

Abstract
Fibrosis results from aberrant wound healing and is characterized by an accumulation of extracellular matrix, impairing the function of an affected organ. Increased deposition of extracellular matrix proteins, disruption of matrix degradation, but also abnormal post‐translational modifications alter the biochemical composition and biophysical properties of the tissue microenvironment – the stroma. Macrophages are known to play an important role in wound healing and tissue repair, but the direct influence of fibrotic stroma on macrophage behaviour is still an under‐investigated element in the pathogenesis of fibrosis. In this review, the current knowledge on interactions between macrophages and (fibrotic) stroma will be discussed from biochemical, biophysical, and cellular perspectives. Furthermore, we provide future perspectives with regard to how macrophage–stroma interactions can be examined further to ultimately facilitate more specific targeting of these interactions in the treatment of fibrosis. © 2021 The Authors. The Journal of Pathology published by John Wiley & Sons, Ltd. on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: polarization, migration, phagocytosis, extracellular matrix, stiffness, stretch, shear stress, microstructure, profibrotic, antifibrotic
Introduction
Fibrosis results from aberrant wound healing and is characterized by excessive deposition of extracellular matrix (ECM). Fibrosis can affect a wide range of organs and tissues, including but not limited to lungs, heart, liver, skin, and kidney. Increased deposition, disruption of degradation, and abnormal post‐translational modifications of the ECM change the biochemical composition, availability of cell attachment domains, stiffness, and other biophysical properties of the tissue microenvironment – also known as the stroma (Figure 1). Stroma is composed of ECM and cells such as fibroblasts, pericytes, tissue‐resident mesenchymal stromal cells (MSCs), adipocytes, mast cells, and macrophages [1]. Although the composition of ECM varies depending on the type of tissue, ECM is generally composed of collagens (mainly fibrillar collagens such as type I, II, III, V), elastin, fibronectin, and various glycosaminoglycan molecules. Mechanical strength is mainly provided by collagen fibres, while elastin fibres provide elasticity [2]. Traditionally, ECM is considered a network for structural support; however, recent findings indicate ECM to be far more bioactive and dynamic – orchestrating biochemical and biomechanical messages that regulate cell behaviour [1, 3]. Dynamic remodelling of ECM is essential to preserve tissue homeostasis and dysregulation of ECM remodelling is an emerging research field, especially in cancer and in fibrosis [4, 5, 6].
Figure 1.
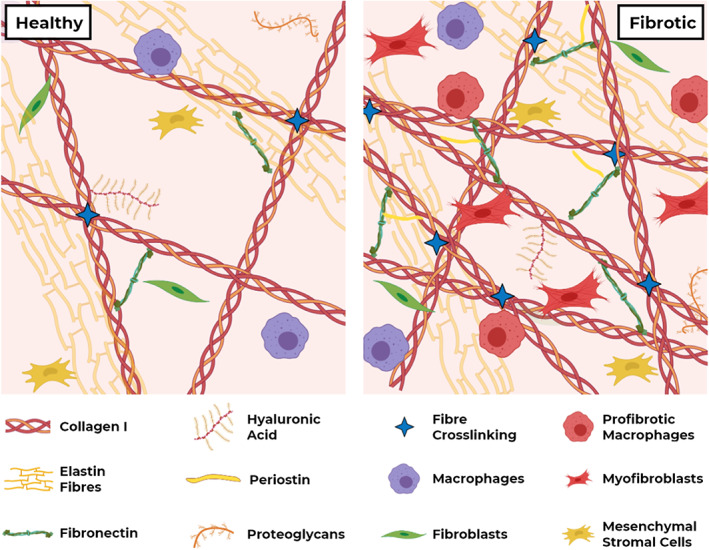
Schematic representation of stroma in healthy and fibrotic conditions. In fibrosis, aberrant ECM deposition and remodelling, as well as higher numbers and altered behaviour of fibroblasts and macrophages, change the biochemical and biophysical properties of stroma, and the consequential interactions with resident cells.
Macrophages are crucial players in the maintenance of tissue homeostasis [7]. Their ability to polarize into a spectrum of phenotypes allows them to execute a wide variety of functions, all necessary to preserve tissue integrity (Figure 2) [8]. Initially, tissue injury will push macrophages towards a pro‐inflammatory phenotype. Subsequent switching of macrophages to a wound healing phenotype that promotes ECM production by myofibroblasts and eventually polarizing to a pro‐remodelling phenotype is required to ensure restoration of normal tissue architecture [9]. These phenotypes can be induced by cytokines such as IFN‐γ and TNF‐α (pro‐inflammatory), IL‐4 and IL‐13 (wound healing), or IL‐10 and TGF‐β (pro‐remodelling), amongst a plethora of other stimuli [10].
Figure 2.
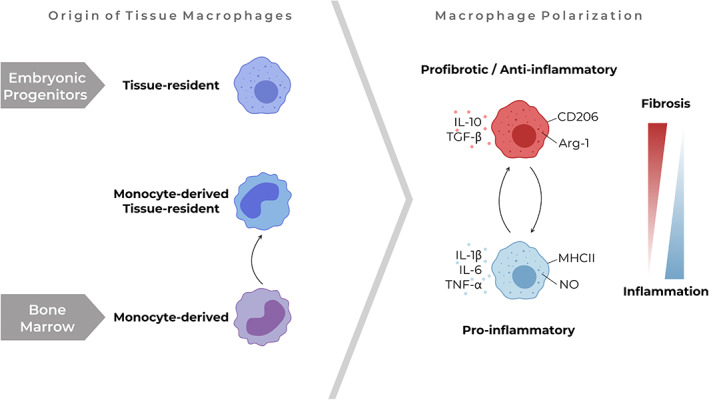
Macrophages in stroma. Tissue macrophages derive from different origins. Despite their differences, macrophages from all origins can change their polarization status to a more pro‐inflammatory or profibrotic/anti‐inflammatory phenotype upon stimuli from their microenvironment, as can be identified by metabolic changes (arginase‐1 versus NO), the expression of surface markers (e.g. MHCII and CD206) or the expression and secretion of cytokines (e.g. TGF‐β, IL‐10, TNF‐α, IL‐1β, and IL‐6). Arg‐1, arginase‐1.
As macrophages contribute to all phases of wound healing, their potential role in the development and progression of fibrotic diseases is emerging [9]. Recent advances in single‐cell RNA‐sequencing have shone light on the heterogeneity of macrophages within and between healthy and fibrotic human tissues and allowed detailed identification of macrophage populations [11, 12, 13]. Macrophages that populate tissues can have two different origins, i.e. tissue‐resident macrophages derived from embryonic progenitors and monocyte‐derived macrophages (Figure 2). Whereas tissue‐resident macrophages mostly arrive on site during organ development and are self‐maintaining, monocytes migrate from blood to a tissue upon injury and can differentiate into macrophages throughout life [14]. In fibrosis, monocyte‐derived macrophages appear to be an important contributor as large numbers of these macrophages have been described in fibrosis and depletion of these macrophages has been shown to attenuate fibrosis in several mouse models [15, 16, 17, 18]. Although these monocyte‐derived macrophages were long thought to be relatively short‐lived, it recently became evident that these monocyte‐derived macrophages can also adopt a phenotype similar to tissue‐resident macrophages, supporting the emerging role of the tissue microenvironment in instructing macrophage behaviour [15, 19]. Furthermore, we now know that microenvironmental cues can have long‐term effects on macrophage function: a concept called trained immunity [20]. These developments suggest that the direct effect of fibrotic stroma on macrophage behaviour may be an under‐recognized element in the pathogenesis of fibrosis.
In this review, we summarize the current knowledge of macrophage–stroma interactions in biochemical, biophysical, and cellular perspectives. First, we focus on the recent advances related to influences by altered stromal composition on macrophages. Next, we outline the contribution of biophysical changes in the fibrotic stroma to macrophage behaviour and describe the influence of fibrotic stroma on macrophage–stromal cell interactions. Lastly, we provide future perspectives on how these interactions can be further examined to generate knowledge of their potential value as targets for the treatment of fibrosis.
Biochemical interactions between fibrotic stroma and macrophages
In fibrosis, aberrant deposition of ECM changes the biochemical composition of the stroma (Figure 1). Increased deposition and altered ratios of ECM proteins, such as fibronectin, collagen, periostin, and glycosaminoglycans, have been shown to influence the profibrotic behaviour of stromal cells [21, 22, 23]. Although studies have indicated effects of individual ECM proteins [24, 25, 26, 27] and solubilized whole ECM on macrophages [28, 29, 30], the exact influence of fibrotic stroma on macrophages is still unknown. Emerging data from preclinical models indicate differential responses of macrophages in fibrosis: monocyte‐derived macrophages were shown to be profibrotic in, for instance, models of peritoneal and hepatic fibrosis [17, 18], while local microenvironmental cues were proposed as the driving force for the in situ transitioning of monocyte‐derived macrophages towards an antifibrotic phenotype in a resolving model of CCl4‐induced liver fibrosis in mice [31]. In addition, CD14+ monocytes and macrophages derived from CD14+ monocytes have been shown to activate ECM‐stored latent TGF‐β, a well‐known factor for inducing the progression of fibrosis, through integrin αvβ8‐mediated pathways [32]. These observations suggest that the altered stroma present in fibrotic tissues may be a driving factor underlying changes in macrophage behaviour in fibrotic disease.
An important player in the altered fibrotic stroma is fibronectin. It regulates collagen organization, and higher deposition of fibronectin has been reported in pulmonary [33], hepatic [34], cardiac [35], and renal [36] fibrosis compared with non‐fibrotic conditions. Although fibroblasts and other stromal cells are the predominant source of fibronectin [37], monocytes have also been shown to secrete fibronectin when pro‐inflammatory cytokines are present in their microenvironment [38] and alternatively‐activated macrophages were found to have high fibronectin expression at both gene and protein levels [39]. Fibronectin‐adsorbed surfaces primed macrophages to a pro‐inflammatory phenotype through activation of β1 integrin–PI3K/Akt signalling [40]. In contrast, a fibronectin‐rich environment has been shown to prime macrophages to adopt an anti‐inflammatory profile in response to pathogen‐associated molecular patterns through engagement of TLR2/4 coupled integrin β1‐signalling in vitro [41]. These differential effects may possibly be caused by alternative effects of fibronectin‐containing extra domain A (FN‐EDA) and plasma fibronectin [42] on macrophages and require more investigation. Fibronectin can also promote the migration of macrophages, as demonstrated in vitro [43]. As infiltrated macrophages can also produce fibronectin in an inflammatory environment, a continuing cycle of inflammation, macrophage influx and priming, and fibronectin deposition may therefore contribute to fibrotic pathobiology.
In vitro systems show that instructions from altered and abnormal composition of fibrotic stroma direct macrophages towards more profibrotic responses, although these systems generally only allow a simplified version of the multitude of functions of macrophages in physiological conditions. Such models are also used to investigate the importance of the presence, number, and availability of cell binding motifs in stroma. Human monocytic THP‐1 cells encapsulated in methacrylated gelatin (GelMA; collagen‐based and therefore cell‐binding) hydrogels had a more anti‐inflammatory profile, with increased secretion of arginase‐1 mediated through integrin α2β1 and possibly STAT6 signalling, compared with synthetic poly(ethylene glycol)dimethacrylate‐based hydrogels, suggesting that the presence of cell binding motifs is linked to macrophage polarization [44]. Similarly, comparison of hyaluronic acid hydrogels to hyaluronic acid–gelatin hydrogels identified more attachment of peripheral blood‐derived monocytes to the hyaluronic acid–gelatin hydrogels, due to the higher number of cell‐binding motifs in the latter [45]. Moreover, coating surfaces with collagen type I induced polarization of alveolar macrophages towards a more anti‐inflammatory MHCIILo‐CD206Hi phenotype compared with macrophages cultured on uncoated surfaces [46]. Similarly, in an in vitro experiment with mouse bone marrow‐derived macrophages, a collagen type I coating yielded more arginase‐1‐producing macrophages compared with fibronectin‐coated surfaces, suggesting differential effects of distinct ECM proteins probably driven by their distinct cell‐binding domains [26]. Post‐translational modifications of collagen, such as fibre crosslinking, are important changes affecting the biochemical composition of stroma during fibrosis. These inevitably lead to changes in binding domain availability, fibre alignment, and other biophysical changes, although the exact influences on macrophages are unknown to date [47, 48, 49, 50, 51]. As with the positive feedback loop between fibrotic ECM and fibroblasts described by Parker et al [22], the above‐mentioned findings indicate a possible profibrotic interchange between the presence, altered numbers, and types of cell‐binding domains found in fibrotic stroma and macrophages.
Periostin, another important ECM matricellular protein/matrikine, is more abundantly expressed in pulmonary [52], hepatic [53], renal [54], and cardiac [55] fibrosis compared with healthy conditions. In an acute kidney injury model, periostin‐overexpressing mice had more tissue‐resident macrophages with a pro‐regenerative phenotype than wild‐type mice [56]. In the same study, periostin overexpression alone did not result in spontaneous kidney fibrosis, while periostin‐KO mice had lower numbers of pro‐regenerative macrophages. Conversely, in a bleomycin model of lung fibrosis, periostin‐KO mice had less collagen in their lungs than did wild‐type mice, in concordance with peripheral blood‐derived monocytes having higher levels of periostin in patients with idiopathic pulmonary fibrosis (IPF) than non‐disease control donors [52]. In vitro models of macrophage migration showed that periostin stimulated the migration of primary murine macrophages, which also had higher expression of integrin αV, one of the periostin receptors, in the lesion site compared with control. Moreover, periostin promoted the secretion of TNF‐α from these macrophages, hypothetically through engagement of the focal adhesion kinase (FAK) pathway [57]. The exact influence of elevated periostin levels on both tissue‐resident and monocyte‐derived macrophages in fibrosis is yet to be understood, but the presented data suggest that periostin may disturb the balance between these two types of macrophages in favour of profibrotic monocyte‐derived macrophages.
Glycosaminoglycans, non‐fibrous components of the ECM, are also expressed at higher levels in fibrotic lungs [58], liver [59], kidney [60], and heart [61], compared with non‐fibrotic conditions. Solubilized urinary bladder‐derived ECM, which has high levels of hyaluronic acid, did not change TNF‐α, NO, and arginase‐1 levels in bone marrow‐derived macrophages in vitro [29]. Once the hyaluronic acid in this ECM was degraded, however, more NO secretion was found while TNF‐α secretion remained unchanged, suggesting that the hyaluronic acid content of the ECM plays a role in fine‐regulating the pro‐inflammatory responses of macrophages. In contrast, hyaluronic acid‐containing collagen type I hydrogels promoted anti‐inflammatory polarization of THP‐1 macrophages through CD44 signalling and the STAT3 pathway compared with unmodified collagen type I hydrogels [62]. Monocyte‐derived macrophages displayed lower expression of pro‐inflammatory markers in hyaluronic acid‐containing collagen type I hydrogels compared with collagen type I hydrogels without hyaluronic acid [63]. In addition, effects of post‐translational modifications, such as additional sulphate groups on high‐molecular‐weight hyaluronan, on macrophage responses were investigated in the same study; however, no additional influence of the sulphate groups was observed. In contrast, another study comparing high sulphate with low sulphate content in hyaluronic acid‐containing collagen type I hydrogels did find altered macrophage responses, as the production of pro‐inflammatory cytokines was lower in high‐sulphated hydrogels [64]. Although these studies had some opposing results, they do collectively suggest that the amount of, and modifications of, glycosaminoglycans could regulate macrophage responses.
In conclusion, differing macrophage responses may be driven by changes in the composition of the ECM and aberrant abundancies of ECM components during fibrosis. Fibrotic ECM–macrophage interactions could promote a profibrotic loop, stimulating further fibrotic responses leading to the recruitment of more profibrotic macrophages. In addition to elements determining the structure of ECM, bioactive factors (for example, growth factors) anchored in the ECM could also contribute to the biochemical interactions between macrophages and stroma. However, the challenge of distinguishing the effects of factors stored in ECM from paracrine effects of factors secreted by cells limits our understanding of the contribution of this reservoir function of (fibrotic) stroma to macrophage regulation. Even though there is increased momentum in the emergence of findings dissecting these elements, more studies are needed to address the following questions: (1) whether and what specific combinations of ECM proteins and their binding domains are responsible for regulating macrophage responses in fibrosis; and (2) which mechanisms underlie the priming of recruited macrophages towards a profibrotic profile by the fibrotic stroma?
Macrophage responses to biophysical changes
The altered biochemical composition of the stroma in fibrosis is also associated with changes in biophysical properties such as tissue stiffness, levels of shear stress, and microstructure [65]. However, knowledge on whether and how macrophages respond to these biophysical changes is still limited [66]. The knowledge available in the public domain is mainly based on biomaterials research, which has aimed at controlling macrophage behaviour to modulate the foreign body response by regulating biophysical properties of substrates or scaffolds [67].
Stiffness
Native tissue stiffness is highly dependent on the organ of interest, ranging from around 100 Pa in the brain to a few GPa in calcified bone [65, 68]. Fibrosis is consistently associated with (local) increases in the stiffness of the tissue, independent of the organ of interest [65]. This increase is predominantly caused by an excess or a different ratio of ECM proteins. Moreover, post‐translational remodelling such as crosslinking of ECM proteins and an increase in cellular stiffness have also been described to contribute to these stiffness alterations [69, 70].
Several studies have indicated an effect of substrate stiffness on macrophage behaviour. Although the studied stiffnesses and compositions of the substrates varied greatly, stiffening of the substrate is generally associated with more macrophage adhesion and a bigger and more flattened phenotype as compared to a smaller and more rounded shape on soft substrates [71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82]. Nonetheless, these observed morphological changes on two‐dimensional (2D) substrates may not be fully translatable to an in vivo situation. For instance, He et al reported that encapsulation of macrophages inside a stiff 3D hydrogel resulted in a more rounded shape compared with a softer hydrogel [83].
Related to these morphological changes, functional changes in the migratory behaviour and phagocytic activity of macrophages have also been observed. Infiltration and accumulation of monocyte‐derived macrophages have been associated strongly with fibrosis, as inhibition of monocyte infiltration dampened the fibrotic response in experimental fibrosis models [18, 84]. Higher substrate stiffness generally tends to stimulate macrophage migration and may therefore exacerbate fibrotic responses [74, 75, 82]. These investigations were all in 2D setups and the influence of substrate stiffness on movement in 3D is yet to be explored thoroughly. In addition to their migratory behaviour, the phagocytic capacity of macrophages is important in tissue homeostasis as well. For instance, impaired phagocytosis has been observed in airway macrophages from patients with IPF [85], whose lungs are stiffer than those of healthy individuals [58, 86]. However, studies investigating whether substrate stiffness has a direct effect on the phagocytic capacity of macrophages reported variable results. Some studies indicated that higher substrate stiffness promoted phagocytosis compared with low stiffness [77, 87], while others described a biphasic effect or no significant effect at all [74, 78].
The effect of substrate stiffness on pro‐inflammatory or profibrotic responses of macrophages has also not yet been shown conclusively. Most studies indicated more pro‐inflammatory behaviour when substrate stiffness increased, based on the expression of markers such as IL‐1β, IL‐6, and TNF‐α [75, 78, 80, 81, 83, 88, 89], although the opposite has also been described [77, 82, 87, 90]. However, these studies all investigated short‐term responses of macrophages to substrates of various stiffnesses. In the context of fibrosis, it would be of interest to study the long‐term effects of stiffness on macrophage polarization.
Recently, several molecular mechanisms underlying macrophage mechanosensing and subsequent mechanotransduction have been elucidated. The mechanosensing abilities of macrophages have been attributed to transient receptor potential vanilloid type 4 (TRPV4), as deletion or inhibition of this mechanosensitive calcium channel abolished the observed effects of substrate stiffness on macrophage behaviour [87, 91]. Interestingly, higher expression of TRPV4 has been shown in human fibrotic tissues and TRPV4‐deficient mice were protected from fibrosis [92]. Furthermore, Previtera and Sengupta showed that stiffer substrates activated the TLR4‐signalling pathway and thereby stimulated macrophage responses to pro‐inflammatory stimuli [88]. Although the exact role of TLR4 in fibrosis still needs to be defined, several studies have indicated that TLR4 plays a role in the development of fibrosis in mice [93, 94]. In addition to a possible role for these two receptors, substrate stiffness has also been associated with higher expression of integrins by macrophages, as well as a higher density of podosomes [75, 78, 81, 82, 95]. Podosomes are versatile, integrin‐mediated adhesion structures that not only have mechano‐sensing and ‐transducing properties, but also have the ability to mediate local recruitment and the release of matrix‐degrading enzymes upon adhesion to a stiff substrate [95, 96]. This direct matrix‐degrading response of macrophages to substrate stiffness especially calls for further investigation into podosome function in fibrosis. Furthermore, in many non‐myeloid cells, Yes‐associated protein (YAP) and transcriptional coactivator with PDZ‐binding motif (TAZ) signalling is well known to be activated by stiff substrates [97]. Only recently, it was demonstrated that YAP signalling also plays a role in macrophage mechanotransduction, as macrophages cultured on stiffer hydrogels showed increased expression and nuclear translocation of YAP [89]. This nuclear translocation of YAP, which requires actin polymerization, has been associated with pro‐inflammatory macrophage responses [89, 98]. However, further elucidation of YAP/TAZ signalling in macrophage polarization is required, as a role in Wnt5a‐ and TGFβ1‐mediated profibrotic macrophage polarization has also been described [99].
In summary, macrophages are able to detect changes in substrate stiffness, but exact responses of macrophages and the role of mechanotransduction in fibrosis remain unclear. Large differences in variables between the existing studies, such as substrate composition, viscoelasticity, porosity, ECM protein coating, macrophage origin, but also the experimental timeline, are likely to explain the inconsistent effects of stiffness on the macrophage responses observed thus far.
Shear stress and (cyclic) stretch
Pathologies like ventilator‐induced lung injury and obstruction‐induced fibrosis models, such as the bile duct ligation‐induced liver fibrosis model, support the hypothesis that mechanical forces can play an important role in the development or progression of fibrosis [100, 101, 102, 103]. However, the presence and magnitude of mechanical forces, such as (cyclic) stretch and shear stress, vary greatly depending on the organ of interest.
Shear stress is mainly caused by fluid flow, especially in the vasculature and ducts such as renal tubules [65]. In the lungs, airflow induces shear stress on the airway wall [104]. Tissue remodelling in fibrosis decreases the compliance of the vasculature, ducts, and airway walls, and can thereby increase shear stress [104]. Another shear stress‐generating phenomenon, although very small, is interstitial flow [65]. Interstitial fluid pressure is highly dependent on factors such as blood pressure, cell density, and composition of the ECM, and has been described to increase during inflammation and wound healing [105, 106]. It may also play a role in the progression of fibrotic disease as interstitial flow can stimulate myofibroblast differentiation, TGF‐β production, and collagen alignment [106]. In macrophages, the application of interstitial flow induced higher expression of anti‐inflammatory markers such as arginase‐1, TGF‐β, and CD206 through β1‐integrin signalling compared with static conditions [107]. Furthermore, interstitial flow stimulated the migration speed of macrophages [107, 108]. It is therefore not unlikely that the higher than normal interstitial fluid flow found in fibrosis contributes to perpetuation of the fibrotic response by stimulating macrophage infiltration and inducing a profibrotic phenotype.
Whereas shear stress is present in each organ, cyclic stretch or strain mainly plays a role in the heart (pulsatile haemodynamic loading) and lungs (respiration) [109, 110]. Fibrosis in these organs results in lower compliance, thereby also reducing the cyclic stretch magnitudes [65]. Additionally, more subtle stretch forces can be generated by contractions of other cells such as myofibroblasts in wound healing and fibrosis [111]. In vitro, uniaxial cyclic stretch induced elongation of macrophages in the direction of the applied stretch [112, 113]. Although macrophage elongation has previously been associated with an anti‐inflammatory phenotype [114], it is not yet clear how cyclic stretch affects macrophage polarization and functional behaviour, as many contradicting results have been reported. Most studies described higher production of pro‐inflammatory mediators upon cyclic stretch [112, 115, 116, 117, 118, 119, 120], while others found lower production [121], higher production of pro‐remodelling factors [122] or a combination thereof [123]. Interestingly, TRPV4 was shown to regulate the secretion of pro‐inflammatory cytokines by macrophages upon mechanical stretch [120]. Again, experimental variables such as macrophage origin, stretching regime, the way that macrophages were stimulated, and the experimental timeline likely contribute to the inconsistent results and complicate the translation of this information to in vivo.
Microstructural changes of the ECM
Abnormalities in the biochemical composition and post‐translational modifications of the ECM in fibrosis result in a changed micro‐ and nano‐structure of the tissue, which can be sensed by cells [70, 124]. However, the number of studies investigating the interactions of these altered structures with macrophages is limited as the post‐translational modifications observed in fibrosis are not well characterized and are difficult to mimic in vitro. From our own studies and studies in the field of biomaterials, we know that the surface or scaffold topography can modulate macrophage behaviour, but translation into a fibrotic context is challenging [46, 67].
To better understand what topography cells can sense in situ in organs, second harmonic generation imaging can be used to analyse the organization and maturity of collagen fibres in tissue. For example, in IPF, thicker and more mature collagen fibres have been observed with this technique compared with control [48] and the effect of fibre diameter on macrophage polarization has been studied in vitro. Electrospun fibres with larger diameters (2–6 μm) were shown to induce a more anti‐inflammatory macrophage phenotype in comparison to smaller diameters (<1 μm) [125, 126]. Furthermore, high collagen density was recently described to induce a more immunosuppressive macrophage phenotype than low collagen density [127]. The high degree of collagen fibre alignment observed in fibrosis could also affect macrophage behaviour, as alignment of electrospun fibres resulted in lower expression levels of pro‐inflammatory IL‐1β by macrophages compared with randomly organized fibres [119, 128]. In addition to the changes in fibre diameter, density, and alignment, a study by Veres et al demonstrated that macrophages recognized mechanically damaged collagen fibres [129]. In fibrosis, higher levels of denatured, unfolded collagen chains have been detected compared with healthy tissues [130, 131]. Binding of macrophages to uncoiled, denatured collagen fibres induced macrophage spreading and ruffling, thereby increasing the contact area with the damaged fibrils [129]. In addition, we have previously shown that alveolar macrophages behave differently on non‐fibrous collagen type I layers compared with fibrous collagen type I layers [46]: macrophages on fibrous collagen layers transmigrated more, while showing an amoeboid‐like phenotype, while non‐fibrous collagen layers induced a more mesenchymal phenotype and higher expression of mannose receptor CD206, involved in collagen degradation and known to be upregulated on alveolar macrophages in patients with IPF.
Together, these studies indicate that macrophages are sensitive to remodelling‐associated (micro)structural changes of collagen fibres in the ECM, although their subsequent responses and the consequences for fibrosis require further investigation.
Stromal regulation of macrophage–cell interactions in fibrosis
The biochemical and biophysical changes of fibrotic ECM alter the macrophage phenotype via stimulation of multiple mechanisms (summarized in Figure 3), but cells are also an important component of the stroma in addition to the ECM. In the fibrotic microenvironment, (activated) fibroblasts are the predominant cell type, although the activation status and the origins of these fibroblasts lead to different gene profiles and properties, increasing the heterogeneity of fibroblasts found in fibrotic tissues [132]. In addition to fibroblasts, endothelial cells, fibrocytes, and tissue‐resident MSCs are found in both healthy and fibrotic stroma. Fibroblast–macrophage interactions in in vitro models of skin [133, 134], hepatic fibrosis [135, 136], and scaffold‐free 3D spheroid models [137], as well as computational models [138], have shown (bidirectional) feedback mechanisms between these cell types. While direct cell–cell communications in fibrotic disease are outside the scope of this review, the influence of fibrotic stroma on these interactions now emerges as another key player in fibrosis.
Figure 3.
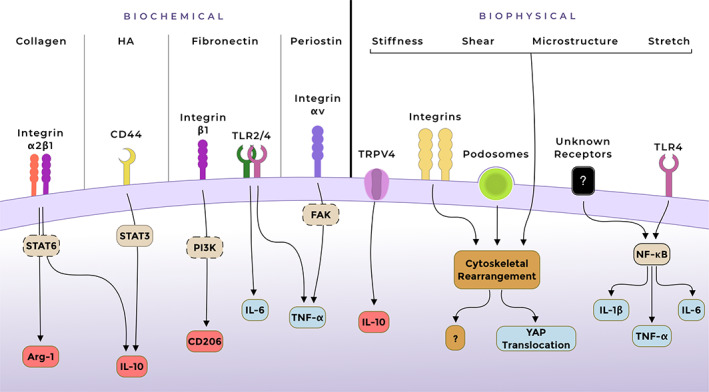
Schematic overview of the most described mechanisms involved in interactions between fibrotic stroma and macrophages. Hypothesised involvement of signalling pathways/molecules is illustrated with dashed frames. Upregulated pro‐inflammatory markers are depicted in blue, and profibrotic/anti‐inflammatory in red. Arg‐1, arginase‐1; FAK, focal adhesion kinase; TRPV4, transient receptor potential vanilloid type 4; YAP, Yes‐associated protein.
Interplay between stromal cells and macrophages through the stroma could also affect the responses of these cells. In a recent study by Shook et al, both macrophages and the local microenvironment were shown to influence the heterogeneity of myofibroblast subpopulations during wound healing [139]. In a fibrocystin knockout mouse model of congenital hepatic fibrosis, depletion of macrophages using clodronate resulted in less accumulation of myofibroblasts and less deposition of collagen compared with non‐treated mice [140]. Similarly, in cardiac fibrosis mouse models, macrophages produced secreted protein, acidic and rich in cysteine (SPARC, also known as osteonectin, an ECM glycoprotein) along with fibroblasts, which in turn promoted collagen production and maturation by fibroblasts [141]. Influences on macrophage behaviour through the modifications on the (alignment of) stroma have also been demonstrated: in a colitis mouse model, a subtype of mesenchymal cells regulated collagen and fibronectin fibre structures, guiding macrophages to profibrotic responses [142]. Also, in prostate cancer, cancer‐associated fibroblasts were found to align fibronectin matrix into parallel fibres to guide the migration of cancer cells [143]. In addition, contraction of the ECM by myofibroblasts has been shown to prime latent TGF‐β for later activation by proteases and integrin‐mediated pathways [144, 145]. For example, integrin αvβ8‐mediated TGF‐β activation by macrophages has been shown to result in perpetuation of the fibrotic response [32, 146]. These findings taken together support the interplay between the cell‐altered stroma and macrophages. Another demonstration of such interplay was in a study by Ford et al, in which mouse macrophages were stationary in dense collagen type I network hydrogels, while co‐culturing them with fibroblasts in the same hydrogels increased their motility [147]. Direct and/or indirect communications between macrophages and fibroblasts in this 3D network resulted in improved directionality and increased speed of migration of macrophages, indicating that fibroblasts modified the microenvironment in favour of macrophages. In concordance, fibroblasts applied greater strains to stiffening collagen fibres [148], while a stiffer matrix promoted the profibrotic activation of fibroblasts [22]. Since macrophage behaviour is guided by both biochemical and biophysical cues in stroma, the above‐mentioned interplay between cell‐modified stroma and macrophages could therefore play an important role in regulating macrophage responses in fibrotic diseases.
The recruitment of monocyte‐derived macrophages and their differentiation to profibrotic macrophages could be a key element in the progression of fibrosis. Alignment or modification of ECM proteins by fibroblasts was found to stimulate migration of profibrotic macrophages and therefore to sustain the fibrotic stroma. Mazur et al have shown that treating collagen surfaces with fibroblast‐activating protein (FAP), a matrix‐modifying enzyme, resulted in more attachment and spread of mouse peritoneal macrophages, compared with non‐treated collagen surfaces [149]. In vitro experiments showed that FAPHi fibroblasts can have both ECM synthetic and proteolytic phenotypes. In line with this, increased FAP expression stimulated the migration of fibroblasts on collagen type I or fibronectin surfaces. Moreover, in vivo data suggest that FAPHi fibroblasts predominantly reside in fibronectin‐rich regions [150, 151]. Although the loss of FAP in different mouse models of lung fibrosis showed different results [152], elevated FAP levels were demonstrated in lung [153] and liver [154] fibrosis patients. These findings suggest that FAP‐mediated regulation of stroma and therefore putative macrophage recruitment could influence the interactions between the fibrotic stroma and macrophages.
Recent studies indicate a contribution of (recruited) macrophages to the fibrotic response by differentiating into myofibroblasts [macrophage‐to‐myofibroblast transition (MMT)]. Differentiation of myeloid cells to fibroblast‐like cells has been demonstrated in a skin wound healing model in mice [155]. In addition, MMT in a mouse renal fibrosis model was proposed as a mechanism for transitioning from an acute inflammatory phase to an active fibrotic phase. In concert, the majority of transitioned macrophages were profibrotic [156]. Similarly, MMT was also demonstrated in human renal allograft biopsy samples [157] as well as in a mouse cardiac fibrosis model, suggesting the involvement of MMT in many types of fibrosis [158]. Direct contribution of MMT was also indicated in a mouse model of myocardial infarction, in which the authors ruled out the contribution of fibrocytes to the new population of (myo)fibroblasts [158, 159]. Furthermore, the lack of fibrocyte‐cell surface marker CD34 on the majority of cells in the granulation tissue of wounds in mice also supports a macrophage origin [155]. Lastly, predominant macrophage markers on these new myofibroblasts found in the chronic renal allograft rejection samples support them being of macrophage origin in contrast to fibrocyte origin [157].
Overall, interactions between macrophages and cells in fibrotic stroma favour the recruitment of (profibrotic) macrophages and/or their profibrotic responses, while profibrotic macrophages in turn support proliferation and survival of activated fibroblasts. Although interactions between macrophages and tissue‐resident MSCs have been shown in preclinical models [160, 161, 162, 163], MSCs affecting macrophages in human fibrosis has yet to be proven. Furthermore, the involvement of MSCs in fibrosis needs deeper understanding. While there are still many unknown factors in the stroma–macrophage–cell interactions in fibrosis, the influence of cell‐modified ECM on macrophage responses warrants more attention. Therefore, more studies are required on (1) the influence of stromal cells on the profibrotic response of newly recruited macrophages; (2) how alignment of ECM fibres in stroma could favour pro‐regenerative macrophage recruitment; and (3) how the ECM composition affects MMT.
Concluding remarks
Based on in vitro studies, macrophages are highly responsive to changes in their microenvironment, regardless of whether these changes are biochemical, biophysical, or indirect via other stromal cells (Figure 4). It is almost inevitable that the altered stroma directly affects macrophage behaviour in fibrosis, but more sophisticated models are required to investigate how macrophages integrate a combination of signals, as biochemical or biophysical changes never come alone in the tissue environment.
Figure 4.
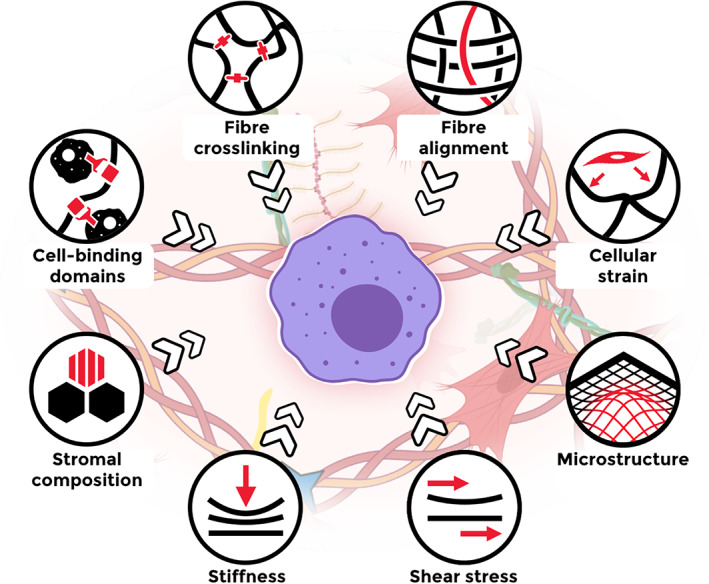
Graphical overview of fibrosis‐related biochemical, biophysical, and cell‐induced changes in stroma, concurrently influencing macrophage behaviour.
The majority of studies that we have discussed were performed in the context of biomaterials and therefore did not aim to mimic the stroma in a healthy or diseased state. Especially when studying the effect of biophysical properties, the chosen biochemical composition of the substrate may affect the response of macrophages in a synergistic or antagonistic manner. It is likely that the various contradictory results found between studies are a consequence of the impact of these kinds of interferences. It is also important to note that the majority of the discussed studies used 2D culture systems, complicating the translation to the 3D microenvironment in vivo. Furthermore, most studies investigated the first 24–48 h of macrophage responses, whereas studying later points in time may be more relevant for understanding the remodelling phase in fibrosis. Thus, although we can learn much from the biomaterials field, models that are more reflective of the organ environment are required to investigate how the fibrotic stroma affects macrophage behaviour.
The high variation in macrophage responses may also be due to different origins of the macrophages studied. Monocyte‐derived macrophages and macrophage cell lines are both commonly used to study macrophage behaviour in response to microenvironmental changes, but their responses can vary, especially when compared with primary organ‐specific macrophages. Given the evidence that myofibroblasts and macrophages isolated from fibrotic patients behave differently than macrophages from non‐fibrotic patients, more studies are needed that investigate whether primary macrophages of fibrotic patients still respond to stimuli comparably to macrophages from non‐fibrotic donors [164, 165]. Such studies would also help towards understanding the contribution of macrophages to both the development and the progression of fibrosis.
Currently, the number of available antifibrotic therapeutics is limited. In 2014, nintedanib and pirfenidone were first approved by the FDA for the treatment of pulmonary fibrosis and their effectiveness in slowing down fibrosis in other organs is currently under investigation [166, 167, 168, 169]. Interestingly, both pirfenidone and nintedanib have been shown to change macrophage behaviour and to interfere with collagen type I fibril formation [170, 171, 172]. Therapeutic strategies that specifically skew profibrotic macrophages towards a (slightly more) pro‐inflammatory phenotype may present as an alternative or addition to existing treatment regimens, as recent studies have shown in mouse models of fibrosis [173, 174]. Stimulating or inhibiting the pathways by which the fibrotic stroma affects macrophage behaviour (Figure 3), such as mechanosensing, may enable us to interfere with the fibrotic interplay between the stroma and macrophages (Figure 5). However, as the described pathways are not specific for macrophages, unexpected systemic effects need to be taken into account or circumvented by specifically targeting macrophages in the fibrotic area. Nonetheless, a better understanding of macrophage–stroma interactions and their role in the development and progression of fibrotic disease is required to specifically target macrophage function for the treatment of tissue fibrosis.
Figure 5.
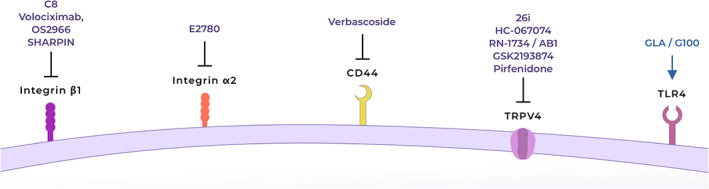
Putative targets and therapeutics to skew profibrotic macrophages to a more pro‐inflammatory phenotype. Antagonists/inhibitors for integrin β1 [175, 176, 177, 178], integrin α2 [179, 180], CD44 [181] or TRPV4 [87, 91, 182, 183, 184]. Agonist for TLR4 [185, 186, 187]. TRPV4: transient receptor potential vanilloid type 4.
Author contributions statement
GFV, MN, JKB and BNM conceived the concept and all the authors designed the manuscript. GFV and MN performed the literature search and drafted the manuscript and the figures. MN designed the figures. All the authors critically revised the manuscript and agreed with the final version.
Acknowledgements
GFV was funded by the Graduate School of Medical Sciences of the University of Groningen. JKB was funded by a Rosalind Franklin Fellowship co‐funded by the EU and the University of Groningen.
Conflict of interest statement: GFV has no conflicts of interest to declare. MN, IHH, JKB, and BNM receive unrestricted research funds from Boehringer Ingelheim GmbH & Co KG. IHH has received research funding from the Netherlands Lung Foundation (outside the submitted work). MS and MJT are employees of Boehringer‐Ingelheim Pharma GmbH & Co KG. PvR is a co‐founder/scientific advisor/shareholder of BiomACS BV.
Contributor Information
Janette K Burgess, Email: j.k.burgess@umcg.nl.
Barbro N Melgert, Email: b.n.melgert@rug.nl.
References
- 1. Gartner L. Connective tissue. In Textbook of Histology (5th edn), Gartner LP (ed). Elsevier, 2020; 103–124. [Google Scholar]
- 2. Burgstaller G, Oehrle B, Gerckens M, et al. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J 2017; 50: 1601805. [DOI] [PubMed] [Google Scholar]
- 3. Burgess JK, Muizer K, Brandsma C‐A, et al. Dynamic reciprocity: the role of the extracellular matrix microenvironment in amplifying and sustaining pathological lung fibrosis. In Fibrosis in Disease, Willis M, Yates CC, Schisler JC (eds). Springer Nature: Switzerland, 2019; 239–270. [Google Scholar]
- 4. Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: a strained relationship. Biochim Biophys Acta Rev Cancer 2020; 1873: 188356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karsdal MA, Nielsen MJ, Sand JM, et al. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol 2013; 11: 70–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol 2018; 68–69: 490–506. [DOI] [PubMed] [Google Scholar]
- 7. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016; 44: 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xue J, Schmidt SV, Sander J, et al. Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014; 40: 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol 2017; 79: 593–617. [DOI] [PubMed] [Google Scholar]
- 10. Adhyatmika A, Putri KSS, Beljaars L, et al. The elusive antifibrotic macrophage. Front Med (Lausanne) 2015; 2: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morse C, Tabib T, Sembrat J, et al. Proliferating SPP1/MERTK‐expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J 2019; 54: 1802441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reyfman PA, Walter JM, Joshi N, et al. Single‐cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am J Respir Crit Care Med 2019; 199: 1517–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramachandran P, Dobie R, Wilson‐Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature 2019; 575: 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta 2013; 1832: 989–997. [DOI] [PubMed] [Google Scholar]
- 15. Misharin AV, Morales‐Nebreda L, Reyfman PA, et al. Monocyte‐derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 2017; 214: 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joshi N, Watanabe S, Verma R, et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M‐CSF/M‐CSFR signalling in monocyte‐derived alveolar macrophages. Eur Respir J 2020; 55: 1900646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen YT, Hsu H, Lin CC, et al. Inflammatory macrophages switch to CCL17‐expressing phenotype and promote peritoneal fibrosis. J Pathol 2020; 250: 55–66. [DOI] [PubMed] [Google Scholar]
- 18. Han J, Zhang X, Lau JKC, et al. Bone marrow‐derived macrophage contributes to fibrosing steatohepatitis through activating hepatic stellate cells. J Pathol 2019; 248: 488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lavin Y, Winter D, Blecher‐Gonen R, et al. Tissue‐resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014; 159: 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Netea MG, Domínguez‐Andrés J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020; 20: 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lau JY, Oliver BG, Baraket M, et al. Fibulin‐1 is increased in asthma – a novel mediator of airway remodeling? PLoS One 2010; 5: e13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 2014; 124: 1622–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu G, Cooley MA, Jarnicki AG, et al. Fibulin‐1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight 2016; 1: e86380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Armstrong JW, Chapes SK. Effects of extracellular matrix proteins on macrophage differentiation, growth, and function: comparison of liquid and agar culture systems. J Exp Zool 1994; 269: 178–187. [DOI] [PubMed] [Google Scholar]
- 25. Correia CR, Gaifem J, Oliveira MB, et al. The influence of surface modified poly(L‐lactic acid) films on the differentiation of human monocytes into macrophages. Biomater Sci 2017; 5: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luu TU, Liu WF. Regulation of macrophages by extracellular matrix composition and adhesion geometry. Regen Eng Transl Med 2018; 4: 238–246. [Google Scholar]
- 27. Ploeger DTA, van Putten SM, Koerts JA, et al. Human macrophages primed with angiogenic factors show dynamic plasticity, irrespective of extracellular matrix components. Immunobiology 2012; 217: 299–306. [DOI] [PubMed] [Google Scholar]
- 28. Slivka PF, Dearth CL, Keane TJ, et al. Fractionation of an ECM hydrogel into structural and soluble components reveals distinctive roles in regulating macrophage behavior. Biomater Sci 2014; 2: 1521–1534. [DOI] [PubMed] [Google Scholar]
- 29. Meng FW, Slivka PF, Dearth CL, et al. Solubilized extracellular matrix from brain and urinary bladder elicits distinct functional and phenotypic responses in macrophages. Biomaterials 2015; 46: 131–140. [DOI] [PubMed] [Google Scholar]
- 30. Huleihel L, Dziki JL, Bartolacci JG, et al. Macrophage phenotype in response to ECM bioscaffolds. Semin Immunol 2017; 29: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 2012; 109: E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelly A, Gunaltay S, McEntee CP, et al. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8‐mediated TGFβ activation. J Exp Med 2018; 215: 2725–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burgess JK, Mauad T, Tjin G, et al. The extracellular matrix – the under‐recognized element in lung disease? J Pathol 2016; 240: 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005; 115: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Dijk A, Niessen HWM, Ursem W, et al. Accumulation of fibronectin in the heart after myocardial infarction: a putative stimulator of adhesion and proliferation of adipose‐derived stem cells. Cell Tissue Res 2008; 332: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Samarakoon R, Dobberfuhl AD, Cooley C, et al. Induction of renal fibrotic genes by TGF‐β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal 2013; 25: 2198–2209. [DOI] [PubMed] [Google Scholar]
- 37. Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell‐mediated matrix assembly process. Matrix Biol 2005; 24: 389–399. [DOI] [PubMed] [Google Scholar]
- 38. Kitamura N, Nishinarita S, Takizawa T, et al. Cultured human monocytes secrete fibronectin in response to activation by proinflammatory cytokines. Clin Exp Immunol 2000; 120: 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gratchev A, Guillot P, Hakiy N, et al. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein βIG‐H3. Scand J Immunol 2001; 53: 386–392. [DOI] [PubMed] [Google Scholar]
- 40. Lv L, Xie Y, Li K, et al. Unveiling the mechanism of surface hydrophilicity‐modulated macrophage polarization. Adv Healthc Mater 2018; 7: e1800675. [DOI] [PubMed] [Google Scholar]
- 41. Fei D, Meng X, Yu W, et al. Fibronectin (FN) cooperated with TLR2/TLR4 receptor to promote innate immune responses of macrophages via binding to integrin β1. Virulence 2018; 9: 1588–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011; 4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Digiacomo G, Tusa I, Bacci M, et al. Fibronectin induces macrophage migration through a SFK‐FAK/CSF‐1R pathway. Cell Adh Migr 2017; 11: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cha BH, Shin SR, Leijten J, et al. Integrin‐mediated interactions control macrophage polarization in 3D hydrogels. Adv Healthc Mater 2017; 6: 1700289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ščigalková I, Bystroňová J, Kovářová L, et al. The effect of healing phenotype‐inducing cytokine formulations within soft hydrogels on encapsulated monocytes and incoming immune cells. RSC Adv 2019; 9: 21396–21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vasse GF, Kühn PT, Zhou Q, et al. Collagen morphology influences macrophage shape and marker expression in vitro . J Immunol Regen Med 2018; 1: 13–20. [Google Scholar]
- 47. Ikenaga N, Peng ZW, Vaid KA, et al. Selective targeting of lysyl oxidase‐like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut 2017; 66: 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tjin G, White ES, Faiz A, et al. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech 2017; 10: 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Philp CJ, Siebeke I, Clements D, et al. Extracellular matrix cross‐linking enhances fibroblast growth and protects against matrix proteolysis in lung fibrosis. Am J Respir Cell Mol Biol 2018; 58: 594–603. [DOI] [PubMed] [Google Scholar]
- 50. González A, López B, Ravassa S, et al. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross‐linking. Biochim Biophys Acta Mol Cell Res 2019; 1866: 1421–1432. [DOI] [PubMed] [Google Scholar]
- 51. Klepfish M, Gross T, Vugman M, et al. LOXL2 inhibition paves the way for macrophage‐mediated collagen degradation in liver fibrosis. Front Immunol 2020; 11: 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Naik PK, Bozyk PD, Bentley JK, et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2012; 303: L1046–L1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang Y, Liu W, Xiao H, et al. Matricellular protein periostin contributes to hepatic inflammation and fibrosis. Am J Pathol 2015; 185: 786–797. [DOI] [PubMed] [Google Scholar]
- 54. Mael‐Ainin M, Abed A, Conway SJ, et al. Inhibition of periostin expression protects against the development of renal inflammation and fibrosis. J Am Soc Nephrol 2014; 25: 1724–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao S, Wu H, Xia W, et al. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J Cardiol 2014; 63: 373–378. [DOI] [PubMed] [Google Scholar]
- 56. Kormann R, Kavvadas P, Placier S, et al. Periostin promotes cell proliferation and macrophage polarization to drive repair after AKI. J Am Soc Nephrol 2020; 31: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yokota K, Kobayakawa K, Saito T, et al. Periostin promotes scar formation through the interaction between pericytes and infiltrating monocytes/macrophages after spinal cord injury. Am J Pathol 2017; 187: 639–653. [DOI] [PubMed] [Google Scholar]
- 58. Booth AJ, Hadley R, Cornett AM, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med 2012; 186: 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. George J, Tsutsumi M, Takase S. Expression of hyaluronic acid in N‐nitrosodimethylamine induced hepatic fibrosis in rats. Int J Biochem Cell Biol 2004; 36: 307–319. [DOI] [PubMed] [Google Scholar]
- 60. Colombaro V, Jadot I, Declèves AE, et al. Lack of hyaluronidases exacerbates renal post‐ischemic injury, inflammation, and fibrosis. Kidney Int 2015; 88: 61–71. [DOI] [PubMed] [Google Scholar]
- 61. Suleiman M, Abdulrahman N, Yalcin H, et al. The role of CD44, hyaluronan and NHE1 in cardiac remodeling. Life Sci 2018; 209: 197–201. [DOI] [PubMed] [Google Scholar]
- 62. Kim H, Cha J, Jang M, et al. Hyaluronic acid‐based extracellular matrix triggers spontaneous M2‐like polarity of monocyte/macrophage. Biomater Sci 2019; 7: 2264–2271. [DOI] [PubMed] [Google Scholar]
- 63. Friedemann M, Kalbitzer L, Franz S, et al. Instructing human macrophage polarization by stiffness and glycosaminoglycan functionalization in 3D collagen networks. Adv Healthc Mater 2017; 6: 1600967. [DOI] [PubMed] [Google Scholar]
- 64. Franz S, Allenstein F, Kajahn J, et al. Artificial extracellular matrices composed of collagen I and high‐sulfated hyaluronan promote phenotypic and functional modulation of human pro‐inflammatory M1 macrophages. Acta Biomater 2013; 9: 5621–5629. [DOI] [PubMed] [Google Scholar]
- 65. Wells RG. Tissue mechanics and fibrosis. Biochim Biophys Acta 2013; 1832: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meli VS, Veerasubramanian PK, Atcha H, et al. Biophysical regulation of macrophages in health and disease. J Leukoc Biol 2019; 106: 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sridharan R, Cameron AR, Kelly DJ, et al. Biomaterial based modulation of macrophage polarization: a review and suggested design principles. Mater Today 2015; 18: 313–325. [Google Scholar]
- 68. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 2011; 4: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jaffar J, Yang SH, Kim SY, et al. Greater cellular stiffness in fibroblasts from patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2018; 315: L59–L65. [DOI] [PubMed] [Google Scholar]
- 70. Kristensen JH, Karsdal MA, Genovese F, et al. The role of extracellular matrix quality in pulmonary fibrosis. Respiration 2014; 88: 487–499. [DOI] [PubMed] [Google Scholar]
- 71. Nemir S, Hayenga HN, West JL. PEGDA hydrogels with patterned elasticity: novel tools for the study of cell response to substrate rigidity. Biotechnol Bioeng 2010; 105: 636–644. [DOI] [PubMed] [Google Scholar]
- 72. Fereol S, Fodil R, Labat B, et al. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil Cytoskeleton 2006; 63: 321–340. [DOI] [PubMed] [Google Scholar]
- 73. Beningo KA, Wang Y. Fc‐receptor‐mediated phagocytosis is regulated by mechanical properties of the target. J Cell Sci 2002; 115: 849–856. [DOI] [PubMed] [Google Scholar]
- 74. Adlerz KM, Aranda‐Espinoza H, Hayenga HN. Substrate elasticity regulates the behavior of human monocyte‐derived macrophages. Eur Biophys J 2016; 45: 301–309. [DOI] [PubMed] [Google Scholar]
- 75. Hsieh JY, Keating MT, Smith TD, et al. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. APL Bioeng 2019; 3: 016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Irwin EF, Saha K, Rosenbluth M, et al. Modulus‐dependent macrophage adhesion and behavior. J Biomater Sci Polym Ed 2008; 19: 1363–1382. [DOI] [PubMed] [Google Scholar]
- 77. Patel NR, Bole M, Chen C, et al. Cell elasticity determines macrophage function. PLoS One 2012; 7: e41024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sridharan R, Cavanagh B, Cameron AR, et al. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater 2019; 89: 47–59. [DOI] [PubMed] [Google Scholar]
- 79. Ullm S, Krüger A, Tondera C, et al. Biocompatibility and inflammatory response in vitro and in vivo to gelatin‐based biomaterials with tailorable elastic properties. Biomaterials 2014; 35: 9755–9766. [DOI] [PubMed] [Google Scholar]
- 80. Zhuang Z, Zhang Y, Sun S, et al. Control of matrix stiffness using methacrylate–gelatin hydrogels for a macrophage‐mediated inflammatory response. ACS Biomater Sci Eng 2020; 6: 3091–3102. [DOI] [PubMed] [Google Scholar]
- 81. Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)‐based hydrogels. J Biomed Mater Res A 2012; 100: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li J, Wang S, Li Y, et al. miRNA‐mediated macrophage behaviors responding to matrix stiffness and ox‐LDL. J Cell Physiol 2020; 235: 6139–6153. [DOI] [PubMed] [Google Scholar]
- 83. He X‐T, Wu R‐X, Xu X‐Y, et al. Macrophage involvement affects matrix stiffness‐related influences on cell osteogenesis under three‐dimensional culture conditions. Acta Biomater 2018; 71: 132–147. [DOI] [PubMed] [Google Scholar]
- 84. Heymann F, Hammerich L, Storch D, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C‐C motif chemokine receptor 8 in mice. Hepatology 2012; 55: 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Allden SJ, Ogger PP, Ghai P, et al. The transferrin receptor CD71 delineates functionally distinct airway macrophage subsets during idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2019; 200: 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. De Hilster RHJ, Sharma PK, Jonker MR, et al. Human lung extracellular matrix hydrogels resemble the stiffness and viscoelasticity of native lung tissue. Am J Physiol Lung Cell Mol Physiol 2020; 318: L698–L704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Scheraga RG, Abraham S, Niese KA, et al. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide‐stimulated macrophage phagocytosis. J Immunol 2016; 196: 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Previtera ML, Sengupta A. Substrate stiffness regulates proinflammatory mediator production through TLR4 activity in macrophages. PLoS One 2015; 10: e0145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Meli VS, Atcha H, Veerasubramanian PK, et al. YAP‐mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv 2020; 6: eabb8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gruber E, Heyward C, Cameron J, et al. Toll‐like receptor signaling in macrophages is regulated by extracellular substrate stiffness and Rho‐associated coiled‐coil kinase (ROCK1/2). Int Immunol 2018; 30: 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Goswami R, Merth M, Sharma S, et al. TRPV4 calcium‐permeable channel is a novel regulator of oxidized LDL‐induced macrophage foam cell formation. Free Radic Biol Med 2017; 110: 142–150. [DOI] [PubMed] [Google Scholar]
- 92. Zhan L, Li J. The role of TRPV4 in fibrosis. Gene 2018; 642: 1–8. [DOI] [PubMed] [Google Scholar]
- 93. Seki E, De Minicis S, Österreicher CH, et al. TLR4 enhances TGF‐β signaling and hepatic fibrosis. Nat Med 2007; 13: 1324–1332. [DOI] [PubMed] [Google Scholar]
- 94. Pulskens WP, Teske GJ, Butter LM, et al. Toll‐like receptor‐4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS One 2008; 3: e3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Labernadie A, Bouissou A, Delobelle P, et al. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun 2014; 5: 5343. [DOI] [PubMed] [Google Scholar]
- 96. Linder S, Wiesner C. Feel the force: podosomes in mechanosensing. Exp Cell Res 2016; 343: 67–72. [DOI] [PubMed] [Google Scholar]
- 97. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011; 474: 179–184. [DOI] [PubMed] [Google Scholar]
- 98. Liu M, Yan M, Lv H, et al. Macrophage K63‐linked ubiquitination of YAP promotes its nuclear localization and exacerbates atherosclerosis. Cell Rep 2020; 32: 107990. [DOI] [PubMed] [Google Scholar]
- 99. Feng Y, Liang Y, Zhu X, et al. The signaling protein Wnt5a promotes TGFβ1‐mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J Biol Chem 2018; 293: 19290–19302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cabrera‐Benitez NE, Laffey JG, Parotto M, et al. Mechanical ventilation‐associated lung fibrosis in acute respiratory distress syndrome: a significant contributor to poor outcome. Anesthesiology 2014; 121: 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Martínez‐Klimova E, Aparicio‐Trejo OE, Tapia E, et al. Unilateral ureteral obstruction as a model to investigate fibrosis‐attenuating treatments. Biomolecules 2019; 9: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Crespo Yanguas S, Cogliati B, Willebrords J, et al. Experimental models of liver fibrosis. Arch Toxicol 2016; 90: 1025–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tag CG, Sauer‐Lehnen S, Weiskirchen S, et al. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp 2015; 96: e52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Xia G, Tawhai MH, Hoffman EA, et al. Airway wall stiffening increases peak wall shear stress: a fluid–structure interaction study in rigid and compliant airways. Ann Biomed Eng 2010; 38: 1836–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Swartz MA, Fleury ME. Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng 2007; 9: 229–256. [DOI] [PubMed] [Google Scholar]
- 106. Ng CP, Hinz B, Swartz MA. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro . J Cell Sci 2005; 118: 4731–4739. [DOI] [PubMed] [Google Scholar]
- 107. Li R, Serrano JC, Xing H, et al. Interstitial flow promotes macrophage polarization toward an M2 phenotype. Mol Biol Cell 2018; 29: 1927–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lee SWL, Seager RJ, Litvak F, et al. Integrated in silico and 3D in vitro model of macrophage migration in response to physical and chemical factors in the tumor microenvironment. Integr Biol (Camb) 2020; 12: 90–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Voorhees AP, Han HC. Biomechanics of cardiac function. Compr Physiol 2015; 5: 1623–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wirtz HR, Dobbs LG. The effects of mechanical forces on lung functions. Respir Physiol 2000; 119: 1–17. [DOI] [PubMed] [Google Scholar]
- 111. Pakshir P, Alizadehgiashi M, Wong B, et al. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat Commun 2019; 10: 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Matheson LA, Fairbank NJ, Maksym GN, et al. Characterization of the Flexcell™ Uniflex™ cyclic strain culture system with U937 macrophage‐like cells. Biomaterials 2006; 27: 226–233. [DOI] [PubMed] [Google Scholar]
- 113. Oya K, Sakamoto N, Sato M. Hypoxia suppresses stretch‐induced elongation and orientation of macrophages. Biomed Mater Eng 2013; 23: 463–471. [DOI] [PubMed] [Google Scholar]
- 114. McWhorter FY, Wang T, Nguyen P, et al. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A 2013; 110: 17253–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Oya K, Sakamoto N, Ohashi T, et al. Combined stimulation with cyclic stretching and hypoxia increases production of matrix metalloproteinase‐9 and cytokines by macrophages. Biochem Biophys Res Commun 2011; 412: 678–682. [DOI] [PubMed] [Google Scholar]
- 116. Pugin J, Dunn I, Jolliet P, et al. Activation of human macrophages by mechanical ventilation in vitro . Am J Physiol 1998; 275: L1040–L1050. [DOI] [PubMed] [Google Scholar]
- 117. Shan S, Fang B, Zhang Y, et al. Mechanical stretch promotes tumoricidal M1 polarization via the FAK/NF‐κB signaling pathway. FASEB J 2019; 33: 13254–13266. [DOI] [PubMed] [Google Scholar]
- 118. Wu J, Yan Z, Schwartz DE, et al. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch‐induced lung inflammation and injury. J Immunol 2013; 190: 3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Schoenenberger AD, Tempfer H, Lehner C, et al. Macromechanics and polycaprolactone fiber organization drive macrophage polarization and regulate inflammatory activation of tendon in vitro and in vivo . Biomaterials 2020; 249: 120034. [DOI] [PubMed] [Google Scholar]
- 120. Pairet N, Mang S, Fois G, et al. TRPV4 inhibition attenuates stretch‐induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS One 2018; 13: e0196055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Maruyama K, Sakisaka Y, Suto M, et al. Cyclic stretch negatively regulates IL‐1β secretion through the inhibition of NLRP3 inflammasome activation by attenuating the AMP kinase pathway. Front Physiol 2018; 9: 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dziki JL, Giglio RM, Sicari BM, et al. The effect of mechanical loading upon extracellular matrix bioscaffold‐mediated skeletal muscle remodeling. Tissue Eng Part A 2018; 24: 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wissing TB, Van Haaften EE, Koch SE, et al. Hemodynamic loads distinctively impact the secretory profile of biomaterial‐activated macrophages – implications for in situ vascular tissue engineering. Biomater Sci 2020; 8: 132–147. [DOI] [PubMed] [Google Scholar]
- 124. Merl‐Pham J, Basak T, Knüppel L, et al. Quantitative proteomic profiling of extracellular matrix and site‐specific collagen post‐translational modifications in an in vitro model of lung fibrosis. Matrix Biol Plus 2019; 1: 100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Garg K, Pullen NA, Oskeritzian CA, et al. Macrophage functional polarization (M1/M2) in response to varying fiber and pore dimensions of electrospun scaffolds. Biomaterials 2013; 34: 4439–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang Z, Cui Y, Wang J, et al. The effect of thick fibers and large pores of electrospun poly(ε‐caprolactone) vascular grafts on macrophage polarization and arterial regeneration. Biomaterials 2014; 35: 5700–5710. [DOI] [PubMed] [Google Scholar]
- 127. Larsen AMH, Kuczek DE, Kalvisa A, et al. Collagen density modulates the immunosuppressive functions of macrophages. J Immunol 2020; 205: 1461–1472. [DOI] [PubMed] [Google Scholar]
- 128. Hinz B. The extracellular matrix and transforming growth factor‐β1: tale of a strained relationship. Matrix Biol 2015; 47: 54–65. [DOI] [PubMed] [Google Scholar]
- 129. Veres SP, Brennan‐Pierce EP, Lee JM. Macrophage‐like U937 cells recognize collagen fibrils with strain‐induced discrete plasticity damage. J Biomed Mater Res A 2015; 103: 397–408. [DOI] [PubMed] [Google Scholar]
- 130. Hwang J, Huang Y, Burwell TJ, et al. In situ imaging of tissue remodeling with collagen hybridizing peptides. ACS Nano 2017; 11: 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Jaramillo C, Guthery SL, Lowichik A, et al. Quantitative liver fibrosis using collagen hybridizing peptide to predict native liver survival in biliary atresia: a pilot study. J Pediatr Gastroenterol Nutr 2020; 70: 87–92. [DOI] [PubMed] [Google Scholar]
- 132. Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214: 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhu Z, Ding J, Ma Z, et al. Alternatively activated macrophages derived from THP‐1 cells promote the fibrogenic activities of human dermal fibroblasts. Wound Repair Regen 2017; 25: 377–388. [DOI] [PubMed] [Google Scholar]
- 134. Ullm F, Riedl P, Machado de Amorim A, et al. 3D scaffold‐based macrophage fibroblast coculture model reveals IL‐10 dependence of wound resolution phase. Adv Biosyst 2020; 4: e1900220. [DOI] [PubMed] [Google Scholar]
- 135. Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013; 58: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Preisser L, Miot C, Le Guillou‐Guillemette H, et al. IL‐34 and macrophage colony‐stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014; 60: 1879–1890. [DOI] [PubMed] [Google Scholar]
- 137. Tan Y, Suarez A, Garza M, et al. Human fibroblast‐macrophage tissue spheroids demonstrate ratio‐dependent fibrotic activity for in vitro fibrogenesis model development. Biomater Sci 2020; 8: 1951–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Adler M, Mayo A, Zhou X, et al. Principles of cell circuits for tissue repair and fibrosis. iScience 2020; 23: 100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shook BA, Wasko RR, Rivera‐Gonzalez GC, et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018; 362: eaar2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Locatelli L, Cadamuro M, Spirlì C, et al. Macrophage recruitment by fibrocystin‐defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 2016; 63: 965–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. McDonald LT, Zile MR, Zhang Y, et al. Increased macrophage‐derived SPARC precedes collagen deposition in myocardial fibrosis. Am J Physiol Heart Circ Physiol 2018; 315: H92–H100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yang JM, Lee HS, Seo JH, et al. Structural environment built by AKAP12+ colon mesenchymal cells drives M2 macrophages during inflammation recovery. Sci Rep 2017; 7: 42723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Erdogan B, Ao M, White LM, et al. Cancer‐associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol 2017; 216: 3799–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Klingberg F, Chow ML, Koehler A, et al. Prestress in the extracellular matrix sensitizes latent TGF‐β1 for activation. J Cell Biol 2014; 207: 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tatler AL, Jenkins G. TGF‐β activation and lung fibrosis. Proc Am Thorac Soc 2012; 9: 130–136. [DOI] [PubMed] [Google Scholar]
- 146. Wipff PJ, Rifkin DB, Meister JJ, et al. Myofibroblast contraction activates latent TGF‐β1 from the extracellular matrix. J Cell Biol 2007; 179: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ford AJ, Orbach SM, Rajagopalan P. Fibroblasts stimulate macrophage migration in interconnected extracellular matrices through tunnel formation and fiber alignment. Biomaterials 2019; 209: 88–102. [DOI] [PubMed] [Google Scholar]
- 148. Hall MS, Alisafaei F, Ban E, et al. Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs. Proc Natl Acad Sci U S A 2016; 113: 14043–14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mazur A, Holthoff E, Vadali S, et al. Cleavage of type I collagen by fibroblast activation protein‐α enhances class A scavenger receptor mediated macrophage adhesion. PLoS One 2016; 11: e0150287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Avery D, Govindaraju P, Jacob M, et al. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol 2018; 67: 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang XM, Yu DMT, McCaughan GW, et al. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX‐2 human stellate cell line. Hepatology 2005; 42: 935–945. [DOI] [PubMed] [Google Scholar]
- 152. Kimura T, Monslow J, Klampatsa A, et al. Loss of cells expressing fibroblast activation protein has variable effects in models of TGF‐β and chronic bleomycin‐induced fibrosis. Am J Physiol Lung Cell Mol Physiol 2019; 317: L271–L282. [DOI] [PubMed] [Google Scholar]
- 153. Acharya PS, Zukas A, Chandan V, et al. Fibroblast activation protein: a serine protease expressed at the remodeling interface in idiopathic pulmonary fibrosis. Hum Pathol 2006; 37: 352–360. [DOI] [PubMed] [Google Scholar]
- 154. Levy MT, McCaughan GW, Marinos G, et al. Intrahepatic expression of the hepatic stellate cell marker fibroblast activation protein correlates with the degree of fibrosis in hepatitis C virus infection. Liver 2002; 22: 93–101. [DOI] [PubMed] [Google Scholar]
- 155. Sinha M, Sen CK, Singh K, et al. Direct conversion of injury‐site myeloid cells to fibroblast‐like cells of granulation tissue. Nat Commun 2018; 9: 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Meng XM, Wang S, Huang XR, et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis 2016; 7: e2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wang YY, Jiang H, Pan J, et al. Macrophage‐to‐myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol 2017; 28: 2053–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Haider N, Boscá L, Zandbergen HR, et al. Transition of macrophages to fibroblast‐like cells in healing myocardial infarction. J Am Coll Cardiol 2019; 74: 3124–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Chong SG, Sato S, Kolb M, et al. Fibrocytes and fibroblasts – where are we now. Int J Biochem Cell Biol 2019; 116: 105595. [DOI] [PubMed] [Google Scholar]
- 160. Fiore E, Malvicini M, Bayo J, et al. Involvement of hepatic macrophages in the antifibrotic effect of IGF‐I‐overexpressing mesenchymal stromal cells. Stem Cell Res Ther 2016; 7: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mansouri N, Willis GR, Fernandez‐Gonzalez A, et al. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019; 4: e128060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Saldaña L, Bensiamar F, Vallés G, et al. Immunoregulatory potential of mesenchymal stem cells following activation by macrophage‐derived soluble factors. Stem Cell Res Ther 2019; 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hou J, Shi J, Chen L, et al. M2 macrophages promote myofibroblast differentiation of LR‐MSCs and are associated with pulmonary fibrogenesis. Cell Commun Signal 2018; 16: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Morimoto K, Janssen WJ, Terada M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir Med 2012; 106: 1800–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ebener S, Barnowski S, Wotzkow C, et al. Toll‐like receptor 4 activation attenuates profibrotic response in control lung fibroblasts but not in fibroblasts from patients with IPF. Am J Physiol Lung Cell Mol Physiol 2017; 312: L42–L55. [DOI] [PubMed] [Google Scholar]
- 166. Liu F, Bayliss G, Zhuang S. Application of nintedanib and other potential anti‐fibrotic agents in fibrotic diseases. Clin Sci 2019; 133: 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Li Z, Liu X, Wang B, et al. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial–mesenchymal transition and renal fibrosis. Nephrol Ther 2017; 22: 589–597. [DOI] [PubMed] [Google Scholar]
- 168. Liu F, Wang L, Qi H, et al. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin Sci 2017; 131: 2125–2143. [DOI] [PubMed] [Google Scholar]
- 169. Öztürk Akcora B, Storm G, Prakash J, et al. Tyrosine kinase inhibitor BIBF1120 ameliorates inflammation, angiogenesis and fibrosis in CCl4‐induced liver fibrogenesis mouse model. Sci Rep 2017; 7: 44545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li Y, Li H, Liu S, et al. Pirfenidone ameliorates lipopolysaccharide‐induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol Immunol 2018; 99: 134–144. [DOI] [PubMed] [Google Scholar]
- 171. Bellamri N, Morzadec C, Joannes A, et al. Alteration of human macrophage phenotypes by the anti‐fibrotic drug nintedanib. Int Immunopharmacol 2019; 72: 112–123. [DOI] [PubMed] [Google Scholar]
- 172. Knuppel L, Ishikawa Y, Aichler M, et al. A novel antifibrotic mechanism of nintedanib and pirfenidone inhibition of collagen fibril assembly. Am J Respir Cell Mol Biol 2017; 57: 77–90. [DOI] [PubMed] [Google Scholar]
- 173. Zhang F, Ayaub EA, Wang B, et al. Reprogramming of profibrotic macrophages for treatment of bleomycin‐induced pulmonary fibrosis. EMBO Mol Med 2020; 12: e12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis 2017; 76: 1941–1948. [DOI] [PubMed] [Google Scholar]
- 175. Ricart AD, Tolcher AW, Liu G, et al. Volociximab, a chimeric monoclonal antibody that specifically binds α5β1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res 2008; 14: 7924–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Peuhu E, Salomaa SI, De Franceschi N, et al. Integrin beta 1 inhibition alleviates the chronic hyperproliferative dermatitis phenotype of SHARPIN‐deficient mice. PLoS One 2017; 12: e0186628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Carbonell WS, Delay M, Jahangiri A, et al. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab‐resistant glioblastoma. Cancer Res 2013; 73: 3145–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Chang Y, Lau WL, Jo H, et al. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in vivo . J Am Soc Nephrol 2017; 28: 1998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Funahashi Y, Sugi NH, Semba T, et al. Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res 2002; 62: 6116–6123. [PubMed] [Google Scholar]
- 180. Mita M, Kelly KR, Mita A, et al. Phase I study of E7820, an oral inhibitor of integrin α‐2 expression with antiangiogenic properties, in patients with advanced malignancies. Clin Cancer Res 2011; 17: 193–200. [DOI] [PubMed] [Google Scholar]
- 181. Wang C, Wang Z, Chen C, et al. A low MW inhibitor of CD44 dimerization for the treatment of glioblastoma. Br J Pharmacol 2020; 177: 3009–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kuronuma K, Wakabayashi M, Yoshioka T, et al. Effects of a TRPV4 antagonist and pirfenidone on idiopathic pulmonary fibrosis. Eur Respir J 2020; 56(suppl 64): 454. [Google Scholar]
- 183. Tsuno N, Yukimasa A, Yoshida O, et al. Pharmacological evaluation of novel (6‐aminopyridin‐3‐yl)(4‐(pyridin‐2‐yl)piperazin‐1‐yl) methanone derivatives as TRPV4 antagonists for the treatment of pain. Bioorg Med Chem 2017; 25: 2177–2190. [DOI] [PubMed] [Google Scholar]
- 184. Rahaman SO, Grove LM, Paruchuri S, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest 2014; 124: 5225–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Bhatia S, Miller NJ, Lu H, et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with Merkel cell carcinoma. Clin Cancer Res 2019; 25: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Fischetti L, Zhong Z, Pinder CL, et al. The synergistic effects of combining TLR ligand based adjuvants on the cytokine response are dependent upon p38/JNK signalling. Cytokine 2017; 99: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Yang HZ, Wang JP, Mi S, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol 2012; 180: 275–292. [DOI] [PubMed] [Google Scholar]