

Abstract
The KV1.3 voltage‐gated potassium ion channel is involved in many physiological processes both at the plasma membrane and in the mitochondria, chiefly in the immune and nervous systems. Therapeutic targeting KV1.3 with specific peptides and small molecule inhibitors shows great potential for treating cancers and autoimmune diseases, such as multiple sclerosis, type I diabetes mellitus, psoriasis, contact dermatitis, rheumatoid arthritis, and myasthenia gravis. However, no KV1.3‐targeted compounds have been approved for therapeutic use to date. This review focuses on the presentation of approaches for discovering new KV1.3 peptide and small‐molecule inhibitors, and strategies to improve the selectivity of active compounds toward KV1.3. Selectivity of dalatazide (ShK‐186), a synthetic derivate of the sea anemone toxin ShK, was achieved by chemical modification and has successfully reached clinical trials as a potential therapeutic for treating autoimmune diseases. Other peptides and small‐molecule inhibitors are critically evaluated for their lead‐like characteristics and potential for progression into clinical development. Some small‐molecule inhibitors with well‐defined structure–activity relationships have been optimized for selective delivery to mitochondria, and these offer therapeutic potential for the treatment of cancers. This overview of KV1.3 inhibitors and methodologies is designed to provide a good starting point for drug discovery to identify novel effective KV1.3 modulators against this target in the future.
Keywords: design of KV1.3 inhibitors, KV1.3 channel, mitochondrial KV1.3, peptide inhibitors, small‐molecule inhibitors
Abbreviations
- Bax
Bcl‐2‐like protein 4
- ChTx
charybdotoxin
- CREB
cyclic AMP‐response element‐binding protein
- diTC
[3H]C20‐29 dihydrocorreolide
- HEK
human embryonic kidney
- HTS
high‐throughput screening
- KTX
scorpion‐derived kaliotoxin
- Kv
voltage‐dependent potassium channels
- NCBI
National Center for Biotechnology Information
- NMR
nuclear magnetic resonance
- NPAA
nonproteinogenic amino acids
- ROS
reactive oxygen species
- ShK
toxin derived from the sea anemone Stichodactyla helianthus
- TEVC
two‐electrode voltage clamp
- α‐KTx
K+‐channel blocker toxin family
1. INTRODUCTION
KV1.3 channels are members of the voltage‐gated potassium ion (K+) channel family. They are widely expressed in the body and participate in multiple physiological processes. Moreover, they are involved in a variety of pathologies, including chronic inflammation and cancer progression. 1 Therefore they have been regarded as relevant therapeutic targets for the development of anti‐inflammatory 2 and anticancer therapies. 3 KV1.3 channels are located at the cell plasma membrane, the inner mitochondrial membrane (mitoKV1.3), the nuclear membrane, and the membrane of the cis‐Golgi apparatus. 4 The plasma membrane KV1.3 channels control the resting potential and action potential in excitable cells while, in nonexcitable tissues, they regulate cell volume and cell proliferation. MitoKV1.3 regulates the mitochondrial membrane potential, mitochondrial volume, and production of reactive oxygen species (ROS). 5
KV1.3 is highly expressed in macrophages, microglia, and effector memory T‐cells, suggesting that KV1.3 plays a role in immune and inflammatory responses in human diseases, such as multiple sclerosis, rheumatoid arthritis, psoriasis, type I diabetes mellitus, and asthma. 6 Aberrant (predominantly high) expression of KV1.3 has been observed in different types of tumors. 7 Overexpression of KV1.3 in cancer cells gives them an advantage by enhancing tumorigenic processes, such as cell proliferation, migration, and metastasis. 8 Expression of mitoKV1.3 in the inner mitochondrial membrane appears to correlate with that at the plasma membrane; while plasma membrane KV1.3 is required for cell proliferation, 4 mitoKV1.3 regulates apoptosis. 5
Despite the apparent potential of this target for a wide range of diseases, and despite the efforts made to develop specific inhibitors, KV1.3 remains underexploited and no KV1.3 inhibitors have reached the market yet. There are many potential reasons for this, including its high degree of homology within the KV channel family, making the design of a selective and safe drug challenging. The availability of a crystal structure of the target and/or a small‐molecule inhibitor in complex with one of the KV1.x isoforms would provide detailed knowledge of the binding interactions and would help to improve the potency and selectivity of KV1.3 inhibitors; however, as such a crystal structure is not yet available, this also represents a limiting factor in the design of potent and selective KV1.3 inhibitors. A final challenge is the design of inhibitors that have the required physicochemical properties to allow them to be effectively administered to patients and to penetrate barriers to the target tissues, to reach KV1.3 in both the plasma membrane and the inner mitochondrial membrane.
Peptides have a larger interaction surface area than small molecules and thus offer the possibility of subtype selectivity. 9 In fact, the only KV1.3 inhibitor currently in clinical trials as a therapeutic for autoimmune diseases is dalatazide, a 37 amino acid synthetic peptide that is a derivative of ShK toxin isolated from the venom of the sea anemone Stichodactyla helianthus. 10 Significant progress has been made in the chemical modifications of ShK, which, together with other strategies, has resulted in peptide analogs with picomolar affinities and more than 1000‐fold selectivity for KV1.3 over other closely related ion channels. 11 The successful approaches to drug design were strategies that modified the flexibility of peptides or examined the evolutionary role of specific toxin residues, considering that animal toxins evolved over millions of years to bind specifically to their targets. 12
This article discusses the current state of research on KV1.3 inhibitors. Information and issues important for medicinal chemists to develop potent and selective KV1.3 inhibitors are highlighted. In this review, we summarize the existing evidence that KV1.3 is involved in the pathology of devastating diseases and is, as such, an attractive target for drug discovery. The structure of KV1.3 is presented, highlighting the channel parts that have been shown to be essential for targeting by peptides and small molecules. In addition, the permeation, gating, and inactivation mechanisms of KV1.3 are described. The review focuses on the presentation and discussion of approaches to the discovery of peptide and small molecule modulators of KV1.3, with emphasis on strategies to increase selectivity. Some recent reviews have reported on the relevance of KV1.3 in pathological processes, supporting the role of KV1.3 as a prospective drug target. Therefore, this review's focus is on the presentation of methodologies for the discovery of new Kv1.3 inhibitors, with an objective review of the literature since 1995, to list the most active and selective compounds to date. This overview of KV1.3 inhibitors and methodologies is designed to provide a good starting point for drug discovery in identifying novel selective KV1.3 inhibitors to facilitate the design of new drugs against this target in the future.
2. PHYSIOLOGICAL AND PATHOLOGICAL FUNCTIONS OF KV1.3
2.1. KV1.3 in the immune system
In the early days of KV1.3 research, KV1.3 inhibitors were believed to be general immunosuppressants, and were therefore used, for example, to prevent transplantation rejection. Nowadays, they are conceived as immunomodulators that can selectively block C‐C chemokine receptor type 7 (CCR7−) effector memory T‐cells. Upon activation by antigens, naïve T‐cells proliferate and differentiate into two types of memory T‐cells: effector memory T‐cells, which immediately translocate to the center of the inflammation, and central memory T‐cells, which have a lower threshold for activation. 13
Two main players, equally expressed in all three types of quiescent T‐cells (at ~300 channels per cell), determine potassium flux in T‐cells: KCa3.1 and KV1.3. In the activated state, KCa3.1 channel expression is upregulated to ~500 channels per cell in naïve and central memory T‐cells, while KV1.3 expression remains constant. Vice versa, no increased expression of KCa3.1 is seen upon effector memory T‐cell activation, while here KV1.3 expression increases to ~1500–2000 channels per cell.2, 4, 13
Activation of T‐cells by antigen‐presenting cells causes rearrangements in the membrane that result in immunological synapses, which take the form of an accumulation of cholesterol‐enriched lipid rafts that can recruit T‐cell receptors. 14 KCa3.1 and KV1.3 are recruited to this immunological synapse, where they can be activated by cell membrane depolarization upon an increase in intracellular Ca2+.15, 16 KV1.3 and KCa3.1 determine the resting membrane potential of T‐cells, as their activation results in membrane hyperpolarization, which provides an increased driving force for Ca2+, which regulates gene transcription and proliferation of T‐cells.17, 18 KV1.3 inhibitors might, therefore, be useful for the management of all T‐cell‐mediated autoimmune and chronic inflammatory diseases (Table 1).13, 19 KV1.3 is also important for other immune cell types, such as in B‐lymphocytes, natural killer cells, macrophages, dendritic cells, and monocytes. 4
Table 1.
T‐cell‐mediated autoimmune and chronic inflammatory diseases where overexpression of KV1.3 has been defined7, 20
| Disease group | Human disease |
|---|---|
| Autoimmune | Multiple sclerosis18, 21 |
| Rheumatoid arthritis 22 | |
| Type I diabetes mellitus20, 23 | |
| Alopecia areata 17 | |
| Systemic lupus erythematosus 24 | |
| Crohn's disease (inflammatory bowel disease) 17 | |
| Rapidly progressive glomerulonephritis 17 | |
| Psoriasis 17 | |
| Chronic inflammation | Chronic renal failure 25 |
| Ulcerative colitis 26 | |
| Atherosclerosis27, 28 | |
| Allergic contact dermatitis 17 | |
| Asthma 29 | |
| Age‐dependent hypertension 17 |
2.2. KV1.3 in the nervous system
In the nervous system, KV1.3 has been identified in some subsets of neurons and all types of glial cells (oligodendrocytes, microglia, and astrocytes). Upon injury, oligodendrocyte precursor cells maturate and can regenerate neuronal myelin sheaths. These precursor cells contain the Shaker‐type K+ channels KV1.1 to KV1.6, all sensitive to tetraethylammonium, whose action inhibits oligodendrocyte precursor cell proliferation. Interestingly, the selective block of KV1.3 in oligodendrocyte precursor cells has similar effects to the general K+ inhibitor tetraethylammonium, suggesting a chief role for KV1.3 in cell‐cycle progression of these oligodendrocyte precursor cells. 30
The role of microglia is to provide a first barrier for “intruders” in the central nervous system. Potassium channel activity changes upon microglia activation. More specifically, in quiescent cells, KV1.5 expression dominates, while KV1.3 is the main K+ channel in proliferating microglial cells. KV1.3 is also involved in microglial migration and cytokine release. Finally, astrocytes show little turnover under normal conditions, but they can proliferate after injury, which is also linked to increased K+ channel expression. 4
2.3. KV1.3 in other systems
KV1.3 is expressed in excitable and nonexcitable cells, such as vascular smooth muscle cells, platelets, kidney and colon epithelial cells, osteoclasts, testes, adipocytes, and skeletal muscle cells. KV1.3 also participates in the pathways that regulate energy homeostasis and body weight, and might thus be a therapeutic target for obesity, although the role of KV1.3 in adipose tissue is still not clear. Furthermore, KV1.3 is a putative pharmacological target for the treatment of type II diabetes mellitus. 17 Inhibition of KV1.1 and KV1.3 channels using the scorpion‐derived toxin kaliotoxin (KTX) in rat models facilitates cognitive processes, such as learning, which indicates a further possible role for KV1.3 in the treatment of memory disorders.19, 31, 32
2.4. KV1.3 channels and cell proliferation
The KV1.3 effects on cell proliferation have several possible explanations (Table 2), which are not necessarily mutually exclusive. In general, the hypotheses to explain the influence of KV1.3 on cell proliferation can be divided into those that link cell proliferation with membrane potential, and those that link cell proliferation with noncanonical functions of KV1.3.4, 17, 33, 34
Table 2.
Proposed therapeutic use of KV1.3 inhibitors, their mechanism of action and strategies to achieve selectivity4, 35, 36, 37, 38
| Target | Localization | Possible therapeutic use | Mechanism of action | Selectivity | ||
|---|---|---|---|---|---|---|
| Proposed | Possible explanation or hypothesis | Strategy to achieve | Possible explanation or hypothesis | |||
| KV1.3 | Plasma membrane of T‐cells | T‐cell‐mediated autoimmune and chronic inflammatory diseases | Inhibition of proliferation in TEM cells | Depolarization results in decreased driving force for Ca2+, which regulates gene transcription and proliferation of T‐cells | Selective suppression of activated TEM cells, but not naive and TCM cells | Naive and TCM cells escape suppression due to upregulated KCa3.1 |
| Changes in membrane potential are required for cell cycle progression | ||||||
| Ion fluxes influence cell volume | ||||||
| Mito‐KV1.3 | Inner mitochondrial membrane | Cancer treatment | Induction of apoptosis in cancer cells | KV1.3 inhibitors mimic interaction of Kv1.3 with Bax and cause apoptosis | Selective apoptosis of cancer cells due to their escalated ROS production | Upregulation of mitoKV1.3 in some cancer cells |
| Elevated basal ROS levels in cancer cells | ||||||
| KV1.3 | Plasma membrane of cancer cells | Cancer treatment | Suppressing proliferation of cancer cells | Changes in membrane potential are necessary for cell cycle progression and Ca2+ signaling | Selective inhibition of proliferation in cancer cells, but not normal cells | Some cancer cells have upregulated channel expression |
| Ion fluxes influence cell volume | ||||||
| Depolarisation causes conformational changes of KV1.3 and leads to proproliferative signaling pathways | ||||||
| Proproliferative role of KV1.3 in the “channelosome” model | ||||||
Cell proliferation is affected by the membrane potential at different levels. Permeation of K+ through the KV1.3 pores influences membrane potential, and therefore influences the cell cycle. More specifically, a rapid hyperpolarization occurs before the initiation of the S phase, while a prolonged depolarization takes place during the G2/M transition. 39 Indirectly, the membrane potential regulates cell volume, as this is a consequence of ion fluxes followed by the movement of water. The cell volume changes through the cell cycle, and this affects both mitosis and apoptosis. 33 On another level, KV1.3 undergoes conformational changes in response to changes in membrane potential. Accessory subunits of KV1.3 can convert these voltage‐dependent conformational changes into signals that affect cell proliferation. Experiments with nonpermeating mutants of KV1.3 suggested that rather than permeation, the gating is the crucial factor for KV1.3 effects on cell proliferation, 40 which corresponds to the nuclear localization of KV1.3 in certain cells. 41
Besides the membrane potential, cell proliferation is affected by the signaling molecules associated to KV1.3. These are also called the “channelosome,” which can interact with proteins that influence multiple signaling cascades. 42
2.5. Role and expression of KV1.3 in different types of cancers
Aberrant expression of KV1.3 has been shown for several types of tumors and cancer cells. In general, the expression of KV1.3 shows no clear patterns (Table 3) and in cancer cells it depends on the type, and stage of disease.7, 43 However, in addition to their role in cell proliferation, migration, and invasion, KV1.3 appears to contribute to the development of cancers. 8 Some tumors induced in experimental models showed high levels of several KV channels, including KV1.3. Therefore KV1.3 channels might serve as novel markers of the metastatic phenotype, and also as potential new therapeutic targets (Table 2), especially in cancer tissues with upregulated KV1.3 expression.7, 43 A recently published review by Tajti et al. 44 focuses more on the therapeutic applications of Kv1.3 in (neuro)inflammatory diseases.
Table 3.
| Cancer tissue | Type of a cancer or cancer cell line | Expression of KV1.3 | Contradictory data |
|---|---|---|---|
| Colon | Colon adenocarcinoma | Altered | |
| Breast | Highly tumorigenic M13SV1R2‐N1, weakly tumorigenic M13SV1R2 cells | Elevateda | Abdul et al.: protein levels vary 45 |
| Grade I and II breast adenocarcinoma | Elevateda | ||
| Grade III breast adenocarcinoma | Decreaseda | ||
| Prostate | Weakly metastatic LNCaP, AT‐2 | Elevateda | In PC3, DU145, LNCaP, MDA‐PCA‐2B cell lines 46 |
| Strongly metastatic PC3 and Mat‐LyLu | Decreaseda | ||
| Benign prostatic hyperplasia, human prostate cancer | Elevated | ||
| Smooth muscle | Leiomyoma | Decreased | |
| Leiomyosarcoma | Elevated | ||
| Skeletal muscle | Embryonal rabdomyosarcoma | Decreased | |
| Alveolar rabdomyosarcoma | Elevated | ||
| Brain | Astrocytoma | Decreased | |
| Oligodendroglioma | Decreased | ||
| Glioblastoma | Decreased | ||
| Lymph node | Follicular B‐cell lymphoma | Decreased | |
| Mantle lymphoma | Decreased | ||
| T‐cell lymphoma | Decreased | ||
| Diffuse large B‐cell lymphoma | Altered | ||
| Anaplastic lymphoma | Decreased | ||
| Chronic lymphocytic leukemia | Increased |
Expression is compared to healthy and control samples.
Significantly reduced expression of KV1.3 was detected in stomach, and brain tumors, kidney and bladder carcinomas, lung and pancreas adenocarcinomas, squamous cell skin carcinomas, and mammary duct carcinomas (Table 3). On the other hand, increased KV1.3 expression was identified in breast, colon, and prostate tumors, smooth muscle, and skeletal muscle cancer, and in mature neoplastic B cells in chronic lymphocytic leukemia.3, 7, 37, 43, 47
Comes et al. 7 showed low KV1.3 expression in the healthy colon, and increased KV1.3 expression in colon adenocarcinoma, whereby 75% of the tumor cells had “moderate expression” of KV1.3, and therefore a clear expression pattern has not been established.
The studies of Abdul on the expression of KV1.3 in breast cancer delivered contradictory data (Table 3). 45 Jang et al. 48 detected significantly higher KV1.3 expression in highly tumorigenic M13SV1R2‐N1 cells, and weakly tumorigenic M13SV1R2 cells, than in normal cells. Brevet et al. 49 showed markedly reduced KV1.3 expression in breast adenocarcinoma specimens compared to normal breast tissue, and an inverse correlation between channel expression and grade of disease.
Abdul et al. indicated high KV1.3 levels in normal human prostate epithelium and variable levels in clinical prostate cancer cell lines (Table 3). 46 The density of KV1.3 appeared to be inversely correlated with the metastatic capacity of human prostate cancer cells, and was significantly higher in weakly metastatic than strongly metastatic cells. 7
Bielanska et al. 50 investigated KV1.3 expression in human smooth muscle tumors. KV1.3 was poorly expressed in the healthy muscle and indolent leiomyoma specimens, whereas the aggressive leiomyosarcoma showed high levels of KV1.3 expression. This increased expression of KV1.3 had close correlation with stage of malignancy, and thus KV1.3 might be a tumorigenic target for aggressive human leiomyosarcoma. Similar data were obtained in the case of aggresive alveolar rhabdomyosarcoma, where the expression of KV1.3 correlated with tumor malignancy. 51
Different glioma samples were investigated for KV1.3 expression and normal glia proliferating cells have increased KV1.3 expression. KV1.3 is expressed in a variety of glial tumors, although it does not show any correlation with glioma subtype or malignancy grade, which correlates well with expression of KV1.3 in proliferative normal glial cells. This indicates that KV1.3 is required for cell proliferation in neoplastic and nonneoplastic tissues. 52
The expression of KV1.3 was also examined in a panel of human non‐Hodgkin's lymphomas. 7 KV1.3 expression was lower in follicular B‐cell, mantle, anaplastic and T‐cell lymphomas in comparison with control lymph nodes (Table 3). There was no clear connection with malignancy or clinical aggressiveness, but their data suggested that KV1.3 acted as a tumor suppressor. As suppression of KV1.3 prevented apoptosis, KV1.3 might be involved in tumor development.7, 53 Pathological B‐cells of chronic lymphocytic leukemia had higher KV1.3 protein expression versus healthy cells, and treatment with the KV1.3 inhibitor clofazimine reduced the tumor size in an orthotopic melanoma mouse model. 54
2.6. Mitochondrial KV1.3 and apoptosis
The localization of KV1.3 at the plasma membrane of lymphocytes defines its roles in setting the membrane potential, and in Ca2+ signaling, cell proliferation, and autoimmune diseases; however, KV1.3 expression has also been observed in the mitochondria of lymphocytes. 55 Although several K+ channels reside in the inner mitochondrial membrane, KV1.3 appears to be partly responsible for the basal K+ conductance in mitochondria. 56 As the orientation and electrophysiological properties of KV1.3 in mitochondria are the same as at the plasma membrane, both forms of the channel are likely the product of the same gene. 35
Mitochondria are centers of energy and ROS production, and they have a role in many signaling cascades. The intrinsic apoptotic pathway, for example, is often enabled by activation of proapoptotic proteins of the Bcl‐2 family, such as Bcl‐2‐like protein 4 (Bax) and Bcl‐2 homologous antagonist (Bak). Interestingly, mitochondrial KV1.3 in lymphocytes is required to induce apoptosis by Bax, where Bax inhibits KV1.3 in a toxin‐like manner. The K+ flow from the cytosol to the mitochondrial matrix is blocked and causes hyperpolarization of the inner mitochondrial membrane, which leads to ROS production and activation of the apoptosome.57, 58 Several types of cancer cells are deficient in proapoptotic proteins, such as Bax, which results in resistance to apoptosis and reduction of the efficiency of chemotherapeutics. Therefore, there is an urgent need to generate compounds that can restore apoptosis in cancer cells. Mitochondrial KV1.3 can be considered as an important therapeutic target here, because inhibition of this channel by either peptide toxins or small molecules will promote apoptosis in cancer cells.59, 60
Finally, KV1.3 expression has also been reported at the cis‐Golgi of astrocytes. KV1.3 is believed to form aggregates or higher‐order assemblies, which prevents trafficking to the cell surface and causes retention in the Golgi apparatus. In the cis‐Golgi, KV1.3 might be directly involved in Golgi activities, such as regulation of the Golgi membrane potential, or it might serve as a back‐up source for KV1.1, which is also found in the plasma membrane of astrocytes. 61
3. KV1.3 CHANNEL AS A DRUG TARGET
3.1. The structure of KV1.3
As indicated, KV1.3 belongs to the voltage‐dependent K+ channels (KV) that are present in all living organisms, and it is known to participate in many cellular events. KV1.3 is a transmembrane protein that forms aqueous pores, that facilitate the passage of K+ through the plasma membrane along its electrochemical gradient. The KV proteins comprise 12 subfamilies (KV1–KV12) that can be classified into three groups.1, 17 The KV1 (Shaker) subfamily is the largest KV subfamily, as it includes eight voltage‐gated K+ channels (KV1.1–KV1.8). 62
X‐ray crystallography, homology modeling and ab‐initio molecular modeling have provided deep insights into the structure of the KV channels to understand their mechanism of ion conduction.63, 64, 65 Kremer et al. 66 revealed the X‐ray structure of the T1 domain from human KV1.3 at 1.2Å resolution following its crystallization under near‐physiological conditions. However, the crystal structure of the whole of KV1.3 has not yet been resolved. Homology models for KV1.3 have been constructed using the crystal structure of KV1.2 from Rattus norvegicus (PDB ID 3LUT, 2A79) and the KV1.2–KV2.1 paddle chimera channel (PDB ID 2R9R) as templates (Figure 1).67, 68, 69
Figure 1.
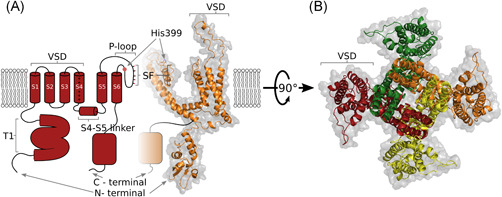
Structural representations of the KV1.3 channel. (A) Hybrid side view as two‐dimensional (left, red) and three‐dimensional homology (right, orange) representations of two opposing domains in (B). (B) Top (extracellular) view of all four domains, with each colour coded, which shows the domain‐swapping architecture of the KV1.3 channel. The homology model was built using Modeller 9.21, Kv1.2 (PDB 3LUT) as a template, Kv1.3 sequance was retrieved from Uniprot (Accession No.: P22001), and the figure was prepared using PyMOL70, 71, 72, 73 [Color figure can be viewed at wileyonlinelibrary.com]
In the National Center for Biotechnology Information (NCBI) protein data bank, there is a 575‐amino‐acid‐long sequence of the KV1.3 ion channel (NCBI: NP_002223.3). A search for similar sequences revealed more than 70% identity with the sequence of KV1.2 from R. norvegicus (NCBI: NP_037102.1). KV1.2 and KV1.3 also share 93% sequence identity for their pore domains. 68 Characterization of specific structural differences between these KV channels has been achieved using both computational methods and mutagenesis studies, with scorpion toxins as molecular probes; for example, ADWX‐1. Sequence alignment of KV1.1, KV1.2, and KV1.3 shows some variations in their amino‐acid sequence, and consequently structural differences in the turret region, pore helix, and filter region.70, 74, 75
As a K+ channel, KV1.3 is an assembly of four identical individual subunits, each of which consists of six transmembrane domains, known as S1–S6 (Figure 1). Helices S5 and S6 and the linker between them, which is known as the P‐loop, assemble together to form a central pore domain; this contains the channel selectivity filter and gates.62, 63, 76 Five conserved signature sequences that function as a selectivity filter are located on the P‐loop (T75, V76, G77, Y78, G79), and these mimic the water structure around the permeating K+ ion. The oxygen cages cannot correctly coordinate the binding of smaller ions at the same location, and this is a basis for the K+ selectivity.64, 77 Below the selectivity filter, there is a wide water‐filled pore that faces into the cytoplasm, and that traverses more than half of the phospholipid bilayer of the membrane. The K+ ions pass from the helical bundle through the wide pore, to reach the selectivity filter at the end.63, 78 Four voltage‐sensing domains, each made up of helices S1 to S4, surround the pore domain of the neighboring subunit, which leads to the domain‐swapping architecture, and which controls its gates.76, 79 At the N‐terminus, preceding the S1 helix, tetramerization or the T1 domain serve as the docking platform for auxiliary subunits.62, 63, 80
3.2. Gating of KV1.3
As for all KV channels, KV1.3 channels open upon membrane depolarization. KV1.3 has two distinctive biophysical properties: C‐type inactivation and cumulative inactivation during repetitive depolarizing pulses. For the Shaker K+ channel family, two types of inactivation have been described. N‐type inactivation, which is also known as ball and chain inactivation, consists of occlusion of the pore with an N‐terminal cytoplasmic particle. KV1.3 channels inactivate via the slower P/C‐type inactivation (Figure 2), which consists of constriction of the selectivity filter on the extracellular side of the pore, which results from two conformational changes. First, the extracellular gate of the channel closes, which leads to the collapse of the selectivity filter (i.e., P‐type inactivation). Second, a further conformational change stabilizes the nonconducting state and the conformation of the voltage sensors (i.e., C‐type inactivation).81, 82 The structural changes that occur during the C‐type inactivation are radical because KV1.3 requires a considerable time to be ready to open again (i.e., 30–60 s), depending on the length of the depolarizing pulses. Consequently, if a channel is subjected to rapid depolarizing pulses, fewer and fewer channels will be ready to open again with every pulse, resulting in the use dependence. 83 C‐type inactivation is considerably delayed by increased extracellular K+ concentration, apparently, because K+ interacts with the residues in the external vestibule that are involved in C‐type inactivation. 84
Figure 2.
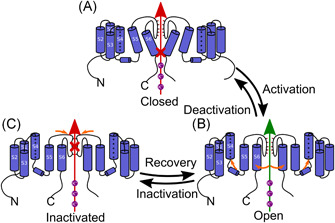
Schematic representation of the P/C‐type channel gating of KV1.3. (A) Closed state of the P/C‐type inactivating channel. (B) Open state of the P/C‐type inactivating channel, where permeation of K+ is possible. Orange arrows, movement of S4 out of the membrane, and movement of S5 and S6 to open the central part of the permeation pathway. (C) Inactivated state of P/C‐type inactivating channel, with collapse of the selectivity filter marked with orange arrows81, 82 [Color figure can be viewed at wileyonlinelibrary.com]
The inactivation of KV1.3 depends on factors such as temperature, phosphorylation, interactions with inhibitors (e.g., tetraethylammonium) and ion composition of the external medium. 85 What makes KV1.3 inactivation unique compared to other channels of the Shaker family is the influence of extracellular pH: inactivation is reduced with lower external pH, contrary to what happens in other KV channels. 81 The H399 residue in the S5‐S6 linker region of KV1.3 has a role in this pH dependence of inactivation. Protonation of H399 at low pH is believed to form a barrier at the outermost binding site for K+, which prevents the exit of K+ from the pore by electrostatic repulsion. 86 As a histidine at this position is unique to KV1.3, compounds that can interact with this residue should show interesting selectivity for KV1.3 over the other closely related channels. 87
3.3. Binding sites of KV1.3 inhibitors
Venom peptides and small molecules can interact with KV1.3 in multiple ways, such as by blocking the ion‐conducting pore from the external side, or more specifically from the external vestibule, or by modifying the channel gating through binding to the voltage sensor domain (Figure 3). 88 From the internal side, small molecules can bind to the selectivity filter or act from the water‐filled cavity under the selectivity filter.
Figure 3.
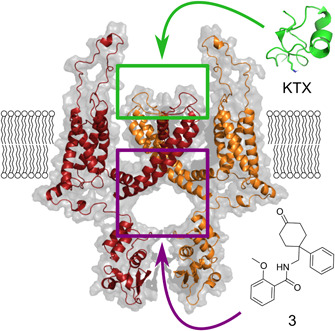
Representation of the binding sites of KV1.3 inhibitors. Green square, extracellular binding site of KTX toxin (green, PDB 1KTX) as a representative toxin; purple square, central cavity preserved in many K+ ion channels as the binding site for small molecules, such as believed for inhibitor 3. The homology model was build using Modeller 9.21, Kv1.2 (PDB 3LUT) as a template, KV1.3 sequence was retrieved from Uniprot (Accession No.: P22001), and the figure was prepared using Pymol70, 71, 72, 73 [Color figure can be viewed at wileyonlinelibrary.com]
An important issue that has to be taken into consideration when we design KV1.3 inhibitors is binding cooperativity,89, 90, 91 which means that binding of the inhibitor to one of the four KV1.3 subunits causes a readjustment in the binding site (induced fit) 92 that triggers a rearrangement in the empty binding site of the neighboring subunit, increasing binding affinity. So, binding of the inhibitor to one subunit could enhance the binding of a second molecule of inhibitor to the adjacent subunit. Binding cooperativity can be identified from the Hill coefficients higher than 1.
Binding of several classical KV channel inhibitors that were originally described to block the Shaker type channel in the early 1980s, such as tetraethylammonium, d‐tubocurarine, and verapamil, has been studied in KV1.3 models. These compounds are organic cations and they can block the open K+ channels by physically occluding the inner pore and inserting their ammonium group into the ion permeation pathway.93, 94 Binding sites of more potent and selective KV1.3 small molecule inhibitors are described in the later sections.
Many scorpion toxins have been described as modulators of K+ channels. These toxins are typically composed of 23–64 amino acids, and they are structurally characterized by an α/β motif that is stabilized by cysteines, with disulfide bridges that form covalent links between the antiparallel β‐sheets (Figure 4). These toxins block K+ flow by binding to and thereby obstructing the external pore with a 1:1 stoichiometry.81, 95 The binding site of the scorpion‐derived toxin KTX has been extensively studied on KV1.3, more specifically for KcsA–KV1.3, a chimera of two channels. Similar to agitoxin2 and charybdotoxin (ChTx), three interface regions show significant changes in their solid‐state nuclear magnetic resonance (NMR) chemical shifts, which have been described as for: the α‐helix (S11, L15), the second β‐strand (M23, R24, K27, M29), and the end of the third β‐strand (T36, P37).96, 97 After the binding of KTX, the internal and external sides of the KcsA–KV1.3 pore remain structurally unchanged. For residues in the KTX‐binding region, however, solid‐state NMR has revealed changes in the chemical shifts in both the pore helix and the selectivity filter. In the vestibule, D64 appears to be a critical residue, as mutation of this residue changes the affinity of KTX by three orders of magnitude. On the other hand, for the selectivity filter, significant changes in the chemical shifts were reported for G77, Y78, and G79. The ion binding site of the K+ channel shows surprising conformational flexibility, as it can adopt different conformations depending on the binding ion. When a solution changes from K+ to Na+ as the main ion, the selectivity filter shows an altered conformation, with changes to its backbone angles, and its subsequently shut down. KTX is believed to insert into the selectivity filter, much deeper than toxins were initially expected to. Its K27 lysine sidechain extends from the interaction surface, making contact with the K+ channel pore near G77. The methylene groups of the lysine sidechain replace the water in the entry region of the pore, which facilitates entry into the selectivity filter. 97 This functional dyad consists of an aromatic residue and a lysine at position 27, and appears to be the crucial structural element to bind and block KV1.x channels for many scorpion toxins, such as agitoxin2 and ChTx (with aromatic residues F25 and Y36, respectively). 98 Interestingly, in the presence of a positive residue at position 27 (i.e., lysine, arginine), the application of an intracellular K+ solution increases the dissociation rate of the toxins, such as ChTx, in a voltage‐dependent manner. When K27 is substituted with a neutral residue, the dissociation rate becomes insensitive to the internal solution or voltage, which argues for a toxin binding site close to a K+ binding site.99, 100
Figure 4.
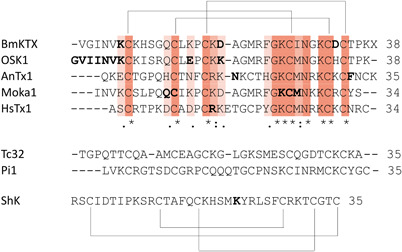
Above: the aligned sequences of BmKTX 101 (1BKT), OSK1 102 (1SCO), AnTx 103 (KAX6C_ANUPH), moka1 104 (2KIR), and HsTx1 105 (1QUZ) are shown together with the pattern of their disulfide bridges. Residues in bold are amino acids of which mutations have been shown to alter Kv1.3 binding. Middle: Sequences of Tc32 106 (2JP6) and Pi1 107 (1WZ5) are shown. Below: Sequence of ShK 108 (1ROO) is shown together with the pattern of its disulfide bridges. Residue in bold is an amino acid of which mutations have been shown to alter Kv1.3 binding [Color figure can be viewed at wileyonlinelibrary.com]
Zhu et al. identified eight conserved residues in the α‐KTx K+‐channel blocker toxin family that are structurally and functionally important for binding to KV channels, and called this the scorpion toxin signature. This scorpion toxin signature constitutes six cysteines with three disulfide bridges and two amino acids (Lys, Asn) in a four‐residue‐long motif around the fourth cysteine (Lys‐Cys4‐Xaa‐Asn; where Xaa is any amino acid). They showed conversion of an insect defensin (navidefensin2‐2) into a neurotoxin (navitoxin) that bound KV channels in the same manner as scorpion α‐KTxs. Two prerequisites for binding are that at least two residues of the scorpion toxin signature are present and that steric hindrance is removed (which is caused by the n‐loop in defensins). Wild‐type navitoxin is active on KV1.1, KV1.2, and KV1.3, while each of the mutants (by mutating either Lys or Asn) lose their activity toward KV1.3, which argues again for the importance of a positively charged residue for the interaction with KV1.3. 109
Stichodactyla toxin is one of the most potent KV1.3 toxins. It is derived from a sea anemone (S. helianthus), and it has a very different structure compared to the scorpion toxins (Figure 4). Three disulfide bridges stabilize the main architecture of ShK in a 1‐6/2‐4/3‐5 paired pattern, compared to the 1‐4/2‐5/3‐6 paired pattern of scorpion toxins. Furthermore, ShK includes two short α‐helices that run from residues 14 to 19 and 21 to 24. The N‐terminus constitutes an extended strand, a loop, and a helix, while the C‐terminus shows several chain reversals, including a type 1 β‐turn at residues 28–31.98, 110 Nevertheless, ShK similarly interacts with Kv1.3, and therefore a common interaction surface that favors KV1.3 binding can be hypothesized. 98 Initially, it was thought that K22 was the critical residue for KV1.3 pore blockage. 111 Zhao et al. 112 however, showed a more complex mechanism in which K22 appears important for the affinity, and it is residue R24 that determines the voltage dependency of KV1.3 blockage.
This theory of a common interaction surface that consists of a central positively charged residue that can protrude into the pore accompanied by aromatic residues that can interact with the selectivity filter has been questioned. This is based on the scorpion toxin Tc32, which does not have this interaction surface but nevertheless binds KV1.3 with high affinity. Moreover, the chemical synthesis of the scorpion toxin Pi1 without its functional dyad has revealed that the presence of K24 and Y33 is not a prerequisite for KV1.2 binding. Removal of this functional dyad resulted in a lesser block of KV1.2 compared to the wild‐type Pi1, but not in a loss of activity, which suggests that more residues are involved, rather than K24 and Y33 being the sole determinants of toxin binding. Therefore, it is suggested that other amino acids would surround the signature sequence and reach out to the turret region of the channel, resulting in a ring of basic residues.81, 98, 113
3.4. Electrophysiological methods
Across the plethora of methods and techniques used, electrophysiological techniques have been shown to be useful in ion‐channel‐related drug discovery. The golden standards in electrophysiology are the two‐electrode voltage clamp (TEVC) technique with Xenopus laevis oocytes, 114 and the patch‐clamp technique with mammalian or X. laevis oocytes cells. 115
As indicated, TEVC involves two electrodes: one to measure the internal potential and one to inject the current. These can be inserted into large cells, such as oocytes. This is an ideal technique for screening as it is relatively fast to perform, and the external medium can be changed multiple times. Additionally, recordings are stable over a long period of time, and there is high sensitivity as the channels in the whole‐cell are measured. The biggest disadvantage is the large membrane capacitance caused by the size of the oocyte and the numerous membrane invaginations, which makes it difficult to obtain high‐quality data during the first 1–2 ms of depolarization. Moreover, when a (hydrophobic) drug has an internal binding site, the IC50 or EC50 values seen using oocytes are sometimes biased compared to mammalian cells, due to the large internal volume of the oocyte. 116
On the other hand, the patch‐clamp technique using mammalian cells in the single‐channel or whole‐cell mode literally creates a patch on the membrane that can be either used as “cell‐attached” or excised. This technique provides better time resolution, but it is generally more time‐consuming to collect sufficient high‐quality data and perform the analysis. 116
Another point that can cause pharmacological differences in drug discovery is the composition of the membrane: Xenopus oocytes should be seen as an amphibian system, whereas mammalian cells, like HEK cells, more closely resemble human biomembranes in different organs. 117 As well as the common TEVC and patch‐clamp techniques, there are also many “sub‐techniques,” such as the cut‐open oocyte voltage clamp, inside‐out patches, and outside‐out patches. Other nonelectrophysiological low‐throughput techniques that are used include biochemical approaches or in‐vivo studies. 116
When preparing the pulse protocol plan for testing KV1.3 inhibitors, it is necessary to keep in mind that the extracellular K+ concentration influences the rate of C‐type inactivation. High levels of K+ depolarize the membrane and slow‐down the C‐type inactivation rate. When using higher K+ concentration, KV1.3 inhibitors, which bind to the C‐type inactivated state, appear less potent under physiological conditions.83, 84
With the rise in high‐throughput screening (HTS), cell‐based assays with fluorescent dyes and ion fluxes have become dominant in ion‐channel drug discovery. These approaches have certainly been shown to be useful, but they remain very indirect measures of ion‐channel activity. 118 The lack of advances in electrophysiological HTS has impeded ion‐channel drug discovery, as this type of assay provides very rich information about the direct activity of an ion channel. Still, the manual execution is very labor‐intensive and requires a high level of expertise. 119 Recent progress has led to the development of automated systems, such as the HiClamp system (Multi Channels System), for example. This is an automated version of TEVC that consists of a 96‐well plate with oocytes and a 96‐well plate with the compounds to be tested. The HiClamp system automatically collects and positions the oocyte in a basket, impales it with the two electrodes, and submerges it in the test compounds. Similar systems are available for patch‐clamp, such as PatchServer (Multi Channel Systems). Combining these new electrophysiology technologies with nonelectrophysiological HTS represents a promising strategy for ion‐channel drug discovery.
4. DISCOVERY AND DEVELOPMENT OF PEPTIDE‐BASED KV1.3 INHIBITORS
4.1. Venom drug‐discovery strategies
Toxins in venoms are defined as compounds that cause dose‐dependent pathophysiological injury to live organisms, resulting in decreased viability. Venoms contain a mixture of salts, small molecules, peptides, and proteins. Most toxins are small peptides or proteins, and they often show high potency, target selectivity, and biological stability. Because of their greater size, peptides have a larger interaction surface than small molecules, which provides better interactions with the target of interest. 9
In the early drug discovery era, the “phenotypic based approach” was predominant. The starting point of this strategy was the venoms that have effects on humans, which were subsequently exploited to understand their mechanisms of action. Advantages of this strategy are that no prior knowledge of the mechanism of action is required, and that there is a high probability of in‐vivo effects and therapeutic relevance. On the other hand, without knowing the mechanism of action, it is also more difficult to optimize the molecular properties of a toxin. 9 Successful application of this strategy was seen for Prialt®, a pain killer derived from the sea snail Conus magus. 120 Low‐throughput approaches were mainly used to unravel the targets and mechanisms of action. For electrophysiological recordings, intracellular measurements have become the method of choice for evaluating the effects of venoms and toxins, as these allow control over and measurement of the transmembrane potential. Cells can be either current clamped, which allows measurement of changes in membrane potential, or voltage‐clamped, which allows measurement of changes in current. 121
From the 1990s onward, drug discovery strategies shifted more to the “target‐based approach.” This strategy starts from a specific hypothesis in which a target that is believed to be important for certain pathologies is screened against many compounds/peptides. In this case, molecular and chemical knowledge can be used to verify the hypothesis, but it is not always certain that the resulting drug will also be relevant in the pathology in vivo.9, 120 To optimally exploit the biodiversity of animal venoms for drug discovery, target‐based screening methodologies advanced more and more to HTS, as described above.
After discovering an active venom, there remains the crucial process of identifying the active peptide. Irrespective of whether the activity of a venom is discovered through phenotypic or target‐based approaches, the process from venom to peptide can be either bioassay‐guided or sequence‐based. The bioassay‐guided approach is commonly used, consisting of purifying a venom and its subsequent fractions based on the observed activity on a target, until a pure peptide is obtained. The sequence‐based approach allows a list of peptides and proteins present in a venom to be obtained by sequencing the transcriptome of the venom gland and identifying toxin‐like genes using bioinformatics tools. 122
BmKTX is a toxin derived from the scorpion Buthus martensi, and its activity on KV1.3 was discovered in the latter, more nonconventional, way (i.e., sequence‐based) (Figure 5). By mass‐fingerprinting of C18 HPLC fractions with matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry, compounds within the 3700–4300 Da range were identified, which is a molecular mass range that is typically observed for scorpion toxins belonging to the α‐subfamily that act on K+ channels. Subsequently, these compounds were tested on mice for their toxicological properties, and on oocytes for their activity on KV1.3. In the latter, BmKTX with a free C‐terminus had an IC50 of 0.6 nM, and C‐amidated BmKTX had an IC50 of 0.2 nM on KV1.3. 101 Another example of a KV1.3‐inhibiting peptide discovered by a sequence‐based approach is purlisin‐NT. This toxin‐like defensin was isolated from the fungus Purpureocillium lilacinum and it inhibits KV1.3 with an IC50 of 0.2 µM. 123
Figure 5.
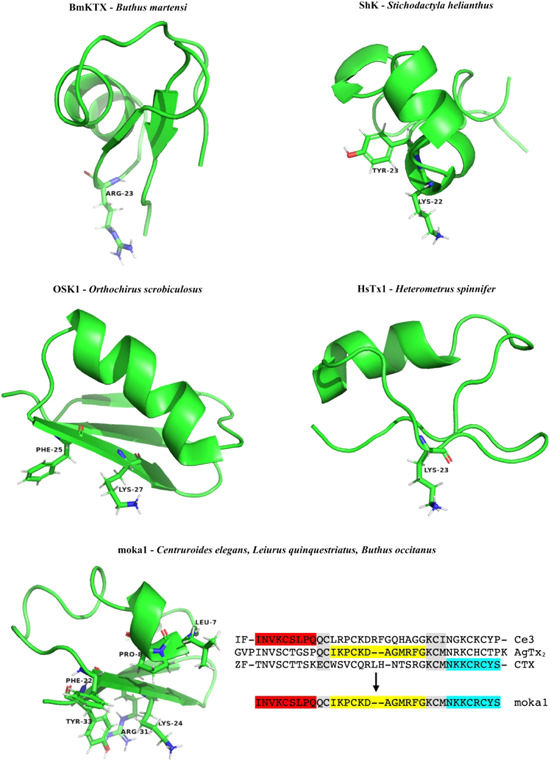
NMR structures of BmKTX 124 (PDB ID code: 1BKT), ShK 110 (PDB ID code: 1ROO), OSK1 125 (PDB ID code: 1SCO), HsTx1 126 (PDB ID code: 1QUZ), and moka1 104 (PDB ID code: 2KIR) with labeling of residues thought to be important for binding to Kv1.3; design of moka1 by scaffold‐/target‐biased strategy [Color figure can be viewed at wileyonlinelibrary.com]
4.2. Venom drug design strategies
Many peptides that act on KV1.3 with excellent potencies have been isolated and described.6, 127 A frequently occurring pitfall, however, is insufficient selectivity for KV1.3 over other KV channels, and especially over KV1.1, because of its high homology to KV1.3 (65%–70%). 73 After isolation of an active peptide, there are different strategies available to improve the active peptide's selectivity toward the target of interest, thereby avoiding potential side effects. Some of these strategies are discussed below, focusing on those that have been applied to KV1.3 toxins. A more comprehensive review was published by Bingham et al., 128 where they provided an excellent overview of the bioengineering strategies for conopeptides.
4.2.1. Acidic‐residue‐function‐guided drug design
Acidic toxin residues have an evolutionary function, as they can shift the binding interface between toxin and channel by changing polarity. Positive charges of basic toxins interact with the negatively charged vestibule of K+ channels, while acidic residues are typically distributed over the nonbinding interface of the toxin, because of the electrical repulsion with the channel vestibule. Therefore, the position of these acidic residues determines the toxin binding interface. 129 BmKTX, for example, contains the two acidic residues D19 and D33. 130 Reorientation of these residues creates two new peptides with distinct interaction surfaces, more specifically known as BmKTX‐19 and BmKTX‐196 (Table 4), with one acid residue (D33) and two acid residues (D6, D33), respectively. Both of these peptides showed less than 50% block against KV1.1, KV1.2, KV7.1, KV11.1, SKCa2, SKCa3, and IKCa at a concentration of 1 µM, while the calculated IC50 values against KV1.3 were 0.4 nM for BmKTX‐19 and 7.3 nM for BmKTX‐196; these are close to the IC50 of wild‐type BmKTX. These peptides only differ from each other and wild‐type BmKTX by a few amino acids, and these all show similar structures. However, they each interact with KV1.3 in different orientations. First, wild‐type BmKTX interacts with KV1.3 through its turn motif between the α‐helix and the antiparallel β‐sheet domain, with R23 as the pore‐blocking residue. For BmKTX‐19, the interaction occurs with the turn motif between the first β‐sheet and the α‐helix domains, in which K8, H9, and K15 are facing the pore, and therefore likely to be important for the interaction. Finally, for BmKTX‐196, the α‐helix domains interact with KV1.3, in which K15, K18, and K19 are oriented toward the pore. 129 This strategy can be used to reorientate the binding interface of the toxins to create potent KV1.3 inhibitors with good selectivity profiles, potentially resulting in more favorable drugs, based on the evolutionary role of these acid toxin residues.
Table 4.
Potencies (as IC50 or K d) of toxins and their analogues for Kv1.3, and their respective selectivity for other ion channels
| Species | Toxin | IC50 or K d for Kv1.3 | IC50/selectivity for other ion channels |
|---|---|---|---|
| Buthus martensi | BmKTX 101 | 0.2 nM | BKCa: no significant inhibitory activity ≤100 nM |

|
BmKTX‐19 129 | 0.4 nM | Kv1.1, Kv1.2, Kv7.1, Kv11.1, SKCa2, SKCa3, and IKCa > 1.0 µM |
| BmKTX‐196 129 | 7.3 nM | ||
| Stichodactyla helianthus | ShK 131 | 11 pM | mKv1.1: 16 pM |

|
rKv1.2: 9000 pM | ||
| mKv1.4: 312 pM | |||
| hKv1.5: >100,000 pM | |||
| hKv1.6: 165 pM | |||
| mKv1.7: 11,500 pM | |||
| mKv3.1: >100,000 pM | |||
| rKv3.4: >100,000 pM | |||
| hKCa4: 28,000 pM | |||
| ShK‐Dap22 131 | 23 pM | mKv1.1: 1800 pM | |
| rKv1.2: 39,000 pM | |||
| mKv1.4: 37,000 pM | |||
| hKv1.5: >100,000 pM | |||
| hKv1.6: 10,500 pM | |||
| mKv1.7: >100,000 pM | |||
| mKv3.1: >100,000 pM | |||
| rKv3.4: >100,000 pM | |||
| hKCa4: >100,000 pM | |||
| Orthochirus scrobiculosus | OSK1 132 | 14 pM | Kv1.1: 0.6 nM |

|
Kv1.2: 5.4 nM | ||
| KCa3.1: 225 nM | |||
| AOSK1 133 | 3.0 pM | Kv1.1: 0.4 nM | |
| Kv1.2: 3.0 nM | |||
| KCa3.1: 228 nM | |||
| [Δ36–38]‐AOSK1 133 | 1.9 nM | Kv1.1: 365 nM | |
| Kv1.2: no effect | |||
| [Δ1–7]‐AOSK 133 | 0.1 nM | Kv1.1: 7.9 nM | |
| Kv1.2: no effect | |||
| Anuroctonus phaiodactylus | AnTx103, 134 | 0.7 nM | Kv1.1: no significant effect at 100 nMKv1.2: 6.1 nM |

| |||
| N17A/F32T‐AnTx 134 | 0.6 nM | Kv1.1: no significant effect at 100 nM | |
| Kv1.2: 9.6 µM | |||
| Centruroides elegans | moka1 104 | 1.0 nM | Kv1.1: 1000‐fold |
| Leiurus quinquestriatus | Kv1.2: 620‐fold | ||
| Buthus occitanus | KCa1.1: no effect | ||
| Heterometrus spinnifer | HsTx1 135 | 29 pM | Kv1.1: 11,330 pM |

| |||
| HsTX1[R14A] 135 | 45 pM | Kv1.1: <20% block at 100 nM | |
| HsTX1[R14Abu] 135 | 50 pM | Kv1.1: >100 nM |
Note: Photographs: Buthus martensi (by Isaac Miller), Stichodactyla helianthus (by Neil DeMaster), Orthochirus scrobiculosus (by Alex Shaffir), Anuroctonus phaiodactylus (by Alice Abela), and Heterometrus spinnifer (by Daniel Maier).
4.2.2. Chemical modification
As well as the naturally occurring amino acids, there are also the nonproteinogenic amino acids (NPAAs). There are approximately 800 NPAAs in nature, while thousands have been synthesized by chemical modification. Due to the great diversity in their physicochemical characteristics, these NPAAs can be used to improve peptide stability, permeability. and potency, thereby contributing to better bioavailability. 136 First of all, stability can be improved by stabilizing the backbone configuration or eliminating the enzyme recognition sites, avoiding proteolysis. For example, glucagon‐like peptide 1 (GLP‐1), a hormone that stimulates insulin secretion, is inactivated by the enzyme dipeptidyl peptidase‐4. Exendin‐4 is a GLP‐1 analog in the saliva of the Gila monster, and its half‐life has been prolonged from 2.4 h 137 to 8 days 138 by fusion of albumin to its K40. Another way to improve stability is to cyclize a peptide using NPAAs, which reduces the protease susceptibility. Also, permeability can be improved by increasing peptide helicity, lipophilicity, intermolecular H‐bond formation and glycosylation, again using NPAAs. Finally, by modulation of protein‐protein interactions, NPAAs can contribute to the potency and selectivity for the target of interest. 136
This can be illustrated by ShK, one of the most potent toxins on KV1.3 (Figure 5). This toxin shows equal potencies in the picomolar range for KV1.3 and KV1.1 (Table 4), and it also shows activity for KV1.2 and KV1.6 at the nanomolar range. 131 By using mutant cycle analysis, a probable docking configuration was established. An analog of ShK, known as ShK‐Dap22, was then created by replacing K22 of the functional dyad with the positively charged NPAA diamino propionic acid. This analog is selective for KV1.3 and retains its potency. To understand this selectivity, the structure was determined by NMR and compared to the structure of ShK via 2D nuclear Overhauser enhancement, which showed clear differences in local structure and dynamics around the substituted residue. Strong energetic contacts between Dap22 and both H404 and D386 were observed, which appear not to be as tight in KV1.1 due to its different architecture. 19 Residues H404 and D386 lie further away from the pore, and ShK‐Dap22 shows only weak interactions with residues in the pore compared to ShK, indicating a reorientation of the toxin binding site by alteration of just one residue. 139
4.2.3. Residue truncation
OSK1 is a 38‐residue toxin derived from the scorpion Orthochirus scrobiculosus, which belongs to the α‐KTx3 family (Figure 5). It is folded according to the common α/β scaffold and contains the conventional three‐disulfide bridges at positions C1–C4, C2–C5, and C3–C6. OSK1 is very positively charged, which is important in its interaction with the negative pore of KV1.3. OSK1 is most potent on KV1.3 (Table 4), with an IC50 of 14 pM, but also interacts with KV1.1 and KV1.2, with IC50 values of 0.6 and 5.4 nM, respectively. 140 It also affects apamin‐insensitive small conductance Ca2+‐activated channels in neuroblastoma‐glioma NG108‐15 hybrid cells, a channel that is believed to be identical to KCa3.1. While KV channels differ from each other mainly for the turret region, the differences with KCa3.1 channels are generally located in the pore helix. The amino acids believed to have key roles in the interaction with OSK1 are located in the turret region and comprise D376, S378, N382, Y400, G401, D402, M403, and H404. 132 A characteristic of OSK1 is its N‐cap and C‐cap in the α‐helix, which are formed by two bifurcated hydrogen bonds. At the N‐end, the carbonyl Q13 forms a hydrogen bond with the amide protons of K9 and I10, while at the C‐end, the carbonyl of C18 forms a hydrogen bond with the amide protons of G22 and M23. Another characteristic of the OSK1 α‐helix is a hydrogen‐bonded salt bridge between the sidechain of R12 and E16. These characteristics do not only stabilize the OSK1 structure and folding, but are also important for toxin specificity. The flexible β‐turn formed by N30 and G31 is important for the toxin activity. 125
Different approaches have been used to improve the selectivity of the OSK1 peptide (Table 4). An amino acid substitution resulted in OSK1‐E16/K20D, also known as AOSK1, which was approximately fivefold more potent than OSK1 and showed higher selectivity toward KV1.1 and KV1.2. 133 Moreover, this peptide was used as a template for domain trimming. Trimming of the C‐terminal region resulted in lower activity on all of the channels tested, with an IC50 of 1.8 nM on KV1.3, probably because this changes the β‐sheet structure. Although less potent, the deletion of these three C‐terminal amino acids resulted in a 200‐fold selectivity for KV1.3 over KV1.1 and a 1000‐fold selectivity for KV1.3 over KV1.2 and KV3.2. Trimming of the N‐terminal region did not cause significant changes in the affinity for KV1.3 and KV1.1; however, it lowered the affinity for KV1.2. This trimming of the N‐terminal region is a more desirable strategy, as it resulted in different affinities for different channel subtypes, rather than lowering the affinity for all of the channel subtypes.11, 133
4.2.4. Reducing conformational flexibility
Anuroctoxin (AnTx) is a scorpion toxin that was isolated from Anuroctonus phaiodactylus. It has high affinity for KV1.3 in human T‐lymphocytes, but no effects on Ca2+‐activated IKCa1 K+ channels (Table 4). AnTx shows little similarity with other scorpion toxins and is blocked at both the N‐ and C‐terminus amino acids by a pyroglutamic acid and an amidated lysine, respectively. It has been suggested that AnTx acts as a classical KV1.3 pore blocker, with a K d of 0.7 nM and approximately sevenfold selectivity for KV1.3 over KV1.2. 103 Sequence alignment shows the functional dyad in AnTx at positions K23 and F32, similar to K27 and Y36 in ChTx. 8 Originally, the functional dyad was considered as crucial for blocking KV channels in general; however, it appears especially important for blocking KV1.2, while it is not essential for KV1.3 blockage. Most high‐affinity KV1.2 blocking toxins contain a tyrosine as the aromatic residue of their functional dyad. 134 Replacing this tyrosine with a more polar residue, such as threonine or asparagine, can theoretically shift the selectivity toward KV1.3. The resulting F32T‐AnTx mutant is indeed selective for KV1.3 over KV1.2, although with lower affinity. The N17, which is positioned between the α‐helix and first β‐strand, appears to be another important residue in AnTx. In KV1.2‐selective toxins, this residue is often a positively charged arginine or a polar glutamine. According to docking simulations, the residues at the corresponding position in Pi4 and CoTx1 form salt bridges with the negatively charged sidechains of the KV1.2 residues, which would facilitate the positioning of the toxin. Replacement of the polar N17 with alanine did not significantly change the affinities toward KV1.2 or KV1.3; however, a combination of both of these mutations resulted in an approximate 16,000‐fold selectivity of N17A/F32T AnTx for KV1.3 over KV1.1, KV1.2, and KCa3.1, while retaining high affinity for KV1.3. 134 The wild‐type and double mutant AnTx have similar α/β folds that are stabilized by cysteines, but structurally the wild‐type is intrinsically less defined than the double mutant. Wild‐type AnTx has greater flexibility, and can adopt different conformations that allow nonselective binding for KV1.2 and KV1.3. On the other hand, N17A/F32T AnTx has a more rigid structure, which results in its selective binding of KV1.3. 11 This strategy allows the transformation of nonselective peptides into selective peptides by stabilizing one of the possible conformations. 134
4.2.5. Scaffold‐/target‐biased strategies
Moka1 is a toxin (Figure 5) that was designed by the scaffold‐/target‐biased strategy. In this strategy, a phage library is screened against a selected target, which results in natural selection and amplification of toxins that show the best interactions with the selected target. To find a specific toxin for the human KV1.3 channel, a combinatorial library was designed with KTX as the lead, as its KV1.3 blocking mechanism is well understood. 104 The library was composed of 31 α‐KTx toxins, as a family of short‐chain K+ channel blocking peptides with three or four disulfide bridges that were derived from scorpions, 141 plus toxin precursor sequences that were highly similar to KTX. To maintain the scaffold architecture and promote the folding of the new toxins, the numbers and positions of the disulfide bridges were kept, and the conserved QC and KCM regions were retained or introduced. This resulted in a library of 11,200 unique toxins and 20 reformed toxins, which were subsequently added to the KcsA‐KV1.3 target (which contained the KV1.3 pore domain). This finally led to the identification of moka1. This toxin contains domains of three different scorpion species, as the A, B, and C domains. Domain A originated from toxin Ce3 of the Central American scorpion Centruroides elegans. Domain B was a combination of the domains of agitoxin‐2 and agitoxin‐3 of the Middle Eastern scorpion Leiurus quinquestriatus and KTX3 of the north African scorpion Buthus occitanus. Domain C originated from ChTx and Lq2 of the Middle Eastern scorpion L. quinquestriatus. 104
In oocytes, moka1 (Table 4) resulted in 50% block of KV1.3 currents at 1 nM, with a rapid on‐rate and a slow off‐rate. Moka1 has different pharmacology compared to its parental toxins and KTX, as it has a 1000‐fold selectivity for KV1.3 over KV1.1, 620‐fold selectivity for KV1.3 over KV1.2, and no measurable effect on KCa1.1. This better selectivity can be explained by the combined presence of five residues that originated from Ce3, AgTx2, and CTX that are important for binding, a combination that does not occur naturally. The shorter N‐terminus, which has a different position, might contribute to this selectivity as it has been shown to influence toxin binding for α‐KTx toxins. This strategy allows the isolation of new toxins based on interactions with their targets, to which they are physically linked by the encoding gene. 104
4.2.6. Computational methods
Another strategy to improve receptor subtype selectivity is through the use of computational methods. Docking methods and molecular dynamics simulations can provide models for interactions between ligands and receptors, which can thus provide insight into the importance of certain residues. On the other hand, free‐energy calculations provide the binding free‐energy of toxins and their analogs by the potential of mean force and can anticipate the effects of mutations by free‐energy perturbations. 142
This strategy was used to understand the KV1.3 selectivity of HsTx1, a toxin consisting of 34 residues folded along the classical α/β scaffold that was isolated from the scorpion Heterometrus spinnifer (Figure 5). HsTx1 belongs to a unique family of scorpion toxins that comprise four disulfide bridges. 105 While most scorpion toxins that act on K+ channels have a free C‐terminus, the K+ channel toxins with four disulfide bridges described to date are C‐terminally amidated. Moreover, the free form of HsTx1 is less potent than the amidated form, which suggests that the presence of a free carboxyl group destabilizes toxin binding by electrostatic repulsion. More specifically, NMR of Pi, another peptide from the α‐KTx6 family with a similarly orientated C‐terminus, showed that the extra disulfide bridge orients the C‐terminus of Pi toward the interface of the toxin that is important for binding to the channel. Contrary to Pi, removal of this extra disulfide bridge abolishes the block for KV1.1 and KV1.3. 143 HsTx1 has an almost 1000‐fold selectivity for KV1.3 over KV1.1 (Table 4). In general, the KV1‐toxin complex is believed to form a hydrogen bond between the pore‐inserting lysine of the toxin and a tyrosine in the selectivity filter. Comparisons of the binding modes of HsTx1 with KV1.1 and KV1.3 using docking and molecular dynamics simulations showed that in the case of the KV1.1‐HsTx1 complex, K23 does not completely fit into the filter to make the hydrogen bond with the tyrosine carbonyl. 135 Three factors related to steric hindrance and charge interactions can explain this preferred binding to KV1.3 over KV1.1. First of all, the interaction surface of HsTx1 consists of a long β‐sheet, which limits interactions with the channel. This is however the case for both KV1.1 and KV1.3. Second, KV1.1 does not form crosslinks between Y379 and D377, which results in protrusion of the side chains out of the pore. In KV1.3, the sidechains of H404 form crosslinks with D402, which avoids the steric hindrance seen for KV1.1. Finally, strong associations between three arginine sidechains with aspartate side chains in the turret region of the channel impedes the toxin from coming closer to the pore. KV1.3 does not have these three arginine sidechains coupling to the turret region, which results in better insertion of the K23 sidechain. 69 Another observation when comparing the binding modes is that R14 in HsTx1 is strongly coupled to E353 in KV1.1, while no interaction with KV1.3 has been observed. Mutation of this R14 residue can therefore increase the KV1.3 selectivity. Potential of mean force calculations have shown that mutation of R14 causes an increase in KV1.3 selectivity by more than 2 kcal/mol. This has been established by functional assays, in which HsTX1[R14A] and HsTX1[R14Abu] showed KV1.3 affinities in the low picomolar range, with >2000‐fold selectivity over KV1.1. 135
5. DRUG DISCOVERY APPROACHES FOR DESIGNING NEW KV1.3 SMALL‐MOLECULE INHIBITORS
Several different approaches have been used for the discovery and design of new small‐molecule KV1.3 inhibitors (Table 5). Extensive HTS campaigns of chemical libraries have produced some hit compounds, but none of these molecules have been optimized to provide appropriate candidates for clinical studies. KV1.3 inhibitors were also discovered through the exploitation of biological information and designed by structure‐based and ligand‐based drug design methodologies. After gaining initial hits from the approaches mentioned above, various analogs were synthesized in a hit‐to‐lead optimization process to improve the pharmacodynamic and pharmacokinetic properties, and mainly the selectivities and potencies, and to explore the structure–activity relationships.
Table 5.
Strategies for the discovery of small‐molecule KV1.3 inhibitors
| Strategy | Assay | Compound |
|---|---|---|
| High‐throughput screening | Ligand binding assays | 1 |
| Ion flux 86Rb assays | 2–4 | |
| Automated electrophysiological assays | 5–8 | |
| Exploitation of biological information | Folk medicine; ethnopharmacology | 9, 9a–9f |
| Drug repurposing | 10 (clofazimine) | |
| Clinical observations of side effects | 11 (diphenoxylate) | |
| Structure‐based drug design methodology | Computer‐aided drug design—virtual screening | 12, 13 |
| Ligand‐based drug design methodology | Computer‐aided ligand‐based three‐dimensional similarity searches | 14, 15 |
| Analogs of natural plant compounds | 16a–16f | |
| Peptidomimetics—synthetic ligands of peptide toxins | 17–20 |
5.1. HTS campaigns
Some specific HTS approaches have been carried out for ion channels with ligand‐binding, flux‐based, and fluorescence‐based assays. The latest HTS strategy extensively used for screening of KV1.3 inhibitors is the automated electrophysiological patch‐clamp assay. 144 A successful HTS ligand binding campaign provided the first nanomolar small‐molecule KV1.3 inhibitor, which was discovered in 1995: dihydroquinoline (Figure 6). This arose through HTS with 125I‐ChTx displacement, and it inhibited the 125I‐ChTx binding to KV1.3 with an IC50 of 83 nM, and suppressed human T‐cell activation. 145
Figure 6.
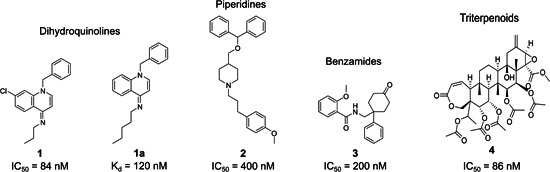
Representative and most studied KV1.3 inhibitors from high‐throughput screening campaigns
Its analog 1a (Figure 6), which inhibited 125I‐ChTx binding with K d of 120 nM, was extensively studied to determine the mechanism of channel block by dihydroquinoline. Here, 1a selectively blocked the C‐type inactivated state of the channel, as indicated by three lines of evidence: studies with a panel of external vestibular KV1.3 mutants, and the facts that changing the holding potential from −80 to −50 mV intensified the sensitivity of the channel to 1a and that exposure to high external K+ reduced its potency. 146 Interestingly, the potency differences of 1a differ between 125I‐ChTx displacement (K d, 120 nM) and patch‐clamp (IC50, 150 nM) assays on the one side, and inhibition of 86Rb‐efflux (IC50, 2.3 µM) on the other. The explanation for this might be that dihydroquinoline blocks KV1.3 through interaction with residues that are exposed during the conformational changes of the C‐type inactivation, which occurs through a cooperative mechanism of four subunits in the tetramer, followed by dynamic rearrangements of the outer mouth. 86Rb‐efflux assays, which require higher levels of K+, might underestimate the potency of KV1.3 inhibitors that inhibit the C‐type inactivated state.82, 84, 147, 148 Compound 1a was 60‐ to 270‐fold more selective against KV1.x family channels but lacked selectivity over the KV1.4 neuronal and cardiac channel (Table 6). 146 Additionally, dihydroquinoline potently inhibited the neuronal Na+ channel, with K i of 9 nM. 149 Thus, the main limitation of dihydroquinolines is the lack of specificity for KV1.3.145, 146, 149
Table 6.
Potencies and selectivities of representative and most‐studied KV1.3 inhibitors from high‐throughput screening approaches
| Potency (nM) | |||||
|---|---|---|---|---|---|
| Structural class | Compound | Test type | IC50 | K d | Selectivity |
| Dihydroquinolines | 1 145, 146, 149 | 125I‐charybdotoxin binding to Jurkat T | 83 | ‐ | Potently inhibited voltage‐gated neuronal Na+ channels in CHO cells (K i = 9 nM) |
| 1a 145, 146, 149 | lymphocytes (n‐type K+ channels) | ‐ | 120 | No selectivity against KV1.4 (IC50 = 300 nM); 60‐ to 270‐fold selectivity against other Kv1 family channels | |
| Piperidines | 2 150, 151 | 86Rb+ efflux in human T cells | 400 | ‐ | Potently inhibited KV1.4 (IC50 = 170 nM); 10‐fold selectivity over KV1.2; 40‐fold selectivity over KV3.1 |
| Benzamides | 3 152, 153, 154 | 86Rb+ efflux in CHO cells, expressing KV1.3 | 200 | ‐ | Inhibited KV1.x family channels with similar potencies (no selectivity) |
| 3f 152, 153, 154 | channels | 50 | ‐ | Sixfold selectivity over KV1.x (heteromultimeric KV1.1/KV1.2 channels) | |
| 3g 152, 153, 154 | 100 | ‐ | No selectivity over KV1.x (heteromultimeric KV1.1/KV1.2 channels) | ||
| Triterpenoids | Correolide 4 155, 156, 157, 158 | 86Rb+ efflux in CHO cells expressing KV1.3 channel | 86 | ‐ | Inhibited KV1x family channels with 4‐ to 14‐fold lower potency |
| Correolide analog 4a 155, 156, 157, 158 | Plasma membranes from CHO/Kv1.3 cells | ‐ | 11 | ||
Ion flux 86Rb assays have been wide and successfully used in the pharmaceutical industry, which prompted the discovery of piperidines150, 151 (Figures 6, 2), benzamides152, 153, 154 (Figures 6, 3), and the natural compound correolide (Figures 6, 4).155, 156, 157, 158 Piperidines were discovered in a library screen using the HTS 86Rb‐efflux assay. Competition experiments revealed that piperidine‐based 2 bound to residues at the inner surface of the channel, with an overlap of the site of action of verapamil. Like dihydroquinolines 1 and 1a, compound 2 inhibited KV1.3 with use dependence by binding to the residues involved in the C‐type inactivation. Mutation of specific residues in the external vestibule that influenced the C‐type inactivation rate altered the channel sensitivity to compound 2, and there was a direct correlation between the inactivation time constants and the IC50 values. 151 Piperidine 2 shows an IC50 of 400 nM and was selected for further optimization due to its at least 10‐fold selectivity over most of the KV1.x family channels, except KV1.4 (IC50, 170 nM) (Table 6). Crucial structural elements for activity were the basic piperidine nitrogen atom and the benzhydryl lipophilic head group with two phenyl rings and a lipophilic tail attached to a basic nitrogen (Figure 7). Removal of the benzhydryl moiety in analog 2a resulted in reduced potency, while compounds 2b–2d that incorporated lipophilic moieties were more potent (Figure 7). The substitution of the central piperidine with pyridine or piperazine was not tolerated, while replacement with a tropane moiety retained activity (Figures 7, 2f). Compound 2e incorporated an aliphatic chain in the tail part of the molecule and had an IC50 of 500 nM, while the potency was increased with the incorporation of a methyl‐substituted phenyl ring (2g; IC50, 200 nM). The development of piperidine‐based compounds was discontinued due to low potencies and selectivities.150, 151
Figure 7.
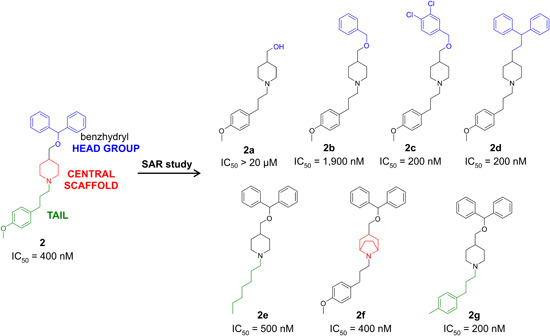
Structure–activity relationships in the exploration of piperidine‐based KV1.3 inhibitors [Color figure can be viewed at wileyonlinelibrary.com]
The next successful example of using HTS was for a series of benzamide compounds that were identified using the 86Rb‐flux assay. The parent benzamide‐based compound 3 (Figure 8) inhibited KV1.3 with an IC50 of 200 nM and had no selectivity for the KV1.x family (Table 6). The structure–activity relationships revealed that modification at position 2 of the 2‐methoxy phenyl ring was tolerated while moving the methoxy group to positions 3 or 4 (Figures 8, 3a, 3b) resulted in reduced potency. The activity was also lost with methylation of the amide nitrogen (Figures 8, 3c), substitution of the amide carbonyl by a sulphonyl group (Figures 8, 3d), and removal of the unsubstituted phenyl ring (Figures 8, 3e). On the other hand, the potency was retained with the ketone reduction to a hydroxy group, separation of the trans and cis diastereoisomers, and further synthesis of the C1 carbamate analogs (Figures 8, 3f, 3g). 154 The most potent and selective benzamide derivative was 3g (sixfold selectivity over KV1.x), as a trans analog with a propyl carbamate group. In general, in comparison to their cis analogs, the trans C1 carbamate analogs exhibited selectivity for KV1.3 over the other KV1x family channels (Table 6).152, 154
Figure 8.
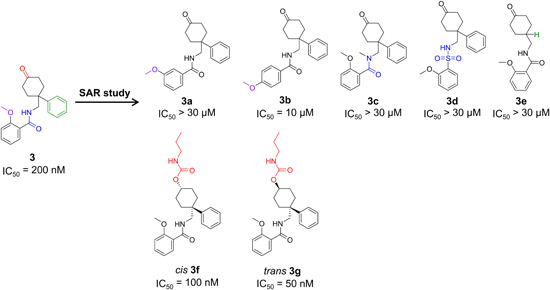
Structure–activity relationships exploration of benzamide‐based KV1.3 inhibitors [Color figure can be viewed at wileyonlinelibrary.com]
Reversible, saturable and time‐dependent binding of benzamide 3g to KV1.3 occurred when two molecules of inhibitor interacted with one channel tetramer between the conformational changes of the C‐type inactivation. The precise binding site of benzamide 3g has not been mapped yet, but it might be located in the water cavity, below the selectivity filter (Figure 3). Interestingly, as this region is structurally identical across the KV1.x family, the selectivity of benzamide 3g for KV1.3 might be explained through its interaction with the C‐type inactivation state. As KV1.3 and KV1.4 undergo profound conformational changes between gating in the outer vestibule, which are absent in other KV1.x family channel, the specificity for KV1.3 might be achieved with the development of inhibitors that bind to the residues, that are significantly engaged in the C‐type inactivation process.152, 153
A natural hit compound that was acquired through the 86Rb ion flux HTS assays was the triterpenoid correolide (Figures 6, 4) from the Costa Rican tree Spachea correae (IC50, 86 nM). However, correolide also inhibited other KV1x family channels with similar potencies (Table 6). 156 The correolide (tritium‐labeled) analog [3H]C20‐29 dihydrocorreolide (diTC; Figures 9, 4a) inhibited KV1.3 with a K d of 11 nM in a specific, saturable, and reversible way, with one binding site per KV1.3 tetramer. Two independent approaches have suggested that inhibitor 4a was bound in the pore, on the cytoplasmic side of the selectivity filter (Figure 9). The molecular model for the docking experiment with diTC was created on the crystal structure of KcsA with the S5‐S6 region of KV1.3. Site‐directed mutagenesis identified unique S5 and S6 residues, that contributed to the 4a high‐affinity interaction with KV1.x family channels, some of the which were involved in conformational changes between gating.157, 158 Higher affinity for KV1.3 over other KV1.x family channels might be connected with the C‐type inactivation state, which is characteristic for KV1.3, and which contributes to the unique shape of the diTC binding site. The important structural elements for this interaction are five polar acetyl groups in the 4a molecule. Correolide‐based inhibitors most likely have state‐dependent interactions with the inactivated form of KV1.3, and therefore have limited toxicity and efficacy in vivo. 158 When pentacyclic correolide was simplified with the removal of its E‐ring to a tetracyclic C18‐analog (Figures 9, 4b), its potency was retained in the nanomolar range (IC50, 37 nM). 159 Future optimization of correolide is precluded due to a limited supply of the natural parent compound, the lack of selectivity for KV1.3 over other KV1.x family channels, and the complex molecular structures of new simplified analogs.155, 156, 157, 158, 159
Figure 9.
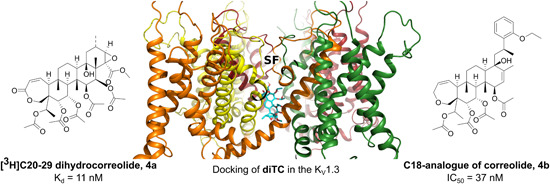
diTC 4a (left), docking of diTC with KV1.3 158 (middle), and simplified correolide analog 4b 159 (right). diTC, [3H]C20‐29 dihydrocorreolide [Color figure can be viewed at wileyonlinelibrary.com]
Correolide was successfully used to demonstrate the involvement of KV1.3 in coronary metabolic dilation. Myocardial blood flow was measured in groups of KV1.3‐null mice and wild‐type mice that has been given correolide. The myocardial blood flow was efficiently reduced during increased cardiac work in both groups when given norepinephrine. 160 Limb‐transplanted rats with intraperitoneal administration of correolide showed significantly higher survival compared to the placebo control and untreated groups. 161
In an automated pipette‐based patch‐clamp screening, some thienopyridines 162 (representative compound 5; Figure 10) and sulfonamides 163 (representative compound 6; Figure 10) were identified that inhibited the KV1.3 currents at 1 µM, while they also very efficiently inhibited KV1.5 currents at 1 µM. Using planar electrode‐based patch clamp screening, some 1,1‐dioxidobenzo[d]isothiazoles164, 165 (representative compound 7; Figure 10) and amine‐based compounds 166 (representative compound 8; Figure 10) were discovered, which were later optimized to provide KV1.3 inhibitors with low nanomolar potencies.
Figure 10.
KV1.3 inhibitors identified using automated electrophysiological patch‐clamp assays
Using an IonWorks patch‐clamp HTS assay, 1,1‐dioxidobenzo[d]isothiazole was discovered (hit compound 7; Figure 11), which moderately inhibited KV1.3 currents (Table 7; IC50, 1000 nM). The benzothiophene (Figures 11, 7a) and benzothiazole (Figures 11, 7b) scaffolds eliminated this activity, and the simplified analog with a cyano group (Figures 11, 7c) resulted in decreased activity (IC50, 4800 nM). An analog with an amide bond (Figures 11, 7f) had approximately twofold higher potency compared to hit compound 7. 164 First generation 1,1‐dioxidobenzo[d]isothiazoles had high clearance rates in vivo, and therefore with the second generation compounds, the intention was to improve in vitro half‐life (t 1/2) values. Insertion of a fluorine atom at position 6 of the 1,1‐dioxidobenzo[d]isothiazole scaffolds (Figures 11, 7g, 7h) yielded compounds with improved in vitro t 1/2; moreover, amides 7g and 7h had the appropriate pharmacokinetic and solubility properties but also showed very efficient inhibition of KV1.5 currents. 165 The limitation of all of these compounds was the lack of detailed selectivity tests and the unknown binding site(s). The most potent amide 7i (Figure 11) had a dimethyl substitution on the propyl linker and showed an IC50 of 160 nM.164, 165
Figure 11.
Structure–activity relationships exploration of 1,1‐dioxidobenzo[d]isothiazole‐based analogs [Color figure can be viewed at wileyonlinelibrary.com]
Table 7.
Potency and selectivity of KV1.3 inhibitors identified in automated electrophysiological patch‐clamp assays
| Structural class | Compound | Cell type | Type of automated electrophysiological assay | IC50 (nM) or % inhibition | Selectivity over KV1.5 |
|---|---|---|---|---|---|
| Thienopyridines | 5 162 | CHO cells expressing KV1.3 | Automated pipette‐based patch‐clamp (autopatch electrophysiology method) | 1 µM: 99% inhibition | 1 µM: 93% inhibition |
| Sulfonamides | 6 163 | CHO cells expressing KV1.3 | Automated pipette‐based patch‐clamp (autopatch electrophysiology method) | 1 µM: 100% inhibition | 1 µM: 91% inhibition |
| 1,1‐[d]‐Dioxidobenzo isothiazoles | 7 164, 165 | CGE22 cells expressing human KV1.3 |
Micro‐fabricated planar electrode‐based patch clamp (ionWorks platform) |
1000 | Not studied |
| 7g 164, 165 | 350 | IC50 = 540 nM | |||
| 7h 164, 165 | 250 | IC50 = 480 nM | |||
| Amine derivates | 8 166 | CHO cells expressing KV1.3 | Planar electrode‐based patch clamp (Nanion Technologies) whole‐cell patch‐clamp | <50 | Not studied |
5.2. Exploitation of biological information (retroactive exploitation of observations made in man)
As the probability of acquiring a potential hit compound from HTS is low, the financial investment is high, and the strategy is time‐consuming, sometimes knowledge‐based approaches have a better likelihood of success.167, 168 Hit compounds from the exploitation of biological information have been obtained from research into folk medicines (e.g., 5‐methoxypsoralen; Figures 12, 9),169, 170, 171, 172 from “new use for old” drugs (e.g., clofazimine; Figures 12, 10),173, 174 and from clinical observations of already registered drugs (e.g., diphenoxylate; Figures 12, 11). 175
Figure 12.
Representative hit compounds from exploitation of biological information approaches
Psoralens (Figure 13) are a representative example of folk medicines as a starting point for successful drug discovery. Initial studies on psoralen activity on ion channels were based on an anecdotal report from South America that long‐term use of tea made from the plant Ruta graveolens alleviated symptoms of multiple sclerosis. Extracts from this medicinal plant were then shown to block delayed‐rectifier K+ currents in nodes of Ranvier. Through extensive screening to find the pharmacologically active compound from the plant, 5‐methoxypsoralen (Figures 13, 9) was shown to be an inhibitor of KV1.2 channels in neurons and KV1.3 channels in T cells.169, 170 Structure–activity relationships revealed that the psoralen scaffold could be substituted only at the 5‐position and the neighboring 4‐position. In addition, alkyl linkers with one to five methylene groups were introduced into the 5‐hydroxypsoralen ring at position 5 (Figures 13, 9a), connecting psoralene with the distal phenyl group. The optimal length for the alkyl linker was four methylene groups (Psora‐4; Figures 13, 9b; IC50, 3 nM), and further increasing the length of the alkyl sidechain reduced potency.
Figure 13.
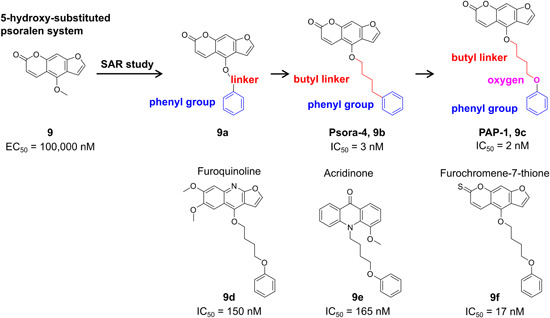
Structure–activity relationships exploration of psoralen derivatives [Color figure can be viewed at wileyonlinelibrary.com]
As Psora‐4 selectively inhibits proliferation in human and rat TEM cells, it might represent a therapy for T‐cell‐mediated autoimmune diseases. 170 Its efficacy was demonstrated in rats with antiglomerular basement membrane disease glomerulonephritis, where the group treated with the inhibitor had less proteinuria compared to the placebo group. Psora‐4 might also be effective in the treatment of glumerulonephritis. 176
With the second‐generation compound PAP‐1 (Figures 13, 9c) 171 with an IC50 of 2 nM, 23‐fold selectivity against KV1.5 was achieved by prolongation of the butyl linker with one oxygen atom, next to the phenyl group. PAP‐1 inhibited KV1.3 with use dependence by binding to the residues exposed in the C‐type inactivated state conformation of the KV1.3 channel.170, 171 PAP‐1 is the most potent and selective small‐molecule KV1.3 inhibitor reported to date, and it shows 33‐ to 125‐fold selectivity over the other KV1.x family channels, and more than 1000‐fold selectivity for the distantly related K+ channels, like KCa3.1, KV2.1, KV3.1, and KV11.1 (hERG) (Table 8). 171
Table 8.
Potencies and selectivities of representative hit compounds from exploitation of biological information approaches
| Potency (nM) | |||||
|---|---|---|---|---|---|
| Structural class | Compound | Cell type | IC50 | EC50 | Selectivity tests |
| 5‐Methoxysoralen | 9 170, 171 | L929 cells stably expressing KV1.3; manual whole‐cell patch‐clamp | ‐ | 100,000 | No selectivity |
| Psora‐4 | 9b 170, 171 | ‐ | 3 | Lacking selectivity over KV1.5 (EC50 = 8 nM), 17‐ to 70‐fold selectivity over Kv1x family channels | |
| PAP‐1 | 9c 170, 171 | ‐ | 2 | 23‐fold selectivity over KV1.5 | |
| Furoquinoline | 9d 172 | L929 cells expressing KV1.3; whole‐cell patch‐clamp | 150 | ‐ | Threefold selectivity over KV1.5 |
| Acridinone | 9e 172 | 165 | ‐ | Threefold selectivity over KV1.5 | |
| Furochromene‐7‐thione | 9f 172 | 17 | ‐ | 22‐fold selectivity over KV1.5 | |
| Clofazimine | 10 173, 174 | Jurkat T cells | 300 | ‐ | Moderate selectivity over KV1.1, KV1.2, KV1.5, and KV3.1 (10 µM: ≤ 50% inhibition) |
| Diphenoxylate | 11 175 | L929 cells expressing KV1.3; manual whole‐cell patch‐clamp | 5000 | ‐ | Not tested |
| Diphenoxylate analog | 11a 175 | 750 | ‐ | ||
The PAP‐1 high‐affinity interaction with KV1.3 was mapped and explained by the model of Zimin et al., which revealed the importance of the coumarin moiety. Carbonyl groups in PAP‐1 chelate potassium ions, and their charge attracts water molecules, which form further hydrogen bonds with nearby coumarin molecules, or more specifically, with ether oxygens. Two or four coumarin rings can interact with potassium ions inside the pore of KV1.3, while the side chains of PAP‐1 protrude out into the intersubunit interfaces between helices S5 and S6. 177 The Marzian et al. model showed why Psora‐4 inhibits KV1.3 with use dependence and reasonable selectivity over KV1.x family channels. As the two molecules simultaneously bind at two different receptor sites in KV1.3, first in the central pore, and second, in the side pockets, this results in an extremely stable nonconducting state of KV1.3 and provides a high‐affinity interaction with KV1.3.178, 179
Application of PAP‐1 offers many therapeutic possibilities, especially in the treatment of T‐cell‐mediated autoimmune diseases. Its efficacy was demonstrated in female Lewis rats, where it dose‐dependently suppressed the delayed type hypersensitivity reaction. 171 PAP‐1 successfully lowered diabetes type I incidence in the experimental group of diseased rats, and efficiently reduced β‐cell destruction. 20 Furthermore, it dose‐dependently reduced interleukin 2 (IL‐2) and interferon‐γ production in in vitro studies, as well as improving the pathology in the severe combined immunodeficient (SCID) mouse psoriasis skin xenograft model. 180 PAP‐1 improved previously manifested Alzheimer's disease symptoms in trangenic mice with cognitive deficiency and amyloid plaques, and, in in vitro studies, PAP‐1 application promoted higher amyloid‐β uptake by microglia. 181 Studies with diseased mice and rats have documented that PAP‐1 can successfully alleviate symptoms of inflammation after ischemic stroke. 182
Later attempts replaced the psoralen scaffold, which formed reactive metabolites, with furoquinoline (Figures 13, 9d) or acridinone (Figures 13, 9e), although new analogs had only threefold selectivity over KV1.5. More successful heterocycle substitution was obtained for furochromene‐7‐thione (Figures 13, 9f), which preserved 22‐fold selectivity for KV1.3 over KV1.5. Compounds 9d and 9f showed use‐dependent inhibition, meaning that they blocked the C‐type inactivated state of KV1.3, similar to 9b and 9c. On the other hand, 9e was an open‐channel blocker, similar to verapamil. 172
The antimycobacterial drug clofazimine (Figures 12, 10) is another promising small‐molecule inhibitor with good potency and a favorable selectivity profile for KV1.3. 183 Clofazimine emerged as a hit in IL‐2 reporter gene bioassay screening of a library of FDA‐approved clinical therapeutics, where it efficiently suppressed Ca2+ signaling and IL‐2 production in human T cells. Patch‐clamp experiments demonstrated that clofazimine specifically inhibited KV1.3 currents in T‐cells, with moderate selectivity over related KV channels and a Hill coefficient of 0.75. 173 Further studies discovered two separate use‐dependent and state‐dependent mechanisms of KV1.3 block, with both significantly depending on KV1.3 translation to the open state. 174 The interesting finding of this investigation was that clofamizine inhibited the opened channel, while it did not interact with the closed or inactivated states. When the open channel block had already occurred, changes in pulse protocols did not result in further inhibition. This unique mode of action might be advantageous over other KV1.3 inhibitors that specifically interact with the inactivated state, and that for efficacious inhibition require high levels of KV1.3 entering the C‐type inactivation states in vivo. Additionally, clofazimine is already approved for medical use, and is generally considered safe for clinical use.173, 174
Psora‐4 (9b), PAP‐1 (9c), and clofazimine (10) inhibited mitochondrial KV1.3 currents and induced apoptosis of different human and mouse cancer cell lines.35, 47, 184, 185 In KV1.3 expressing mouse CTLL‐2 cells, these compounds invoked classical apoptosis pathways, with mitochondrial depolarization, elevated ROS production, cytochrome c release, and caspase‐3, caspase‐9, and poly(ADP‐ribose) polymerase cleavage. The cells that underwent apoptosis were only the cancer cells (KV1.3 expressing human SAOS2, mouse B16F10 melanoma, and human B‐CLL cells), and not normal cells (HEK293 and K562 cells, and T and B cells from healthy subjects).47, 184 Clofazimine was also tested in vivo on C57BL/6 mice, with injected cultured B16F10 cells, which grow as tumors. This treatment with clofazimine reduced the tumor size by 90%, and the B16F10 cells isolated from tumors had comparable KV1.3 expression to the cultured cells. 184 Clofazimine induced apoptosis in pancreatic ductual adenocarcinoma (PDAC) cell lines via its specific inhibitory action on the potassium channel KV1.3. It significantly and strongly reduced the primary tumor weight in an orthotopic PDAC xenotransplantation model in spontaneous mutant beige mouse model (SCID beige mouse). 185
The starting points in medicinal chemistry are sometimes clinical observations (e.g., side effects) of already registered drugs, which can promote biological testing against other targets. Diphenoxylate (Figures 12, 11) is a well‐known opioid agonist and antidiarrhea agent, and it was found to inhibit KV1.3 K+ channels with an IC50 of 5 µM. Diphenoxylate had been shown to successfully treat psoriasis and other inflammatory skin conditions in a small clinical trial, and later it was recognized that it also inhibited KV1.3 currents. Several simplified analogs of diphenoxylate were prepared, and compound 11a (Figure 12) was even more potent for inhibition of the KV1.3 channel (Table 8, IC50, 750 nM). 175
5.3. Structure‐based drug design methods
Structure‐based design has become a state‐of‐the‐art method in hit identification, hit‐to‐lead expansion, and lead optimization. However, in the development of drugs that target voltage‐gated K+ channels, this approach has been very limited due to the lack of crystal structures of representatives of the KV family isoforms in different conformational states (i.e., open and closed). Moreover, there is no crystal structure of a ligand–KV channel complex available. Therefore, the binding sites and the specific interaction patterns of KV1.3 inhibitors in these binding sites remain unknown. The situation is further complicated because some of KV1.3 inhibitors bind to the channel with 2:1 stoichiometry, as reflected by their Hill coefficients, which means the simultaneous binding to two, not necessarily identical, binding sites. Taken together, structure‐based design approaches have currently shown limited success for voltage‐gated K+ channels; however, based on increasing structural data, including cryo‐electron microscopy and crystal structures of ion channels, this will hopefully change soon.
A starting point for the discovery of new KV1.3 inhibitors can be based on the three‐dimensional structure of the target. 186 As an X‐ray structure of the KV1.3 channel is not available, a homology model was constructed on the basis of the crystal structure of the bacterial KcsA channel. This bacterial channel shares around 45% homology in the inner vestibule with KV1.3 and IK‐1. Virtual screening of a large compound library against the potential binding site in the internal cavity below the K+‐selectivity filter defined dihydrophenanthridines (Figures 14, 12) and bridged biaryls (Figures 14, 13) as potential hit compounds. Analog 12a (Figure 14) was designed with structural variations at N5 and C6 of the 5,6‐dihydrophenanthridine scaffold, which resulted in a higher potency (IC50, 710 nM). In the bridged biaryl hit, the aromatic C6 substituent was eliminated, and a carboxamide derivative 13a was synthesized (Figure 14), which was substituted at position N5, which provided 7.5‐fold higher potency (IC50, 4000 nM). This dihydrophenanthridine 12a weakly inhibited IK‐1 currents at 20 µM, while the bridged biaryl 13a showed only fourfold selectivity over IK‐1 (Table 9). Both structural classes require further studies for confirmation of the binding sites and their selectivities. 186
Figure 14.
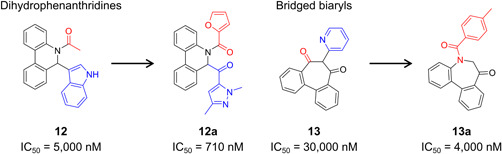
KV1.3 inhibitors from structure‐based drug design methods and structural optimization of hit compounds 12 and 13 [Color figure can be viewed at wileyonlinelibrary.com]
Table 9.
Activity and selectivity of KV1.3 inhibitors gained from structure‐based drug design methods
| Structural class | Compound | IC50 (nM) | Selectivity over IK‐1 channel |
|---|---|---|---|
| Dihydrophenanthridines | 12 186 | 5000 | Not studied |
| 12a 186 | 710 | 20 µM: 12% inhibition of IK‐1 | |
| Bridged biaryls | 13 186 | 30,000 | Not studied |
| 13a 186 | 4000 | Fourfold |
Note: Cell systems used according to Grissmer et al. 187
5.4. Ligand‐based drug design methods and peptidomimetics
The absence of a KV1.3 crystal structure and the lack of information on the exact binding sites of the known KV1.3 inhibitors are the main limitations for the structure‐based discovery of novel KV1.3 inhibitors. In contrast, ligand‐based computational approaches can be successfully used to identify new KV1.3 inhibitors, as they do not require knowledge of the ligand‐binding site. However, the structure of a potent KV1.3 inhibitor, ideally as a selective ligand against a related isoform, is needed as a template for ligand‐based similarity searching as a method of virtual screening of compound libraries.
A ligand‐based virtual screening methodology using KV1.3 inhibitor 3 (Figure 8) as a query was successfully used for the identification of a novel structural class of KV1.3 inhibitors. After the three‐dimensional similarity search of commercially available compounds was performed, hit compounds were categorized according to their shape (molecular geometry) and color (presence of relevant pharmacophores) similarity scores to give the so‐called TanimotoCombo score. According to this score, five highly ranked compounds were purchased and tested for KV1.3 inhibition. Hit compound 14 showed an IC50 of 920 nM (Figure 15) as the most potent of these KV1.3 inhibitors, but also significantly inhibited KV1.1, KV1.4, KV2.1, and KV3.1 currents (Table 10). On the other hand, hit compound 15 with an IC50 of 17.4 µM was a less potent inhibitor of KV1.3, but had no effect on the other KV1.x family channels and more distantly related channels. 188
Figure 15.
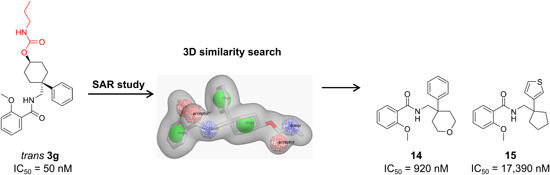
Three‐dimensional similarity search for the discovery of new KV1.3 inhibitors. LBDD, ligand‐based drug design [Color figure can be viewed at wileyonlinelibrary.com]
Table 10.
Potencies and selectivities of KV1.3 inhibitors discovered by ligand‐based drug design methodologies, along with peptidomimetics
| Potency (nM) | |||||||
|---|---|---|---|---|---|---|---|
| Structural class | Compound | Cell type | IC50 | K i | K d | EC50 | Selectivity |
| Benzamides | 14 188 | Applied ex vivo to KV1.3 expressing oocytes | 920 | ‐ | ‐ | ‐ | 10 µM: 76% inhibition of KV1.1; 51% inhibition of KV1.4; 79% inhibition of KV2.1; 46% inhibition of KV3.1 |
| 15 188 | 17,390 | ‐ | ‐ | ‐ | 10 µM: no significant effects on other channels | ||
| Khellinone | 16 189, 190 | L929 cells expressing KV1.3; whole‐cell patch‐clamp | 45,000 | ‐ | ‐ | ‐ | |
| 16a 189, 190 | 400 | ‐ | ‐ | ‐ | Threefold less active on KV1.1; 20‐fold over KV1.5, KV1.7; no effect on KV1.2, KV1.4, hERG, IKCa1 | ||
| 16b 189, 190 | 280 | ‐ | ‐ | ‐ | 10‐fold over KV1.1, KV1.2 | ||
| 16c 189, 190 | 400 | ‐ | ‐ | ‐ |
Threefold over KV1.1; fivefold over KV1.2; 10‐fold over KV1.4; twofold over KV1.5; 10‐ to 20‐fold over hERG |
||
| 16d 189, 190 | 480 | ‐ | ‐ | ‐ | |||
| Benzofurans | 16e 191 | Planar patch clamp, semi‐automated in human Jurkat cells expressing KV1.3 | 8 | ‐ | ‐ | ‐ | Not determined |
| Aryls | 16f 192 | <50 | ‐ | ‐ | ‐ | 45‐fold over KV1.1; 37‐fold over KV1.5 | |
| Tetra‐phenylporphyrin peptidomimetics | 17 193, 194 | Competitive binding assays; 125I‐HgTX1A19Y/Y37F; membranes from HEK293 cells transfected with human KV1.3 | ‐ | 20 | ‐ | ‐ | Not determined |
| 1H‐benzo[d] imidazol‐7‐yl peptidomimetics | 18 195 | Patch‐clamp, on L929 cells expressing murine KV1.3 | ‐ | ‐ | 95,000 | ‐ | Not determined |
| N‐alkylated indole‐7‐carboxamido peptidomimetics | 19 196 | Patch‐clamp, on L929 cells expressing murine KV1.3 | ‐ | ‐ | ‐ | 75,000 | Not determined |
| Star polymer mimetics | 20 197 | HEK293 cells expressing mammalian KV1.3 | ‐ | ‐ | 300 | ‐ | 10 µM: 51% inhibition of KV1.1; 61% inhibition of hERG |
Different series of KV1.3 inhibitors have been based on the natural ligand khellinone (Figures 16, 16), which was extracted and isolated from the medicinal plant, Ammi visnaga. Chalcone (Figures 16, 16a) inhibited KV1.3 with use dependence (Hill coefficient, 2), with an acceleration of the inactivation of KV1.3, thus resembling the inhibition mechanism of benzamide (Figures 8, 3). Khellinone dimers (Figures 16, 16b) integrated two molecules of 16, joined by a hydrophobic linker. The most potent compound from this series was 16b, which contained a p‐xylyl linker (IC50, 280 nM). Conversely, 16b did not affect the inactivation kinetics, and its Hill coefficient was close to 1. The suggested binding site for 16a and 16b was the hydrophobic pocket on the intracellular site, most likely as in the water‐filled cavity below the selectivity filter. The further design of khellinone dimers did not proceed as they had insufficient aqueous solubility and selectivity between KV1.1 and KV1.2. 189
Figure 16.
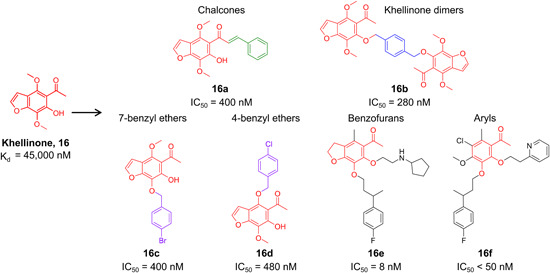
Design of compounds based on natural compound khellinone (16) [Color figure can be viewed at wileyonlinelibrary.com]
Two further series with analogs of 7‐benzyl ethers (Figures 16, 16c) and 4‐benzyl ethers (Figures 16, 16d) were obtained by replacing either the 4‐methoxy or 7‐methoxy substituents in khellinone with a substituted benzyloxy group. Compound 16c inhibited KV1.3 with mixed kinetics and a Hill coefficient of 1.8; the first molecule appeared to bind to the open state, and the second to the C‐type inactivated state. Conversely, analog 16d induced an open‐channel block, with an acceleration of the KV1.3 inactivation, similar to verapamil (Hill coefficient 1.2), although the precise binding site for 16c and 16d remains unknown. 190 The disadvantage of the benzyl ether series was the lack of selectivity over the KV1.x family channels and hERG. In contrast, chalcones were selective over hERG, IKCa1, and the KV1.x family channels, except for only threefold over KV1.1.189, 190 According to several patents filed, to achieve selectivity for KV1.3 over the KV1x family channels, new khellinone analogs were screened using the first semiautomated planar patch‐clamp device (Port‐a‐Patch): benzofurans (Figures 16, 16e) 191 and aryls (Figure 16). 192 The aryl analog 16f showed 45‐fold selectivity over KV1.1, and 37‐fold over KV1.5. 192
Another ligand‐based approach was used for synthetic ligands for K+ channels: the tetraphenylporphyrins (Figures 17, 17). These were primarily designed to mimic the structures of peptide toxins and to interact with all four channel subunits simultaneously. Compound 17 inhibited the 125I‐HgTX1A19Y/Y37F binding to KV1.3 in a dose‐dependent manner, and it was hypothesized that the positive charge was important for this interaction. 193 Furthermore, solid‐phase NMR was carried out with compound 17 and KcsA‐KV1.3 channel preparations in liposomes. After compound 17 bound to the channel, the residues that showed significant changes in their chemical shifts were detected, which indicated likely involvement in the conformational changes of the selectivity filter, as associated with the inhibitor binding. When compound 17 bound to KcsA‐KV1.3, it transferred the selectivity filter into a nonconductive conformation (similar to its nonconductive conformation at pH 4) and shifted the channel into an inactivated state. Subsequently, in silico molecular‐dynamics‐based ligand‐protein docking studies predicted that the porphyrin ring of 17 inserted its positively charged amine arm deep into the selectivity filter of KcsA‐KV1.3, which prevented the binding of K+ to the binding sites of S2 or S3 and therefore shifted the selectivity filter into a nonconductive conformation. Thus, with these tetraphenylporphyrins, an additional condition (apart from pH 4) that could induce the nonconductive conformation of the selectivity filter was discovered. 194 However, this structural class has too great a molecular complexity and does not have confirmed specificity for KV1.3, and it is therefore not promising for new drug discovery.193, 194
Figure 17.
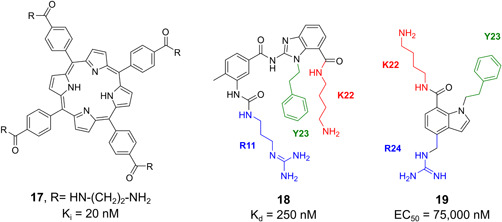
Peptidomimetics, based on the structural elements of peptide toxins [Color figure can be viewed at wileyonlinelibrary.com]
The ShK toxin 195 (Table 4) is a 35‐residue polypeptide with a defined binding epitope that consists of discontinuous (R11, F27) and continuous (K22‐Y23‐R24) elements that show relatively weak binding. Rational design and synthesis of type‐III mimetics 198 of the ShK binding surface revealed first‐generation compound 18 (Figure 17) that mimicked the arginine (R11), lysine (K22), and tyrosine (Y23) residues. Analogs with several basic scaffolds were also developed, and compound 18 with the 1H‐benzo[d]imidazol‐7‐yl scaffold was identified as a relatively weak binder to KV1.3 (K d, 95 µM). As the phenolic OH of the tyrosine (Y23) in the mimetics was not necessary for activity, phenethyl substituent was included instead. 195 The second‐generation compound 19 (Figure 17) mimicked the ShK residues of lysine (K22), tyrosine (Y23), and arginine (R24), and had slightly higher potency (EC50, 75 µM). The basic scaffold of N‐alkylated indole‐7‐carboxamido was selected due to the ease of synthesis and its drug‐like features. Furthermore, indole‐7‐carboxamido was substituted at position 4 with a methyleneguanidinyl group, which mimicked the arginine (R24) at position 7 with an aminobutyl group, representing the lysine (K22), and at position 1 with a phenethyl, to mimic the tyrosine (Y23) of the side chain of ShK. 196
ChTx 197 is a 37‐amino‐acid peptide that is positively charged at neutral pH and is therefore attracted to the negatively charged outer vestibular wall of KV1.3, which has several rings of acidic residues, such as D433 and D449. The key ChTx residue K27 protrudes into the extracellular end of the channel filter and physically occludes the ion permeation pathway, while the toxin basic residues also form hydrogen bonds with the acidic residues in the vestibular wall of the channel. Star polymers (Figures 18, 20) were specifically de novo designed to mimic the lysine residue (K27) of ChTx, and therefore its activity on KV1.3. The inner core of 20 links the four arms, each of which comprises three ethylene glycol repeat units and a lysine‐like terminus. Furthermore, molecular dynamics simulations identified two significant electrostatic interactions between 20 and the KV1.3 channel. The first was a hydrogen bond between the terminal ammonium group of the lysine mimetic 20 and the carbonyl groups of Y447 in the selectivity filter, and thus the lysine side chain of the mimetic 20 physically occluded the pore of KV1.3, in a similar manner to ChTx. The second crucial contact was between the amine group of a different arm of polymer 20 and the acidic residue D422 in the vestibular wall of the channel. Compound 20 inhibited KV1.3 currents with a submicromolar affinity (K d, 300 nM); however, 20 also effectively blocked hERG and KV1.1 at 10 µM. 197
Figure 18.
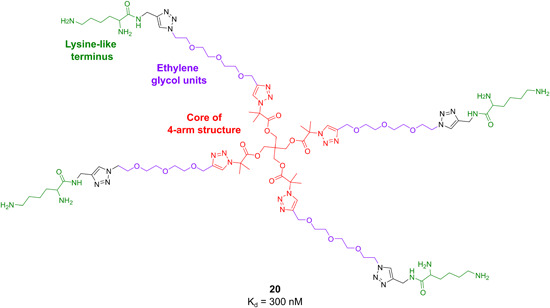
Star polymer, based on the structural elements of peptide toxins [Color figure can be viewed at wileyonlinelibrary.com]
6. SPECIFICALLY TARGETING MITOCHONDRIAL CHANNELS WITH KV1.3 ION CHANNEL MODULATORS
The mitochondria of cancerous cells are an emerging oncological target, as they require specific characteristics and functions for bioenergetic processes, such as the Krebs cycle, and the production of ROS for cancer cells. Recently, a prominent role of mitoKV1.3 in the mitochondria of some types of cancers was defined. 199 Expression of the KV1.3 channel was altered in the cancer cells versus healthy cells, but not with any unique pattern.3, 37, 60
Selectively targeting cancer cells is challenging because they tend to proliferate uncontrollably and are often resistant to apoptotic stimuli. Previous review articles have described that the mitoKV1.3 channel is directly involved in apoptosis of normal and cancer cells, and that KV1.3 activity is required for the proliferation of KV1.3 channel‐expressing cells. Inhibition of mitoKV1.3 channels can selectively activate apoptosis pathways in cancer cells if there is elevated basal ROS production in the cancer cell mitochondria, although the KV1.3 channels need to be upregulated in cancer tissues at the same time. On the other hand, apoptosis does not occur in normal cells with upregulated KV1.3 expression and in cancer cells with downregulated KV1.3 expression. Some compounds have already been tested in vitro, ex vivo, and in vivo, which demonstrated that they could selectively kill cancer cells while sparing healthy cells.3, 35, 37, 60
There are two general strategies to deliver a drug specifically to the mitochondria. The first depends on structural modifications, with the attachment of a mitochondria‐targeting ligand to the pharmacologically active compound. The second strategy uses nanocarriers, which can selectively transfer active compounds to mitochondrial compartments. 199
The first approach has been used to create specific mitochondria‐targeting KV1.3 inhibitors (Figure 19).200, 201 Mitochondria‐targeting ligands are lipophilic and membrane‐permeant cations conjugated to the active KV1.3 channel inhibitor through a stable or labile linker. If a stable linker is selected, the KV1.3 channel inhibitor remains attached to the cation when in solution. Conversely, with the incorporation of a “bioreversible” amide or ester bond as a linker, the initial prodrug is hydrolyzed to the pharmacologically active compound. The type and length of the included spacer significantly affect the chemical and spatial properties of these conjugates in terms of their optimal binding affinity, specificity for the desired mitochondrial channel, and water solubility. Additionally, the position of the cation needs to be optimized through structure–activity relationship studies. Conjugates carry positive charge and are accumulated in the matrix or inner mitochondrial membrane at negative electrochemical potential. 199
Figure 19.
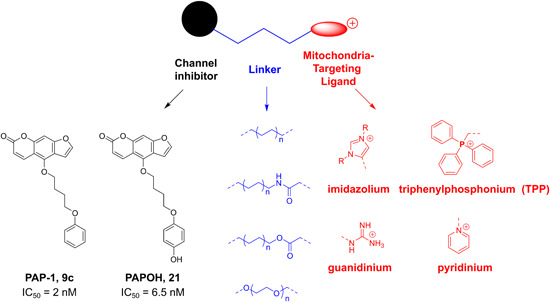
Creation of specific mitochondria‐delivering KV1.3 inhibitors by attachment of membrane‐permeant cations [Color figure can be viewed at wileyonlinelibrary.com]
As PAP‐1 (Figures 19, 9c) was the most potent and selective KV1.3 inhibitor, it was an ideal option for the channel inhibitor part, and also as its 4‐hydroxy analog, PAPOH (Figure 19, 21) as the prodrug. The psoralen analogs PCARBTP (22), PAPTP (23), and PCTP (24) (Figure 20) were also designed by linking scaffold 9c or 21 (Figure 19) to the mitochondria‐targeting cation triphenylphosphonium.200, 201 The prodrug PCARBTP contained a labile carbamate linker, and it was hydrolyzed to PAPOH in mouse blood, although part of it reached the inner mitochondrial membrane intact in vivo. Analog PAPTP incorporated 9c and triphenylphosphonium associated with a stabile propyl linker. 200 Conjugation of scaffold 21 and the triphenylphosphonium with a carbonate linker gave the prodrug PCTP, which went through slow hydrolysis in Dulbecco's modified Eagle medium (culture medium for mammalian cells) to produce PAPOH (Tables 11) and 4‐triphenylphosphonium‐butan‐1‐ol (t 1/2, 17 h). 201
Figure 20.
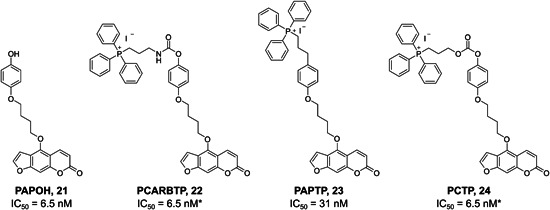
KV1.3 inhibitors specifically delivered to the mitochondria
Table 11.
Potencies of compounds specifically delivered to the mitochondria
| Structural class | Compound | IC50 (nM) |
|---|---|---|
| Psoralen | PAPOH, 21 200 | 6.5 |
| Psoralen prodrugs | PCARBTP, 22 | 6.5 (for product ofhydrolysis: PAPOH, 21) |
| PCTP, 24 200, 201 | ||
| Stable psoralen | PAPTP, 23 200 | 31 |
Note: All systems used Jurkat T lymphocytes and whole‐cell patch‐clamping.
In KV1.3‐expressing Jurkat leukemia T cells and in mouse melanoma B16F10 cells, the psoralens PCARBTP, PAPTP, and PCTP induced apoptosis and mitochondrial swelling, decreased membrane potential, and increased ROS production; however, these were not efficient when the cells showed low expression of KV1.3. They resulted in cell death for the four human pancreatic ductal adenocarcinoma cell types (Bx‐PC3, PANC‐1, As‐PC1, CAPAN‐1 cells).200, 201 Compounds PCARBTP and PAPTP were tested in vivo using an orthotopic mouse B16F10 melanoma model, and these greatly reduce the B16F10 tumor volume, where PAPTP co‐applied with cisplatin reduced the tumor volume by >90%. Treating mice with PCARBTP and PAPTP resulted in significant reductions in human pancreatic Colo357 tumors. 200
7. CHALLENGES AND PERSPECTIVES IN THE DESIGN OF KV1.3 MODULATORS
KV1.3 has a very attractive profile in terms of expression, accessibility, and pathophysiology, but its targeting requires exquisite specificity that can only be achieved by drug design that encompasses a number of critical factors. The design of potent, selective, and safe small‐molecule inhibitors of KV1.3 is challenging, for reasons that are both specific to KV1.3 and common to the design of ion channel inhibitors in general. These are mainly the technical difficulties in ion channel HTS, 144 the lack of three‐dimensional structures that enable structure‐based design,67, 68, 69 the challenges of achieving subtype selectivity 68 also taking into account stoichiometry of heteromeric channels and possible interactions with auxiliary subunits, 202 and ideally state/conformation selectivity.
One of the main obstacles for rational design is the absence of the crystal structure of the KV1.3 and/or an inhibitor in a complex with KV1.3 in a closed and/or open conformation. Homology models of KV1.3 with inner‐pore inhibitors have been built based on the available X‐ray structures of KV1.2 70 and KcsA,203, 204 data from mutational studies, and electrophysiological and ligand‐binding experiments. However, usually, it is unclear which X‐ray structure provides the best template for a homology model, because they differ in sequence similarity and structure quality, as well as conformation or state of the channel. Also, it should be noted that, for example, the inner pore dimensions between X‐ray structures of channels used for building a model can vary widely and, consequently, homology models should be interpreted with caution.
Now, the design of new compounds is more promising since for several voltage‐ and ligand‐gated ion channels there are cryo‐EM structures available.205, 206 These offer new templates for the building of higher quality KV1.3 homology models in open and closed conformations. Ligand binding studies should be coupled with mutational studies at the predicted binding site of the channel to guide the molecular docking to obtain a putative binding mode of the ligand. Then, the channel–ligand complex can be extensively studied by long molecular dynamics simulations, to obtain further insight into the interactions formed. In parallel, structure–activity relationships for a series of active and inactive ligands (based on a single scaffold, to exclude possible binding to different binding sites of the channel) should be derived and then linked to the results of molecular dynamics simulations. If there is a fit between the molecular dynamics and structure–activity relationships data, the identified binding mode should be further explored, for the design of new compounds.
In the absence of structural information for the KV1.3 channel, ligand‐based drug design methodology provides a good alternative. Pharmacophore models can be created based on active and inactive ligand datasets and used in virtual screening to identify new structural classes of KV1.3 inhibitors. 188 However, in the creation of the pharmacophore models, only ligands binding to the same binding site should be selected to get meaningful results. A set of ligand‐based pharmacophore models can be built for different KV1.x isoforms and used in silico to predict the selectivity of the designed compounds, which could then guide the further design of KV1.3‐selective ligands. Thus far, many efforts have focused on small molecules, and medicinal chemistry approaches have aimed at synthesizing optimized compounds with improved potency, but in many cases, the selectivity remains an issue.
An important issue that needs to be addressed in the design of KV1.3 inhibitors are the two particular biophysical properties of KV1.3: C‐type inactivation and cumulative inactivation during repetitive depolarizing pulses. While C‐type inactivation confers high‐affinity interactions with KV1.3 for selected inhibitors, and provides a selective feature, it also presents an additional challenge for the design of inhibitors. When selecting the most promising binding mode of the ligand for rational drug design, the large and not fully identified conformational changes of the ligand binding sites that can occur during C‐type inactivation81, 82 need to be considered. Because of this, structure–activity relationships across a series of ligands rather than the predicted free energy of the ligand–receptor complex should be a decisive criterion for choosing the appropriate binding mode. 83
The state‐dependent inhibition of KV1.3 channels that results in use‐dependent inhibition in excitable cells is another issue that needs to be considered. Accordingly, when planning inhibitor testing, this increase in the number of blocked KV1.3 channels with time should be taken into account, and therefore functional assays rather than binding assays should be selected. 83
A most recent approach in cancer drug discovery is to target the mitochondrial process of apoptosis in cancer cells, which is connected with ROS production.207, 208 As the peptides cannot cross mitochondrial membranes, it is essential to discover new small molecules with high potency for KV1.3. Mitochondriotropic derivates of the KV1.3 inhibitors show great therapeutic potential, but only very few analogs have been developed so far. A further challenges is to attach the adequate transport groups to small molecules, and to optimize the length of the linker. Selective transport of inhibitors to mitochondria improves target specificity and efficacy in vitro and in vivo, as has already been demonstrated with PAP‐1 mitochondriotropic analogs.200, 201 Another advantage of this strategy is the possibility to selectively induce apoptosis in cancer cells with abberant KV1.3 expression, which might be useful as a new cancer therapy with fewer side effects.
Even though larger molecules, for example, peptides and antibodies, can have their own well‐known shortcomings, such as poor membrane permeability and the risk of triggering adverse immune reactions, they constitute an attractive alternative to small molecules. First‐of‐all, because their much higher binding selectivity (which can approach true specificity), both with respect to subtypes and off‐target effects, than what is generally possible to achieve with small molecules. Indeed, from the metabolism of peptides and antibodies, the drug–drug interactions and metabolism‐mediated toxicities that can often be seen with the metabolism of small molecules are not expected. The feasibility of targeting KV1.3 with peptides and toxins has been well demonstrated. In particular, as illustrated above, venomous animals have developed a myriad of highly potent and selective peptides that can both inhibit and activate ion channels, and that have been incredibly useful to probe the ion channel structure–function relationships. 209
8. CONCLUSION
One of the hurdles for successful KV1.3 drug development is the lack of a KV1.3 crystal structure to enable the structure‐based drug design of new molecules. The other general technical issue in the ion channel drug discovery field were the difficulties in ion channel HTS. This has been addressed by the development of ultra‐high‐throughput membrane potential and flux assay systems and automated electrophysiology platforms, with the requirement for specialized biophysical expertise to carry out the screening and to analyze the results. However, the foremost challenge in the design of KV1.3 inhibitors is the need to achieve ion channel subtype selectivity. Obtaining selectivity within the KV1 family is difficult because the subtypes are highly homologous. This is especially challenging for small‐molecule compounds, owing to their small size, and therefore their limited number of interaction points with the targets. Another aspect of interest that is highly challenging is the considerable domain homology that exists between even remotely related ion channel families. Even highly subtype‐selective compounds can therefore have off‐target effects on other ion channels.210, 211
Animal venoms represent alternative sources of KV1.3 ion‐channel‐selective modulators, and the venoms of several species are known to be rich sources of KV1.3 ion‐channel‐modulating peptides. These peptides often bind to the poorly conserved extracellular loops of KV1.3, which offers the potential for significant subtype selectivity. Natural venom peptides, however, do not usually have all of the necessary properties for successful pharmaceutical development. Their membrane permeability might not be optimal, and their plasma half‐lives might not be sufficient for therapeutic applications. 212 These are, however, disadvantages that can often be conquered by chemical modification.213, 214 Venom peptides can therefore provide suitable starting scaffolds for further modifications. In addition, medicinal chemistry approaches in the discovery of KV1.3 inhibitors will undoubtedly remain very important for future breakthroughs at the level of the different modalities of small molecules, peptides, antibodies and hybrid molecules.
ACKNOWLEDGMENTS
Jan Tytgat was supported by grants G0E7120N, GOC2319N, and GOA4919N (FWO‐Vlaanderen), and grant CELSA/17/047 (KU Leuven). Steve Peigneur was supported by grant PDM/19/164 (KU Leuven). This project has received funding from the Max‐Planck Society and the European Union through Horizon 2020 Research and Innovation Programme under the Marie Skłodowska‐Curie grant agreement No.: 813834‐PHIONIC‐H2020‐MSCA‐ITN‐2018. Lucija P. Mašič was supported by grants J1‐9192, N1‐0098, P1‐0208 (ARRS; Slovenian Research Agency), and grant CELSA 005‐1/2017 (University of Ljubljana).
Biographies
Špela Gubič, MPharm, received her Master's degree in Pharmacy from the Faculty of Pharmacy, University of Ljubljana. She is a PhD student at the Chair of Pharmaceutical Chemistry at the Faculty of Pharmacy, University of Ljubljana. She is involved in the studies of ion channel modulators. Her research is focused mainly on the design and synthesis of novel KV1.3 and KV10.1 small molecule inhibitors.
Louise A. Hendrickx is a PhD‐student at the Laboratory Toxicology & Pharmacology at the KU Leuven. She holds a Bachelor's degree in Pharmaceutical Sciences and a Master's degree in Drug Development (KU Leuven). In 2017, she participated in the Erasmus exchange program and stayed at the University of Padova for 4 months for her Master's thesis. In 2018, she performed an internship of 6 months at the Quality Assurance Department of Novartis Pharma and also took part in the extracurricular activity “the Honors Program of Transdisciplinary Insights” at the KU Leuven. In this program, she brainstormed together with a team about strategies for nonjudgement counseling in prenatal screening for Down syndrome, resulting in her first joint publication. Since 2019, Louise is doing research in the Laboratory Toxicology & Pharmacology, focusing on ligands, both peptides and small molecules, for voltage‐gated potassium channels involved in cancer. In 2020, she published her first scientific paper as a PhD‐student.
Žan Toplak is a PhD student at the Chair of Pharmaceutical Chemistry at the Faculty of Pharmacy, University of Ljubljana. He received Master's degree in Pharmacy from the Faculty of Pharmacy, University of Ljubljana. His research interests involve design, synthesis and in silico studies of novel ligands for ion channel modulation, with focus on voltage‐gated potassium channels as potential anti‐cancer drug targets.
Maša Sterle, MPharm, received her Master's degree in Pharmacy from the Faculty of Pharmacy, University of Ljubljana. She is a PhD student at the Chair of Pharmaceutical Chemistry at the Faculty of Pharmacy, University of Ljubljana. She is involved in the studies on how to overcome antimicrobial drug resistance. Her research is focused mainly on the design and synthesis of novel DNA gyrase B inhibitors with antibacterial activity.
Steve Peigneur is a Postdoctoral researcher at the lab of Toxicology & Pharmacology at the Catholic University of Leuven (KU Leuven), Belgium. He holds a Bachelor's degree in Biomedical Lab Techniques from the University Colleges Leuven‐Limburg (UCLL). In 2019, he obtained his PhD in Biochemistry at the Federal University of Minas Gerias in Belo Horizonte, Brazil. He has published over 150 articles in peer reviewed journals. His research in the field of drug discovery and development mainly focusses on the characterisation of novel ligands for ion channels and receptors, starting from natural sources such as venomous animals and poisonous plants.
Tihomir Tomašič received his degree in pharmacy in 2006 and his PhD in pharmaceutical sciences in 2011 from the University of Ljubljana, Faculty of Pharmacy, Ljubljana, Slovenia. He did his post‐doctoral research at Inte:Ligand, Vienna, Austria, under the supervision of Prof. Dr. Sharon D. Bryant. At present he is an associate professor of medicinal chemistry at the Faculty of Pharmacy, University of Ljubljana. His main research interests concern computer‐aided design, synthesis and evaluation of biologically active compounds, particularly with anticancer and antibacterial activity. His research focus at present is on the development of Hsp90 and ion channel inhibitors displaying anticancer activities.
Luis A. Pardo's work has been focused on the structure and function of ion channels, mainly potassium channels. After the discovery of the implication of KV10 channels in cancer in the late 1990s, the interest of the group is centered on understanding the mechanisms ad roles of potassium channels during tumor progression and on the design of diagnostic and therapeutic tools targeting them. Luis Pardo is a Max‐Planck research group leader at the Max Planck Institute of Experimental Medicine, Göttingen, Germany, since 2008. He was affiliated with the Department of Molecular Biology of Neuronal Signals, led by Prof. Walter Stühmer since 1996. Between 2001 and 2004, he served as CSO at iOnGen AG, Göttingen Germany, a company he founded, together with Walter Stühmer and the Max‐Planck Society in 2001. From 1993 to 1996, he was a scientist at the Biochemistry Department, University of Oviedo, Spain. Previously, he had received post‐doctoral training (1991–1993) in the Department led by Prof. E: Neher at the Max‐Planck Institute for Biophysical Chemistry (Göttingen, Germany), after completing his PhD. degree at the Biochemistry and Molecular Biology Department at University of Oviedo (Spain). He also received his MD from this University in 1986.
Jan Tytgat is full professor and Head of the Laboratory Toxicology & Pharmacology at the Catholic University of Leuven (KU Leuven) in Belgium. He holds a Bachelor's and Master's degree in Pharmaceutical Sciences (Faculty of Pharmacy, KU Leuven) and obtained a PhD in Physiology (Faculty of Medicine, KU Leuven). From 1990–1992 he stayed at the Harvard Medical School (Boston, USA) for a post‐doctoral training. Jan Tytgat's bibliometric data are: h index 50, citations 11,431 and publications >300. He has received several scientific prizes, among which the prize of the Research Council in 1994 (KU Leuven) and in 2008 the international prize Dr. E. Delcroix (the Flemish Marine Institute, for his research in the area of Cnidaria intoxications and drug discovery starting from marine systems and organisms). In 2013, he received the prize for best scientific dissemination from the Flemish Academy of Arts and Sciences. Jan Tytgat is a member of the Superior Health Council of Belgium and a former member of the scientific committee of the Federal Agency for the Safety of the Food Chain in Belgium. He has been President of the European Section of the International Society on Toxinology (IST) and was also appointed as Established Investigator (Pesquisador Visitante Especial) from 2013 to 2015, in the frame of Science without Borders (Ciências sem Fronteras), from the CNPq agency in Brazil. Jan Tytgat is also leading a forensic toxicology laboratory in Belgium at the request of the Ministry of Justice in his country. He is an expert in the field of drugs, alcohol, doping, and other xenobiotic substances, causing harm to human health, the environment and relevant to sport ethics. In this scope, he is also often contacted by the Belgian and Flemish authorities, national and international agencies/institutes, press and media for an expert's opinion, appointed for toxicological reports and risk evaluations. He is also Director of the division “Biopharmaceutical Sciences.”
Anamarija Zega received her PhD in pharmaceutical sciences in 2002 from the University of Ljubljana (Faculty of Pharmacy, Ljubljana, Slovenia). She did her postdoctoral research in Dr. Michel Arthur's group at Laboratoire de Recherche Moléculaire sur les Antibiotiques, Inserm in Paris. At present she is associate professor of Medicinal Chemistry at the Faculty of Pharmacy (University of Ljubljana). Her main research interests concern synthesis and biological evaluation of biologically active compounds, particularly with antibacterial and anticancer activity, NMR spectroscopy and investigation of compound promiscuity. Her research focus at present is on the development of DNA gyrase inhibitors targeting resistant ESKAPE pathogens and Mycobacterium tuberculosis and identification of new inhibitors of KV1.3 channels as potential anticancer compounds.
Lucija P. Mašič is a full professor of Medicinal Chemistry and assistant professor of Toxicological Chemistry. She is internationally recognized expert in the field of Medicinal chemistry and Toxicology. She did a post‐doctoral education in 2009 in AstraZeneca, Mölndal, Sweden, Safety Assessment Department in a group of Dr. Scott Boyer. She has more than 80 SCI publications, author of two US patents, more than 1350 citations, her h‐index is 31 (Google Scholar), organizer of several international meetings and member of the International Scientific Committee of ISMC (international Symposiums on Medicinal Chemistry) symposiums: ISMC Berlin 2012, ISMC Lisbon 2014 and ISMC Ljubljana 2018. She has been involved in three EU FP6 and 7 projects (FP6 INTAFAR, FP7 MAREX, INTEGRATE Marie Curie Educational Training Network (ETN)) and IMI Enable project. Her research activities are: Structure and ligand based drug design; Design and synthesis of voltage‐gated potassium ion channels modulators (KV1.3 and hEAG1); Design and synthesis of novel antibacterial compounds (DNA Gyrase); Toxicological testing in the early drug discovery process; In vitro studies of drug metabolism; Testing compounds on nuclear receptors; Metabolism of xenobiotics and activity studies of their metabolites; and Bimolecular mechanism studies of toxicity.
Gubič Š, Hendrickx LA, Toplak Ž, et al. Discovery of KV1.3 ion channel inhibitors: Medicinal chemistry approaches and challenges. Med Res Rev. 2021;41:2423‐2473. 10.1002/med.21800
Contributor Information
Jan Tytgat, Email: jan.tytgat@kuleuven.be.
Anamarija Zega, Email: Anamarija.Zega@ffa.uni-lj.si.
Lucija P. Mašič, Email: lucija.peterlin@ffa.uni-lj.si.
REFERENCES
- 1. Serrano‐Albarrás A, Estadella I, Cirera‐Rocosa S, Navarro‐Pérez M, Felipe A. KV1.3: a multifunctional channel with many pathological implications. Expert Opin Ther Targets. 2018;22:101‐105. [DOI] [PubMed] [Google Scholar]
- 2. Chandy KG, Norton RS. Peptide blockers of KV1.3 channels in T cells as therapeutics for autoimmune disease. Curr Opin Chem Biol. 2017;38:97‐107. [DOI] [PubMed] [Google Scholar]
- 3. Teisseyre A, Palko‐Labuz A, Sroda‐Pomianek K, Michalak K. Voltage‐gated potassium channel KV1.3 as a target in therapy of cancer. Front Oncol. 2019;9:933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pérez‐García MT, Cidad P, López‐López JR. The secret life of ion channels: KV1.3 potassium channels and proliferation. Am J Physiol Cell Physiol. 2018;314:C27‐C42. [DOI] [PubMed] [Google Scholar]
- 5. Checchetto V, Azzolini M, Peruzzo R, Capitanio P, Leanza L. Mitochondrial potassium channels in cell death. Biochem Biophys Res Commun. 2018;500:51‐58. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Li G, Guo J, et al. KV1.3 channel as a key therapeutic target for neuroinflammatory diseases: state of the art and beyond. Front Neurosci. 2020;13:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Comes N, Bielanska J, Vallejo‐Gracia A, et al. The voltage‐dependent K+ channels KV1.3 and KV1.5 in human cancer. Front Physiol. 2013;4:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pardo LA, Contreras‐Jurado C, Zientkowska M, Alves F, Stühmer W. Role of voltage‐gated potassium channels in cancer. J Membr Biol. 2005;205:115‐124. [DOI] [PubMed] [Google Scholar]
- 9. Herzig V, Cristofori‐Armstrong B, Israel MR, Nixon SA, Vetter I, King GF. Animal toxins—nature's evolutionary‐refined toolkit for basic research and drug discovery. Biochem Pharmacol. 2020;181:114096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Castro FB K, Cologna CT, Fornari‐Baldo EC, et al. From animal poisons and venoms to medicines: achievements, challenges and perspectives in drug discovery. Front Pharmacol. 2020;11:1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen B, Cao Z, Li W, Sabatier JM, Wu Y. Treating autoimmune disorders with venom‐derived peptides. Expert Opin Biol Ther. 2017;17:1065‐1075. [DOI] [PubMed] [Google Scholar]
- 12. Holford M, Daly M, King GF, Norton RS. Venoms to the rescue. Science. 2018;361:842‐844. [DOI] [PubMed] [Google Scholar]
- 13. Wulff H, Pennington M. Targeting effector memory T‐cells with KV1.3 blockers. Curr Opin Drug Discov Devel. 2007;10:438‐445. [PubMed] [Google Scholar]
- 14. Gaus K, Chklovskaia E, Groth BF, De S, Jessup W, Harder T. Condensation of the plasma membrane at the site of T lymphocyte activation. J Cell Biol. 2005;171:121‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pérez‐Verdaguer M, Capera J, Serrano‐Novillo C, Estadella I, Sastre D, Felipe A. The voltage‐gated potassium channel KV1.3 is a promising multitherapeutic target against human pathologies. Expert Opin Ther Targets. 2016;20:577‐591. [DOI] [PubMed] [Google Scholar]
- 18. Rangaraju S, Chi V, Pennington MW, Chandy KG. KV1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin Ther Targets. 2009;13:909‐924. [DOI] [PubMed] [Google Scholar]
- 19. Zhao Y, Huang J, Yuan X, et al. Toxins targeting the KV1.3 channel: potential immunomodulators for autoimmune diseases. Toxins. 2015;7:1749‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beeton C, Wulff H, Standifer NE, et al. KV1.3 channels are a therapeutic target for T cell‐mediated autoimmune diseases. Proc Natl Acad Sci USA. 2006;103:17414‐17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wulff H, Calabresi PA, Allie R, et al. The voltage‐gated KV1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toldi G, Bajnok A, Dobi D, et al. The effects of KV1.3 and IKCa1 potassium channel inhibition on calcium influx of human peripheral T lymphocytes in rheumatoid arthritis. Immunobiology. 2013;218:311‐316. [DOI] [PubMed] [Google Scholar]
- 23. Toldi G, Vásárhelyi B, Kaposi A, et al. Lymphocyte activation in type 1 diabetes mellitus: the increased significance of KV1.3 potassium channels. Immunol Lett. 2010;133:35‐41. [DOI] [PubMed] [Google Scholar]
- 24. Nicolaou SA, Neumeier L, Takimoto K, et al. Differential calcium signaling and KV1.3 trafficking to the immunological synapse in systemic lupus erythematosus. Cell Calcium. 2010;47:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kazama I, Maruyama Y, Endo Y, et al. Overexpression of delayed rectifier K+ channels promotes in‐situ proliferation of leukocytes in rat kidneys with advanced chronic renal failure. Int J Nephrol. 2012;2012:581581‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koch Hansen L, Sevelsted‐Møller L, Rabjerg M, et al. Expression of T‐cell KV1.3 potassium channel correlates with pro‐inflammatory cytokines and disease activity in ulcerative colitis. J Crohns Colitis. 2014;8:1378‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Wang Y‐F, Yang X‐F, et al. Specific KV1.3 blockade modulates key cholesterol‐metabolism‐associated molecules in human macrophages exposed to ox‐LDL. J Lipid Res. 2013;54:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Somodi S, Balajthy A, Szilágyi O, et al. Analysis of the K+ current in human CD4+ T lymphocytes in hypercholesterolemic state. Cell Immunol. 2013;281:20‐26. [DOI] [PubMed] [Google Scholar]
- 29. Koshy S, Huq R, Tanner MR, et al. Blocking KV1.3 channels inhibits Th2 lymphocyte function and treats a rat model of asthma. J Biol Chem. 2014;289:12623‐12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vautier F, Belachew S, Chittajallu R, Gallo V. Shaker‐type potassium channel subunits differentially control oligodendrocyte progenitor proliferation. GLIA. 2004;48:337‐345. [DOI] [PubMed] [Google Scholar]
- 31. Das P, Parsons A, Scarborough J, et al. Electrophysiological and behavioral phenotype of insulin receptor defective mice. Physiol Behav. 2005;86:287‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kourrich S, Mourre C, Soumireu‐Mourat B. Kaliotoxin, a KV1.1 and KV1.3 channel blocker, improves associative learning in rats. Behav Brain Res. 2001;120:35‐46. [DOI] [PubMed] [Google Scholar]
- 33. Habela CW, Sontheimer H. Cytoplasmic volume condensation is an integral part of mitosis. Cell Cycle. 2007;6:1613‐1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Urrego D, Tomczak AP, Zahed F, Stühmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prosdocimi E, Checchetto V, Leanza L. Targeting the mitochondrial potassium channel KV1.3 to kill cancer cells: drugs, strategies, and new perspectives. SLAS Discov. 2019;24:882‐892. [DOI] [PubMed] [Google Scholar]
- 36. Comes N, Serrano‐Albarrás A, Capera J, et al. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim Biophys Acta BBA‐Biomembr. 2015;1848:2477‐2492. [DOI] [PubMed] [Google Scholar]
- 37. Teisseyre A, Gąsiorowska J, Michalak K. Voltage‐gated potassium channels KV1.3 ‐ potentially new molecular target in cancer diagnostics and therapy. Adv Clin Exp Med. 2015;24:517‐524. [DOI] [PubMed] [Google Scholar]
- 38. Serrano‐Novillo C, Capera J, Colomer‐Molera M, Condom E, Ferreres J, Felipe A. Implication of voltage‐gated potassium channels in neoplastic cell proliferation. Cancers. 2019;11:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pardo LA. Voltage‐gated potassium channels in cell proliferation. Physiology. 2004;19:285‐292. [DOI] [PubMed] [Google Scholar]
- 40. Cidad P, Jiménez‐Pérez L, García‐Arribas D, et al. KV1.3 channels can modulate cell proliferation during phenotypic switch by an ion‐flux independent mechanism. Arterioscler Thromb Vasc Biol. 2012;32:1299‐1307. [DOI] [PubMed] [Google Scholar]
- 41. Jang SH, Byun JK, Jeon WI, et al. Nuclear localization and functional characteristics of voltage‐gated potassium channel KV1.3. J Biol Chem. 2015;290:12547‐12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee A, Fakler B, Kaczmarek LK, Isom LL. More than a pore: ion channel signaling complexes. J Neurosci. 2014;34:15159‐15169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bielanska J, Hernandez‐Losa J, Perez‐Verdaguer M, et al. Voltage‐dependent potassium channels KV1.3 and KV1.5 in human cancer. Curr Cancer Drug Targets. 2009;9(8):904‐914. [DOI] [PubMed] [Google Scholar]
- 44. Tajti G, Wai DCC, Panyi G, Norton RS. The voltage‐gated potassium channel KV1.3 as a therapeutic target for venom‐derived peptides. Biochem Pharmacol. 2020;181:114146. [DOI] [PubMed] [Google Scholar]
- 45. Abdul M, Santo A, Hoosein N. Activity of potassium channel‐blockers in breast cancer. Anticancer Res. 2003;23:3347‐3351. [PubMed] [Google Scholar]
- 46. Abdul M, Hoosein N. Expression and activity of potassium ion channels in human prostate cancer. Cancer Lett. 2002;186:99‐105. [DOI] [PubMed] [Google Scholar]
- 47. Leanza L, Trentin L, Becker KA, et al. Clofazimine, Psora‐4 and PAP‐1, inhibitors of the potassium channel KV1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia. 2013;27:1782‐1785. [DOI] [PubMed] [Google Scholar]
- 48. Jang SH, Kang K‐S, Ryu PD, Lee SY. KV1.3 voltage‐gated K(+) channel subunit as a potential diagnostic marker and therapeutic target for breast cancer. BMB Rep. 2009;42:535‐539. [DOI] [PubMed] [Google Scholar]
- 49. Brevet M, Haren N, Sevestre H, Merviel P, Ouadid‐Ahidouch H. DNA methylation of KV1.3 potassium channel gene promoter is associated with poorly differentiated breast adenocarcinoma. Cell Physiol Biochem. 2009;24:25‐32. [DOI] [PubMed] [Google Scholar]
- 50. Bielanska J, Hernandez‐Losa J, Moline T, et al. Increased voltage‐dependent K+ channel KV1.3 and KV1.5 expression correlates with leiomyosarcoma aggressiveness. Oncol Lett. 2012;4:227‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bielanska J, Hernández‐Losa J, Moline T, et al. Differential expression of KV1.3 and KV1.5 voltage‐dependent K+ channels in human skeletal muscle sarcomas. Cancer Invest. 2012;30:203‐208. [DOI] [PubMed] [Google Scholar]
- 52. Preußat K, Beetz C, Schrey M, et al. Expression of voltage‐gated potassium channels KV1.3 and KV1.5 in human gliomas. Neurosci Lett. 2003;346:33‐36. [DOI] [PubMed] [Google Scholar]
- 53. Vallejo‐Gracia A, Bielanska J, Hernández‐Losa J, et al. Emerging role for the voltage‐dependent K+ channel KV1.5 in B‐lymphocyte physiology: expression associated with human lymphoma malignancy. J Leukoc Biol. 2013;94:779‐789. [DOI] [PubMed] [Google Scholar]
- 54. Leanza L, Biasutto L, Managò A, Gulbins E, Zoratti M, Szabò I. Intracellular ion channels and cancer. Front Physiol. 2013;4:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szabò I, Bock J, Jekle A, et al. A novel potassium channel in lymphocyte mitochondria. J Biol Chem. 2005;280:12790‐12798. [DOI] [PubMed] [Google Scholar]
- 56. Laskowski M, Augustynek B, Kulawiak B, et al. What do we not know about mitochondrial potassium channels? Biochim Biophys Acta. 2016;1857:1247‐1257. [DOI] [PubMed] [Google Scholar]
- 57. Szabo I, Bock J, Grassme H, et al. Mitochondrial potassium channel KV1.3 mediates Bax‐induced apoptosis in lymphocytes. Proc Natl Acad Sci USA. 2008;105:14861‐14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leanza L, Venturini E, Kadow S, Carpinteiro A, Gulbins E, Becker KA. Targeting a mitochondrial potassium channel to fight cancer. Cell Calcium. 2015;58:131‐138. [DOI] [PubMed] [Google Scholar]
- 59. Wrzosek A, Augustynek B, Żochowska M, Szewczyk A. Mitochondrial potassium channels as druggable targets. Biomolecules. 2020;10:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Checchetto V, Prosdocimi E, Leanza L. Mitochondrial KV1.3: a new target in cancer biology? Cell Physiol Biochem. 2019;53:52‐62. [DOI] [PubMed] [Google Scholar]
- 61. Zhu J, Yan J, Thornhill WB. The KV1.3 potassium channel is localized to the cis‐Golgi and KV1.6 is localized to the endoplasmic reticulum in rat astrocytes. FEBS J. 2014;281:3433‐3445. [DOI] [PubMed] [Google Scholar]
- 62. Rudy B, Maffie J, Amarillo Y, et al. Voltage Gated Potassium Channels: Structure and Function of Kv1 to Kv9 Subfamilies. In: Squire LR, ed. Encyclopedia of Neuroscience. 1st ed. Oxford, UK: Oxford Academic Press; 2009:397‐425. [Google Scholar]
- 63. Yellen G. The voltage‐gated potassium channels and their relatives. Nature. 2002;419:35‐42. [DOI] [PubMed] [Google Scholar]
- 64. Kuang Q, Purhonen P, Hebert H. Structure of potassium channels. Cell Mol Life Sci. 2015;72:3677‐3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kremer W, Weyand M, Winklmeier A, Schreier C, Kalbitzer HR. 1.2 Å X‐ray structure of the renal potassium channel KV1.3 T1 domain. Protein J. 2013;32:533‐542. [DOI] [PubMed] [Google Scholar]
- 67. Rashid MH, Kuyucak S. Affinity and selectivity of ShK toxin for the KV1 potassium channels from free energy simulations. J Phys Chem B. 2012;116:4812‐4822. [DOI] [PubMed] [Google Scholar]
- 68. Chen R, Robinson A, Gordon D, Chung S‐H. Modeling the binding of three toxins to the voltage‐gated potassium channel (KV1.3). Biophys J. 2011;101:2652‐2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rashid MH, Kuyucak S. Free energy simulations of binding of HsTx1 toxin to KV1 potassium channels: the basis of KV1.3/KV1.1 selectivity. J Phys Chem B. 2014;118:707‐716. [DOI] [PubMed] [Google Scholar]
- 70. Chen X, Wang Q, Ni F, Ma J. Structure of the full‐length Shaker potassium channel KV1.2 by normal‐mode‐based X‐ray crystallographic refinement. Proc Natl Acad Sci USA. 2010;107:11352‐11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2016;54:5.6.1‐5.6.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schrodinger LLC. The PyMOL Molecular Graphics System, Version 2.3.0. 2019.
- 73.The UniProt Consortium [database online]. Cambridgeshire, UK: EMBL‐EBI, Lozana, CH: SIB, Washington, DC: PIR; 2002. Updated July 23, 2020.
- 74. Yin S‐J, Jiang L, Yi H, et al. Different residues in channel turret determining the selectivity of ADWX‐1 inhibitor peptide between KV1.1 and KV1.3 channels. J Proteome Res. 2008;7:4890‐4897. [DOI] [PubMed] [Google Scholar]
- 75. Zhao Y, Chen Z, Cao Z, Li W, Wu Y. Diverse structural features of potassium channels characterized by scorpion toxins as molecular probes. Molecules. 2019;24:2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tombola F, Pathak MM, Isacoff EY. How does voltage open an ion channel? Annu Rev Cell Dev Biol. 2006;22:23‐52. [DOI] [PubMed] [Google Scholar]
- 77. Kim DM, Nimigean CM. Voltage‐gated potassium channels: a structural examination of selectivity and gating. Cold Spring Harb Perspect Biol. 2016;8:a029231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gouaux E, MacKinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461‐1465. [DOI] [PubMed] [Google Scholar]
- 79. Barros F, Pardo LA, Domínguez P, Sierra LM, de la Peña P. New structures and gating of voltage‐dependent potassium (KV) channels and their relatives: a multi‐domain and dynamic question. Int J Mol Sci. 2019;20:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage‐dependent Shaker family K+ channel. Science. 2005;309:897‐903. [DOI] [PubMed] [Google Scholar]
- 81. Panyi G. Biophysical and pharmacological aspects of K+ channels in T lymphocytes. Eur Biophys J. 2005;34:515‐529. [DOI] [PubMed] [Google Scholar]
- 82. Panyi G, Sheng Z, Tu L, Deutsch C. C‐type inactivation of a voltage‐gated K+ channel occurs by a cooperative mechanism. Biophys J. 1995;69:896‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wulff H, Zhorov BSK. Channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem Rev. 2008;108:1744‐1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Levy DI, Deutsch C. Recovery from C‐type inactivation is modulated by extracellular potassium. Biophys J. 1996;70:798‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choi KL, Aldricht RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage‐activated K+ channels. Proc Natl Acad Sci USA. 1991;88:5092‐5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Somodi S, Varga Z, Hajdu P, et al. pH‐dependent modulation of KV1.3 inactivation: role of His399. Am J Physiol Cell Physiol. 2004;287:C1067‐C1076. [DOI] [PubMed] [Google Scholar]
- 87. Chandy KG, Strong M, Aiyar J, Gutman GA. Structural and biochemical features of the KV1.3 potassium channel: an aid to guided drug design. Cell Physiol Biochem. 1997;7:135‐147. [Google Scholar]
- 88. Wulff H, Castle NA, Pardo LA. Voltage‐gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Krůsek J. Allostery and cooperativity in the interaction of drugs with ionic channel receptors. Physiol Res. 2004;53:569‐579. [PubMed] [Google Scholar]
- 90. Qiu F, Rebolledo S, Gonzalez C, Larsson HP. Subunit interactions during cooperative opening of voltage‐gated proton channels. Neuron. 2013;77:288‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tytgat J, Hess P. Evidence for cooperative interactions in potassium channel gating. Nature. 1992;359:420‐423. [DOI] [PubMed] [Google Scholar]
- 92. Koshland DE. Correlation of structure and function in enzyme action. Science. 1963;142:1533‐1541. [DOI] [PubMed] [Google Scholar]
- 93. Rossokhin A, Teodorescu G, Grissmer S, Zhorov B. Interaction of d‐tubocurarine with potassium channels: molecular modeling and ligand binding. Mol Pharmacol. 2006;69:1356‐1365. [DOI] [PubMed] [Google Scholar]
- 94. Dreker T, Grissmer S. Investigation of the phenylalkylamine binding site in hKV1.3 (H399T), a mutant with a reduced C‐type inactivated state. Mol Pharmacol. 2005;68:966‐973. [DOI] [PubMed] [Google Scholar]
- 95. Goldstein SA, Miller C. Mechanism of charybdotoxin block of a voltage‐gated K+ channel. Biophys J. 1993;6:1613‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ranganathan R, Lewis JH, MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle‐based structure analysis. Neuron. 1996;16:131‐139. [DOI] [PubMed] [Google Scholar]
- 97. Lange A, Giller K, Hornig S, et al. Toxin‐induced conformational changes in a potassium channel revealed by solid‐state NMR. Nature. 2006;440:959‐962. [DOI] [PubMed] [Google Scholar]
- 98. Dauplais M, Lecoq A, Song J, et al. On the convergent evolution of animal toxins. J Biol Chem. 1997;272:4302‐4309. [DOI] [PubMed] [Google Scholar]
- 99. Park CS, Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 1992;9:307‐313. [DOI] [PubMed] [Google Scholar]
- 100. Park CS, Miller C. Mapping function to structure in a channel‐blocking peptide: electrostatic mutants of charybdotoxin. Biochemistry. 1992;31:7749‐7755. [DOI] [PubMed] [Google Scholar]
- 101. Romi‐Lebrun R, Lebrun B, Martin‐Eauclaire MF, et al. Purification, characterization, and synthesis of three novel toxins from the Chinese scorpion Buthus martensi, which act on K+ channels. Biochemistry. 1997;36:13473‐13482. [DOI] [PubMed] [Google Scholar]
- 102. Grishin EV, Korolkova YV, Kozlov SA, et al. Structure and function of the potassium channel inhibitor from black scorpion venom. Pure Appl Chem. 1996;68:2105‐2109. [Google Scholar]
- 103. Bagdáany M, Batista CVF, Valdez‐Cruz NA, et al. Anuroctoxin, a new scorpion toxin of the alpha‐KTx 6 subfamily, is highly selective for KV1.3 over IKCa1 ion channels of human T lymphocytes. Mol Pharmacol. 2005;67:1034‐1044. [DOI] [PubMed] [Google Scholar]
- 104. Takacs Z, Toups M, Kollewe A, et al. A designer ligand specific for KV1.3 channels from a scorpion neurotoxin‐based library. Proc Natl Acad Sci USA. 2009;106:22211‐22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lebrun B, Romi‐lebrun R, Yasuda A, et al. A four‐disulphide‐bridged toxin, with high affinity towards voltage‐gated K+ channels, isolated from Heterometrus spinnifer (Scorpionidae) venom. Biochem J. 1997;328:321‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Stehling EG, Sforca ML, Pignatelli A, et al. Structural and functional characterization of the recombinant form of the KV1.3 channel blocker Tc32. 2008. 10.2210/pdb2JP6/pdb [DOI] [PubMed]
- 107. Olamendi‐Portugal T, Gómez‐Lagunas F, Gurrola GB, Possani LD. A novel structural class of K+‐channel blocking toxin from the scorpion Pandinus imperator . Biochem J. 1996;315:977‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Castañeda O, Sotolongo V, Amor AM, et al. Characterization of a potassium channel toxin from the Caribbean Sea anemone Stichodactyla helianthus . Toxicon. 1995;33:603‐613. [DOI] [PubMed] [Google Scholar]
- 109. Zhu S, Peigneur S, Gao B, Umetsu Y, Ohki S, Tytgat J. Experimental conversion of a defensin into a neurotoxin: implications for origin of toxic function. Mol Biol Evol. 2014;31:546‐559. [DOI] [PubMed] [Google Scholar]
- 110. Tudor JE, Pallaghy PK, Pennington MW, Norton RS. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat Struct Biol. 1996;3:317‐320. [DOI] [PubMed] [Google Scholar]
- 111. Rauer H, Pennington M, Cahalan M, Chandy KG. Structural conservation of the pores of calcium‐activated and voltage‐gated potassium channels determined by a sea anemone toxin. J Biol Chem. 1999;274:21885‐21892. [DOI] [PubMed] [Google Scholar]
- 112. Zhao R, Dai H, Mendelman N, Chill JH, Goldstein SAN. Tethered peptide neurotoxins display two blocking mechanisms in the K+ channel pore as do their untethered analogs. Sci Adv. 2020;6:eaaz3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mouhat S, Mosbah A, Visan V, et al. The “functional” dyad of scorpion toxin Pi1 is not itself a prerequisite for toxin binding to the voltage‐gated KV1.2 potassium channels. Biochem J. 2004;377:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Guan B, Chen X, Zhang H. Two‐electrode voltage clamp. Methods Mol Biol. 2013;998:79‐89. [DOI] [PubMed] [Google Scholar]
- 115. Rubaiy HN. A short guide to electrophysiology and ion channels. J Pharm Pharm Sci. 2017;20:48‐67. [DOI] [PubMed] [Google Scholar]
- 116. Goldin AL. Expression of Ion Channels in Xenopus oocytes. In: Clare JJ, Trezise DJ, eds. Expression and Analysis of Recombinant Ion Channels: From Structural Studies to Pharmacological Screening. 1st ed. Weinheim, DE: Wiley‐VCH; 2006:1‐25. [Google Scholar]
- 117. Tapper AR, George AL. Heterologous expression of ion channels. Methods Mol Biol. 2003;217:285‐294. [DOI] [PubMed] [Google Scholar]
- 118. Dunlop J, Bowlby M, Peri R, et al. Ion channel screening. Comb Chem High Throughput Screen. 2008;11:514‐522. [DOI] [PubMed] [Google Scholar]
- 119. Dabrowski M, Dekermendjian K, Lund P‐E, Krupp J, Sinclair J, Larsson O. Ion channel screening technology. CNS Neurol Disord Drug Targets. 2008;7:122‐128. [DOI] [PubMed] [Google Scholar]
- 120. Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507‐519. [DOI] [PubMed] [Google Scholar]
- 121. Vetter I, Hodgson WC, Adams DJ, McIntyre PD. VenomS‐Based Drug Discovery: Bioassays, Electrophysiology, High‐Throughput Screens and Target Identincation. In: King GF, ed. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics. 1st ed. London, UK: Royal Society of Chemistry; 2015:97‐128. [Google Scholar]
- 122. Prashanth JR, Hasaballah N, Vetter I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology. 2017;127:4‐19. [DOI] [PubMed] [Google Scholar]
- 123. Shen B, Cao Z, Wu Y, et al. Purlisin, a toxin‐like defensin derived from clinical pathogenic fungus Purpureocillium lilacinum with both antimicrobial and potassium channel inhibitory activities. FASEB J. 2020;34:15093‐15107. [DOI] [PubMed] [Google Scholar]
- 124. Renisio JG, Romi‐Lebrun R, Blanc E, Bornet O, Nakajima T, Darbon H. Solution structure of BmKTX, a K+ blocker toxin from the Chinese scorpion Buthus Martensi . Proteins. 2000;38:70‐78. [DOI] [PubMed] [Google Scholar]
- 125. Jaravine VA, Nolde DE, Reibarkh MJ, et al. Three‐dimensional structure of toxin OSK1 from Orthochirus scrobiculosus scorpion venom. Biochemistry. 1997;36:1223‐1232. [DOI] [PubMed] [Google Scholar]
- 126. Savarin P, Romi‐Lebrun R, Zinn‐Justin S, et al. Structural and functional consequences of the presence of a fourth disulfide bridge in the scorpion short toxins: solution structure of the potassium channel inhibitor HsTX1. Protein Sci. 1999;8:2672‐2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Oliveira IS, Ferreira IG, Alexandre‐Silva GM, et al. Scorpion toxins targeting KV1.3 channels: insights into immunosuppression. J Venom Anim Toxins Trop Dis. 2019;25:e148118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bingham JP, Andrews EA, Kiyabu SM, Cabalteja CC. Drugs from slugs. Part II ‐ conopeptide bioengineering. Chem Biol Interact. 2012;200:92‐113. [DOI] [PubMed] [Google Scholar]
- 129. Chen Z, Hu Y, Hong J, et al. Toxin acidic residue evolutionary function‐guided design of de novo peptide drugs for the immunotherapeutic target, the KV1.3 channel. Sci Rep. 2015;5:18‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chen Z, Hu Y, Hu J, et al. Unusual binding mode of scorpion toxin BmKTX onto potassium channels relies on its distribution of acidic residues. Biochem Biophys Res Commun. 2014;447:70‐76. [DOI] [PubMed] [Google Scholar]
- 131. Kalman K, Pennington MW, Lanigan MD, et al. Shk‐Dap22, a potent KV1.3‐specific immunosuppressive polypeptide. J Biol Chem. 1998;273:32697‐32707. [DOI] [PubMed] [Google Scholar]
- 132. Mouhat S, Visan V, Ananthakrishnan S, et al. K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom. Biochem J. 2005;385:95‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mouhat S, Teodorescu G, Homerick D, et al. Pharmacological profiling of Orthochirus scrobiculosus toxin 1 analogs with a trimmed N‐terminal domain. Mol Pharmacol. 2006;69:354‐362. [DOI] [PubMed] [Google Scholar]
- 134. Bartok A, Fehér K, Bodor A, et al. An engineered scorpion toxin analogue with improved KV1.3 selectivity displays reduced conformational flexibility. Sci Rep. 2015;5:18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Rashid MH, Huq R, Tanner MR, et al. A potent and KV1.3‐selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci Rep. 2014;4:4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ding Y, Ting JP, Liu J, Al‐Azzam S, Pandya P, Afshar S. Impact of non‐proteinogenic amino acids in the discovery and development of peptide therapeutics. Amino Acids. 2020;52:1207‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bray GM, Exenatide Am. Exenatide. J Health Syst Pharm. 2006;63:411‐418. [DOI] [PubMed] [Google Scholar]
- 138. Lorenz M, Evers A, Wagner M. Recent progress and future options in the development of GLP‐1 receptor agonists for the treatment of diabesity. Bioorg Med Chem Lett. 2013;23:4011‐4018. [DOI] [PubMed] [Google Scholar]
- 139. Lanigan MD, Kalman K, Lefievre Y, Pennington MW, Chandy KG, Norton RS. Mutating a critical lysine in ShK toxin alters its binding configuration in the pore‐vestibule region of the voltage‐gated potassium channel, KV1.3. Biochemistry. 2002;41:11963‐11971. [DOI] [PubMed] [Google Scholar]
- 140. Bhuyan R, Seal A. Molecular dynamics of KV1.3 ion channel and structural basis of its inhibition by scorpion toxin‐OSK1 derivatives. Biophys Chem. 2015;203‐204:1‐11. [DOI] [PubMed] [Google Scholar]
- 141. Tytgat J, Chandy KG, Garcia ML, et al. A unified nomenclature for short‐chain peptides isolated from scorpion venoms: α‐KTx molecular subfamilies. Trends Pharmacol Sci. 1999;20:444‐447. [DOI] [PubMed] [Google Scholar]
- 142. Kuyucak S, Norton RS. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med Chem. 2014;6:1645‐1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Carrega L, Mosbah A, Ferrat G, et al. The impact of the fourth disulfide bridge in scorpion toxins of the α‐KTx6 subfamily. Proteins. 2005;61:1010‐1023. [DOI] [PubMed] [Google Scholar]
- 144. Yu H, Li M, Wang W, Wang X. High throughput screening technologies for ion channels. Acta Pharmacol Sin. 2016;37:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hill RJ, Grant AM, Volberg W, et al. WIN 17317‐3: novel nonpeptide antagonist of voltage‐activated K+ channels in human T lymphocytes. Mol Pharmacol. 1995;48:98‐104. [PubMed] [Google Scholar]
- 146. Nguyen A, Kath JC, Hanson DC, et al. Novel nonpeptide agents potently block the C‐type inactivated conformation of KV1.3 and suppress T cell activation. Mol Pharmacol. 1996;50:1672‐1679. [PubMed] [Google Scholar]
- 147. Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C‐type inactivation of K channels. Biophys J. 1995;69:2449‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chandy KG, Wulff H, Beeton C, Calabresi PA, Gutman GA, Pennington M. KV1.3 Potassium Channel: Physiology, Pharmacology and Therapeutic Indications. In: Triggle DJ, Gopalakrishnan M, Rampe D, Zheng W, eds. Voltage‐Gated Ion Channels as Drug Targets. 1st ed. Weinheim, DE: Wiley‐VCH; 2006:214‐274. [Google Scholar]
- 149. Wanner SG, Glossmann H, Knaus H‐G, et al. WIN 17317‐3, a new high‐affinity probe for voltage‐gated sodium channels. Biochemistry. 1999;38:11137‐11146. [DOI] [PubMed] [Google Scholar]
- 150. Burgess LE, Koch K, Cooper K, et al. The SAR of UK‐78,282: a novel blocker of human T cell KV1.3 potassium channels. Bioorg Med Chem Lett. 1997;7:1047‐1052. [Google Scholar]
- 151. Hanson DC, Nguyen A, Mather RJ, et al. UK‐78,282, a novel piperidine compound that potently blocks the KV1.3 voltage‐gated potassium channel and inhibits human T cell activation. Br J Pharmacol. 1999;126:1707‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Schmalhofer WA, Bao J, McManus OB, et al. Identification of a new class of inhibitors of the voltage‐gated potassium channel, KV1.3, with immunosuppressant properties. Biochemistry. 2002;41:7781‐7794. [DOI] [PubMed] [Google Scholar]
- 153. Schmalhofer WA, Slaughter RS, Matyskiela M, et al. Di‐substituted cyclohexyl derivatives bind to two identical sites with positive cooperativity on the voltage‐gated potassium channel, KV1.3. Biochemistry. 2003;42:4733‐4743. [DOI] [PubMed] [Google Scholar]
- 154. Miao S, Bao J, Garcia ML, et al. Benzamide derivatives as blockers of KV1.3 ion channel. Bioorg Med Chem Lett. 2003;13:1161‐1164. [DOI] [PubMed] [Google Scholar]
- 155. Goetz MA, Hensens OD, Zink DL, et al. Potent nor‐triterpenoid blockers of the voltage‐gated potassium channel KV1.3 from Spachea correae . Tetrahedron Lett. 1998;39:2895‐2898. [Google Scholar]
- 156. Felix JP, Bugianesi RM, Schmalhofer WA, et al. Identification and biochemical characterization of a novel nortriterpene inhibitor of the human lymphocyte voltage‐gated potassium channel, KV1.3. Biochemistry. 1999;38:4922‐4930. [DOI] [PubMed] [Google Scholar]
- 157. Hanner M, Schmalhofer WA, Green B, et al. Binding of correolide to KV1 family potassium channels: mapping the domains of high affinity interactions. J Biol Chem. 1999;274:25237‐25244. [DOI] [PubMed] [Google Scholar]
- 158. Hanner M, Green B, Gao Y‐D, et al. Binding of correolide to the KV1.3 potassium channel: characterization of the binding domain by site‐directed mutagenesis. Biochemistry. 2001;40:11687‐11697. [DOI] [PubMed] [Google Scholar]
- 159. Bao J, Miao S, Kayser F, et al. Potent KV1.3 inhibitors from correolide—modification of the C18 position. Bioorg Med Chem Lett. 2005;15:447‐451. [DOI] [PubMed] [Google Scholar]
- 160. Ohanyan V, Yin L, Bardakjian R, et al. KV1.3 channels facilitate the connection between metabolism and blood flow in the heart. Microcirculation. 2017;24:e12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hautz T, Krapf C, Grahammer J, et al. Targeting the KV1.3 potassium channel for immunosuppression in vascularized composite allotransplantation—a pilot study. Transpl Int. 2013;26:552‐561. [DOI] [PubMed] [Google Scholar]
- 162. Ford J, Palmer NJ, Atherall JF, inventors; Xention Limited, assignee. Thieno[2,3‐b]pyridines as potassium channel inhibitors. US patent 7,576,212 B2. August 18, 2009.
- 163. Hamlyn RJ, Mulla M, John DE, inventors; Xention Limited, assignee. Potassium channel blockers. US patent 8,372,840 B2. February 12, 2013.
- 164. Haffner CD, Thomson SA, Guo Y, et al. N‐{3‐[(1,1‐dioxido‐1,2‐benzothiazol‐3‐yl)(phenyl)amino]propyl}benzamide analogs as potent KV1.3 inhibitors. Part 1. Bioorg Med Chem Lett. 2010;20:6983‐6988. [DOI] [PubMed] [Google Scholar]
- 165. Haffner CD, Thomson SA, Guo Y, et al. Substituted N‐{3‐[(1,1‐dioxido‐1,2‐benzothiazol‐3‐yl)(phenyl)amino]propyl}benzamide analogs as potent KV1.3 ion channel blockers. Part 2. Bioorg Med Chem Lett. 2010;20:6989‐6992. [DOI] [PubMed] [Google Scholar]
- 166. Harvey A, Bombrun A, Cooke R, inventors; Merck Patent GmbH, Bionomic Limited, assignee. Amine derivatives as potassium channel blockers. AU patent 2012255690 B2. November 22, 2012.
- 167. Mayr LM, Bojanic D. Novel trends in high‐throughput screening. Curr Opin Pharmacol. 2009;9:580‐588. [DOI] [PubMed] [Google Scholar]
- 168. Macarron R, Banks MN, Bojanic D, et al. Impact of high‐throughput screening in biomedical research. Nat Rev Drug Discov. 2011;10:188‐195. [DOI] [PubMed] [Google Scholar]
- 169. Bohuslavizki KH, Hänsel W, Kneip A, Koppenhôfer E, Niemôller E, Sanmann K. Mode of action of psoralens, benzofurans, acridinons, and coumarins on the ionic currents in intact myelinated nerve fibres and its significance in demyelinating diseases. Gen Physiol Biophys. 1994;13:309‐328. [PubMed] [Google Scholar]
- 170. Vennekamp J, Wulff H, Beeton C, et al. KV1.3‐blocking 5‐phenylalkoxypsoralens: a new class of immunomodulators. Mol Pharmacol. 2004;65:1364‐1374. [DOI] [PubMed] [Google Scholar]
- 171. Schmitz A, Sankaranarayanan A, Azam P, et al. Design of PAP‐1, a selective small molecule KV1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol. 2005;68:1254‐1270. [DOI] [PubMed] [Google Scholar]
- 172. Bodendiek SB, Mahieux C, Hänsel W, Wulff H. 4‐phenoxybutoxy‐substituted heterocycles—a structure‐activity relationship study of blockers of the lymphocyte potassium channel KV1.3. Eur J Med Chem. 2009;44:1838‐1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Ren YR, Pan F, Parvez S, et al. Clofazimine inhibits human KV1.3 potassium channel by perturbing calcium oscillations in T lymphocytes. PLOS One. 2008;3:e4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Faouzi M, Starkus J, Penner R. State‐dependent blocking mechanism of KV1.3 channels by the antimycobacterial drug clofazimine: state‐dependent block of KV1.3 by clofazimine. Br J Pharmacol. 2015;172:5161‐5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Nguyen W, Howard BL, Jenkins DP, Wulff H, Thompson PE, Manallack DT. Structure‐activity relationship exploration of KV1.3 blockers based on diphenoxylate. Bioorg Med Chem Lett. 2012;22:7106‐7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hyodo T, Oda T, Kikuchi Y, et al. Voltage‐gated potassium channel KV1.3 blocker as a potential treatment for rat anti‐glomerular basement membrane glomerulonephritis. Am J Physiol Renal Physiol. 2010;299:F1258‐F1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Zimin PI, Garic B, Bodendiek SB, Mahieux C, Wulff H, Zhorov BS. Potassium channel block by a tripartite complex of two cationophilic ligands and a potassium ion. Mol Pharmacol. 2010;78:588‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Marzian S, Stansfeld PJ, Rapedius M, et al. Side pockets provide the basis for a new mechanism of KV channel–specific inhibition. Nat Chem Biol. 2013;9:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wulff H, Yarov‐Yarovoy V. Sticking to nooks and crannies. Nat Chem Biol. 2013;9:473‐474. [DOI] [PubMed] [Google Scholar]
- 180. Kundu‐Raychaudhuri S, Chen Y‐J, Wulff H, Raychaudhuri SP. KV1.3 in psoriatic disease: PAP‐1, a small molecule inhibitor of KV1.3 is effective in the SCID mouse psoriasis—xenograft model. J Autoimmun. 2014;55:63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Maezawa I, Nguyen HM, Di Lucente J, et al. KV1.3 inhibition as a potential microglia‐targeted therapy for Alzheimer's disease: preclinical proof of concept. Brain J Neurol. 2018;141:596‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Chen Y‐J, Nguyen HM, Maezawa I, Jin L‐W, Wulff H. Inhibition of the potassium channel KV1.3 reduces infarction and inflammation in ischemic stroke. Ann Clin Transl Neurol. 2018;5:147‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. Clofazimine: current status and future prospects. J Antimicrob Chemother. 2012;67:290‐298. [DOI] [PubMed] [Google Scholar]
- 184. Leanza L, Henry B, Sassi N, et al. Inhibitors of mitochondrial KV1.3 channels induce Bax/Bak‐independent death of cancer cells. EMBO Mol Med. 2012;4:577‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zaccagnino A, Managò A, Leanza L, et al. Tumor‐reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget. 2016;8:38276‐38293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Pegoraro S, Lang M, Dreker T, et al. Inhibitors of potassium channels KV1.3 and IK‐1 as immunosuppressants. Bioorg Med Chem Lett. 2009;19:2299‐2304. [DOI] [PubMed] [Google Scholar]
- 187. Grissmer S, Nguyen AN, Aiyar J, et al. Pharmacological characterization of five cloned voltage‐gated K+ channels, types KV1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227‐1234. [PubMed] [Google Scholar]
- 188. Hendrickx LA, Dobričić V, Toplak Ž, et al. Design and characterization of a novel structural class of KV1.3 inhibitors. Bioorganic Chem. 2020;98:103746. [DOI] [PubMed] [Google Scholar]
- 189. Baell JB, Gable RW, Harvey AJ, et al. Khellinone derivatives as blockers of the voltage‐gated potassium channel KV1.3: synthesis and immunosuppressive activity. J Med Chem. 2004;47:2326‐2336. [DOI] [PubMed] [Google Scholar]
- 190. Harvey AJ, Baell JB, Toovey N, Homerick D, Wulff H. A new class of blockers of the voltage‐gated potassium channel KV1.3 via modification of the 4‐ or 7‐position of khellinone. J Med Chem. 2006;49:1433‐1441. [DOI] [PubMed] [Google Scholar]
- 191. Flynn BL, Baell JB, Harvey AJ, inventors; Bionomics Limited, assignee. Novel benzofuran potassium channel blockers and uses thereof. WO patent 2008/040057 A1. April 10, 2008.
- 192. Flynn BL, Baell JB, Chaplin JH, inventors; Bionomics Limited, assignee. Novel aryl potassium channel blockers and uses thereof. WO patent 2009/043117 A1. April 9, 2009.
- 193. Gradl SN, Felix JP, Isacoff EY, Garcia ML, Trauner D. Protein surface recognition by rational design: nanomolar ligands for potassium channels. J Am Chem Soc. 2003;125:12668‐12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Ader C, Schneider R, Hornig S, et al. A structural link between inactivation and block of a K+ channel. Nat Struct Mol Biol. 2008;15:605‐612. [DOI] [PubMed] [Google Scholar]
- 195. Baell JB, Harvey AJ, Norton RS. Design and synthesis of type‐III mimetics of ShK toxin. J Comput Aided Mol Des. 2002;16:245‐262. [DOI] [PubMed] [Google Scholar]
- 196. Harvey AJ, Gable RW, Baell JB. A three‐residue, continuous binding epitope peptidomimetic of ShK toxin as a KV1.3 inhibitor. Bioorg Med Chem Lett. 2005;15:3193‐3196. [DOI] [PubMed] [Google Scholar]
- 197. Chen R, Lu D, Xie Z, et al. Peptidomimetic star polymers for targeting biological ion channels. PLOS One. 2016;11:e0152169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ripka AS, Rich DH. Peptidomimetic design. Curr Opin Chem Biol. 1998;2:441‐452. [DOI] [PubMed] [Google Scholar]
- 199. Parrasia S, Mattarei A, Furlan A, Zoratti M, Biasutto L. Small‐molecule modulators of mitochondrial channels as chemotherapeutic agents. Cell Physiol Biochem. 2019;53:11‐43. [DOI] [PubMed] [Google Scholar]
- 200. Leanza L, Romio M, Becker KA, et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo . Cancer Cell. 2017;31:516‐531. [DOI] [PubMed] [Google Scholar]
- 201. Mattarei A, Romio M, Managò A, et al. Novel mitochondria‐targeted furocoumarin derivatives as possible anti‐cancer agents. Front Oncol. 2018;8:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Vacher H, Trimmer JS. Diverse roles for auxiliary subunits in phosphorylation‐dependent regulation of mammalian brain voltage‐gated potassium channels. Pflugers Arch. 2011;462:631‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Uysal S, Vasquez V, Tereshko V, et al. Crystal structure of full‐length KcsA in its closed conformation. Proc Natl Acad Sci USA. 2009;106:6644‐6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cuello LG, Cortes DM, Perozo E. The gating cycle of a K+ channel at atomic resolution. eLife. 2017;6:e28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Wu J, Yan Z, Li Z, et al. Structu re of the voltage‐gated calcium channel CaV1.1 complex. Science. 2015;350:aad2395. [DOI] [PubMed] [Google Scholar]
- 206. Wang W, MacKinnon R. Cryo‐EM structure of the open human ether‐à‐go‐go‐related K+ channel hERG. Cell. 2017;169:422‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Nguyen C, Pandey S. Exploiting mitochondrial vulnerabilities to trigger apoptosis selectively in cancer cells. Cancers. 2019;11:916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Leanza L, Managò A, Zoratti M, Gulbins E, Szabo I. Pharmacological targeting of ion channels for cancer therapy: in vivo evidences. Biochim Biophys Acta. 2016;1863:1385‐1397. [DOI] [PubMed] [Google Scholar]
- 209. Wulff H, Christophersen P, Colussi P, Chandy GK, Yarov‐Yarovoy V. Antibodies and venom peptides: new modalities for ion channels. Nat Rev Drug Discov. 2019;18:339‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Sanguinetti MC, Tristani‐Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463‐469. [DOI] [PubMed] [Google Scholar]
- 211. Vandenberg JI, Perozo E, Allen TW. Towards a structural view of drug binding to hERG K+ channels. Trends Pharmacol Sci. 2017;38:899‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide‐based drugs. Chem Biol Drug Des. 2013;81:136‐147. [DOI] [PubMed] [Google Scholar]
- 213. Tanner MR, Tajhya RB, Huq R, et al. Prolonged immunomodulation in inflammatory arthritis using the selective KV1.3 channel blocker HsTX1[R14A] and its PEGylated analog. Clin Immunol. 2017;180:45‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Tarcha EJ, Olsen CM, Probst P, et al. Safety and pharmacodynamics of dalazatide, a KV1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLOS One. 2017;12:e0180762. [DOI] [PMC free article] [PubMed] [Google Scholar]