the q‐value method (R package q‐value, version 2.14; Storey, 2003) and deemed significant if q <.01 under at least one level of K.
2.9. Annotation analysis of heat stress‐associated SNPs
For each of the three studied species, we investigated the molecular functions of genes located in proximity of heat stress‐associated SNPs (i.e., SNPs associated with one of the two BAF variables, BAF5 or BAF1). To facilitate the comparisons between the different species, we first re‐annotated genes using the Uniprot/swissprot protein database as common reference (metazoa entries, release 2020_01; Boeckmann et al., 2003). We decided to use Uniprot/swissprot because it features standardized annotations of protein functions that are manually curated. The first step of the re‐annotation procedure was extraction of the predicted protein sequences for the genes in the original annotation of the two reference genomes (Cunning et al., 2018; Fuller et al., 2020). These protein sequences were used to perform a similarity search (blastp; version 2.71; Madden & Coulouris, 2008) against the Uniprot/swissprot database. Each gene was re‐annotated with the best significant hit (E‐value <0.01) and inherited protein name and gene ontology (GO) terms describing molecular functions (Ashburner et al., 2000).
We then proceeded in two stages to characterize the molecular functions of genes located in proximity of SNPs associated with heat stress. In a first step, we investigated the putative functions of the SNPs that are strongly associated with heat stress. We therefore focused only on the SNPs significantly (q <.01) associated with BAF5 or BAF1 and manually investigated the annotation of the closest gene with a functional annotation. Since we applied a conservative cut‐off threshold, only a limited number of genes was found and we therefore could not apply any enrichment analyses of gene functions (Maleki et al., 2020).
In a second step, we evaluated the redundancy in the annotations of groups of SNPs that are less strongly associated with heat stress. In this case we used the gene‐set enrichment method of the R package setrank (version 1.1; Simillion et al., 2017). SetRank features a “ranked analysis” where genes are first ranked based on a biologically meaningful criterion, and subsequently an enrichment test uncovers the GO annotations that are overrepresented among genes with the highest ranks. In our case, we retrieved genes located ±5 kb from every SNP and ranked them based on the (ascending) q‐value of the genotype–environment associations with BAF5 and BAF1. GO terms were deemed significant when the SetRank p‐value was <.01 and the adjusted p‐value (corrected for multiple testing and overlap between gene‐set categories) was <.05.
These two steps made it possible to identify molecular functions that recurred in proximity of SNPs associated with heat stress in the three different species.
3. RESULTS
The DArT‐seq analytical pipeline resulted in the genotyping of 188 samples by 57,374 bi‐allelic SNPs for Acropora millepora, and 128 and 150 samples by 70,640 SNPs for Pocillopora damicornis and Pocillopora acuta, respectively (Table 1). After filtering for similarity (E‐value <10−6) of the sequences surrounding SNPs against the reference genome, 47,529 SNPs were retained in A. millepora and 48,049 in the Pocillopora species. Before imputation of the genotype matrices, the percentage of missing genotypes per SNP was larger in A. millepora (average ± SD: 0.2 ± 0.15) than in P. damicornis (0.14 ± 0.15) and P. acuta (0.16 ± 0.15). After imputation, this percentage was comparable across the three species (A. millepora: 0.009 ± 0.05; P. damicornis: 0.002 ± 0.02; P. acuta: 0.001 ± 0.02).
TABLE 1.
Workflow of the analysis
| A. millepora | Pocillopora | ||
|---|---|---|---|
| P. damicornis | P. acuta | ||
| Sampling | 370 ind. | 360 ind. | |
| Microsatellite | – | 128 ind. | 150 ind. |
| DArT‐seq | 188 ind. × 57,374 SNPs | 128 ind. × 70,640 SNPs | 150 ind. × 70,640 SNPs |
| BLAST against reference | 188 ind. × 47,529 SNPs | 127 ind. × 48,049 SNPs | 145 ind. × 48,049 SNPs |
| Filtering (Missing values, MAF, MGF, LD, clonality) | 167 ind. × 11,935 SNPs | 118 ind. × 7,895 SNPs | 110 ind. × 8,343 SNPs |
For each of the species of interest (Acropora millepora, Pocillopora damicornis, Pocillopora acuta), we report the number of individuals (ind.) and single nucleotide polymorphisms (SNPs) obtained or retained after each of the various steps of the workflow.
MAF=minor allele frqeuency, MGF=major genotype frequency, LD=linkage disequilibrium.
After filtering for rare variants, missing values and clonality, we obtained a final genotype matrix of 167 individuals by 11,935 SNPs for A. millepora, of 118 individuals by 7,895 SNPs for P. damicornis and of 110 individuals by 8,343 SNPs for P. acuta (Table 1). The A. millepora genotyped samples distributed across all the 20 sampling sites (18 of having five samples or more), while genotyped samples of P. damicornis and P. acuta were distributed across 17 sites each (both with 10 sites having five samples or more). Samples from the Pocillopora species were found in sympatry at 15 sites, although P. damicornis appeared to be more frequent on sites on the west coast of Grande Terre, whereas P. acuta was more frequnt on the east coast (Figure 1; Figure S1).
3.1. Neutral genetic structure
We first ran a genome scan to distinguish neutral SNPs from outlier SNPs in each of the studied species. The analysis identified 346 outlier SNPs in A. millepora, 278 in P. damicornis and 265 in P. acuta (Table S4). The neutral SNPs (11,589 SNPs in A. millepora, 7,617 in P. damicornis and 8,078 in P. acuta) were retained for the analysis of the neutral genetic structure.
The analysis of population structure was performed running a DAPC of neutral genotype matrices. We first tested the analysis without a priori information on groups of samples to be discriminated. For the three studied species, the optimal number of clusters according to the Bayesian Inference Criterion was equal to one (Figure S3), and DAPC could therefore not be computed.
As we used sampling locations as a priori information on groups of samples, DAPC results showed that sampling locations that were spatially close tended to display similar values across the first two discriminant functions (Figure 2). This observation was particularly visible in the first discriminant function of the two Pocillopora species: in P. damicornis we observed a genetic cluster of sampling locations from the southwestern coast of Grande Terre (Figure 2b). In addition, the first two discriminant functions in P. damicornis highlighted the separation of a sampling site located in the northern part of the west coast. In P. acuta, the first discriminant function displayed a genetic cluster of sampling locations on the east coast of Grande Terre (Figure 2c).
FIGURE 2.
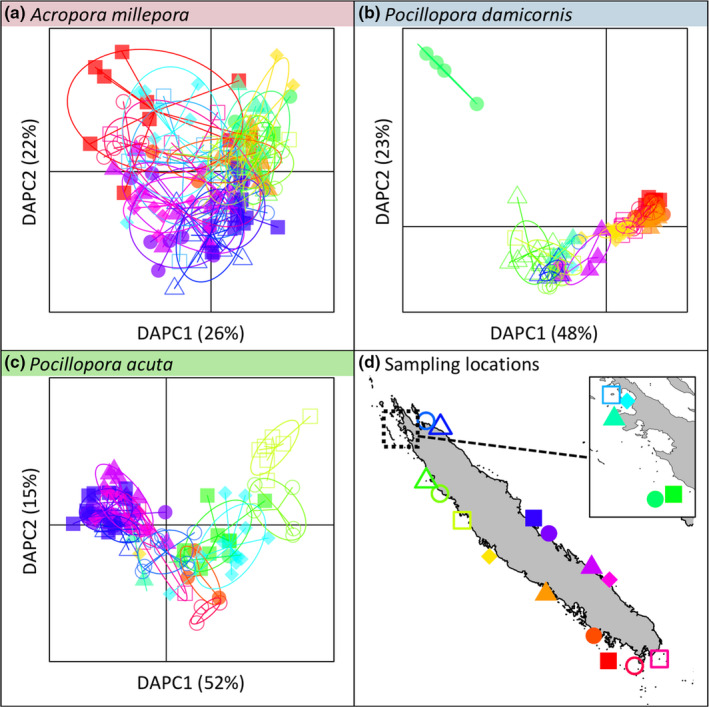
Discriminant analysis of principal components (DAPC) of the genotype matrices for three species studied: Acropora millepora (a), Pocillopora damicornis (b) and Pocillopora acuta (c). Each scatterplot displays the distribution of samples across the first two discriminant functions of the DAPC (with the percentage of variance explained, in parentheses). DAPC was performed with a priori information on sampling locations. Symbols correspond to the sampling location of samples and the position of these locations are displayed in (d)
Of note, the first discriminant function explained roughly half of the total variance of the genotype matrix in the Pocillopora species (48% in P. damicornis; 52% in P. acuta). In contrast, the percentage of variance explained by the first discriminant function in A. millepora was lower (26%) and comparable to the percentage of the second discriminant function (22%). Indeed, DAPC results for A. millepora suggested a larger genetic variation within sampling location (Figure 2a), compared to the Pocillopora species (Figure 2b,c).
The average (± SD) minor allele frequency (MAF) was 0.260 ± 0.16 in A. millepora, 0.262 ± 0.158 in P. damicornis and 0.279 ± 0.155 in P. acuta. In the studied species, the distribution of MAF across the different sampling locations did not show any particular spatial structure (Figure S4).
3.2. Local adaptation
In total, 20 SNPs were found significantly associated (q <.01) with heat stress variables (BAF5 or BAF1) for A. millepora, 25 for P. damicornis and nine for P. acuta (Table 2a). In comparison, all the other environmental descriptors of SST were found associated with 44 SNPs in A. millepora, 18 in P. damicornis and 18 in P. acuta (Table S5). For the other types of environmental descriptors, sea surface salinity was generally the one with the highest number of associated SNPs (79 associations across the three studied species), followed by chlorophyll concentration (65 associations), suspended particulate matter (13 associations) and sea current velocity (11 associations; Table S5).
TABLE 2.
Significant genotype–environment associations with heat stress in Acropora millepora, Pocillopora damicornis and Pocillopora acuta
| (a) Number of significant genotype–environment associations | |||
|---|---|---|---|
| ENV | collinear ENV | # SNPs | #uSNPs |
| Acropora millepora | |||
| BAF5 | SST5.osd, SST5.hsd | 10 | 20 |
| BAF1 | – | 14 | |
| Pocillopora damicornis | |||
| BAF5 | SST5.hsd, SST5.osd | 18 | 25 |
| BAF1 | – | 10 | |
| Pocillopora acuta | |||
| BAF5 | SST5.hsd, SSS.om, SSS.hm | 4 | 9 |
| BAF1 | – | 7 | |
| (b) Top five significant genotype–environment associations | |||||
|---|---|---|---|---|---|
| Closest gene | |||||
| Chr/Contig | Position | Name | Distance | ENV | q‐value |
| Acropora millepora | |||||
| chr2 | 15023481 | MICOS complex subunit MIC60 | 14044 BFE | BAF5 | 4.08E‐11 |
| Sc0000070 | 588818 | Low‐density lipoprotein receptor‐related protein 6 | 7929 BFS | BAF1 | .0001 |
| chr12 | 3092569 | Adipose triglyceride lipase | 1902 BFS | BAF1 | .0001 |
| chr3 | 16787176 | Enoyl‐[acyl‐carrier‐protein] reductase | CDS | BAF1 | .0002 |
| chr11 | 5209122 | Hemicentin‐2 | 16601 BFE | BAF1 | .0007 |
| Pocillopora damicornis | |||||
| NW_020846863.1 | 50886 | Melanocyte‐stimulating hormone receptor | 43840 BFE | BAF1 | 7.43E‐07 |
| NW_020844635.1 | 120593 | Omega‐3 fatty acid desaturase fat‐1 | 8241 BFS | BAF5 | 1.92E‐05 |
| NW_020846229.1 | 44431 | Protocadherin Fat 3 | CDS | BAF1 | .0001 |
| NW_020847397.1 | 2088 | Tumour necrosis factor receptor superfamily member 27 | 2079 BFS | BAF1 | .0005 |
| NW_020846232.1 | 92716 | Glutamine and serine‐rich protein 1 | 3792 BFE | BAF5 | .0007 |
| Pocillopora acuta | |||||
| NW_020845243.1 | 143820 | Quinone oxidoreductase PIG3 | Intronic | BAF1 | .0003 |
| NW_020847027.1 | 161943 | Multidrug resistance‐associated protein 4 | CDS | BAF1 | .0004 |
| NW_020845228.1 | 63533 | Receptor‐type tyrosine‐protein phosphatase delta | CDS | BAF1 | .0011 |
| NW_020845314.1 | 124 | — | — | BAF1 | .0013 |
| NW_020847378.1 | 374764 | Golgin subfamily A member 1 | Intronic | BAF1 | .0040 |
Table (a) displays the number of SNPs significantly associated (#SNPs; q < .01) with the environmental variables (ENV) describing bleaching alert frequency (BAF5 and BAF1, computed out of sea surface temperature data at 5 and 1 km of resolution, respectively). The ‘collinear ENV’ column displays the identifiers of environmental variables highly correlated (R > .9) with ENV. The #uSNPs column shows the number of SNPs found significant for at least one of the two BAF variables. Table (b) displays the five significant SNPs more strongly associated with BAF5 and BAF1. For every SNP, the table shows the genomic position (chromosome or contig, and position), the name of the closest known gene with the distance from the SNP (number of nucleotides before start—BFS—or before end—BFE, intronic or in coding sequence—CDS), the environmental variable involved in the association (ENV) and the q‐value of the association. The panel in top right position displays the key of the abbreviations for the identifiers of the environmental variables. Table S5 displays the count of significant SNPs for all the other (non‐collinear) environmental variables; Table S6 shows the complete list of SNPs significantly associated with BAF5 and BAF1. Environmental variables identifiers key: BAF5 = bleaching alert frequency (5‐km resolution) BAF1 = bleaching alert frequency (1‐km resolution) SST5 = sea surface temperature (5‐km resolution) SSS = sea surface salinity; .om = overall mean .hm = highest monthly mean .osd = overall standard deviation .hsd = standard dev. of month with highest mean.
3.3. Functional annotations of heat stress‐associated SNPs
For each of the studied species, we analysed the genome annotations to identify the genes located in proximity of SNPs significantly (q <.01) associated with heat stress. Such genes were generally located within 10 kb of the significant SNPs (15 out of 20 in A. millepora, 23 out of 25 in P. damicornis and all nine SNPs in P. acuta; Table S6). Among these, we found genes coding for the MICOS complex subunit MIC60 (q = 4.08E‐11; Figure 3), Low‐density lipoprotein receptor‐related protein 6 (q =.0001) and Enoyl‐[acyl‐carrier‐protein] reductase (q = 0.0002) in A. millepora; Melanocyte‐stimulating hormone receptor (q = 7.43E‐07), HMG‐CoA reductase (q =.002) and Malonyl‐CoA decarboxylase (q =.003) in P. damicornis; Quinone oxidoreductase PIG3 (q =.0003), Receptor‐type tyrosine‐protein phosphatase delta (q =.0011) and Golgin subfamily A member 1 (q =.004) in P. acuta (Table 2b, Table S6). Of note, we did not find any gene that recurred in proximity of significant SNPs from different species (Table S6).
FIGURE 3.
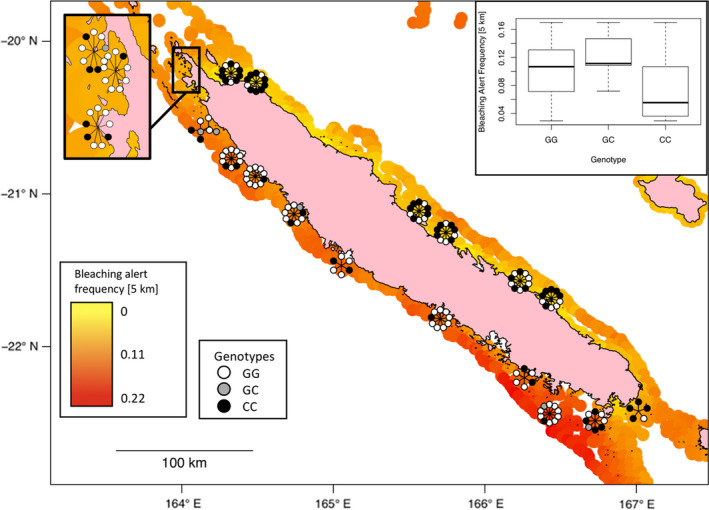
Example of significant genotype–environment associations. The map displays the superposition between environmental gradient (here the frequency of bleaching alert since 1981, measured at 5‐km resolution) and the distribution of an associated (q <.01) SNP of Acropora millepora. Every circle corresponds to the SNP genotype for an individual colony. For illustrative reasons, genotypes are radially distributed around the sampling locations. The boxplot in the top‐right corner shows how the environmental variable distributes within each genotype. The SNP represented here is located on chromosome 2 (position 15023481) of the A. millepora genome, and the closest annotated gene codes for the mitochondrial MICOS complex subunit MIC60
We then ranked all SNPs based on the q‐value describing the associations with BAF variables (BAF5 and BAF1), and investigated whether SNPs with the highest ranks (i.e., those more strongly associated with heat stress) were located in proximity (± 5 kb) of genes sharing identical molecular functions. We uncovered 18 GO terms of molecular functions as over‐represented (p <.01, adjusted p <.05) among such genes in A. millepora, 35 in P. damicornis and 34 in P. acuta (Table S7). Among these, we found terms referring to “chaperone binding” (GO: 0051087), “FAD binding” (GO: 0071949), “trans‐2‐enoyl‐CoA reductase (NADPH) activity” (GO: 0019166) and “p53 binding” (GO: 0002039) in A. millepora; “malonyl‐CoA decarboxylase activity” (GO: 0050080), “mitogen‐activated protein kinase binding” (GO: 0051019), “chaperone binding” (GO: 0051087), “Hsp70 protein binding” (GO: 0030544) and “Hsp90 protein binding” (GO: 0051879) in P. damicornis; “quinone binding” (GO: 0048038), “NADPH:quinone reductase activity” (GO: 0003960), “DNA helicase activity” (GO: 0003678), “p53 binding” (GO: 0002039) and “Hsp70 protein binding” (GO: 0030544) in P. acuta (Table S7). In total, five of these functions recurred in different species: “p53 binding,” “5–3 exoribonuclease activity,” “chaperone binding,” “Hsp70 protein binding” and “opioid receptor” (Table 3).
TABLE 3.
Functional annotations of heat stress‐associated SNPs
| GO term | GO term description | AM | PD | PA | |||
|---|---|---|---|---|---|---|---|
| BAF5 | BAF1 | BAF5 | BAF1 | BAF5 | BAF1 | ||
| GO:0002039 | p53 binding | x | x | ||||
| GO:0004534 | 5–3 exoribonuclease activity | x | x | ||||
| GO:0030544 | Hsp70 protein binding | x | x | ||||
| GO:0031628 | opioid receptor binding | x | x | ||||
| GO:0051087 | chaperone binding | x | x | ||||
For each of the studied species, Acropora millepora (AM; pink), Pocillopora damicornis (PD; blue) and Pocillopora acuta (PA; green), the table displays GO terms describing molecular functions that are overrepresented (p <.01, Adj. p <.1) in genes located in proximity of SNPs associated with the bleaching alert frequency variables (BAF5 and BAF1, computed from sea surface temperature data at 5‐km and 1‐km resolution, respectively). This table shows the GO terms observed as overrepresented in at least two of the studied species, while the complete list of all the GO terms is shown in Table S7.
4. DISCUSSION
4.1. Population structure
Prior to the analysis of local adaptation, we evaluated the population structure of the three studied species from New Caledonia. Such preliminary analysis is crucial, since strong structure of neutral genetic variation, for instance due to cryptic species or isolated populations, can cause bias in the investigation of adaptive genetic variants (Rellstab et al., 2015; Selmoni, Vajana, et al., 2020). However, the fact that we could not identify clear genetic clusters for running the DAPC without a priori information suggested the absence of genetically isolated groups in the three studied populations. This view was supported by the lack of clear differences in the frequency of minor alleles observed across the different sampling locations.
The DAPC using sampling locations as the grouping factor indicated the presence of a spatial structure in each of the three studied populations. In Acropora millepora, we observed a weak structure, with substantial variation within sampling locations. This observation was consistent with the weak structure recently observed in an A. millepora population from a section of the Australian Great Barrier Reef with spatial extent similar to the one of New Caledonia (Fuller et al., 2020).
In comparison, the population structure of the two Pocillopora species appeared to be more stressed. Corroborating these observations, previous work in New Caledonia and northwestern Australia suggested high levels of population differentiation in Pocillopora damicornis (Oury et al., 2020; L. Thomas et al., 2014), and in Pocillopora acuta in New Caledonia (Gélin et al., 2018).
Of note, we observed a higher frequency of P. damicornis along the west coast of Grande Terre, and a higher frequency of P. acuta along the east coast. These disproportions are probably due to sampling bias, rather than to a divergent ecological specialization of the two species. Indeed, we often found the two species in sympatry, and previous studies systematically found both species along both coasts of Grande Terre (Gélin, Pirog, et al., 2018; Oury et al., 2020).
4.2. Different types of local adaptation
In each of the studied species, we detected genotype–environment associations that might underpin local adaptation. These associations rarely involved outlier SNPs, but this was not surprising since genotype–environment association methods are more sensitive to small shifts in allele frequencies, compared to outlier tests (Rellstab et al., 2015).
In general, we observed that the environmental variables associated with the largest number of SNPs were those describing SST averages and standard deviations (80 SNPs across the three species), sea surface salinity (79 SNPs), chlorophyll concentration (65 SNPs) and BAF (54 SNPs). These similar numbers are explainable by the fact that all these variables are partially collinear to each other and some of them might therefore be involved in false associations with SNPs. Furthermore, the number of variables per environmental descriptor is likely to drive the numbers of significant SNPs. For instance, SST and salinity have the largest numbers of environmental descriptors (up to six noncollinear environmental variables per species), while BAF has the lowest (two per species, i.e., BAF1 and BAF5).
In fact, BAF is the environmental descriptor with the highest average number of significant SNPs per environmental variable (nine). Coral bleaching is a major threat for coral survival, and bleaching conditions emerge when SST variation exceeds seasonal averages (Hughes et al., 2017; Liu et al., 2003). BAF descriptors account precisely for this selective constraint (SST variation over average), and this might explain why genotype–environment associations with BAFs were on average more frequent. Previous work on coral seascape genomics also reported a predominance of adaptive signals related to BAF (Selmoni, Rochat, et al., 2020).
4.3. Candidate molecular targets for heat stress adaptation
Genes located in proximity of the SNPs associated with heat stress displayed some molecular functions that are known to be implicated in coral heat stress responses. In some cases, such functions were found in proximity of the few SNPs significantly associated with heat stress. More often, however, we found such functions as over‐represented among groups of SNPs that are associated with heat stress, but necessarily significantly associated.
One of the main examples of such molecular functions concerns molecular chaperones. These are proteins, such as heat shock proteins (Hsp), that intervene in cellular responses to heat stress to assist the folding or unfolding of proteins in the endoplasmic reticulum (Oakley et al., 2017). In corals, the role of these proteins in the heat response, as well as their up‐regulation under thermal stress, have been reported in several studies (Desalvo et al., ,2008, 2010; Maor‐Landaw & Levy, 2016; Oakley et al., 2017; Rosic et al., 2011). Here we found the GO terms “molecular chaperones,” “Hsp70 protein binding” and “Hsp90 protein binding” as over‐represented in proximity of SNPs associated with heat stress in the three studied species. Of note, we found one of the SNPs most strongly associated (q =.08) with heat stress in A. millepora in the coding sequence of chaperone Sacsin, which was recently proposed to be involved in bleaching resistance in A. millepora on the Great Barrier Reef (Fuller et al., 2020).
Another cellular signature of heat stress is the accumulation of ROS in the cytoplasm (Patel et al., 2018). Previous studies showed that corals exposed to heat stress respond to this accumulation by activating the molecular pathways of the oxidative stress response (Louis et al., 2017; Nielsen et al., 2018; Oakley et al., 2017; Voolstra et al., 2009, 2011). In the three studied species, we found SNPs associated with heat stress in proximity of molecular actors of the oxidative homeostasis, such as Quinone oxidoreductase PIG3, Malonyl‐CoA decarboxylase, HMG‐CoA reductase and Enoyl‐[acyl‐carrier‐protein] reductase. One of the proposed sources of ROS accumulation is leakage from the host mitochondrion, even though the underlying mechanisms are poorly known (Dunn et al., 2012). It is noteworthy that in A. millepora, the SNP most strongly associated with heat stress was in proximity of the MICOS complex subunit MIC60 gene. MICOS is a key protein in maintenance of the mitochondrial inner membrane architecture, through which ROS are produced, and the outer membrane, through which ROS diffuse into the cytoplasm (Muñoz‐Gómez et al., 2015; Zhao et al., 2019).
Additional molecular signatures of coral heat response were found in proximity of SNPs associated with heat stress, even though such observations were scattered across the three studied species. For instance, oxidative stress is known to trigger inflammatory responses and apoptosis (Courtial et al., 2017; Patel et al., 2018; Yuan et al., 2019), and we observed the over‐representation of GO terms implicated in these functions such as “mitogen‐activated protein kinase binding” (in P. damicornis) and “p53 binding” (in A. millepora and P. acuta). Another example concerns the GO term “DNA helicase activity” (P. acuta), as this function was previously found as a potential target for heat stress adaptation in a coral population from Japan (Selmoni, Rochat, et al., 2020).
4.4. Limitations and future directions
In the “Population structure” section we discussed the potential confounding role that neutral genetic variation can have on seascape genomics studies. There are, however, other elements that should be considered when assessing the statistical power of the study.
The main element is sample size, as previous work suggested working with sample sizes of at least 200 individuals to secure sufficient statistical power under any demographic scenario (Selmoni, Vajana, et al., 2020). We compensated for this potential lack of statistical power with a sampling design strategy maximizing the environmental contrasts between the sampling locations. Nevertheless, partial collinearity persisted between different environmental descriptors and this might have led the false discoveries in the genotype–environment association study (Leempoel et al., 2017). A possible solution could be the extension of the study area to the reefs of the neighbouring islands, as this might introduce new combinations of environmental gradients and reduce collinearity.
We also encountered important trade‐offs related to the genotyping technique. Compared to traditional RAD‐seq approaches, DArT‐seq loci appeared indeed to be enriched in functional regions of the genome and this facilitated the interpretation of the results (Gawroński et al., 2016; Lowry et al., 2017). However, some of the genetic variants required substantial imputation (missing rate >20%), and such variants appeared less likely to be detected as associated wityh the environment (Table S8). This is not surprising, since rare genotypes (such as adaptive ones) are known to be more difficult to impute (Hoffmann & Witte, 2015). An increase in sequencing depth would reduce the need for imputation and consequently increase the statistical power of the study.
The next step in the characterization of corals’ adaptive potential is experimental validation. Our work found several genetic variants that might confer selective advantages against thermal stress. For each of the studied species, we can now define multiple‐loci genotypes of heat stress‐resistant colonies and test their fitness under experimental heat stress conducted in aquaria (Krueger et al., 2017). As a result, this analysis will allow us to (i) further investigate the role of different heat stress‐associated genotypes and molecular pathways and (ii) provide a concrete measure of the thermal ranges that these coral populations might sustain in the years to come. This information is of paramount importance, as it will allow us to predict the reefs that are expected to already carry heat‐tolerant colonies and to define conservation strategies accordingly (Selmoni, Rochat, et al., 2020). For instance, marine protected areas could be established to preserve reefs with higher adaptive potential against heat stress, where such reefs could provide the breeding stock to restore damaged reefs (Baums, 2008; van Oppen et al., ,2015, 2017).
5. CONCLUSIONS
In this study, seascape genomics allowed us to uncover genetic variants potentially implicated in adaptive processes against different types of heat stress in three coral species of New Caledonia. These variants were located next to genes coding for molecular actors that participate in well‐understood cellular reactions against thermal stress. Of note, some of these potential targets for adaptation recurred in the analyses of different species, supporting the robustness and the power of the seascape genomics. Future studies will focus on performing experimental assays to validate the implication of potentially adaptive genotypes and newly identified genes in the heat stress response and to measure the thermal ranges tolerated by the diverse adaptive genotypes.
AUTHOR CONTRIBUTIONS
O.S., G.L., H.M., L.V., S.J. and V.B.L. initiated the project and organized the sampling campaign. O.S., G.L., C.P. and V.B.L. collected coral samples during the field campaign. F.B. and H.M. contributed to the species identification. O.S. performed the DNA purification, H.M. and N.O. performed the microsatellite analysis. O.S. performed the seascape genomics analysis, supported by G.L., H.M., S.J. and V.B.L. O.S. wrote the first version of the manuscript, which was the critically revised by all other authors. All authors read and approved the final manuscript.
Supporting information
Fig S1‐S4
Table S1‐S8
Stéphane Joost and Véronique Berteaux‐Lecellier contributed equally.
DATA AVAILABILITY STATEMENT
Data and scripts used in this work are available on Zenodo (https://doi.org/10.5281/zenodo.4545056). Raw sequencing reads are available on NCBI (BioProject identifier: PRJNA702071). Benefit generated: this research was led in collaboration with scientists from the countries providing the genetic samples, and all collaborators are included as co‐authors. Indigenous communities and local authorities were informed and granted the sampling permits for this study (details in the Material and Methods section). This study addresses a major conservation concern for the studied organisms. All data resulting from this research are publicly available as described above.
REFERENCES
- Ashburner, M. , Ball, C. A. , Blake, J. A. , Botstein, D. , Butler, H. , & Cherry, J. M. … Consortium, G. O. (2000). Gene Ontology: Tool for The Unification of Biology. Nature Genetics, 25(1), 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baums, I. B. (2008). A restoration genetics guide for coral reef conservation. Molecular Ecology, 17(12), 2796–2811. 10.1111/j.1365-294X.2008.03787.x. [DOI] [PubMed] [Google Scholar]
- Bay, R. A. , & Palumbi, S. R. (2014). Multilocus adaptation associated with heat resistance in reef‐building corals. Current Biology, 24(24), 2952–2956. 10.1016/j.cub.2014.10.044. [DOI] [PubMed] [Google Scholar]
- Bellwood, D. R. , Hughes, T. P. , Folke, C. , & Nyström, M. (2004). Confronting the coral reef crisis. Nature, 429, 827–833. 10.1038/nature02691. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B. , Bairoch, A. , Apweiler, R. , Blatter, M. C. , Estreicher, A. , Gasteiger, E. , & Schneider, M. (2003). The SWISS‐PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research, 31(1), 365–370. 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury, P. J. , Zhang, Z. , Kroon, D. E. , Casstevens, T. M. , Ramdoss, Y. , & Buckler, E. S. (2007). TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 23(19), 2633–2635. 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: an analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, Y. , Li, Z. , Farrara, J. D. , & Hung, P. (2009). Blending Sea surface temperatures from multiple satellites and in situ observations for coastal oceans. Journal of Atmospheric and Oceanic Technology, 26(7), 1415–1426. 10.1175/2009JTECHO592.1. [DOI] [Google Scholar]
- Chin, T. M. , Vazquez‐Cuervo, J. , & Armstrong, E. M. (2017). A multi‐scale high‐resolution analysis of global sea surface temperature. Remote Sensing of Environment, 200, 154–169. 10.1016/j.rse.2017.07.029. [DOI] [Google Scholar]
- Courtial, L. , Picco, V. , Grover, R. , Cormerais, Y. , Rottier, C. , Labbe, A. , & Ferrier‐Pagès, C. (2017). The c‐Jun N‐terminal kinase prevents oxidative stress induced by UV and thermal stresses in corals and human cells. Scientific Reports, 7(1), 45713. 10.1038/srep45713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunning, R. , Bay, R. A. , Gillette, P. , Baker, A. C. , & Traylor‐Knowles, N. (2018). Comparative analysis of the Pocillopora damicornis genome highlights role of immune system in coral evolution. Scientific Reports, 8(1), 16134. 10.1038/s41598-018-34459-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance, A. (2019, November 28). These corals could survive climate change — and help save the world’s reefs. Nature, 575, 580–582. 10.1038/d41586-019-03629-7. [DOI] [PubMed] [Google Scholar]
- Darling, E. S. , Alvarez‐Filip, L. , Oliver, T. A. , McClanahan, T. R. , & Côté, I. M. (2012). Evaluating life‐history strategies of reef corals from species traits. Ecology Letters, 15(12), 1378–1386. 10.1111/j.1461-0248.2012.01861.x. [DOI] [PubMed] [Google Scholar]
- Desalvo, M. K. , Sunagawa, S. , Voolstra, C. R. , & Medina, M. (2010). Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Marine Ecology Progress Series, 402, 97–113. 10.3354/meps08372. [DOI] [Google Scholar]
- Desalvo, M. K. , Voolstra, C. R. , Sunagawa, S. , Schwarz, J. A. , Stillman, J. H. , Coffroth, M. A. , & Medina, M. (2008). Differential gene expression during thermal stress and bleaching in the Caribbean coral Montastraea faveolata. Molecular Ecology, 17(17), 3952–3971. 10.1111/j.1365-294X.2008.03879.x. [DOI] [PubMed] [Google Scholar]
- Dunn, S. R. , Pernice, M. , Green, K. , Hoegh‐Guldberg, O. , & Dove, S. G. (2012). Thermal stress promotes host mitochondrial degradation in symbiotic Cnidarians: are the batteries of the reef going to run out? PLoS One, 7(7), e39024. 10.1371/journal.pone.0039024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EU Copernicus Marine Service (2017). Global ocean – in‐situ‐near‐real‐time observations. Retrieved February 2, 2017, from http://marine.copernicus.eu. [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925–929. 10.1111/2041-210X.12382. [DOI] [Google Scholar]
- Frichot, E. , Schoville, S. D. , Bouchard, G. , & François, O. (2013). Testing for associations between loci and environmental gradients using latent factor mixed models. Molecular Biology and Evolution, 30(7), 1687–1699. 10.1093/molbev/mst063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, Z. L. , Mocellin, V. J. L. , Morris, L. A. , Cantin, N. , Shepherd, J. , Sarre, L. , & Przeworski, M. (2020). Population genetics of the coroal Acropora millepora: Toward genomic prediction of bleaching. Science, 369(6501), 10.1126/science.aba4674. [DOI] [PubMed] [Google Scholar]
- Gawroński, P. , Pawełkowicz, M. , Tofil, K. , Uszyński, G. , Sharifova, S. , Ahluwalia, S. , & Bolibok‐Brągoszewska, H. (2016). DArT Markers effectively target gene space in the rye genome. Frontiers in Plant Science, 7, 1600. 10.3389/fpls.2016.01600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gélin, P. , Fauvelot, C. , Bigot, L. , Baly, J. , & Magalon, H. (2018). From population connectivity to the art of striping Russian dolls: The lessons from Pocillopora corals. Ecology and Evolution, 8(2), 1411–1426. 10.1002/ece3.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gélin, P. , Pirog, A. , Fauvelot, C. , & Magalon, H. (2018). High genetic differentiation and low connectivity in the coral Pocillopora damicornis type β at different spatial scales in the Southwestern Indian Ocean and the Tropical Southwestern Pacific. Marine Biology, 165(10), 10.1007/s00227-018-3428-6. [DOI] [Google Scholar]
- Gélin, P. , Postaire, B. , Fauvelot, C. , & Magalon, H. (2017). Reevaluating species number, distribution and endemism of the coral genus Pocillopora Lamarck, 1816 using species delimitation methods and microsatellites. Molecular Phylogenetics and Evolution, 109, 430–446. 10.1016/j.ympev.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Highsmith, R. (1982). Reproduction by fragmentation in corals. Marine Ecology Progress Series, 7, 207–226. 10.3354/meps007207. [DOI] [Google Scholar]
- Hijmans, R. J. (2016). raster: Geographic data analysis and modeling. Retrieved from, https://cran.r‐project.org/package=raster . [Google Scholar]
- Hoffmann, T. J. , & Witte, J. S. (2015). October 1). Strategies for imputing and analyzing rare variants in association studies. Trends in Genetics, 31, 556–563. 10.1016/j.tig.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, T. P. , Kerry, J. T. , Álvarez‐Noriega, M. , Álvarez‐Romero, J. G. , Anderson, K. D. , Baird, A. H. , & Wilson, S. K. (2017). Global warming and recurrent mass bleaching of corals. Nature, 543(7645), 373–377. 10.1038/nature21707. [DOI] [PubMed] [Google Scholar]
- Hughes, T. P. , Kerry, J. T. , Baird, A. H. , Connolly, S. R. , Dietzel, A. , Eakin, C. M. , & Torda, G. (2018). Global warming transforms coral reef assemblages. Nature, 556(7702), 492–496. 10.1038/s41586-018-0041-2. [DOI] [PubMed] [Google Scholar]
- Hughes, T. P. , Kerry, J. T. , Connolly, S. R. , Baird, A. H. , Eakin, C. M. , Heron, S. F. , & Torda, G. (2019). January 10). Ecological memory modifies the cumulative impact of recurrent climate extremes. Nature Climate Change, 9, 40–43. 10.1038/s41558-018-0351-2. [DOI] [Google Scholar]
- Johnston, E. C. , Forsman, Z. H. , Flot, J. F. , Schmidt‐Roach, S. , Pinzón, J. H. , Knapp, I. S. S. , & Toonen, R. J. (2017). A genomic glance through the fog of plasticity and diversification in Pocillopora. Scientific Reports, 7(1), 1–11. 10.1038/s41598-017-06085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405. 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , Devillard, S. , & Balloux, F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genetics, 11(1), 94. 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian, A. , Wenzl, P. , Huttner, E. , Carling, J. , Xia, L. , Blois, H. , & Uszynski, G. (2012). Diversity arrays technology: A generic genome profiling technology on open platforms. Methods in Molecular Biology, 888, 67–89. 10.1007/978-1-61779-870-2_5. [DOI] [PubMed] [Google Scholar]
- Krueger, T. , Horwitz, N. , Bodin, J. , Giovani, M. E. , Escrig, S. , Meibom, A. , & Fine, M. (2017). Common reef‐building coral in the northern red sea resistant to elevated temperature and acidification. Royal Society Open Science, 4(5), 170038. 10.1098/rsos.170038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leempoel, K. , Duruz, S. , Rochat, E. , Widmer, I. , Orozco‐terWengel, P. , & Joost, S. (2017). Simple rules for an efficient use of geographic information systems in molecular ecology. Frontiers in Ecology and Evolution, 5, 33. 10.3389/fevo.2017.00033. [DOI] [Google Scholar]
- Liu, G. , Strong, A. E. , & Skirving, W. (2003). Remote sensing of sea surface temperatures during 2002 Barrier Reef coral bleaching. Eos, Transactions American Geophysical Union, 84(15), 137–141. 10.1029/2003EO150001. [DOI] [Google Scholar]
- Logan, C. A. , Dunne, J. P. , Eakin, C. M. , & Donner, S. D. (2014). Incorporating adaptive responses into future projections of coral bleaching. Global Change Biology, 20(1), 125–139. 10.1111/gcb.12390. [DOI] [PubMed] [Google Scholar]
- Louis, Y. D. , Bhagooli, R. , Kenkel, C. D. , Baker, A. C. , & Dyall, S. D. (2017). January 1). Gene expression biomarkers of heat stress in scleractinian corals: Promises and limitations. Comparative Biochemistry and Physiology Part ‐ C: Toxicology and Pharmacology, 191, 63–77. 10.1016/j.cbpc.2016.08.007. [DOI] [PubMed] [Google Scholar]
- Lowry, D. B. , Hoban, S. , Kelley, J. L. , Lotterhos, K. E. , Reed, L. K. , Antolin, M. F. , & Storfer, A. (2017). Breaking RAD: an evaluation of the utility of restriction site‐associated DNA sequencing for genome scans of adaptation. Molecular Ecology Resources, 17(2), 142–152. 10.1111/1755-0998.12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loya, Y. , Sakai, K. , Yamazato, K. , Nakano, Y. , Sambali, H. , & van Woesik, R. (2001). Coral bleaching: The winners and the losers. Ecology Letters, 4(2), 122–131. 10.1046/j.1461-0248.2001.00203.x. [DOI] [Google Scholar]
- Luu, K. , Bazin, E. , & Blum, M. G. B. (2017). pcadapt: An R package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources, 17(1), 67–77. 10.1111/1755-0998.12592. [DOI] [PubMed] [Google Scholar]
- Madden, T. , & Coulouris, G. (2008). BLAST command line applications user manual BLAST command line applications user manual – BLAST ® … (pp. 1–28). pp. 1–28. Retrieved from http://www.ncbi.nlm.nih.gov/books/NBK279690/
- Maleki, F. , Ovens, K. , Hogan, D. J. , & Kusalik, A. J. (2020). June 30). Gene set analysis: Challenges, opportunities, and future research. Frontiers in Genetics, 11, 654. 10.3389/fgene.2020.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor‐Landaw, K. , & Levy, O. (2016). Gene expression profiles during short‐term heat stress; branching vs. massive Scleractinian corals of the Red Sea. PeerJ, 2016(3), 10.7717/peerj.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz, M. V. , Treml, E. A. , Aglyamova, G. V. , & Bay, L. K. (2018). Potential and limits for rapid genetic adaptation to warming in a Great Barrier Reef coral. PLoS Genetics, 14(4), e1007220. 10.1371/journal.pgen.1007220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Money, D. , Gardner, K. , Migicovsky, Z. , Schwaninger, H. , Zhong, G. Y. , & Myles, S. (2015). LinkImpute: Fast and accurate genotype imputation for nonmodel organisms. G3: Genes, Genomes . Genetics, 5(11), 2383–2390. 10.1534/g3.115.021667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz‐Gómez, S. A. , Slamovits, C. H. , Dacks, J. B. , & Wideman, J. G. (2015). The evolution of MICOS: Ancestral and derived functions and interactions. Communicative and Integrative Biology, 8(6), 1–5. 10.1080/19420889.2015.1094593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydlarz, L. D. , McGinty, E. S. , & Harvell, C. D. (2010). What are the physiological and immunological responses of coral to climate warming and disease? The Journal of Experimental Biology, 213(6), 934–945. 10.1242/jeb.037580. [DOI] [PubMed] [Google Scholar]
- Nielsen, D. A. , Petrou, K. , & Gates, R. D. (2018). Coral bleaching from a single cell perspective. ISME Journal, 12(6), 1558–1567. 10.1038/s41396-018-0080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley, C. A. , Durand, E. , Wilkinson, S. P. , Peng, L. , Weis, V. M. , Grossman, A. R. , & Davy, S. K. (2017). Thermal shock induces host proteostasis disruption and endoplasmic reticulum stress in the model symbiotic Cnidarian Aiptasia. Journal of Proteome Research, 16(6), 2121–2134. 10.1021/acs.jproteome.6b00797. [DOI] [PubMed] [Google Scholar]
- Oury, N. , Gélin, P. , & Magalon, H. (2020). Cryptic species and genetic connectivity among populations of the coral Pocillopora damicornis (Scleractinia) in the tropical southwestern Pacific. Marine Biology, 167(10), 1–15. 10.1007/s00227-020-03757-z. [DOI] [Google Scholar]
- Palumbi, S. R. , Barshis, D. J. , Traylor‐Knowles, N. , & Bay, R. A. (2014). Mechanisms of reef coral resistance to future climate change. Science, 344(6186), Retrieved from http://science.sciencemag.org/content/344/6186/895. [DOI] [PubMed] [Google Scholar]
- Patel, R. , Rinker, L. , Peng, J. , & Chilian, W. M. (2018). Reactive oxygen species: The good and the bad. In Reactive Oxygen Species (ROS) in Living Cells. doi: 10.5772/intechopen.71547 [Google Scholar]
- Penin, L. , Vidal‐Dupiol, J. , & Adjeroud, M. (2013). Response of coral assemblages to thermal stress: are bleaching intensity and spatial patterns consistent between events? Environmental Monitoring and Assessment, 185(6), 5031–5042. 10.1007/s10661-012-2923-3. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QGIS development team (2009). QGIS geographic information system. Open source geospatial foundation project. Retrieved from. http://www.qgis.org/.
- R Core Team (2016). R: A language and environment for statistical computing. Retrieved from https://www.r‐project.org/. [Google Scholar]
- Rellstab, C. , Gugerli, F. , Eckert, A. J. , Hancock, A. M. , & Holderegger, R. (2015). A practical guide to environmental association analysis in landscape genomics. Molecular Ecology, 24(17), 4348–4370. 10.1111/mec.13322. [DOI] [PubMed] [Google Scholar]
- Riginos, C. , Crandall, E. D. , Liggins, L. , Bongaerts, P. , & Treml, E. A. (2016). Navigating the currents of seascape genomics: How spatial analyses can augment population genomic studies. Current Zoology, 62, 10.1093/cz/zow067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, R. C. , Williams, G. J. , & Turner, J. R. (2018). March 1). Towards developing a mechanistic understanding of coral reef resilience to thermal stress across multiple scales. Current Climate Change Reports, 4, 51–64. 10.1007/s40641-018-0087-0. [DOI] [Google Scholar]
- Roesti, M. , Salzburger, W. , & Berner, D. (2012). Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evolutionary Biology, 12(1), 94. 10.1186/1471-2148-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosic, N. N. , Pernice, M. , Dove, S. , Dunn, S. , & Hoegh‐Guldberg, O. (2011). Gene expression profiles of cytosolic heat shock proteins Hsp70 and Hsp90 from symbiotic dinoflagellates in response to thermal stress: Possible implications for coral bleaching. Cell Stress and Chaperones, 16(1), 69–80. 10.1007/s12192-010-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansaloni, C. , Petroli, C. , Jaccoud, D. , Carling, J. , Detering, F. , Grattapaglia, D. , & Kilian, A. (2011). Diversity Arrays Technology (DArT) and next‐generation sequencing combined: Genome‐wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proceedings, 5(S7), P54. 10.1186/1753-6561-5-S7-P54.22373051 [DOI] [Google Scholar]
- Schmidt‐Roach, S. , Lundgren, P. , Miller, K. J. , Gerlach, G. , Noreen, A. M. E. , & Andreakis, N. (2013). Assessing hidden species diversity in the coral Pocillopora damicornis from Eastern Australia. Coral Reefs, 32(1), 161–172. 10.1007/s00338-012-0959-z. [DOI] [Google Scholar]
- Schmidt‐Roach, S. , Miller, K. J. , Lundgren, P. , & Andreakis, N. (2014). With eyes wide open: A revision of species within and closely related to the Pocillopora damicornis species complex (Scleractinia; Pocilloporidae) using morphology and genetics. Zoological Journal of the Linnean Society, 170(1), 1–33. 10.1111/zoj.12092. [DOI] [Google Scholar]
- Selmoni, O. , Rochat, E. , Lecellier, G. , Berteaux‐Lecellier, V. , & Joost, S. (2020). Seascape genomics as a new tool to empower coral reef conservation strategies: An example on north‐western Pacific Acropora digitifera. Evolutionary Applications, 588228, 10.1101/588228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmoni, O. , Vajana, E. , Guillaume, A. , Rochat, E. , & Joost, S. (2020). Sampling strategy optimization to increase statistical power in landscape genomics: A simulation‐based approach. Molecular Ecology Resources, 20(1), 10.1111/1755-0998.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simillion, C. , Liechti, R. , Lischer, H. E. L. , Ioannidis, V. , & Bruggmann, R. (2017). Avoiding the pitfalls of gene set enrichment analysis with SetRank. BMC Bioinformatics, 18(1), 151. 10.1186/s12859-017-1571-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, J. D. (2003). The positive false discovery rate: A bayesian interpretation and the q‐value. Annals of Statistics, 31(6), 2013–2035. [Google Scholar]
- Sully, S. , Burkepile, D. E. , Donovan, M. K. , Hodgson, G. , & van Woesik, R. (2019). A global analysis of coral bleaching over the past two decades. Nature Communications, 10(1), 1–5. 10.1038/s41467-019-09238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, L. , Kendrick, G. A. , Stat, M. , Travaille, K. L. , Shedrawi, G. , & Kennington, W. J. (2014). Population genetic structure of the Pocillopora damicornis morphospecies along Ningaloo Reef, Western Australia. Marine Ecology Progress Series, 513, 111–119. 10.3354/meps10893. [DOI] [Google Scholar]
- Thomas, L. , Kennington, W. J. , Evans, R. D. , Kendrick, G. A. , & Stat, M. (2017). Restricted gene flow and local adaptation highlight the vulnerability of high‐latitude reefs to rapid environmental change. Global Change Biology, 23(6), 2197–2205. 10.1111/gcb.13639. [DOI] [PubMed] [Google Scholar]
- Thompson, D. M. , & van Woesik, R. (2009). Corals escape bleaching in regions that recently and historically experienced frequent thermal stress. Proceedings. Biological Sciences/The Royal Society, 276(1669), 2893–2901. 10.1098/rspb.2009.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNEP‐WCMC, WorldFish‐Center, WRI, & TNC . (2010). Global distribution of warm‐water coral reefs, compiled from multiple sources including the Millennium Coral Reef Mapping Project. Version 1.3. Retrieved May 9, 2017, from http://data.unep‐wcmc.org/datasets/1
- Van Hooidonk, R. , Maynard, J. A. , & Planes, S. (2013). Temporary refugia for coral reefs in a warming world. Nature Climate Change, 3(5), 508–511. 10.1038/nclimate1829. [DOI] [Google Scholar]
- van Oppen, M. J. H. , Gates, R. D. , Blackall, L. L. , Cantin, N. , Chakravarti, L. J. , Chan, W. Y. , & Putnam, H. M. (2017). September 1). Shifting paradigms in restoration of the world’s coral reefs. Global Change Biology, 23, 3437–3448. 10.1111/gcb.13647. [DOI] [PubMed] [Google Scholar]
- van Oppen, M. J. H. , & Lough, J. M. (2009). Coral bleaching — patterns, processes, causes and consequences. doi: 10.1007/978‐3‐540‐69775‐6_11
- van Oppen, M. J. H. , Oliver, J. K. , Putnam, H. M. , & Gates, R. D. (2015). February 24). Building coral reef resilience through assisted evolution. Proceedings of the National Academy of Sciences of the United States of America, 112, 2307–2313. 10.1073/pnas.1422301112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voolstra, C. R. , Schnetzer, J. , Peshkin, L. , Randall, C. J. , Szmant, A. M. , & Medina, M. (2009). Effects of temperature on gene expression in embryos of the coral Montastraea faveolata. BMC Genomics, 10, 627. 10.1186/1471-2164-10-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voolstra, C. R. , Sunagawa, S. , Matz, M. V. , Bayer, T. , Aranda, M. , Buschiazzo, E. , & Medina, M. (2011). Rapid evolution of coral proteins responsible for interaction with the environment. PLoS One, 6(5), 10.1371/journal.pone.0020392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, C. C. (1999). Staghorn corals of the world : A revision of the coral genus Acropora (Scleractinia; Astrocoeniina; Acroporidae) worldwide, with emphasis on morphology, phylogeny and biogeography. CSIRO. [Google Scholar]
- Wang, L. , Zhang, W. , Li, Q. , & Zhu, W. (2017). AssocTests: Genetic Association Studies.
- Yuan, J. , Liu, L. , Zhang, Y. , Shen, C. , & Lin, S. (2019). Molecular processes and hub genes of Acropora Palmata in response to thermal stress and bleaching. Journal of Coastal Research, 35(1), 26. 10.2112/JCOASTRES-D-18-00053.1. [DOI] [Google Scholar]
- Zhao, R. Z. , Jiang, S. , Zhang, L. , & Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). International Journal of Molecular Medicine, 44, 3–15. 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28(24), 3326–3328. 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1‐S8
Data Availability Statement
Data and scripts used in this work are available on Zenodo (https://doi.org/10.5281/zenodo.4545056). Raw sequencing reads are available on NCBI (BioProject identifier: PRJNA702071). Benefit generated: this research was led in collaboration with scientists from the countries providing the genetic samples, and all collaborators are included as co‐authors. Indigenous communities and local authorities were informed and granted the sampling permits for this study (details in the Material and Methods section). This study addresses a major conservation concern for the studied organisms. All data resulting from this research are publicly available as described above.