

Abstract
Carbon nanodots (CDs) originating from different biomass result in different activities to sensitize photo‐ATRP and photo‐CuAAC reaction protocols with visible light. Free radical polymerization of tri(propylene glycol)diacrylate also exhibited a good efficiency using CDs in combination with an iodonium salt employing LEDs emitting either at 405 nm, 525 nm or 660 nm. Photo‐ATRP experiments confirmed controlled polymerization conditions using CuII at the ppm scale resulting in dispersities between 1.06 to 1.10. Chain end fidelity was successfully provided by chain extension and block copolymerization additionally approving the living feature of polymerization using a CD synthesized from lac dye comprising olefinic moieties in the originating biomass. By global analysis, time resolved fluorescence measurements indicated the appearance of several emitting species contributing to the reactivity of the excited states. Different cytotoxic response appeared following the answer of MCF‐10A cells in a flow cytometry assay; that is 400 μg mL−1. Thus, cell viability was greater 80 % in the case of CD‐2–CD‐5 while that of CD‐1 was close to 70 %.
Keywords: photo-ATRP, photo-CuAAC, radical photopolymerization, sustainable carbon dots
Sustainable carbon dots are prepared as light‐sensitive materials originating from different biomass. Based on their implemented moieties they enable to initiate free radical polymerization choosing LEDs at different wavelength and tailor‐made polymer synthesis facilitating photo‐ATRP and photo click chemistry. CDs take the offer to compete with traditional ATRP based on CuI regarding the dispersity of the polymeric material.
Introduction
Light as a reagent and tool[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 ] has received gained interest because it interdisciplinary connects different fields of chemistry, biology and physics resulting in emerging applications related to display technologies based on holography, [12] 2D[ 13 , 14 ] and 3D printing,[ 15 , 16 , 17 , 18 ] photopharmacology, [19] dentistry [20] or environmental sciences to clean water and/or air from microbial ballast [21] —just to name a few emerging applications. It requires knowledge about the properties of the excited state such as lifetime, redox potentials and excitation energy to design systems working with appropriate light sources such as LEDs or lasers resulting in generation of initiating radicals or conjugate acid to initiate radical or cationic polymerization, respectively.[ 22 , 23 , 24 ] Photochemical sciences also connect well with material synthesis where polymers depict one major field.[ 2 , 8 , 9 , 10 , 11 , 25 , 26 ] From this point of view, controlled radical photopolymerization (CRP) has received a key position occurring based on ATRP[ 26 , 27 ] or RAFT[ 3 , 25 ] polymerization. It enables the tailor‐made photoinduced synthesis at the nanoscale and brings therefore material science to some of the aforementioned applications. Therefore, questions have arisen to connect suitable initiator systems with modern light sources emitting in the UV, [28] visible[ 4 , 27 ] or NIR[ 29 , 30 ] range. New regulations with focus to replace older mercury light sources have accelerated these activities. [31] It additionally addresses the necessity to develop photopolymerization systems comprising more sustainable components. CDs have received form this point of view remarkable interest as light sensitive material.[ 3 , 21 , 27 , 32 , 33 , 34 , 35 , 36 ]
Photosensitizers needed for photopolymerization mostly base on petrochemicals [2] while only a few systems derive from natural resources.[ 3 , 37 , 38 ] Demands of the society have also questioned whether issues such as toxicity of reaction components shall move in the focus to develop greener systems. Photopolymerization as a green technology requests therefore to develop chemical components resulting in high green label of the entire system. This technology possesses the potential to replace energy wasting oven techniques, which has addressed many technological switches.[ 24 , 39 ]
From this point of view, it does not surprise that carbon nanodots (CDs) have gained an increasing interest in material research to design photoactive systems derived from biomass.[ 34 , 36 ] Bottom‐up and Top‐down procedures facilitate synthesis of such materials[ 36 , 40 , 41 ] exhibiting a size of a few nanometers.
Photophysical investigations of CDs indicated fluorescence quantum yields of in the range of 20–40 %. [36] Furthermore, lifetime of the excited state and the spin multiplicity of the participating excited state also affects photochemical pathways and consecutive reaction steps of the excited state. This enables photoredox systems where oxidative and/or reductive processes result in short‐living species such as radicals. [27] Thus, it does not surprise that photoredox systems based on CDs have also become of interest to generate hydrogen in a catalytic cycle [35] used for the cleanest technology to generate energy.
This idea can be adopted to several fields in chemistry including material synthesis. [2] In macromolecular and materials science, mechanistic ideas have received attention for living radical polymerization based on a reaction protocol for RAFT using sunlight as excitation source [3] and photo‐ATRP in combination with blue light LEDs [27] where the CuII concentration operated in the ppm range. Particular the latter reaction possesses huge potential. Moreover, Cu‐catalyzed Azide‐Alkyne Cycloaddition Reaction (CuAAC) complements the availability of reactions used for tailor made synthesis of block copolymers. [42] Several methods for generation of the CuI catalyst are available for both ATRP and CuAAC reactions. They involve the in situ reduction of CuII to generate CuI by 1)various reducing agents, 2)photochemical processes, 3)electrochemical redox processes, and 4)copper‐comprising nanoparticles.[ 5 , 26 , 43 , 44 ] Particularly, the in situ photolytic generation of CuI complexes from CuII under irradiation to initiate controlled radical polymerization facilitates CRP with a copper concentration residing at the ppm‐scale has received remarkable attention.[ 27 , 29 , 30 ] This level of CuII appears as less harmful. From this point of view, light driven reactions have been successfully introduced in this field requesting either UV, visible or NIR light.[ 6 , 29 ] Furthermore, metal free ATRP systems have moved in the focus as well.[ 2 , 43 ]
Although a lot of progress have been achieved in photoinduced ATRP and CuAAC reaction vide supra, there remain some challenges. Many photosensitizers or photocatalysts used for these photoreactions have based on synthetic materials whose synthesis often requires operating with no sustainable feedback. Moreover, complicated synthesis/preparation of these photosensitizers/photocatalysts interfered their applications at large‐scale. Certainly, an urgent demand exists to develop a sustainable and easy to manufacture photocatalyst or photosensitizer with low toxicological response as well. Particularly, CDs may move in this field.[ 21 , 34 , 35 , 36 ] Their interesting electrochemical and photochemical properties enable photoredox systems to initiate radical polymerization. [27] Cytotoxic response of such materials appears at low scale. [27] Inspired by these points, we used products derived from diverse biomass to fabricate CDs for triggering photochemistry such as the photo‐ATRP and photo‐CuAAC reaction. They were made according to a reaction protocol based on solvothermal and hydrothermal methods. [21] Some of them still comprise unsaturated moieties in the originating biomass (laccaic acid) while others do not have it (sodium alginate, citric acid). This contribution focusses on CDs originating from different sources related to biomass resulting in distinct structural patterns as studied by elemental analysis, XPS data, and time resolved fluorescence. They cause different efficiencies to initiate free radical polymerization (FRP), controlled radical polymerization (CRP) and CuACC caused by the different structures generated either by solvothermal or hydrothermal method [21] and therefore properties of the excited state of the CDs.
Results and Discussion
Synthesis, General Properties and Polymerization Efficiency of CDs
CDs originating from biomass comprising aromatic patterns (CD‐1–CD‐3) and aliphatic structural moieties (CD‐4 and CD‐5) are compared regarding their capability to sensitize initiation of radical photopolymerization with visible light emitting LEDs. Solvothermal (CD‐1–CD‐3) and hydrothermal (CD‐4, CD‐5) reaction protocols [27] resulted in different amounts of carbon and nitrogen depending on the amount of reactant, Table 1. The amount on C‐N, C‐O and C=O moieties differed from each other as concluded by XPS analysis (Figures S2–S5 in SI). CD‐1–CD‐3 base on different concentrations of amine used for synthesis. CDs investigated mostly comprise sp2‐carbon connected by moieties carrying the heterocyclic moieties vide supra. Working with CDs also requires a rethinking of the design for the experiments because material properties such as oxidation and reduction potentials, excitation and emission properties, and cytotoxic response represent tunable properties as controlled by the synthetic procedure. Therefore, one mainly knows the response upon an addressed chemical question of a heterogenous material synthesized accordingly [21] vide supra. For this reason, it is very important to know if such materials cause any toxicological issue. Cytotoxicity depicts as one quantity the response of materials considering the answer of MCF‐10A cells in a flow cytometry assay. CD‐2–CD‐5 investigated exhibit a viability of greater 90 % while CD‐1 still exhibits an acceptable quantity, Table 1. They did not obviously inhibit the cellular growth up to a concentration of 400 μg mL−1 (Figure S6 in SI, incubation time 4 h). The low cytotoxic response enables the design of new green photopolymerization systems with sustainable components. In addition, rough estimation of manufacturing costs showed more economic potential for CDs compared to photoinitiating components related to fossil resources. [27]
Table 1.
Summary of CD properties regarding the composition as shown by the sum formula, viability of cells after addition of 400 μg mL−1 CD (incubation time 4 h, see SI for more details), size, optical data such absorption maximum (λ max abs) and fluorescence maximum (λ max fluo) with their respective decay times (τ 1–4) determined by global analysis (globally fitted data appear bold and underlined), [46] and redox potential such as oxidation (E ox) and reduction potential (E ox). It also gives information about the synthetic method applied for synthesis of the CDs that based on solvo‐thermal or hydro‐thermal treatment. Available is also a summary of reactions successfully pursued with each CD. SI gives more details about determination of data. Decay times that were not globally linked change with wavelength. SI gives data of each CD determined at different wavelengths.
|
Entry |
Synthetic methods |
Sum Formula |
cell viability [%] |
Size [≈nm] |
λ max abs [nm] |
λ max flu [nm] |
τ1 [ns] |
τ2 [ns] |
τ3 [ns] |
τ4 [ns] |
E ox [V] |
E red [V] |
Reaction |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
CD‐1 |
Solvothermal |
C80N4O16 |
69 |
4.4 |
488 |
588 |
0.28 |
4.6 |
13.9 |
1.4 |
0.29 |
−0.41 |
photo‐ATRP, FRP, photo‐CuAAC |
|
CD‐2 |
Solvothermal |
C77N8O15 |
92 |
6.1 |
484 |
656 |
0.75 |
3.4 |
11.1 |
|
0.28 |
−0.44 |
FRP, photo‐CuAAC |
|
CD‐3 |
Solvothermal |
C73N12O15 |
92 |
4.2 |
488 |
654 |
0.81 |
3.3 |
9.8 |
|
0.29 |
−0.43 |
photo‐ATRP, FRP, photo‐CuAAC |
|
CD‐4 [a] |
Hydrothermal |
C69N16O12Na3 |
83 |
6.3 |
360 |
476 |
0.12 |
3.1 |
9.5 |
0.57 |
0.26 |
−0.37 |
photo‐ATRP, FRP, photo‐CuAAC |
|
CD‐5 |
Hydrothermal |
C64N13O23 |
92 |
3.7 |
366 |
460 |
0.04 |
5.8 |
11.7 |
36.8 |
0.25 |
−0.42 |
FRP, photo‐CuAAC |
[a] Reported results [27] except the fluorescence data.
Table 1 and the XPS spectra (see supporting information Figures S2–S5) show that CDs comprise the necessary structural elements of Type II photoinitiators. [2] This relates to the larger amount on sp2‐carbon, carbonyl groups and amino functions facilitating together participation in PET where redox potentials and excitation energy are important parameters for the free reaction enthalpy of photoinduced electron transfer (ΔG el) considering either a reductive or oxidation mechanism. [45] From this point of view, onium compounds such as iodonium or sulfonium salts have moved into the focus of an oxidative mechanism to initiate free radical polymerization (FRP). [45] Moreover, excitation energy and electrochemical potential might also enable CDs to sensitize controlled radical polymerization (CRP) using CuII complex in catalytic amount at the ppm scale. This moves the traditional ATRP system [5] towards in a greener direction because CuII shall not be anymore cause a toxicological issue at this concentration. In addition, the photo‐CuACC reaction [42] should also work in the case of CDs. The formation of CuI by oxidation of photoexcited carbon nanodots may lead to the requested triazole structures, which occurred only for some CDs vide infra. This surprises since oxidation potentials may favor the reduction of CuII complex with all CDs according to their redox potentials.
The photo‐ATRP worked in the case of CD‐1, CD‐3 and CD‐4 while the remaining materials CD‐2 and CD‐5 showed no response, Table 1. Only CD‐3 initiated photopolymerization according to a reaction protocol based on metal‐free photo‐ATRP but the isolated polymer indicated occurrence of FRP and no control of polymerization (Đ=2.02, M n=240 kD). Again, all CDs possess similar oxidation potential but a brief survey of their XPS‐spectra in Figures S2‐S5 indicate different structural moieties that might affect their photochemistry. Global analysis of fluorescence decays clearly evidences that each CD resulted in distinct decay dynamics vide infra approving again the different origin of emission observed. This provides a clearer pattern about photochemistry of the singlet state. Fluorescence quantum yields reported for other CDs (20–40 %) [36] may draw the conclusion that the excited singlet state might uptake one key function to describe photochemical processes in such materials. Thus, global analysis of fluorescence decays recorded at different wavelengths can bring new viewpoints about complex photochemical and photophysical events occurring in these new materials. [46]
Photophysics of Carbon Dots
Figures 1 a–c depict absorption (a), fluorescence excitation (b) and fluorescence emission (c) spectra. Comparison between absorption and fluorescence excitation spectra shows differences between them indicating that not all components contributing to absorption result in fluorescence. The excitation spectra exhibited different maxima. Their intersection with the emission spectrum shows an excitation band gap for the S0→S1 transition of 2.1–2.2 eV, and 2.6 eV in the case of CD‐1–CD‐3, and CD‐4 and CD‐5, respectively. Interestingly, the lower gap relates to the lac dye‐based materials while the remaining compounds refer to alginate (CD‐4) and citric acid (CD‐5) based materials carrying no conjugated carbon in the substrate used to make them. Nevertheless, both groups; that is CD‐1–CD‐3, and CD‐4 and CD‐5, exhibit only a small difference between them although both originate from biomass with different origin. Assuming that these emitting species are part of the photoinitiating system, one can draw the conclusions based on their redox potentials residing between 0.25–0.29 V (Table 1) that the energetic level of the HOMO appears large enough that after excitation electron transfer can proceed with an acceptor such as an iodonium salt (E red=−0.6 V [47] ) or the CuII complex (E red=−0.24 V [6] ). This generates initiating species such as radicals and CuI complex needed to initiate FRP and CRP, respectively. Thus, ΔG el appears always significantly below zero. However, results obtained indicate different activities to initiate FRP, CRP, and CuACC reaction. Internal activation barriers caused by individual shape of the potential curves between the substrates may explain these findings.[ 22 , 48 ]
Figure 1.
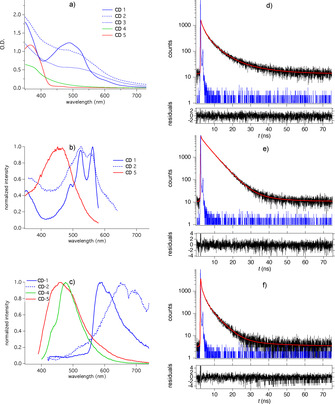
Absorption (a), fluorescence excitation (b), and fluorescence emission (c) spectra obtained in DMSO using different CDs. Excitation occurred at 376 nm to record emission spectra while the emission was kept at 700 nm taking excitation spectra complemented with fluorescence decay curves obtained in the case of CD‐1 recorded at d) 520 nm, e) 600 nm, and f) 720 nm applying 376 nm diode laser excitation. The residuals base on an exponential fit with two linked species, see Table 1.
Furthermore, a structure appears in the excitation spectrum of CD‐1. It exhibits bathochromic shift compared to lac dye [49] from where it originates as caused by the procedure to make CD‐1. Formation of additional unsaturated moieties in the solvothermal procedure may be discussed as one reason for this phenomenon. Additional treatment with amine to synthesize CD‐2 and CD‐3 results in a loss of the fine structure. This also affects the maximum of fluorescence shifting bathochromically after treatment with amine. A drop of emission intensity also occurred by a factor of 4–5 comparing the emission of CD‐1 with respect to CD‐2 and CD‐3. Thus, the treatment of biomass related material for synthesis caused formation of different species that impacts initiation of FRP, CRP and CuACC reaction, Table 1. Furthermore, CD‐4 and CD‐5 exhibit hypsochromic shifted fluorescence and absorption. Although the basis material did not comprise any unsaturated carbon moieties, the applied synthesis procedure resulted in formation of CDs possessing the capability to initiate FRP, CRP and CuACC.
Figures 1 d–f depict the fluorescence decay traces of CD‐1 measured at the three positions related in the emission spectrum to the left, the middle and the right part. Supporting information provides data of the respective decays considering CD‐1–CD‐5. They were globally analyzed in 20 nm intervals over the entire emission spectrum. Data in Table 1 indicate the emission of two (CD‐1, CD‐3, CD‐4) and three components (CD‐2, CD‐5) in the global analysis contributing to the overall emission (see bold and underlined data). It shows the complex mechanism occurring in the excited state, Scheme 1. Global analysis has received an important tool to explore complex photophysics related to respective reaction routes. [46] One of the components exhibits sometimes a negative amplitude meaning that this species definitively functions as a precursor in an equilibrium in the excited state. The average lifetime of the excited state resides in nanoseconds. It appears sufficient to react with further components such as onium salts or CuII complex resulting in reduction of them. Supporting information provides the amplitudes of each component contributing to the decay. Interestingly, all CDs exhibit fluorescence decays in the sub‐nanosecond, a further between 3–6 ns, and an additional appearing around 10–14 ns. The components, which were not linked in the global fit Scheme, additionally fluorescence. One still keeps in mind that CDs depict a pattern of an emitting composition. From this point of view, it surprises that two or three components result in an acceptable global fluorescence decay fit. SI gives more information about the results obtained in the global fit of fluorescence data.
Scheme 1.
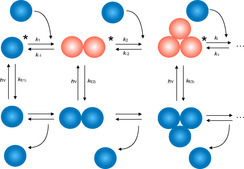
Proposed reaction Scheme for the processes occurring in both aggregation in the ground and excited state, respectively. Emission may proceed by one or aggregated species existing in an equilibrium for both the ground state and the excited state. [50] Global analysis patterns the contribution of each species. [46]
Based on the results shown in Figure 1, one can draw the general reaction Scheme shown in Scheme 1. Aggregation can occur in both the ground and excited state with more reactants as supported by previous discussions on aggregation induced emission. [34] It also explains the short decay component in Figure 1 f in the red part of the emission spectrum caused by excitation of aggregates. We can assume that the probability to react with either an onium salt or CuII complex increases with longer lifetime, and therefore component two and/or three could be the participating species in the initiation protocol. Overall, one can conclude that the decay time appears large enough for the excited state to facilitate the reaction with the mentioned co‐initiating components. There appears no issue related to intramolecular photophysical processes causing a significant shorter decay time resulting in less interaction with the coinitiator. Such intramolecular photo processes can relate for example to proton transfer or intramolecular charge transfer (ICT) diminishing the overall efficiency of the photoinitiating system. Nevertheless, the triplet state might also need more attention for inclusion into the reaction mechanism. This will be the focus of future studies.
Interestingly, fluorescence investigations of dry CDs as powder resulted in much shorter decay times of the emission (see data in Supporting information). Therefore, a solvent certainly results in less interactions and therefore less fluorescence quenching while intermolecular interactions between CDs in the dry material cause the observed quenching as shown by the much shorter decay time. SI information shows the results in Figure S8.
Photoinduced FRP using CDs as Sensitizer
Photoinduced FRP occurred with all CDs using an iodonium salt comprising (t‐C4H9‐Ph)2I+ cation (see Scheme 2 for the structure) carrying three different anions (see Scheme 2 for the respective mechanism). The anions selected do not participate in the redox mechanism but their weak coordination ability (WCA) [51] favors their use in higher viscous monomers in photopolymerization studies. Particular anions b and c derive from super acids. They exhibit outstanding dissolution properties as quantified in the case of anion b approaching up to 577 g L−1 in the monomer TPGDA. [52] This appeared as important to achieve high reactivity.
Scheme 2.
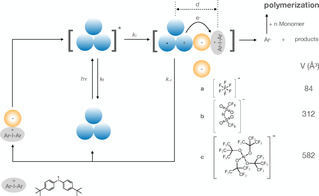
Proposal for generation of initiating radicals based on PET based on photoexcited CDs including molecular aggregates. Excitation results in formation of the excitated state, which transfers an electron to the acceptor that is a diaryl iodonium cation. This proceeds in the singlet state where the size of the anion [48] separates CD +. and the iodyl radical to diminish electron back transfer resulting in a higher yield of initiating radicals for polymerization.
This additionally explains the reactivity differences of the salts. Herein, photoexcited CDs transfer an electron to the iodonium cation 1 resulting in reduction and consecutive decomposition into initiating aryl radicals. Obviously, the anion uptakes a key function here, which was underestimated for a long time. The better ability to dissolve in materials caused by the anions possesses a key function in this scenario. According to Scheme 2, photoexcited CDs (CDs*) result in formation of oxidized CDs (CD +.) while the Ar2I. radical formed decomposes fast into initiating Ar. radical. Competitively, CD +. can also react with Ar2I. that may yield back the CD and 1. Thus, the anion of the iodonium salt must uptake a certain function here. After electron transfer of CD to 1, the charge can move back from 1 −. to CD +. where the latter also possesses a big capability to receive an electron back resulting in a decrease of initiation efficiency.
Scheme 2 summarizes the reaction steps. Thus, one needs to decrease this path to receive a higher yield of Ar.. The weak coordinating anion a, b or c facilitates to solvate CD +. easier, which can diffuse out of the solvent cage within its lifetime. Otherwise, the electron back transfer forms back CD. This also depends on the nature of the anion exhibiting the larges size in the case of c. [48] The back electron transfer plays a certain role in photoexcited CDs, since the fluorescence measurements show that the photochemistry based on the singlet state, plays a certain role vide supra. The larger the radius (d) of the anion, the higher the separation distance of the intermediates CD +. and Ar2I. formed. Thus, both intermediates possess a better probability to escape from solvent cage wherein they were formed if the anion generates a distance d between them being large enough to diminish the rate for electron back transfer, Scheme 2. The results obtained for FRP shown in the Tables S2–S5 in the SI support these findings. Polymerization rate achieves the largest values in case of the anion c using TPGDA as monomer for polymerization. This also agrees with previous findings reported in the case of CD‐4. [27] Interesting appears also the fact that the selection of different CDs also enables higher wavelengths for excitation. This appears of practical interest for applications where scattering and penetration depth of excitation light can be seen as an important point. As a matter of fact, longer wavelength radiation scatters less and compared to these released at shorter wavelengths. [53] Thus, future synthetic work may focus to fabricate CDs exhibiting significant bathochromic shifted absorption to enable applications where scattering can be seen as an issue.
Photoinduced ATRP using CDs as Photocatalyst
Capability of CDs to sensitize generation of initiating radicals in FRP extends their exploration as catalyst for tailor‐made polymer synthesis according to a photo‐ATRP protocol. [6] Both, the metal‐free approach [43] and conditions where CuII resided at the ppm‐scale[ 27 , 29 ] were applied to approve their function to work in controlled radical polymerization. This connects to preliminary work where alginate‐based CDs (CD‐4) demonstrated the feasibility to work in such systems. [27] Thus, controlled polymerization of methyl methacrylate (MMA) in DMSO was studied using ethyl α‐bromophenylacetate (EBPA) as alkyl halide activator, CuBr2 as the deactivator, tris (2‐pyridylmethyl) amine (TPMA) as the ligand and CDs as photocatalyst. Different kinds of CDs were tested for the polymerization of MMA under blue‐light LED irradiation (λ=405 nm, P=100 mW cm−2). Among the CDs used, CD‐1, CD‐3 and CD‐4[ 27 , 29 ] showed good efficiency for the photoinitiated ATRP in conjunction with CuBr2/TPMA catalyst at the ppm scale. Even though CD‐3 also resulted in formation of polymer with very good controlled molecular weight characteristics, this system resulted only in monomer conversion of <3 % after 24 h exposure under 405 nm LED irradiation (entry 8, Table 2). Thus, the brutto‐polymerization constant k pol exhibits in the case of CD‐3 a significant lower value (k pol=0.13×10−2 min−1) compared to CD‐1 (k pol=1.79×10−2 min−1). Comparison with CD‐4 (k pol=11.3×10−2 min−1) also approves that k pol is lower in the case of CD‐3. This may explain the differences obtained for the dispersity Đ, which approaches values obtained for the classical ATRP using an amount of 1000 ppm of CuI catalyst. [4] Thus, less reactivity means higher selectivity: this also nicely fits for these reaction conditions. The higher reactivity of CD‐4 also connects to higher contribution of FRP explaining the higher dispersity (Đ) reported. [27] Moreover, no polymerization was observed in the absence of CuBr2/TPMA catalyst (entry 1, Table 2) or EPBA initiator (entry 2, Table 2) and when the reaction mixture was kept in the dark for 48 h (entry 3, Table 2). These results indicate the importance of adding an ATRP initiator and the photocatalyst as well to interact with the light source resulting in successful controlled polymerization.
Table 2.
Photoinduced Cu‐catalyzed ATRP of MMA using CDs in different experimental conditions[a] exposed at 405 nm.
|
Entry |
CD |
[MMA]/[EBPA]/[CuBr2]/[TPMA] |
t [h] |
Conv. [%] |
M n,th [kDa] |
M n,GPC [kDa] |
Đ |
|---|---|---|---|---|---|---|---|
|
1 |
CD‐1 |
300/1/0/0.135 |
24 |
0 |
|
|
|
|
2 |
CD‐1 |
300/0/0.03/0.135 |
24 |
0 |
|
|
|
|
3[b] |
CD‐1 |
300/1/0.03/0.135 |
48 |
0 |
|
|
|
|
4 |
CD‐1 |
300/1/0.03/0.135 |
24 |
35.3 |
10.7 |
12.8 |
1.10 |
|
5 |
CD‐1 |
300/1/0.00/0.135 |
24 |
0 |
|
|
|
|
6 |
CD‐2 |
300/1/0.03/0.135 |
24 |
0 |
|
|
|
|
7 |
CD‐2 |
300/1/0.00/0.135 |
24 |
0 |
|
|
|
|
8 |
CD‐3 |
300/1/0.03/0.135 |
24 |
<3 |
10.9 |
9.7 |
1.14 |
|
9 |
CD‐3 |
300/1/0.00/0.135 |
24 |
83 |
|
240 |
|
|
10 |
CD‐4 [27] |
300/1/0.03/0.135 |
0.5 |
5.5 |
10.9 |
23.0 |
1.30 |
|
11 |
CD‐4 |
300/1/0.00/0.135 |
1 |
<1 |
|
500 |
2.40 |
|
12 |
CD‐5 |
300/1/0.03/0.135 |
24 |
0 |
|
|
|
|
13 |
CD‐5 |
300/1/0.00/0.135 |
24 |
0 |
[a] Reactions were conducted in 50 vol % DMSO, mCDs=3 mg, and irradiated under blue light LED (λ=405 nm, 100 mW cm−2). Conversion was calculated gravimetrically. Theoretical molecular weight (M n,th) values were calculated based on conversions M n,th=MEBPA+ [MMA]/[EBPA]×conversion×MMMA. Number‐average molecular weight (M n) and disperty (Đ) were obtained by gel permeation chromatography (GPC) in THF based on poly(methyl methacrylate) standards. [b] Reaction was kept in the dark.
Table 2 shows increase of molecular weight and conversion upon exposure evidencing the expected property for the ATRP process applying CD‐1 under blue LED irradiation. Dispersity obtained with this process appears very close to the ideal case; that is Đ tries to approach unity, being comparable with traditional ATRP process requesting very high amount of CuI catalyst (>1000 ppm). However, our approach requests amount ≤100 ppm. Excellent control was achieved with this system being a unique process in photoinduced ATRP. Moreover, only CD‐3, and CD‐4 resulted in polymer formation applying a metal‐free photo‐ATRP approach (entries 9 and 11 in Table 2) while CD‐1, CD‐2, and CD‐5 failed as shown by the entries 5, 7, and 13, respectively. However, polymer analysis approved that this did not follow a controlled polymerization Scheme as shown by the much larger M n and Đ. Thus, one definitively needs CuII to succeed but the remaining concentration at the ppm scale does not seen to appear as harmful. CD‐3 originated from biomass comprising aromatic moieties while sodium alginate responsibly led to CD‐4. Thus, the manufacturing conditions may lead to material that may show an initiating efficiency in the case of metal‐free photo‐ATRP conditions because the amine concentration used for synthesis was the highest in the case of CD‐3 comparing the conditions between CD‐1–CD‐3. Thus, it can certainly generate free radicals following rather FRP approach resulting in 83 % conversion.
Following these results, CD‐1 exhibited the best performance in the photo‐ATRP protocol. Additional studies confirm kinetic data in Figure 2 a, and linear increase of M n and Đ appearing around 1.1 (Figure 2 b). Only 100 ppm of CuBr2/TPMA catalyst were needed to achieve these results with CD‐1 as light sensitive component. The almost symmetrical shape of the curves in Figure 2 c again shows the dominance of the conditions for controlled radical polymerization.
Figure 2.
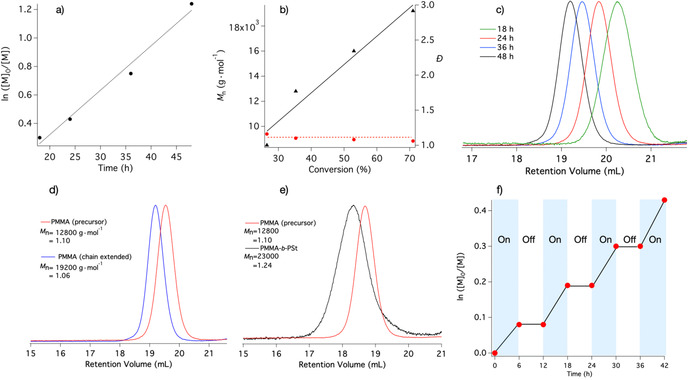
Photoinduced Cu‐catalyzed ATRP of MMA under blue light LED irradiation (λ=405 nm, 100 mW cm−2, reaction condition: [MMA]/[EBPA]/[CuBr2]/[TPMA]=300/1/0.03/0.135 in DMSO 50 vol % (mCD‐1=3 mg). a) Kinetics of the polymerization, b) Number‐average molecular weight (M n, triangle) and dispersity (Đ, point) as a function of monomer conversion, c) GPC of molecular weight distribution for the PMMA synthesized with different exposure time, d) GPC chromatograms of PMMA chain extended, e) Block copolymer (PMMA‐b‐PSt) with polystyrene (PSt), and f) Temporal control of the photoinduced Cu‐catalyzed ATRP using CD‐1 under blue light LED irradiation in the photoinduced Cu‐catalyzed ATRP). Chain extension conditions: ([MMA]0/[Precursor]0/[CuBr2]0/[TPMA]=300/1/0.03/0.135), Block copolymerization conditions: ([St]0/[Precursor]0/[CuBr2]0/[TPMA]=300/1/0.03/0.135). Both reactions are done in 50 vol % DMF at room temperature with 405 nm LED.
Chain‐end functionality of the PMMA obtained by photoinduced Cu‐catalyzed ATRP using CD‐1 was confirmed by chain extension experiments and block copolymerization. The PMMA macroinitiator was synthesized under conditions [MMA]/[EBPA]/[CuBr2]/[TPMA]=300/1/0.03/0.135 in DMSO 50 vol % (mCD‐1=3 mg) facilitating to expose with a 405 nm LED (M n=12 800 g mol, Đ=1.10). Gel permeation chromatography (GPC) of the polymers in Figure 2 d,e exhibited a clear shift toward lower retention volumes without any detectable shoulder or tailing caused by lower molecular weight components in both cases.
Temporal control experiments were performed by switching the blue light ON/OFF intermittently. Kinetic results showed the polymerization of MMA in the presence of CuBr2/TPMA proceeding under blue light whereas the reaction almost stopped by turning off the LED (OFF‐mode). Polymer formation only occurred after turning ON the blue light periodically resulting in higher conversions as shown in Figure 2 f.
Photoinduced CuAAC Reactions Using CDs
A mixture of phenyl acetylene and benzyl azide in the presence of CuBr2/PMDETA complex in DMSO was irradiated with a blue‐emitting LED (λ=405 nm, 100 mW cm−2) applying 1H‐NMR to follow the reaction. Figure 3 shows the conversion degrees obtained for the photoinduced CuAAC reaction between phenyl acetylene and benzyl azide using different CDs and CuBr2/PMDETA. CD‐4 and CD‐5 exhibited good reactivity with almost quantitative yields while CD‐1, CD‐2, and CD‐3 showed less reactivity within an exposure window of 4 h. This again demonstrates the influence of the biomass’ origin relating to aromatic comprising materials in the case of CD‐1–CD‐3, and aliphatic based biomass in the case of CD‐4 and CD‐5. This reaction may receive additional attention in the future to synthesize tailor made polymers with greener components applying a new photocatalyst. It complements preliminary studies applying either UV[ 54 , 55 ] or NIR [42] light to initiate CuACC reaction. Nevertheless, CDs offer the opportunity to direct synthesis towards a route with more green components. Figure 3 exhibits the 1H‐NMR spectral changes during click reaction with CD‐5 as photosensitizer. Triazole proton b′ appearing at 8.7 ppm evidences the success of photo‐CuACC with CDs.
Figure 3.
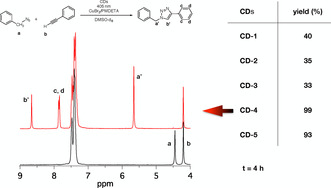
Photoinduced CuAAC reaction between benzyl azide and phenyl acetylene in DMSO under inert atmosphere (λ=405 nm, 100 mW cm−2). Left side: 1H‐NMR spectra of the reaction media before (black) and after (red) the photoinduced click reaction using CD‐4 under blue light LED irradiation (λ=405 nm, 100 mW cm−2). Right side: Conversions determined by 1H‐NMR spectroscopy using different CDs.
Conclusion
CD‐s with distinct origin show different performance to sensitize/initiate all three types of reactions applying a blue‐light emitting LED; that is photo‐ATRP, photoinitiated FRP, and photo‐CuAAC. The latter quantitatively proceeded with CD‐4 and CD‐5 related to biomass comprising mainly aliphatic moieties while those with aromatic origin (CD‐1–CD‐3) did not perform well. The procedure applied for synthesis results therefore in different structural moieties resulting in different reaction efficiencies.
Furthermore, the structures formed based on amine (CD‐2 and CD‐3) did not bring progress in order to receive polymer with narrow molecular weight distribution. Overall, CD‐1 can be seen as the best candidate to make polymers approaching a dispersity close to that of the traditional ATRP using CuI. Our studies evidenced moving towards greener conditions where CuII resided at the ppm‐range. In addition, the use of carbon nanodots also enables such systems to more green systems because they derive from biomass but their origin also differently influences the performance in photo‐ATRP. Future research shall focus on alternative ligands to complex CuII originating from biomass. The search for biomass as a source of ligands for complexing CuII would be of interest although the redox properties and the catalytic activity should remain similar to sensitizers from fossil resources.
Free radical polymerization studies showed well performance of anion c comprising an aluminate pattern followed by b as concluded by comparison with anion a. This can be seen rather as a compromise and challenges future research to enable the use of anions, which i)appear less harmful, and ii) base on biomass.
Comparison of the absorption also indicates different photonic properties concluded by comparison of CD‐1, CD‐2, and CD‐3 with CD‐4, and CD‐5. Future research shall focus on biomass comprising moieties with significant bathochromic shifted absorption. This will enable alternative light sources based on LEDs with emission in the visible and NIR, and of course, sunlight might be an option as well.
Attributed to the nice biocompatibility of sustainable CDs, their assisted photoreaction can become big potential in future biological applications. Before that, the IC 50 influence of these CDs on the cellular morphology and antimicrobial activity should be of interest in future work.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Z.C. acknowledges the excellent young scholar sponsorship program by National Forestry and Grassland Administration of China Funding (2019132611), and the Young Elite Scientists Sponsorship Program by CAST (2018QNRC001). BS acknowledges the Project D‐NL‐HIT carried out in the framework of the INTERREG‐Program Deutschland‐Nederland, which is co‐financed by the European Union, the MWIDE NRW, the Ministerie van Economische Zaken en Klimaat and the provinces of Limburg, Gelderland, Noord‐Brabant und Overijssel. BS additionally thanks the county of North Rhine‐Westphalia for funding of the project REFUBELAS (grant 005‐1703‐0006), and the BMWi for funding the Project Innovative UV‐LED Coatings for financial support of research at Niederrhein University of Applied Sciences (ZF4288703WZ7). Open access funding enabled and organized by Projekt DEAL.
C. Kütahya, Y. Zhai, S. Li, S. Liu, J. Li, V. Strehmel, Z. Chen, B. Strehmel, Angew. Chem. Int. Ed. 2021, 60, 10983.
Contributor Information
Prof. Dr. Zhijun Chen, Email: chenzhijun@nefu.edu.cn.
Prof. Dr. Bernd Strehmel, Email: bernd.strehmel@hsnr.de.
References
- 1. Bonfield H. E., Knauber T., Lévesque F., Moschetta E. G., Susanne F., Edwards L. J., Nat. Commun. 2020, 11, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dadashi-Silab S., Doran S., Yagci Y., Chem. Rev. 2016, 116, 10212–10275. [DOI] [PubMed] [Google Scholar]
- 3. Jiang J., Ye G., Wang Z., Lu Y., Chen J., Matyjaszewski K., Angew. Chem. Int. Ed. 2018, 57, 12037–12042; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12213–12218. [Google Scholar]
- 4. Konkolewicz D., Schröder K., Buback J., Bernhard S., Matyjaszewski K., ACS Macro Lett. 2012, 1, 1219–1223. [DOI] [PubMed] [Google Scholar]
- 5. Matyjaszewski K., Macromolecules 2012, 45, 4015–4039. [Google Scholar]
- 6. Ribelli T. G., Fantin M., Daran J.-C., Augustine K. F., Poli R., Matyjaszewski K., J. Am. Chem. Soc. 2018, 140, 1525–1534. [DOI] [PubMed] [Google Scholar]
- 7. Schmitz C., Oprych D., Kutahya C., Strehmel B., in Photopolymerisation Initiating Systems (Eds.: Lalevée J., Fouassier J.-P.), Royal Society of Chemistry, London, 2018, pp. 431–478. [Google Scholar]
- 8. Wu Z., Jung K., Boyer C., Angew. Chem. Int. Ed. 2020, 59, 2013–2017; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2029–2033. [Google Scholar]
- 9. Zhang L., Wu C., Jung K., Ng Y. H., Boyer C., Angew. Chem. Int. Ed. 2019, 58, 16811–16814; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16967–16970. [Google Scholar]
- 10. Yeow J., Chapman R., Gormley A. J., Boyer C., Chem. Soc. Rev. 2018, 47, 4357–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu S., Yeow J., Boyer C., ACS Macro Lett. 2018, 7, 1376–1382. [DOI] [PubMed] [Google Scholar]
- 12. Bruder F.-K., Hagen R., Rölle T., Weiser M.-S., Fäcke T., Angew. Chem. Int. Ed. 2011, 50, 4552–4573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4646–4668. [Google Scholar]
- 13. Strehmel B., Ernst S., Reiner K., Keil D., Lindauer H., Baumann H., Z. Phys. Chem. 2014, 228, 129–153. [Google Scholar]
- 14. Baumann H., Hoffmann-Walbeck T., Wenning W., Lehmann H.-J., Simpson C. D., Mustroph H., Stebani U., Telser T., Weichmann A., Studenroth R., Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015, pp. 1–51. [Google Scholar]
- 15. Zhu J., Zhang Q., Yang T., Liu Y., Liu R., Nat. Commun. 2020, 11, 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ligon S. C., Liska R., Stampfl J., Gurr M., Mülhaupt R., Chem. Rev. 2017, 117, 10212–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee K., Corrigan N., Boyer C. A. J. M., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202016523; [DOI] [Google Scholar]; Angew. Chem. 2021, 10.1002/ange.202016523. [DOI] [Google Scholar]
- 18. Zhang L., Shi X., Zhang Z., Kuchel R. P., Namivandi-Zangeneh R., Corrigan N., Jung K., Liang K., Boyer C., Angew. Chem. Int. Ed. 2021, 60, 5489–5496; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 5549–5556. [Google Scholar]
- 19. Crespi S., Simeth N. A., König B., Nat. Rev. Chem. 2019, 3, 133–146. [Google Scholar]
- 20. Dawood A., Marti Marti B., Sauret-Jackson V., Darwood A., Br. Dent. J. 2015, 219, 521–529. [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Jiang M., Niu N., Chen Z., Li S., Liu S., Li J., ChemSusChem 2018, 11, 11–24. [DOI] [PubMed] [Google Scholar]
- 22. Strehmel B., Schmitz C., Kütahya C., Pang Y., Drewitz A., Mustroph H., Beilstein J. Org. Chem. 2020, 16, 415–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schubert E. F., Kim J. K., Science 2005, 308, 1274.15919985 [Google Scholar]
- 24. Strehmel B., Schmitz C., Cremanns K., Göttert J., Chem. Eur. J. 2019, 25, 12855–12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Z., Corrigan N., Bagheri A., Jin J., Boyer C., Angew. Chem. Int. Ed. 2019, 58, 17954–17963; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18122–18131. [Google Scholar]
- 26. Corrigan N., Yeow J., Judzewitsch P., Xu J., Boyer C., Angew. Chem. Int. Ed. 2019, 58, 5170–5189; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5224–5243. [Google Scholar]
- 27. Kütahya C., Wang P., Li S., Liu S., Li J., Chen Z., Strehmel B., Angew. Chem. Int. Ed. 2020, 59, 3166–3171; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3192–3197. [Google Scholar]
- 28. Tasdelen M. A., Uygun M., Yagci Y., Macromol. Chem. Phys. 2011, 212, 2036–2042. [Google Scholar]
- 29. Kütahya C., Schmitz C., Strehmel V., Yagci Y., Strehmel B., Angew. Chem. Int. Ed. 2018, 57, 7898–7902; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8025–8030. [Google Scholar]
- 30. Kütahya C., Meckbach N., Strehmel V., Gutmann J. S., Strehmel B., Chem. Eur. J. 2020, 26, 10444–10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.J. Buzek, E. Győri, Richtlinie 2011/65/EU des Europäischen Parlaments und des Rates zur Beschränkung der Verwendung bestimmter gefährlicher Stoffe in Elektro- und Elektronikgeräten, 2011, Bruxelles, EU, https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:174:0088:0110:de:PDF.
- 32. Wang P., Zheng D., Liu S., Luo M., Li J., Shen S., Li S., Zhu L., Chen Z., Carbon 2021, 171, 946–952. [Google Scholar]
- 33. Wang P., Liu C., Tang W., Ren S., Chen Z., Guo Y., Rostamian R., Zhao S., Li J., Liu S., Li S., ACS Appl. Mater. Interfaces 2019, 11, 19301–19307. [DOI] [PubMed] [Google Scholar]
- 34. Liu M. L., Chen B. B., Li C. M., Huang C. Z., Green Chem. 2019, 21, 449–471. [Google Scholar]
- 35. Liu J., Liu Y., Liu N., Han Y., Zhang X., Huang H., Lifshitz Y., Lee S. T., Zhong J., Kang Z., Science 2015, 347, 970–974. [DOI] [PubMed] [Google Scholar]
- 36. Li H., Kang Z., Liu Y., Lee S.-T., J. Mater. Chem. 2012, 22, 24230–24253. [Google Scholar]
- 37. Sautrot-Ba P., Malval J.-P., Weiss-Maurin M., Paul J., Blacha-Grzechnik A., Tomane S., Mazeran P.-E., Lalevée J., Langlois V., Versace D.-L., ACS Sustainable Chem. Eng. 2018, 6, 104–109. [Google Scholar]
- 38. Al Mousawi A., Garra P., Schmitt M., Toufaily J., Hamieh T., Graff B., Fouassier J. P., Dumur F., Lalevee J., Macromolecules 2018, 51, 4633–4641. [Google Scholar]
- 39. Schmitz C., Strehmel B., J. Coat. Technol. Res. 2019, 1–15. [Google Scholar]
- 40. Wang S., Gao L. in Industrial Applications of Nanomaterials: Micro and Nano Technologies (Eds.: Thomas S., Grohens Y., Pottathara Y. B.), Elsevier, Amsterdam, 2019, pp. 181–203. [Google Scholar]
- 41. Sagbas S., Sahiner N., in Nanocarbon and its Composites (Eds.: Khan A., Jawaid M., A. M. Asiri Inamuddin,), Woodhead Publishing, Sawston, 2019, pp. 651–676. [Google Scholar]
- 42. Kütahya C., Yagci Y., Strehmel B., ChemPhotoChem 2019, 3, 1180–1186. [Google Scholar]
- 43. Treat N. J., Sprafke H., Kramer J. W., Clark P. G., Barton B. E., Read de Alaniz J., Fors B. P., Hawker C. J., J. Am. Chem. Soc. 2014, 136, 16096–16101. [DOI] [PubMed] [Google Scholar]
- 44. Magenau A. J., Strandwitz N. C., Gennaro A., Matyjaszewski K., Science 2011, 332, 81–84. [DOI] [PubMed] [Google Scholar]
- 45. Kocaarslan A., Kütahya C., Keil D., Yagci Y., Strehmel B., ChemPhotoChem 2019, 3, 1127–1132. [Google Scholar]
- 46. Strehmel B., Seifert H., Rettig W., J. Phys. Chem. B 1997, 101, 2232–2243. [Google Scholar]
- 47. Schmitz C., Halbhuber A., Keil D., Strehmel B., Prog. Org. Coat. 2016, 100, 32–46. [Google Scholar]
- 48. Pang Y., Shiraishi A., Keil D., Popov S., Strehmel V., Jiao H., Gutmann J. S., Zou Y., Strehmel B., Angew. Chem. Int. Ed. 2021, 60, 1465–1473; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1486–1495. [Google Scholar]
- 49. Pattanayak S., Chakraborty S., Mollick M. M. R., Roy I., Basu S., Rana D., Gauri S. S., Chattopadhyay D., Chakraborty M., New J. Chem. 2016, 40, 7121–7131. [Google Scholar]
- 50.X. Zhang, H. Wang, C. Ma, N. Niu, Z. Chen, S. Liu, J. Li, S. Li, Mater. Chem. Front. 2018, Ahead of Print.
- 51. Krossing I., Raabe I., Angew. Chem. Int. Ed. 2004, 43, 2066–2090; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2116–2142. [Google Scholar]
- 52. Brömme T., Oprych D., Horst J., Pinto P. S., Strehmel B., RSC Adv. 2015, 5, 69915–69924. [Google Scholar]
- 53. Uo M., Kudo E., Okada A., Soga K., Jogo Y., J. Photopolym. Sci. Technol. 2009, 22, 551–554. [Google Scholar]
- 54. Tasdelen M. A., Yagci Y., Angew. Chem. Int. Ed. 2013, 52, 5930–5938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6044–6053. [Google Scholar]
- 55. Tasdelen M. A., Yilmaz G., Iskin B., Yagci Y., Macromolecules 2012, 45, 56–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary