

Abstract
The generation of bioactive molecules from inactive precursors is a crucial step in the chemical evolution of life, however, mechanistic insights into this aspect of abiogenesis are scarce. Here, we investigate the protein‐catalyzed formation of antivirals by the 3C‐protease of enterovirus D68. The enzyme induces aldol condensations yielding inhibitors with antiviral activity in cells. Kinetic and thermodynamic analyses reveal that the bioactivity emerges from a dynamic reaction system including inhibitor formation, alkylation of the protein target by the inhibitors, and competitive addition of non‐protein nucleophiles to the inhibitors. The most active antivirals are slowly reversible inhibitors with elongated target residence times. The study reveals first examples for the chemical evolution of bio‐actives through protein‐catalyzed, non‐enzymatic C−C couplings. The discovered mechanism works under physiological conditions and might constitute a native process of drug development.
Keywords: aldol condensations, antivirals, chemical evolution, protease inhibition, protein-templated reactions
Self‐organized virus killing: Antivirals are generated through aldol condensations catalyzed by the 3C protease of enterovirus D68 and inhibit viral proliferation in infected human cells. Best inhibitors are slow and sticky. The process demonstrates a potentially native mechanism for the evolution of functional bioactive molecules.

Introduction
According to Oparin and Haldane, the chemical evolution of life began with the formation of its chemical building blocks, [1] and considerable progress has been made in explaining the prebiotic generation of sugars, [2] amino acids, [3] and nucleosides. [4] Formation of life's building blocks, however, does not rationalize the generation and selection of functional, bioactive molecules, which are required to develop complex features of living systems including metabolism, compartmentalization, and replication. [5] Understanding the mechanisms yielding functionally bioactive molecules will provide insights into the emergence of the complex features of life and thus is worth pursuing.
We hypothesize here that bioactive molecules can be generated by a mechanism of chemical evolution involving templated reactions embedded within a multidimensional network of chemical reactivity or “dynamic reaction system”. Reversible and irreversible protein‐templated reactions have been studied extensively, mostly in order to identify protein‐binding ligands. [6] Initially, proteins were demonstrated to stabilize imines and hydrazones formed reversibly from aldehydes, amines, and hydrazines. [7] These products have been useful in dynamic ligation assays for the sensitized and site‐directed detection of low‐affinity fragments and bioactive fragment combinations. [8] Protein ligands were also formed via irreversible, templated reactions using dipolar cycloadditions of azides with alkynes, or thioacids. [9] For protein‐catalyzed amidations, the mechanism and saturation kinetics were established yielding fragment combinations with super‐additive affinity. [10] Recently, three‐component Mannich ligations catalyzed by the human transcription factor STAT5 were discovered furnishing anti‐leukemic drugs active in mice. [11] Further templated multicomponent reactions were investigated.[ 12 , 13 ] There have been, however, no reports of protein‐templated, native C−C couplings yet, although these reactions are essential for the construction of many bioactive molecules found in Nature.
In this paper, we study the chemical evolution of antivirals through Knoevenagel condensations, [14] native C−C couplings of the aldol type, catalyzed by the 3C protease of enterovirus EV D68. We envisioned that the protease would be able to activate aldehydes via hydrogen bonds inducing aldol reactions with suitable C‐nucleophiles. To test this hypothesis, potential C‐nucleophiles are screened in an enzyme activity assay. Kinetics and thermodynamics of protein‐templated reactions are analyzed revealing a mechanism for the chemical evolution of bioactive molecules in a dynamic reaction system and providing insights into the mode of action of cellularly active antivirals obtained by this approach.
Results and Discussion
Enteroviral 3C proteases are validated drug targets in enteroviral and rhinoviral infections, [15] being strictly selective for substrates with glutamine (Gln) in P1‐position. The same applies to the 3C‐like coronavirus main proteases. [16] Peptidomimetics such as rupintrivir (AG‐7404) were developed as potent inhibitors of 3C proteases, however, failed in advanced clinical studies so far. [17] This failure might be due to the comparably high molecular weight, limited bioavailability, and reduced ability to cross biological membranes of peptide‐based inhibitors. Fragment‐based inhibitors might overcome these limitations and have shown broad‐band activity toward 3C proteases. [8c] The 3C protease of enterovirus D68 (EV D68) cleaves amide bonds at P1 by attacking the electrophilic carbonyl carbon with the nucleophilic cysteine residue (Cys147) of the active site. This attack is enabled through the activation of the amide carbonyl by H‐bond‐donors providing positive partial charge in the oxyanion hole.
Discovery of Protein‐Catalyzed C−C Couplings
3‐Formyl benzamide 1 has been reported as a moderately active inhibitor of rhinovirus 3C protease, presumably due to efficient biomimicry of the native glutamine residue. [18] We hypothesized that binding of aldehyde 1 through an H‐bonding network might activate the electrophilic aldehyde carbonyl for the addition of C‐nucleophiles resulting in C−C coupling reactions. This should be possible, if the active site favors formation of the enol‐tautomer of an enolizable carbonyl fragment F and thus stabilizes a protein‐bound Zimmerman–Traxler transition state (Figure 1 a). [19] Using the crystal structure 3ZVG, [20] aldehyde 1 was docked into the active site of EV D68 3C protease and a binding model was derived (Figure 1 b). In this, the primary amide of 1 bound to the protease's S1‐pocket via two H‐bonds with Thr142 and one with the imidazole ring of His161. The aldehyde functionality of 1 docked to the oxyanion hole, accepting H‐bonds from the backbone amides of Gly145 and Cys147. The thiol of Cys147 was located closely to the carbonyl‐carbon, ready to attack forming a hemithioacetal intermediate.
Figure 1.

Discovery of protein‐catalyzed C−C couplings. a. Postulated Zimmerman–Traxler transition state of the aldol reaction of aldehyde 1 with the E‐enol tautomer of fragment F bound to EV D68 3C protease. b. Binding hypothesis of 3‐formyl‐benzamide 1 to EV D68 3C protease (3ZVG). c. Enolizable fragments F1–F13. d. Fragment ligation assay of EV D68 3C protease with aldehyde 1 (19 μM) and fragments F1–F13 (19 μM) for 2 h. Activity of protease was determined using a fluorogenic FRET substrate (see text). e. Binding simulation of the presumed anti‐aldol addition product of 1 and F1. f. Covalent binding mode of the Knoevenagel condensation product 2. Color code: white balls—protein carbon atoms, grey sticks—ligand carbon atoms, red balls and sticks—oxygen atoms, blue balls and sticks—nitrogen atoms, yellow balls—protein sulfur atom, red and green arrows—hydrogen bond acceptors and donors, respectively, yellow spheres—lipophilic contacts.
For the testing of this hypothesis, 13 carbonyl fragments (F1–F13) were selected for their potential to form enoles as potent C‐nucleophiles (Figure 1 c). All 13 fragments were inactive as inhibitors (Table S1). Then 2‐formyl‐benzamide 1 (IC50=48 μM) was incubated with EV D68 3C‐protease at a concentration resulting in 75 % residual activity of the enzyme. One of the carbonyl fragments was added per well and the formation of active inhibitors was assayed with the fluorogenic FRET substrate (Dabcyl‐KTLEALFQGPPVY‐E(Edans)‐amide) for read‐out (Figure 1 d). After 2 h incubation, five of the 13 carbonyl fragments (F1, F4, F5, F6, and F8) showed significantly increased inhibition of the protease (p<0.05) in combination with aldehyde 1 compared to the aldehyde alone. HPLC‐QTOF‐MS analysis confirmed the formation of Knoevenagel condensation products in all assays displaying significantly increased inhibition. Relative product formation was quantified after 1 h reaction time at pH values of 4, 6, 8, and 10 with and without the protein (10 μM) present in the assay solution (Figure S1). The highest yields of C−C coupling products were observed for fragment F1 at pH 8.0 with almost 50 %. The aldol addition product with fragment F1, O‐ethyl 2‐cyano‐acetate, fitted into the oxyanion hole of the protease being coordinated by three bifurcated H‐bonds from the backbone amide NH groups of Gly145, Gln146, and Cys147 to the beta‐hydroxy and carbonyl‐functionalities (Figure 1 e). H‐bonds to the secondary hydroxy group might stimulate the elimination of water from the aldol addition product and thus furnish the alpha‐beta‐unsaturated ester as condensation product 2 (Figure 1 f), which might react further under Michael addition of the thiolate of Cys147 to the double bond. Ketone fragments F4–F8 displayed protein‐dependent formation of the Knoevenagel products with a maximum yield of ca. 20 % for F4. pH‐optimum was 8.0 for F4, F6, and F8, pH 6.0 for F5 and F7. Traces of product (ca. 1 %) were formed for F2 and F3 (at pH 10) and for F12 (at all pH values). As Knoevenagel products—according to our initial hypothesis—were expected to be responsible for the increased inhibition, they were re‐synthesized and tested as inhibitors of the EV D68 protease (Table 1). Indeed, most condensation products of aldehyde 1 with enolizable carbonyl compounds yielded potent inhibitors of the 3C protease.
Table 1.
Inhibitors of the 3C protease of enterovirus D68 formed through Knoevenagel condensations of 1 with F1‐F11, their antiviral effect in cells (EC50) and relative antiviral efficiency (AV Eff.). Raw data and S.D. in the Supporting Information.

|
|

The most potent inhibitors 2–6 with IC50 values between 0.46 and 2 μM were more than 100 times more potent than the starting benzaldehyde 1. The strongest effect in the fragment ligation assay was observed for fragment F4, 3‐oxo‐butane nitrile. Thus, the kinetics of the protein‐catalyzed formation of the inhibitor from F4 and cyano‐ketone 5 were determined. At first, 100 μM aldehyde 1 and 100 μM of 3‐oxo‐butane nitrile F4 were incubated with 10 μM of EV D68 protease or without protein at 25 °C for 60 min of 5 was quantified by HPLC‐QTOF‐MS (Figure 2 a). Formation of 5 followed saturation kinetics—typical for a protein‐catalyzed and product‐inhibited reaction. Product concentrations over time were fitted to an exponential saturation function c(t)=c 0+(c max−c 0)(1−e^(−Kt)). The maximum concentration c max was 12.6 μM (with c 0=0) with a half reaction time (t 1/2=ln2/K) of 17 min. The reaction rate k 0=dc/dt at t 0 was 0.51 μM min−1 indicating a 5‐fold rate acceleration in comparison to the uncatalyzed, non‐templated background reaction (0.1 μM min−1). Reaction rates were reduced at lower concentrations of the cyano‐ketone fragment F4 (10 or 50 μM) yielding 12.2 μM (9.2 μM) of 5 with t 1/2 of 115 min (25 min), both with 10 μM protein (Figure 2 b,c). A lower aldehyde concentration (5 μM), 50 μM protein and 100 μM F4 generated 4.8 μM of product 5 with a t 1/2 of 100 min and a background of <0.5 μM (Figure 2 d).
Figure 2.
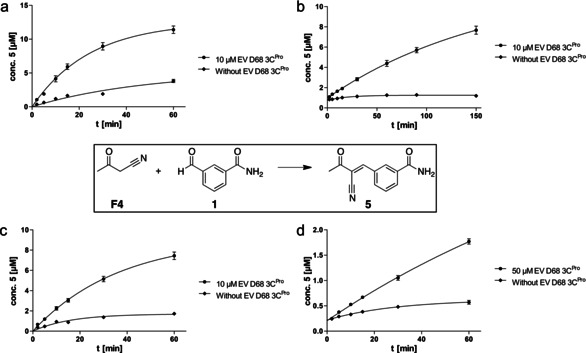
Kinetic analysis of protein‐catalyzed C−C couplings. Time‐dependent formation of 5 at r.t. for 60–150 min with different concentrations of F4, 1 and EV D68 3C. Quantification of 5 was conducted by HPLC‐QToF‐MS using a freshly recorded calibration curve. Time‐dependent concentration of 5 was fitted to the exponential saturation function c(t)=c 0+(c max−c 0)(1−e^(−Kt)). a. F4: 100 μM, 1: 100 μM, c max=12.6 μM and t 1/2=17 min. b. 1: 100 μM, F4: 10 μM, c max=12.2 μM and t 1/2=115 min. c. 1: 100 μM, F4: 50 μM, c max=9.2 μM and t 1/2=25 min. d. 1: 5 μM, F4: 100 μM, c max=4.8 μM and t 1/2=100 min.
The antiviral activity of compounds 2–12 was determined in HeLa cells infected with EV D68 (Table 1, Supporting Information). [21] All compounds tested in the cellular assay were non‐toxic toward the host cells (CC50 >100 μM) and most of them were active as antivirals. Interestingly, for several of the compounds, the antiviral activity in cells (EC50 value) deviated strongly from the inhibition in the enzyme assay (IC50). To assign these deviations the relative antiviral efficiency IC50/ EC50 was calculated for all inhibitors. Values for the inhibition ratio ranged from 0.014 (low antiviral efficiency) for compound 5 to 8.2 (high antiviral efficiency) for inhibitor 12.
Investigating the Chemical and Biochemical Reactivity of Antivirals
As the tested 3‐vinyl‐benzamides 2–13 displayed drastically different relative bioactivities, the next goal was to understand, why this was the case. Realizing that several chemical and biochemical reactions were involved in the generation and variation of bioactivity, we decided to investigate the dynamic reaction system of three representative inhibitors, 2, 5, and 12. For rationalization of their binding see Figure S2. After studying the protein‐catalyzed formation of antivirals, we investigated the reversibility of the C−C couplings with and without protein. While inhibitors 2 and 5 were stable in assay buffer with DTT, decomposition was observed, if DTT was omitted from the buffer (Figure S3). Retro‐reactions of inhibitors 2 and 5 (20 μM) in buffer (pH 8) with and without 10 μM of the protein were monitored by HPLC‐MS, recording the release of 3‐formyl benzamide 1. Cleavage of ketone 5 was significantly faster than that of acrylate ester 2, displaying ca. 30 % decomposition of 5 after 30 min, while only 8 % of 2 were cleaved at the same time. In both cases, the retro‐aldol reaction was pH‐dependent and there was no reaction at pH 4, suggesting the addition of water as the primary step of the cleavage. Decomposition was not accelerated by the protease, excluding a protein‐catalyzed retro‐aldol reaction. Instead the protein reduced the rate of the cleavage reaction slightly, however, without efficiently protecting the inhibitors against decomposition. Even more rapid decomposition was observed for the Knoevenagel products from stronger acidic C‐nucleophiles including barbituric acid, 2‐(methyl‐sulfonyl)‐acetonitrile, and 2‐nitro‐acetonitrile. In these cases, addition of water and retro‐aldol reaction occurred already during synthesis and work‐up. Other inhibitors including diester 12 and cyano‐amides (3,4) did not decompose via a retro‐aldol mechanism.
Next, reactions of the inhibitors with the protease were investigated. Cyano‐ester 2 displayed a linear turnover of the substrate enzyme inhibition assay typical for reversible inhibitors (Figure 3 a). In contrast, cyano‐ketone 5 and diester 12 showed small but visible time‐dependent rate reduction at higher inhibitor concentrations, which could indicate irreversible inhibition of the substrate turnover. [22] Dilution assays were conducted to analyze the subtle differences more clearly (Figure 3 b). For this, the enzyme was inhibited completely at the 20‐fold IC50‐concentration of the inhibitor and then diluted by the factor of 100 prior to addition of the substrate. In the case of inhibitor 2 the activity of the enzyme was instantaneously and fully restored, for inhibitors 5 and 12 the recovery was also rapid but only partial (ca. 80 %). These results suggest that all three inhibitors 2, 5, and 12 act reversibly, although 5 and 12 seem to inhibit the protease with slower kinetics compared to inhibitor 2.
Figure 3.

Reversibility of antiviral protease inhibitors. a. Linear conversion of the FRET substrate over time suggested reversibility of inhibitors 2, 5 and 12. b. Dilution of assays of 2, 5 and 12 with protease from 20× IC50 to 0.2× IC50 indicated reversible inhibition. c. Native protein MS of EV D68 3C protease (7.5 μM) with inhibitors 2 and 5 (100 μM) in 50 mM ammonium bicarbonate buffer with 100 μM DTT, pH 8 showing protein‐inhibitor complex. d. Denaturing protein MS of EV D68 3C protease (8.2 μM) with inhibitor 12 (6.5 μM) in 100 mM HEPES buffer with 1 mM EDTA and 10 μM DTT, pH 8, using a water‐acetonitrile gradient with 0.1 % formic acid. e. Saturation of protein‐ligand complex with inhibitor 2 determined by native MS. Apparent K D: 16 +/− 2.4 μM. For experimental details see Supporting Information.
As these kinetic differences were suspected to be the key to explain the observed cellular antiviral and enzymatic activities, stabilities of the covalent enzyme‐ligand complexes were investigated by employing protein‐mass spectrometry (Figure 3 c–e, Figure S4, S5). Protein‐MS of EV D68 3C protease was recorded and deconvoluted both in native MS in ammonium formate buffer and under denaturing conditions from acetonitrile‐water. Inhibitors 2 and 5 showed no masses of the protein‐ligand complex in denaturing MS, suggesting the rapid decomposition of the protein‐ligand complex. Reversible formation of the protein‐inhibitor complexes with 2 and 5 was confirmed by native protein MS (Figure 3 c,e). Increasing concentrations of inhibitor 2 incubated with a fixed protein concentration yielded a deconvoluted mass signal of the protein‐inhibitor complex with increasing intensity, which was saturated with an apparent dissociation constant (K D) of 16 (+/−2.4) μM. In contrast, inhibitor 12 displayed a protein‐ligand signal with >80 % intensity in denaturing protein‐MS suggesting the slow decomposition of the protein‐ligand complex and a lower k off‐rate (Figure 3 d). In addition, the protein‐inhibitor complex of irreversible inhibitor O‐ethyl 3‐carbamoyl‐cinnamoyl ester 13 was digested with trypsin and the binding site was identified by MS showing the complete alkylation of the 13mer peptide containing Cys147 with inhibitor 13 (Figure S6).
Another factor, which could have an impact on the activity of the generated antivirals, are reactions of the inhibitors with competing, non‐protein nucleophiles in solution. Initially, IC50 values were recorded with dithiothreitol (DTT), a reductant to maintain the activity of the 3C protease. Reducing the concentration of the dithiol DTT increased the activity of some inhibitors considerably including 5 and 12, while the activity of other inhibitors such as 2 was hardly affected (see Table 1). HPLC‐MS analysis of inhibitors 2, 5, and 12 with added DTT showed peaks of the unreacted inhibitor and of inhibitor‐DTT addition products. Quantification indicated 7 % of DTT‐adducts with inhibitor 2, 23 % with inhibitor 5, and 87 % with inhibitor 12 (Figure S7). These results correlated with the observed weak deactivation of compound 2 by DTT and the stronger deactivation of compound 12.
For thermodynamic analysis, binding of inhibitors 2, 5, and 12 was studied by isothermal titration calorimetry (ITC) (Figure 4 a,b). In order to obtain a saturated binding curve of the inhibitors to the protein, buffer and DTT‐concentration needed to be identical in the enzyme contained in the measurement cell and in the syringe used for adding the concentrated ligand solution. While no binding of 12 was observed, inhibitors 2 and 5 displayed similar free binding energies (ΔG) of −28.5 for 2 and −27.2 kJ mol−1 for 5, with apparent affinities of 6.5 and 12.5 μM, respectively. Protein binding of inhibitor 2 was dominated by a large negative enthalpic contribution of ΔH=−50.2 kJ mol−1 and counteracted by a smaller positive entropic contribution of −TΔS=20.9 kJ mol−1. This entropic loss is typical for the reversible binding of small molecules to proteins and reflects the reduced degrees of freedom of the bound versus the free ligand. Inhibitor 5, however, showed different, atypical binding thermodynamics with a negative binding enthalpy of ΔH=−10.5 kJ mol−1 accompanied by a negative entropy term (and, thus positive binding entropy) of −TΔS=−16.7 kJ mol−1. We suspected that this positive binding entropy might result from the release of DTT from inhibitor‐DTT adducts formed at high DTT (10 mM) as detected above in MS. To substantiate this argument, ITC was also conducted at low DTT concentration (0.1 mM) resulting now in a slightly positive entropy term (+0.84 kJ mol−1, see Figure S8) and thus indicating a shift to negative binding entropy with low DTT and less inhibitor‐DTT adduct.
Figure 4.

Thermodynamics and kinetics of protease‐interactions for covalent inhibitors 2, 5, and 12. a. Isothermal titration calorimetry of a solution of 2 (800 μM) added dropwise to EV D68 3C protease (60 μM) in buffer with 10 mM DTT. Binding of 2 was driven by binding enthalpy; ΔG=−28.5 kJ mol−1, N=1, K D=6.5 μM. b. ITC of 5 showed a strong entropic contribution, which was attributed to the release of DTT from the inhibitor‐DTT adduct; ΔG=−27.2 kJ mol−1, N=0.93, K D=12.5 μM. c. Biolayer interferometry (BLI) with EV D68 3C protease immobilized on an NTA‐agarose chips indicated the complete reversibility of the binding for 2 with k on‐rates in the range of 100–200 M−1 s−1 and k off‐rates around 0.015 s−1. d. Binding for 12 was characterized by significantly smaller off‐rates and only partial dissociation resulting in a stronger deviation of the fit using a reversible 1‐to‐1 binding model. e. Symbolic representation of the biolayer interferometry experiment: EV D68 3C protease (grey ribbon) is attached to the sensor (grey balls and plane indicate nickel—nitrilotriacetic acid agarose) via its His‐tag (indicated as imidazole rings). The association and dissociation of inhibitor 2 (indicated as ball and stick model) association—dissociation is recorded as a shift in the interference pattern of visible light as a result of an alteration of the biolayer by small molecule binding. For further details see Supporting Information.
Finally, reversibility and kinetics of inhibitors 2 and 12 with the protease were studied using biolayer interferometry (BLI, Figure 4 c,d). [23] For this, the histidine‐tagged 3C protease was immobilized on a sensor tip modified with nickel‐NTA. Loading of the protein and binding of inhibitors 2 and 12 were then monitored by the time‐dependent shift of the interference pattern of partially reflected light. Binding of both inhibitors was saturated within 120 s with association rates (k on) in the same order of magnitude (100–200 M−1 s−1). Dissociation of both compounds, however, was very different, if monitored in a buffer solution without additional DTT. While inhibitor 2 dissociated completely with a k off around 0.015 s−1, only partial dissociation of inhibitor 12 was observed. The deviation from a reversible binding mode coincided with a reduced correlation of the data fitting with the reversible 1‐to‐1 binding model as reported before. [23] Thus, binding kinetics observed by BLI confirm the high stability of the protease‐ligand complex of 12 corresponding to a significantly longer residence time of 12 compared to inhibitor 2. These findings agree with the high stability of the protease‐inhibitor complex of 12 observed in denaturing MS and with the rapid ligand exchange rate of 2 recorded in native protein MS and in ITC. The relative kinetic stability of the protease‐inhibitor complex of 12 and its elongated residence time [24] corresponds with higher antiviral activity of this compound in infected cells suggesting a robust strategy for the development of fragment‐based inhibitors of viral proteases.
Conclusion
In synopsis, we have studied the chemical evolution of antivirals in a dynamic reaction network, in which the self‐organization of building blocks led to the formation of biochemically and cellularly active inhibitors of EV D68 3C protease (Figure 5). Aldehyde 1 and fragment F bound reversibly to the protein presumably forming a Zimmerman–Traxler transition state intermediate (reaction A) undergoing protein‐catalyzed aldol addition (B) and Knoevenagel condensation (C). The aldol product was released (D) and eliminated water in solution, or the condensation product dissociated into solution (E). The observed protein‐catalyzed aldol condensations constitute the first examples of protein‐templated C−C couplings leading to the formation of potent enzyme inhibitors and antivirals. Little protein‐independent background reactions in solution were detected (A′–C′). Rate and efficiency of inhibitor formation depended on the pH, the CH‐acidity of the C nucleophiles, the binding affinity of the formed C−C coupling products and on the stability of the latter toward hydration and retro‐aldol reactions. Highest rate acceleration and reactivity were found for CH‐acidic starting materials yielding inhibitors with highest affinity.
Figure 5.

Chemical evolution of antivirals in a dynamic reaction system. Formation and reactivity of antivirals through protein‐catalyzed (A,B) or solution‐based (A′,B′) reversible aldol reactions of aldehyde 1 and enolizable carbonyl fragments F1–F12. Aldol additions were followed by Knoevenagel condensations (C,C′). Either aldol addition products or the aldol condensation products were released from the protein (reactions D,E). Condensation products subsequently reacted either with protein‐based thiols (reaction F) or thiol nucleophiles in solution (reaction F′). Michael additions in solution (F′) can reduce the antiviral activity of Knoevenagel products (E,F) but also can disfavor the hydration reaction (reaction C′) leading to decomposition of inhibitors via the retro‐aldol reaction (B′). EWG=electron‐withdrawing group.
In contrast, hydration and retro‐aldol reactions were observed in solution (A′–C′ backwards) with no protein‐catalyzed acceleration (A–C backward). The formed Knoevenagel condensation products reacted reversibly as Michael acceptors with thiol nucleophiles, either with the protein‐thiol of Cys147 (F) or with thiols in solution (F′). The competition between protein thiol and DTT in solution (F versus F′) reduced the inhibitory activity of the condensation products in several cases (most remarkably for the diester 12). Formation of DTT adducts in solution altered the thermodynamics of ligand binding by reducing binding enthalpy and enhancing the binding entropy as found for the ketone inhibitor 5. Competition between the addition of thiol and that of water (i.e. the hydration reaction) (F′ versus C′), however, also inhibited the retro‐aldol reaction and thus stabilized some of the inhibitors (most remarkably the ketone inhibitors such as 5).
The antiviral activity of the formed inhibitors depended critically on the kinetics of the Michael addition reactions of inhibitors with the protein target and with competing nucleophiles present in solution. The most potent antivirals in the cellular infection model were characterized by a thermodynamically stable protein‐inhibitor complex with a slow off‐rate for the release of the antiviral from the protein‐bound state.
The observed formation of bioactive compounds in a dynamic reaction network consisting of target‐catalyzed and competing reactions in solution demonstrates a mechanism for the chemical evolution of drug‐like molecules, which might be exemplary for the emergence of functional molecules in the early development of life. For the chemical evolution of antivirals investigated here, the most efficient formation of inhibitors and the highest yields were observed for the cases of reversible C−C coupling reactions. These findings suggest to search for dynamic reaction systems also for the chemical evolution of other molecules with biological activity and relevance including peptides, heterocycles and nucleic acids.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Freie Universität Berlin, the Deutsche Forschungsgemeinschaft (funds of the Excellence initiative, CRC 765, CRC 1349, Project‐ID 387284271—SFB 1349, and the core facility BIOSUPRAMOL) and by the European Commission (Cooperation Project SILVER, GA No. 260644). We are grateful to Prof. Rolf Hilgenfeld, Lübeck, for providing us with the vector of the EV D68 3C protease. Open access funding enabled and organized by Projekt DEAL.
C. Tauber, R. Wamser, C. Arkona, M. Tügend, U. B. Abdul Aziz, S. Pach, R. Schulz, D. Jochmans, G. Wolber, J. Neyts, J. Rademann, Angew. Chem. Int. Ed. 2021, 60, 13294.
References
- 1.
- 1a. Oparin A. I., The origin of life (transl. from Proiskhozhdenie zhizny. Izd. Moskovhii RabochiI, Moscow 1924), The Macmillan Company, New York, 1938; [Google Scholar]
- 1b. Haldane J. B. S., The Rationalist Annual 1929, 148, 3–10; [Google Scholar]
- 1c. Kitadai N., Maruyama S., Geosci. Front. 2018, 9, 1117–1153; [Google Scholar]
- 1d. Krishnamurthy R., Hud N. V., Chem. Rev. 2020, 120, 4613–4615. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Butlerow A., Liebigs Ann. Chem. 1861, 120, 295–298; [Google Scholar]
- 2b. Ritson D., Sutherland J. D., Nat. Chem. 2012, 4, 895–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller S. L., Science 1953, 117, 528–529. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sutherland J. D., Whitfield J. N., Tetrahedron 1997, 53, 11595–11626; [Google Scholar]
- 4b. Fuller W. D., Sanchez R. A., Orgel L. E., J. Mol. Biol. 1972, 67, 25–33; [DOI] [PubMed] [Google Scholar]
- 4c. Fuller W. D., Orgel L. E., Sanchez R. A., J. Mol. Evol. 1972, 1, 249–257; [DOI] [PubMed] [Google Scholar]
- 4d. Becker S., Thoma I., Deutsch A., Gehrke T., Mayer P., Zipse H., Carell T., Science 2016, 352, 833–836. [DOI] [PubMed] [Google Scholar]
- 5. Szostak J. W., Angew. Chem. Int. Ed. 2017, 56, 11037–11043; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11182–11189. [Google Scholar]
- 6. Jaegle M., Wong E. L., Tauber C., Nawrotzky E., Arkona C., Rademann J., Angew. Chem. Int. Ed. 2017, 56, 7358–7378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7464–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Huc I., Lehn J. M., Proc. Natl. Acad. Sci. USA 1997, 94, 2106–2110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Mondal M., Hirsch A. K., Chem. Soc. Rev. 2015, 44, 2455–2488. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Schmidt M. F., Isidro-Llobet A., Lisurek M., El-Dahshan A., Tan J., Hilgenfeld R., Rademann J., Angew. Chem. Int. Ed. 2008, 47, 3275–3278; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3319–3323; [Google Scholar]
- 8b. Burda E., Rademann J., Nat. Commun. 2014, 5, 5170; [DOI] [PubMed] [Google Scholar]
- 8c. Becker D., Kaczmarska Z., Arkona C., Schulz R., Tauber C., Wolber G., Hilgenfeld R., Coll M., Rademann J., Nat. Commun. 2016, 7, 12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Manetsch R., Expert Opin. Drug Discovery 2006, 1, 525–538; [DOI] [PubMed] [Google Scholar]
- 9b. Bosc D., Camberlein V., Gealageas R., Castillo-Aguilera O., Deprez B., Deprez-Poulain R., J. Med. Chem. 2020, 63, 3817–3833; [DOI] [PubMed] [Google Scholar]
- 9c. Whiting M., Muldoon J., Lin Y. C., Silverman S. M., Lindstrom W., Olson A. J., Kolb H. C., Finn M. G., Sharpless K. B., Elder J. H., Fokin V. V., Angew. Chem. Int. Ed. 2006, 45, 1435–1439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1463–1467. [Google Scholar]
- 10. Jaegle M., Steinmetzer T., Rademann J., Angew. Chem. Int. Ed. 2017, 56, 3718–3722; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3772–3776. [Google Scholar]
- 11. Wong E. L., Nawrotzky E., Arkona C., Kim B. G., Beligny S., Wang X., Wagner S., Lisurek M., Carstanjen D., Rademann J., Nat. Commun. 2019, 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gladysz R., Vrijdag J., Van Rompaey D., Lambeir A.-M., Augustyns K., De Winter H., Van der Veken P., Chem. Eur. J. 2019, 25, 12380–12393. [DOI] [PubMed] [Google Scholar]
- 13. Mancini F., Unver M. Y., Elgaher W. A. M., Jumde V. R., Alhayek A., Lukat P., Herrmann J., Witte M. D., Köck M., Blankenfeldt W., Müller R., Hirsch A. K. H., Chem. Eur. J. 2020, 26, 14585–14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Tietze L. F., Beifuss U., in Comprehensive Organic Synthesis (Ed.: Trost B. M.), Pergamon Press, Oxford, 1991, pp. 341–394; [Google Scholar]
- 14b. Jones G., in Organic Reactions (Ed.: Cope A. C.), John Wiley & Sons, New York, 2011, pp. 205–273; [Google Scholar]
- 14c. Kulchat S., Meguellati K., Lehn J.-M., Helv. Chim. Acta 2014, 97, 1219–1236; [Google Scholar]
- 14d. Balakrishnan B., Chen C. C., Pan T. M., Kwon H. J., Tetrahedron Lett. 2014, 55, 1640–1643; [Google Scholar]
- 14e. Garrabou X., Wicky B. I. M., Hilvert D., J. Am. Chem. Soc. 2016, 138, 6972–6974. [DOI] [PubMed] [Google Scholar]
- 15. Norder H., De Palma A. M., Selisko B., Costenaro L., Papageorgiou N., Arnan C., Coutard B., Lantez V., De Lamballerie X., Baronti C., Solà M., Tan J., Neyts J., Canard B., Coll M., Gorbalenya A. E., Hilgenfeld R., Antiviral Res. 2011, 89, 204–218. [DOI] [PubMed] [Google Scholar]
- 16. Al-Gharabli S. I., Shah S. T. A., Weik S., Schmidt M. F., Mesters J. R., Kuhn D., Klebe G., Hilgenfeld R., Rademann J., ChemBioChem 2006, 7, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayden F. G., Turner R. B., Gwaltney J. M., Chi-Burris K., Gersten M., Hsyu P., Patick A. K., Smith G. J., Zalman L. S., Antimicrob. Agents Chemother. 2003, 47, 3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reich S. H., Johnson T., Wallace M. B., Kephart S. E., Fuhrman S. A., Worland S. T., Matthews D. A., Hendrickson T. F., Chan F., Meador J., Ferre R. A., Brown E. L., DeLisle D. M., Patick A. K., Binford S. L., Ford C. E., J. Med. Chem. 2000, 43, 1670–1683. [DOI] [PubMed] [Google Scholar]
- 19. Zimmerman H. E., Traxler M. D., J. Am. Chem. Soc. 1957, 79, 1920–1923. [Google Scholar]
- 20. Tan J., George S., Kusov Y., Perbandt M., Anemüller S., Mesters J. R., Norder H., Coutard B., Lacroix C., Leyssen P., Neyts J., Hilgenfeld R., J. Virol. 2013, 87, 4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun L., Delang L., Mirabelli C., Neyts J., Bio-protocol 2017, 7, e2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Copeland R. A., Evaluation of Enzyme Inhibitors in Drug Discovery : A Guide for Medicinal Chemists and Pharmacologists, Wiley-Interscience, Hoboken, 2005. [PubMed] [Google Scholar]
- 23. Wartchow C. A., Podlaski F., Li S. L., Rowan K., Zhang X. L., Mark D., Huang K. S., J. Comput.-Aided Mol. Des. 2011, 25, 669–676. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Bradshaw J. M., McFarland J. M., Paavilainen V. O., Bisconte A., Tam D., Phan V. T., Romanov S., Finkle D., Shu J., Patel V., Ton T., Li X., Loughhead D. G., Nunn P. A., Karr D. E., Gerritsen M. E., Funk J. O., Owens T. D., Verner E., Brameld K. A., Hill R. J., Goldstein D. M., Taunton J., Nat. Chem. Biol. 2015, 11, 525; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Lu H., Tonge P. J., Curr. Opin. Chem. Biol. 2010, 14, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary