

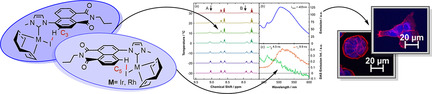
Abstract
Complexes of RhI and IrI of the [M(COD)(NHC)X] type (where M=Rh or Ir, COD=1,5‐cyclooctadiene, NHC=N‐heterocyclic carbene, and X=halide) have recently shown promising cytotoxic activities against several cancer cell lines. Initial mechanism of action studies provided some knowledge about their interaction with DNA and proteins. However, information about their cellular localization remains scarce owing to luminescence quenching within this complex type. Herein, the synthesis of two rare examples of luminescent RhI and IrI [M(COD)(NHC)I] complexes with 1,8‐naphthalimide‐based emitting ligands is reported. All new complexes are comprehensively characterized, including with single‐crystal X‐ray structures. Steric crowding in one derivative leads to two distinct rotamers in solution, which apparently can be distinguished both by pronounced NMR shifts and by their respective spectral and temporal emission signatures. When the photophysical properties of these new complexes are exploited for cellular imaging in HT‐29 and PT‐45 cancer cell lines, it is demonstrated that the complexes accumulate predominantly in the endoplasmic reticulum, which is an entirely new finding and provides the first insight into the cellular localization of such IrI(NHC) complexes.
Keywords: bioorganometallic chemistry, cellular imaging, iridium, luminescence lifetime, rhodium
Anticancer NHC complexes: RhI and IrI N‐heterocyclic carbene (NHC) complexes provide an auspicious class of potential new anticancer agents; however, cellular imaging with luminescent derivatives of these complex types is often hampered by luminescence quenching. Building on an analysis of the parameters for metal‐induced quenching processes, new examples of this complex class are designed with properties suitable for cellular imaging. Their main localization in the endoplasmic reticulum is not only an unprecedented finding but also represents the first ever insight into the cellular localization of Ir(NHC) complexes in the oxidation state +I.
Introduction
The field of metal‐based anticancer agents has been on the rise ever since the discovery of the anticancer properties of cisplatin in 1965, which is in daily clinical use against various types of cancer.[ 1 , 2 , 3 ] In this context, metal complexes of iridium(I) and rhodium(I) that are structurally and electronically similar to cisplatin were investigated in the 1970s and successfully used to cure mice bearing Ehrlich ascites sarcoma.[ 4 , 5 ] However, research in the area quickly ceased and interest shifted towards the investigation of the more stable complexes of Rh and Ir in the oxidation state +III. Today these comprise the vast majority of Rh and Ir complexes under investigation for their anticancer properties.[ 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 ] It is only in the early 2000s that the potential anticancer properties of RhI and IrI were rediscovered. In this respect, complexes of the overall formula [M(COD)(NHC)X] (where M=Rh or Ir, COD=1,5‐cyclooctadiene, NHC=N‐heterocyclic carbene, and X=halide) represent the majority of complexes investigated in the field. The literature provides several examples of this complex class that have shown promising cytotoxic activities against several cancer cell lines[ 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 ] with IC50 values ranging between 600 nm [21] and more than 100 μm. [29] A direct comparison of the general trend predicts that complexes with a RhI center are more active than their IrI counterparts, [18] and that the activity is strongly influenced by the structure of the NHC ligand, potentially accounting for differences in the cellular uptake of the respective complex.[ 25 , 29 ] However, the mechanism of action of this class of compounds is under investigation. Although the RhI complexes have been reported to interact with free DNA[ 19 , 21 ] and DNA model bases, [23] the nuclear Rh content of cells treated with a respective complex has been reported to reach not more than 1–2 % of the total cellular Rh content. [21] In addition, this type of RhI complexes has been reported to interact with enzymes such as albumin [21] and TrxR,[ 25 , 26 ] which was related to a disruption in the cellular metabolic machinery. [26] Taking into account these results, it has been proposed that the mechanism of action of the rhodium complexes proceeds through the generation of reactive oxygen species (ROS), which lead to DNA damage and consequently to cell cycle arrest. [26] A similar generation of ROS was observed for the respective [Ir(COD)(NHC)X] complexes.[ 27 , 29 ] Additionally, a detailed mechanism of action study showed that the interaction of the IrI complexes with the proteins cytochrome C and lysozyme proceeded by the exchange of their COD and halide ligands along with an oxidation of the metal center to the oxidation state +III.[ 28 , 29 , 30 ] However, in contrast to the Rh complexes, interactions with model oligonucleotides were not observed. [29]
A way to complement this knowledge about cytotoxic activities and biomolecular interactions of a specific compound is to investigate its cellular localization by luminescence cell imaging. Cyclometalated complexes of IrIII have been widely exploited for this purpose mainly because of their excellent photophysical suitability for this application.[ 32 , 33 ] The IrIII metal center in combination with a high‐field ligand facilitates a metal‐to‐ligand charge transfer (MLCT) leading to complexes with large Stokes’ shifts and long luminescence lifetimes. [33] This general feature of cyclometalated IrIII complexes can be used in combination with luminescent ligand systems, for example, by creating a luminescent NHC ligand.[ 34 , 35 ] As systems of IrI and RhI lack this natural suitability for metal‐based photophysical applications, introducing luminescent co‐ligands (e.g., emitting NHC ligands in the [M(COD)(NHC)X] type complexes) becomes the next‐best possibility to investigate their cellular localization by luminescence cell imaging.
To create such a luminescent NHC ligand, the most attractive parts for modifications are the heterocycle nitrogen atoms and the imidazolium backbone carbon atoms, offering the possibility to introduce an imidazolium moiety into virtually every possible structure.[ 36 , 37 ] Indeed, the attachment of a luminescent dye to an imidazolium core and its subsequent use for the synthesis of [M(COD)(NHC)X] complexes creates luminescent compounds as reported for, for example, BODIPY[ 38 , 39 ] and coumarin [40] functionalized [M(COD)(NHC)X] complexes. However, although the free NHC ligands showed good luminescence quantum yields, the values dropped significantly after metal ion complexation.
This loss of luminescence was explained by a metal‐related photoinduced electron transfer (PET) quenching process.[ 38 , 39 , 40 ] To the best of our knowledge, there are only two successful reports of luminescent [Rh(COD)(NHC)Cl] complexes used for cellular imaging within cancer cells,[ 24 , 25 ] which revealed mitochondrial localization of the respective complexes. [25] In one of these reports, the NHC ligand is a fusion product of an imidazolium moiety and a 1,8‐naphthalimide luminescent core, [24] which is not only known for its luminescent properties but also for its potential to act as a cytotoxic agent by DNA intercalation.[ 41 , 42 , 43 ] Therein, functionalization of the naphthalimide core with a thioether moiety was the key to obtain a luminescent complex for the application within cells. [24] In contrast, the cellular localization of the respective [Ir(COD)(NHC)X] complexes has not been investigated so far. Also, in contrast to M(+III) complexes, the photophysical details and exact requirements for Rh or Ir complexes in the oxidation state +I, which are suitable for cellular imaging, remain elusive.
Herein, four new luminescent 1,8‐naphthalimide functionalized Rh and Ir complexes of the [M(COD)(NHC)X] type are reported. We report their photophysical properties, their application for cellular imaging as well as insights into their cellular localization to extend the knowledge of this compound class and to reveal similarities and differences between the two different metal centers IrI and RhI. In the course of these investigations, critical structural features for suitable luminescent properties are identified.
Results and Discussion
Synthesis and characterization
The imidazolium‐functionalized 1,8‐naphthalimide derivative 4 was inspired by literature examples of similar AuI[44] and IrIII[34–35] complexes, and synthesized based on modified literature procedures starting from 1,8‐naphthalic anhydride 1 (Scheme 1).[ 34 , 35 , 44 , 45 ] Therein, the first step comprised formation of amide 2 by treatment of 1 with propylamine. After amide formation, functionalization of the aromatic ring system was performed by nucleophilic aromatic substitution with imidazole leading to the intermediate product 3, which was converted to the final imidazolium salt 4 through nucleophilic substitution with methyliodide. The nature of the final product was confirmed by 1H NMR spectroscopy, which showed the presence of the additional alkyl chain at 4.11 ppm, 1.74 ppm, and 1.00 ppm in addition to the signals of the imidazolium heterocycle at 8.13 ppm and the heterocycle methyl group at 4.05 ppm as well as the imidazolium proton at 9.75 ppm. Additionally, single crystals suitable for X‐ray crystallography were obtained, which unambiguously proved the nature of the ligand (Figure S5 in the Supporting Information).
Scheme 1.
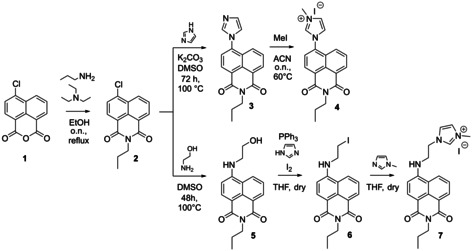
Synthesis of the imidazolium‐functionalized 1,8‐naphthalimide derivatives 4 and 7.
Literature reports about BODIPY‐functionalized IrIII and RhIII Cp* complexes stated that luminescence quenching was highly dependent on the distance between the actual luminescent moiety and the metal center. [46] Short distances resulted in significant luminescence quenching, whereas those complexes with longer linkers connecting the metal center with the luminescent moiety showed enhanced luminescence quantum yields. [46] Taking into account the aforementioned phenomenon of luminescence quenching in respective RhI or IrI [M(COD)(NHC)X] complexes,[ 38 , 39 , 40 ] the second imidazolium‐functionalized 1,8‐naphthalimide derivative 7 with an ethyl linker between the two moieties was designed to reduce the probability of luminescence quenching. The desired ligand was obtained based on a literature procedure. [47] Amide 2 was first functionalized by nucleophilic aromatic substitution with 2‐aminoethanol to yield the intermediate alcohol 5, which was converted to the respective iodide 6 by treatment with triphenylphosphine and iodine. The iodide 6 was then converted to the imidazolium salt 7 by nucleophilic substitution with 1‐methylimidazole, which yielded the final ligand. In comparison to ligand 4, the 1H NMR showed additional signals for the ethyl linker at 3.71 ppm and 3.46 ppm, as well as the expected imidazolium proton at 9.15 ppm, proving the successful formation of the desired ligand.
The ligands 4 and 7 were subsequently used for the synthesis of the respective RhI and IrI [M(COD)(NHC)X] complexes following a literature procedure for similar complexes with an NHC ligand. [48] To obtain the desired complexes, the imidazolium precursors 4 and 7 were treated with DIPEA (N,N‐diisopropylethylamine) and subsequently the respective Rh or Ir precursor [M(COD)Cl]2 was added under inert conditions (Scheme 2). After purification, complexes 8 a and 8 b were obtained as bright‐red solids in yields of 71 % and 66 %, respectively. In contrast to that, complex 9 a was obtained as a yellow solid in a yield of 29 %, whereas complex 9 b could not be obtained by this procedure. To obtain complex 9 b, a classical Ag2O transmetalation [49] procedure was chosen as an alternative, which has been excessively exploited by our group for the synthesis of [Ir(COD)(NHC)Cl] complexes.[ 27 , 28 , 29 , 30 ] This route successfully yielded complex 9 b as a yellow solid in a yield of 22 %. The successful synthesis of complexes 9 a and 9 b was confirmed by signals in the 1H NMR spectrum stemming from the COD ligand (in the areas of 5.25 ppm, 3.53 ppm, 2.30 ppm, and 1.76 ppm for the rhodium complex 9 a and 4.67 ppm, 3.04 ppm, 2.22 ppm, and 1.74 ppm for the iridium analog 9 b) as well as the disappearance of the signal assigned to the imidazolium proton, which indicated formation of a direct metal–carbon bond. This was further indicated in the 13C{1H} NMR spectrum of complex 9 a, which showed a significant shift of the signal assigned to the imidazolium carbon from 136.9 ppm in the free ligand to 181.8 ppm after complexation. The splitting of this signal into a doublet with a coupling constant of 48.8 Hz is caused by the typical 103Rh/13C coupling seen upon direct Rh−C binding [50] and provides another proof for the formation of a direct metal–carbon bond and thus the desired complex structure.
Scheme 2.
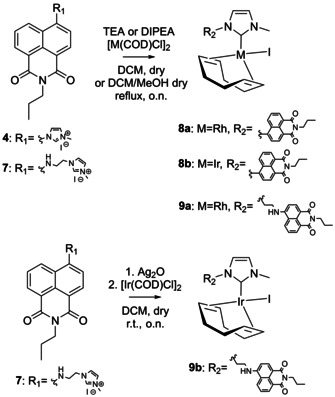
Synthesis of the RhI and IrI complexes 8 a and 8 b as well as 9 a and 9 b.
In contrast to this, the 1H NMR spectra of complexes 8 a and 8 b could not be as unambiguously assigned. Although the spectra showed the expected disappearance of the imidazolium proton at 9.75 ppm and additional signals assigned to the COD ligand in the area of 5.03 ppm, 3.46 ppm, 2.36 ppm, 2.20 ppm, and 1.64 ppm for the rhodium complex 8 a and at 4.52 ppm, 2.91 ppm, 1.91 ppm, 1.42 ppm, and 1.04 ppm for the iridium complex 8 b, the shape of the signals assigned to the aromatic protons at 9.05 ppm and 7.90 ppm in complex 8 a and 8.73 ppm and 7.98 ppm in complex 8 b was unexpectedly broad. Additionally, some of the signals expected from the free ligand 4 were missing in the 13C{1H} NMR spectra of the complexes, especially in the aromatic region (Figure S1 in the Supporting Information), thus casting doubt on the nature of the respective compound. To clarify the nature of the compounds, the 1H NMR spectra of complex 8 a were recorded at decreasing temperatures between 303 K and 243 K. The experiment showed that the initially very broad signals at 9.05 ppm and 7.90 ppm slowly sharpened with decreasing temperature, leading to the final formation of two clearly distinguishable signals at 243 K (Figure 1 a). A 13C{1H} NMR spectrum recorded at this temperature additionally gave rise to the expected signals in the aromatic region that were previously missing (see Figure S2 in the Supporting Information for comparative NMR spectra). For complex 8 a, the signal assigned to the carbene carbon atom directly bound to the rhodium metal center showed the formation of a doublet at 184.9 ppm with a coupling constant of 49.6 Hz comparable to that seen in complex 9 a. Likewise, this is a clear indication for the presence of a direct metal–carbon bond. When similar low‐temperature NMR spectra were recorded for complex 8 b, a comparable effect in the 1H‐ and 13C{1H} NMR spectra became visible (Figure S3 in the Supporting Information); however, the peak sharpening effect on the signals at 8.73 ppm and 7.98 ppm at 243 K was less pronounced (see Figure S3 in the Supporting Information). Altogether, these results proved the nature of the compounds to be as expected; however, this left us with the question of which molecular process within the complexes caused this unusual behavior at ambient temperature.
Figure 1.
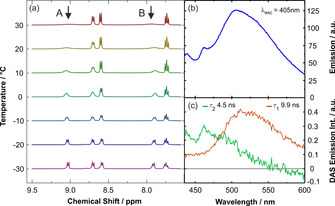
(a) Temperature‐dependent 1H NMR spectra (400 MHz in CDCl3) recorded from 30 °C to −30 °C of complex 8 a. The signals A and B (aromatic protons marked with an arrow) change their shape from a very broad signal to a sharp doublet, indicating the presence of two distinct rotamers 8 a‐A and 8 a‐B. (b) The steady‐state emission spectrum of complex 8 a dissolved in acetone after excitation at 405 nm. (c) The decay‐associated spectra (DAS) from a global biexponential fit to the time‐resolved emission of complex 8 a after excitation at 405 nm, denoting the existence of two distinct species in room‐temperature solution.
To shed further light on this, an in‐depth analysis of the two complexes 8 a and 8 b by 2D NMR spectroscopy (1H,1H‐COSY,13C{1H},1H‐HMBC and 13C{1H},1H‐HSQC) revealed that the shape changing signals stem from the protons at the C3 and C5 carbon atoms in the aromatic ring system directly adjacent to the imidazolium substituent. From this, it was concluded that the change in signal shape most probably originates from a rotational process present at ambient, but significantly slowed down at low temperatures, which is a known feature of the metal–carbon bond in this type of complex.[ 49 , 51 ] However, metal–carbon bond rotation is reported to go along with the formation of two distinct signal sets when the 1H NMR spectra are recorded at low temperatures owing to the formation of two distinct rotamers, [51] which is not the case for the present complexes. The only other position present in the structure available for such a rotational process is the C4−N bond between the aromatic ring system and the imidazolium moiety. This, in contrast, would lead to the formation of the more favorable structure at low temperatures. In the present case, this structure represents either of the two structures 8 a/b‐A or 8 a/b‐B, where the respective C3 or C5 proton is in a suitable position to form a halide bridge with the respective iodo ligand (see Figure 2), a feature that has been reported for complexes of a similar [M(COD)(NHC)X] type. [48]
Figure 2.
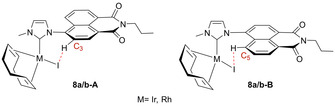
Potential formation of a halide bridge between the hydrogen atoms at carbon C3 or C5 and the iodide ligand in complexes 8 a and 8 b resulting in the respective structures A or B.
To shed further light on the respective molecular structures, slow evaporation of solutions containing the complexes 8 a and 9 a afforded single crystals suitable for diffraction experiments. The obtained structural data unambiguously confirm the expected molecular structures in all cases. Complex 8 a crystallizes in the triclinic space group P with two molecules of 8 a and two molecules of CH2Cl2 in the unit cell, whereas 9 a crystallized in the monoclinic space group P21/n with four molecules of 9 a in the unit cell. Molecular structures are shown in Figures 3. In both cases, the Rh centers adopt a distorted square‐planar coordination geometry, which is typically observed for RhI‐COD complexes. The lengths of the M−NHC bonds were determined to be 2.027 Å (8 a) and 2.034 Å (9 a; Table 1), which is in good agreement with RhI(COD)‐NHC complexes reported in the literature.[ 25 , 52 ] Importantly, the Rh‐bonded halogenide was confirmed to be iodine in both cases. As expected, the aromatic NHC substituents do not interact with the metal centers but participate in extensive intermolecular π‐stacking, which constitutes the dominant packing motif in their solid‐state structures. Owing to its proximity to the NHC ligand as well as the metal‐COD moiety in 8 a, the planar aromatic N‐substituent is rotated by a torsion angle of 53.9° with respect to the NHC ligand, giving rise to the observed isomeric behavior discussed previously. The obtained structure herein (see Figure 3) corresponds to 8 a‐A in Figure 2. However, although the observed torsion angle shows a clear influence of the metal‐bound halide in the formation of structure 8 a‐A, the presence of a distinct halide bridge between the C3‐bound proton and the iodide was not confirmed.
Figure 3.
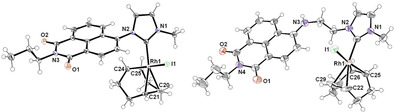
X‐ray crystal structures of complexes 8 a and 9 a (ellipsoid probability 30 %).
Table 1.
Respective bond lengths and angles present in complexes 8 a and 9 a.
|
Complex |
Bond |
Length [Å] |
|
|
Complex |
|
[Å] |
|
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
8 a |
I(1)−Rh(1) |
2.6837(3) |
C(1)‐Rh(1)‐I(1) |
87.09(9) |
9 a |
I(1)−Rh(1) |
2.6558(3) |
C(1)‐Rh(1)‐I(1) |
87.53(10) |
|||||
|
Rh(1)−C(1) |
2.027(3) |
C(1)‐Rh(1)‐C(20) |
159.40(12) |
Rh(1)−C(1) |
2.035(4) |
C(1)‐Rh(1)‐C(22) |
163.4(2) |
|||||||
|
Rh(1)−C(20) |
2.210(3) |
C(1)‐Rh(1)‐C(21) |
164.23(12) |
Rh(1)−C(22) |
2.202(4) |
C(1)‐Rh(1)‐C(29) |
161.0(2) |
|||||||
|
Rh(1)−C(21) |
2.229(3) |
C(1)‐Rh(1)‐C(24) |
91.58(12) |
Rh(1)−C(29) |
2.204(5) |
C(1)‐Rh(1)‐C(25) |
92.81(15) |
|||||||
|
Rh(1)−C(24) |
2.119(3) |
C(1)‐Rh(1)‐C(25) |
94.21(12) |
Rh(1)−C(25) |
2.122(3) |
C(1)‐Rh(1)‐C(26) |
91.73(15) |
|||||||
|
Rh(1)−C(25) |
2.125(3) |
Photophysical behavior
After synthesis of the complexes 8 a–9 b, their photophysical properties were investigated to analyze the potential for use in luminescence cell imaging. The absorption spectra of complexes 8 a and 8 b in acetone display two individual absorption bands. Whereas the high‐energy band around 340 nm coincides with that of the corresponding free NHC ligand 4 (Figure S7 in the Supporting Information), the low‐frequency absorption in the 405–410 nm region is characteristic for the metal complexes (M=Rh, Ir).
In analogy to similar metal–ligand systems, [46] the 340 nm absorption band presumably corresponds to the ligand‐centered π–π* transition (compare Figure S7 in the Supporting Information and closely related species [53] ), as the metalation of the NHC ligand does not affect the spectral position of the absorption maximum significantly (Table 2). Furthermore, the low‐frequency absorption above 400 nm is indicative of a metal‐to‐ligand charge transfer (MLCT) transition, as known, for example, for IrIII complexes.[ 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 ] Excitation of the free ligand 4 at 340 nm displays an emission band with its maximum at 440 nm. Coordination to the metal center results in effective luminescence quenching of the blue emission band, the effect of which is more pronounced for the rhodium compound (Figure S8 a in the Supporting Information). The photochemical behavior of similar NHC ligand systems has been thoroughly studied,[ 24 , 46 , 62 , 63 ] revealing low emission intensities and small quantum yields, owing to a PET‐related quenching mechanism. However, excitation at 405 nm of the complexes 8 a and 8 b shows a significantly different emission behavior. The emission spectrum of 8 a shows three maxima peaking at 440 nm, 460 nm, and 510 nm (Figure 1 b), whereas that of 8 b consists of only one broad emission feature asymmetrically shaped towards the red‐edge (see Figure S8 b in the Supporting Information).
Table 2.
Absorption and emission properties of the investigated complexes 8 a/b and 9 a/b in comparison with the corresponding free ligands 4 and 7 in acetone.
|
Complex |
|
( =340 nm) |
( =405 nm) |
Emission decay constants (streak camera at =405 nm) |
( =405 nm) |
|||
|---|---|---|---|---|---|---|---|---|
|
|
[nm] |
[nm] |
[nm] |
[ps] |
[ns] |
[ns] |
[%] |
|
|
4 |
331 |
433 |
[a] |
|
|
[a] |
[a], 1.78[b] |
|
|
7 |
420 |
– |
502 |
|
– |
9.6 |
74.5 |
|
|
8 a |
334/400 |
441 |
438/462/505 |
|
4.5 |
9.9 |
0.14 |
|
|
8 b |
335/422 |
447 |
458 |
|
– |
7.5 |
0.45 |
|
|
9 a |
427 |
– |
504 |
60/175 |
|
8.7 |
1.0 |
|
|
9 b |
425 |
– |
506 |
<37 |
|
8.7 |
0.9 |
|
[a] Not enough fluorescence at =405 nm. [b] Measured at =340 nm.
To investigate the distinct emission processes of the different metal complexes, time‐resolved luminescence spectroscopy measurements were carried out, using a ps‐streak camera.[ 64 , 65 ]
The emission after 405 nm excitation of the Rh‐metal complex 8 a consists of two spectrally overlapping radiative decays. Their respective lifetimes amount to 4.5 ns ( and 9.9 ns ( ). The corresponding analysis in terms of decay‐associated spectra[ 66 , 67 ] (DAS, Figure 1 c) discloses that the short‐lived emission comprises the emission with maxima at 440 nm and 460 nm, whereas the decay corresponding to peaks at 510 nm. In accordance with the results found by temperature‐dependent NMR spectroscopy, the two emission processes of complex 8 a potentially originate from the distinct rotamers 8 a‐A and 8 a‐B. However, for the Ir‐based complex 8 b only one long‐lived emission species with a lifetime of 7.5 ns was found, possibly indicating that only one of the rotamers deactivates by a radiative relaxation or that just one of them is present in room‐temperature solution.
In contrast to the complexes 8 a/b, the absorption spectra of 9 a and 9 b in acetone consist of only one absorption band in the 440 nm region, which is in agreement with that of the corresponding non‐metalated ligand 7 (Figure S9 in the Supporting Information). Based on studies of similar NHC derivatives,[ 62 , 63 ] it is suggested that the absorption maximum belongs to the typical π–π* transition of the substituted naphthalimide. The electron‐deficient nature of the naphthalimide in combination with the electron‐donating amine side chain enables an internal charge transfer (ICT). The emission spectra of 7 and the metalated complexes 9 a/b contain one emission band around 505 nm, which is in good agreement with the values reported for similar rhodium complexes. [24] As expected, the luminescence of the metal complexes is strongly quenched in comparison to the high‐emissive free ligand (Figure S10 in the Supporting Information). Although the luminescence quantum yield of the NHC ligand 7 amounts to 0.745, those of 9 a/b are drastically decreased to 0.01 and 0.009, respectively, probably caused by a PET mechanism, as already discussed above for 8 a/b.[ 24 , 46 , 62 , 63 ] However, the emission intensities are still significantly higher than those observed for complexes 8 a and 8 b. It appears that in complexes 9 a and 9 b, a longer distance between the metal center and the luminescent dye significantly reduces metal‐related quenching processes. A similar effect has been reported for comparable Ir and Rh Cp* complexes with BODIPY‐derived luminescent ligands. [46] Additionally, time‐resolved luminescence spectroscopy measurements were performed with a streak camera after excitation with laser pulses spectrally centered at 405 nm on a ps–ns timescale. NHC ligand 7 decays in a monoexponential deactivation process with an emission lifetime of 9.6 ns. This prolongated deactivation is typical for such NHC ligands exhibiting chromophores that are more distant from the coordination site. [68] Complexation with the Rh‐/Ir‐metal center results in emission decays, which comprise a sub‐ns as well as a long‐lived component (Figure S11 in the Supporting Information). The deactivation of the Rh complex 9 a is dominated by the rapidly decreasing signal, which can be modeled by contributions with decay times of 60 ps and 175 ps, whereas the slow emission signal is described by a decay constant of 8.7 ns. For compound 9 b, the same decay behavior with a time constant of 8.7 ns was detected, accompanied by one very short emitting deactivation process, falling under the streak camera's IRF of approximately 37 ps (FWHM). In both cases, the long‐lasting emission can be assigned to the luminescence of the ligand system, quenched by interactions with the metal center.
Finally, both complex pairs 8 a/b and 9 a/b are excitable at 405 nm, which was a necessary requirement for the subsequent cellular luminescence experiments performed by confocal microscopy. Owing to the higher emission intensities, complexes 9 a and 9 b were chosen for the following cellular imaging experiments.
Luminescence cell imaging
For luminescence cell imaging by confocal microscopy, HT‐29 colon cancer and PT‐45 pancreatic cancer cells were treated with the respective complexes and an additional marker for the endoplasmic reticulum (ER), mitochondria, F‐actin filaments in the cytoskeleton, and lysosomes, respectively. An analysis of the colocalization of the complexes excited at 405 nm and the respective marker excited at 561 nm by visual observation is shown in Figures S12–S19 (in the Supporting Information). By calculation of Pearson's correlation coefficient (PCC, Table 3), the complexes revealed positive colocalization with the endoplasmic reticulum and with mitochondria, whereas the lysosomal colocalization was less distinct. In contrast to this, no cytoskeletal colocalization was observed.
Table 3.
PCC values calculated for HT‐29 and PT‐45 cells treated with complex 9 a or 9 b and the respective counterstain.
|
Complex |
Cell line |
Alexa Fluor 555 Phalloidin (Cyto‐skeleton) |
ER‐ID red |
Mito‐ID red (Mitochondria) |
LysoTracker Red (Lysosomes) |
|---|---|---|---|---|---|
|
9 a |
HT‐29 |
−0.15 |
0.66 |
0.38 |
0.27 |
|
PT‐45 |
0.08 |
0.78 |
0.50 |
0.21 |
|
|
9 b |
HT‐29 |
0.01 |
0.48 |
0.34 |
0.25 |
|
|
PT‐45 |
−0.12 |
0.71 |
0.23 |
0.30 |
When the two metal centers are compared, the highest degree of colocalization was obtained with the endoplasmic reticulum for both complexes. The rhodium complex showed a slightly higher degree of mitochondrial colocalization than its iridium counterpart, whereas the iridium complex showed a slightly higher degree of lysosomal colocalization. When the two cell lines are compared, the degree of endoplasmic reticulum localization is higher in PT‐45 compared with HT‐29 cells. Interestingly, the mitochondrial localization shows a metal center and cell line dependent behavior: although the rhodium complex shows a higher mitochondrial distribution in PT‐45 cells, the iridium counterpart shows a higher degree of mitochondrial localization in the HT‐29 cells. However, the general metal and cell line related differences in cellular localization are small and the distribution of the complexes can generally be considered independent from the metal center and the respective cancer cell line. The degree of luminescence observed in the cells treated with Ir complex 9 b was slightly lower than of those treated with the Rh congener 9 a, which might be related to a lower degree of cellular uptake of complex 9 b and provides an explanation for differences observed in the cellular distribution patterns of complexes 9 a and 9 b (Figure 4).
Figure 4.
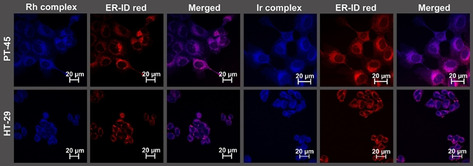
Images obtained by confocal microscopy for PT‐45 and HT‐29 cells incubated with a 2 μm concentration of complex 9 a or 9 b, respectively, in an incubator at 37 °C and 10 % CO2 for 30 min. The cells were subsequently aldehyde fixed and the endoplasmic reticulum was stained with ER‐ID red. Natural luminescence when excited at 405 nm (blue) and 561 nm (red) is shown. The blue luminescence representing complexes 9 a or 9 b shows a good overlay with the red luminescence representing the endoplasmic reticulum. This indicates localization of complexes 9 a and 9 b in the endoplasmic reticulum.
Conclusion
Herein, we report four new [M(COD)(NHC)I] complexes of rhodium and iridium with an NHC ligand derived from 1,8‐naphthalimide, which differ only in the distance between the luminescent 1,8‐naphthalimide core and the respective metal center. Their analysis by 1H NMR spectroscopy and X‐ray crystallography not only revealed the successful formation of the complexes 8 a–9 b but also showed the presence of a dynamic rotational process within complexes 8 a and 8 b. This process is most probably driven by the temporary formation of a halide bridge between the aromatic ring system of the naphthalimide and the iodide ligand.
All of the complexes were investigated for their photophysical behavior. In comparison to the free ligands, the complexes show a significant decrease in luminescence quantum yield. This is interpreted as a PET quenching mechanism in the complexes,[ 38 , 39 , 40 ] which is found to be more pronounced in the case of the rhodium metal center. When the complexes 8 a/b are compared with 9 a/b, the quantum yield is significantly higher in complexes 9 a/b as a consequence of the longer distance between the metal center and the respective 1,8‐naphthalimide core. The aforementioned rotational process present in complex 8 a is reflected in the luminescent properties of the complex, as it shows two spectrally overlapping radiative decays, which most likely stem from the two respective rotamers present in solution at room temperature. Owing to their more favorable luminescence intensities, complexes 9 a/b were chosen for luminescence cell imaging in HT‐29 and PT‐45 cancer cell lines. Within this experiment, the complexes clearly showed cellular uptake, as well as a main localization in the endoplasmic reticulum and mitochondria, whereas only a minor fraction was observed in the lysosomes and almost no cytoskeletal colocalization could be detected. Most remarkably, the complexes do not show any significant metal‐related differences in cellular localization although a slightly lower degree of cellular uptake was present in case of the iridium congener 9 b in comparison to its rhodium counterpart 9 a.
The compounds represent rare examples of iridium(I) and rhodium(I) complexes with a luminescent NHC ligand, which provide emission intensities high enough to be used for cellular imaging. When this feature is exploited within cancer cells, mainly localization of the complexes in the endoplasmic reticulum is observed, which has not been reported so far. Furthermore, the first ever insight into the cellular localization of an iridium(I) NHC complex is provided herein. These findings constitute a valuable addition to the knowledge about the cellular interaction sites of this compound class. The present results further suggest that both types have similar localizations inside of cancer cells, which are largely independent of their metal center. When seen in connection with the established ability of the respective RhI[20–21, 25–26] and IrI[27–30] complexes studied here to bind to proteins, their localization in the endoplasmic reticulum seems to be another clue to contribute to their antiproliferative activity: for some PtII[69–71] and ReI[72] complexes, it was already established that their ability to bind to proteins may trigger ER stress, which ultimately leads to apoptosis, likely via the unfolded protein response mechanism. On balance, a related mechanism may well be in place for our rhodium(I) and iridium(I) NHC complexes, which paves the way for further exploitations of the cytotoxic mechanism of action of this compound class. Future in‐depth studies of these complexes should concentrate on their propensity to induce ER stress and subsequently apoptotic cell death with the aim to establish a more precise cytotoxic mechanism of action beyond the known ability of these compounds to generate ROS.[ 25 , 26 , 29 ]
Experimental Section
Synthesis procedures
Chemicals and materials: All chemicals were purchased from the commercial suppliers Alfa Aesar, Sigma–Aldrich, TCI, or Iris Biotech and used without further purification unless otherwise stated. Air‐ or moisture‐sensitive reactions were performed under a dry and inert nitrogen atmosphere by using Schlenk techniques with application of heavy silicon grease to prevent oxygen or moisture leakages. Solvents were purchased in analytical reagent grade or HPLC grade and used without further purification. Solvents for air‐ and moisture‐sensitive reactions were dried prior use with an MBRAUN MB SPS‐800. After initial distillation, the solvents were dried over 3 Å molecular sieves, and finally dried over an AlOx column and stored under inert nitrogen atmosphere. Methanol was dried over Mg, distilled, and stored under inert nitrogen atmosphere.
6‐Chloro‐2‐propyl‐1 H ‐benzo[de]isoquinoline‐1,3(2 H )‐dione (2): The starting material 1 (5.0 g, 21.49 mmol, 1 equiv) was suspended in EtOH (150 mL). Triethylamine (4.47 mL, 3.26 g, 32.24 mmol, 1.5 equiv) as well as propylamine (2.65 mL, 1.91 g, 32.24 mmol, 1.5 equiv) were added and the reaction mixture was heated at reflux overnight. Cooling of the reaction mixture to room temperature led to precipitation of a yellow solid, which was filtered off and dried. Owing to the color of the leftover reaction mixture, which was anticipated to contain more of the product, the reaction mixture was evaporated to dryness under reduced pressure leading to a yellow solid, which was washed with cold EtOH. Both bright yellow solids were characterized independently and identified as the pure product. The yield was calculated from the combined solids. Yield: 4.95 g (18.08 mmol, 84 %). Analytical data: 1H NMR (300 MHz, chloroform‐d): δ=8.60 (dd, J=7.3, 1.2 Hz, 1 H, arom. CH), 8.51 (dd, J=8.5, 1.2 Hz, 1 H, arom. CH), 8.43 (d, J=7.9 Hz, 1 H, arom. CH), 7.78 (m, 2 H, arom. CH), 4.11 (m, 2 H, N‐CH2), 1.74 (m, 2 H, middle CH2), 1.00 ppm (t, J=7.4 Hz, 3 H, CH3); 13C{1H} NMR (75 MHz, chloroform‐d): δ=163.6 (d, J=19.2 Hz, 2 CO), 139.0 (arom. C‐Cl), 132.0 (arom. CH), 131.1 (arom. CH), 130.6 (arom. CH), 129.3 (arom. CH), 129.1 (arom. CH), 127.9 (arom. C), 127.4 (arom. C), 123.2 (arom. C), 121.7 (arom. C), 42.2 (N‐CH2), 21.5 (middle CH2), 11.6 ppm (CH3).
6‐(1 H ‐Imidazol‐1‐yl)‐2‐propyl‐1 H ‐benzo[de]isoquinoline‐1,3(2 H )‐dione (3): The starting material 2 (1.0 g, 3.65 mmol, 1 equiv), imidazole (323.3 mg, 4.75 mmol, 1.3 equiv) and K2CO3 (1.01 g, 7.31 mmol, 2 equiv) were dissolved in wet DMSO (50 mL) under N2 atmosphere in a Schlenk flask and heated at reflux overnight. The reaction mixture was cooled to room temperature and approximately 150 mL of 1 m NaOH were added, leading to the precipitation of a green, shiny solid, which was filtered off, washed with H2O, and dried under reduced pressure. The product was obtained as a green, shiny solid. Yield: 770 mg (2.52 mmol, 69 %). 1H NMR (300 MHz, [D6]DMSO): δ=8.56 (m, 2 H, 2× arom. CH), 8.14 (t, J=1.1 Hz, 1 H, arom. CH), 8.08 (dd, J=8.6, 1.2 Hz, 1 H, arom. CH), 7.93 (dd, J=9.3, 7.6 Hz, 2 H, arom. CH), 7.73 (t, J=1.3 Hz, 1 H, N‐CH=CH‐N), 7.28 (t, J=1.1 Hz, 1 H, N‐CH=CH‐N), 4.02 (m, 2 H, N‐CH2), 1.67 (m, 2 H, CH2), 0.93 ppm (t, J=7.5 Hz, 3 H, CH3); 13C{1H} NMR (101 MHz, [D6]DMSO): δ=163.2 (CO), 162.7 (CO), 138.9 (arom. C‐N), 138.5 (arom. CH), 131.4 (arom. CH), 130.8 (N‐CH=CH‐N), 129.7 (arom. CH), 129.0 (arom. CH), 128.6 (arom. CH), 128.3 (arom. C), 126.7 (arom. C), 124.4 (arom. C), 122.5 (arom. C), 122.0 (N‐CH=CH‐N), 41.3 (N‐CH2), 20.8 (CH2), 11.3 ppm (CH3); ESI‐MS: 305.9 [M+H]+.
1‐(1,3‐Dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)‐3‐methyl‐1 H ‐imidazol‐3‐ium iodide (4): In a flame‐dried Schlenk flask under N2 atmosphere, the starting material 3 (250 mg, 0.818 mmol, 1 equiv) was dissolved in dry MeCN (25 mL) and methyl iodide (510 μL, 1.16 g, 8.19 mmol, 10 equiv) was added. The reaction mixture was stirred at 65 °C overnight. The reaction mixture was evaporated to dryness under reduced pressure and taken up in a little CH2Cl2 (approx. 3 mL). Addition of the solution to a mixture of hexane/Et2O (45 mL, 1:1) led to the precipitation of a brownish solid, which was filtered off. The solid was dissolved in CH2Cl2 (15 mL) and extracted with 10 % Na2S2O4 (15 mL) repeatedly until the brownish color was completely removed to remove the remaining I2. The organic phase was dried over MgSO4 and evaporated to dryness under reduced pressure, leading to a red‐brown solid, which was characterized as the pure product. The aqueous phase was separated, evaporated to dryness, and a little water was added to dissolve the salts. The water phase was then extracted with MeCN (15 mL), and the organic phase was dried over MgSO4, filtered, and evaporated to dryness under reduced pressure. The remaining red‐brownish solid was as well characterized as the desired product and both solids were combined. Yield: 165 mg (0.370 mmol, 45 %). Analytical data: 1H NMR (200 MHz, [D6]DMSO): δ=9.75 (d, J=2.0 Hz, 1 H, N‐C‐N), 8.65 (m, 2 H, 2× arom. CH), 8.13 (m, 5 H, 3× arom. CH, N‐CH=CH‐N), 4.05 (s, 5 H, 1× N‐CH3, 1× N‐CH2), 1.69 (h, J=7.5 Hz, 2 H, CH2), 0.94 ppm (t, J=7.4 Hz, 3 H, CH3); 13C{1H} NMR (75 MHz, [D6]DMSO): δ=163.0 (CO), 162.5 (CO), 135.9 (N‐C‐N), 131.8 (arom. CH), 130.4 (arom. CH), 129.2 (arom. CH), 128.5 (arom. CH), 128.0 (arom. CH), 126.3 (arom. CH), 125.9 (arom. C), 124.4 (arom. C), 124.2 (arom. C), 124.1 (N‐CH=CH‐N), 122.6 (N‐CH=CH‐N), 41.4 (N‐CH2), 36.4 (N‐CH3), 20.8 (CH2), 11.3 ppm (CH3); HR ESI‐MS: calcd: 320.1394 [M−I−]+; found: 320.1368 [M−I−]+.
6‐((2‐Hydroxyethyl)amino)‐2‐propyl‐1 H ‐benzo[de]isoquinoline‐1,3(2 H )‐dione (5): The starting material 4 (1.0 g, 3.65 mmol, 1 equiv) was dissolved in wet DMSO (50 mL) under N2 atmosphere in a Schlenk flask. Triethylamine (2.53 mL, 2.23 g, 36.53 mmol, 10 equiv) and aminoethanol (2.53 mL, 1.85 g, 18.27 mmol, 5 equiv) were added and the reaction mixture was stirred at 120 °C for 96 h. The reaction mixture was left to cool to room temperature and 1 m NaOH (500 mL) was added, which led to the precipitation of a yellow solid. After filtration of the solid, it was washed with H2O and characterized as the pure product. Yield: 867 mg (2.91 mmol, 80 %). Analytical data: 1H NMR (200 MHz, [D6]DMSO): δ=8.66 (m, 1 H, arom. CH), 8.41 (dd, J=7.4, 1.0 Hz, 1 H, arom. CH), 8.24 (d, J=8.6 Hz, 1 H, arom. CH), 7.66 (m, 2 H, 2× arom. CH), 6.80 (d, J=8.6 Hz, 1 H, NH), 4.88 (t, J=5.5 Hz, 1 H, OH), 4.07–3.86 (m, 2 H, N‐CH2), 3.71 (q, J=5.7 Hz, 2 H, O‐CH2), 3.46 (q, J=5.7 Hz, 2 H, O‐CH2), 1.62 (h, J=7.4 Hz, 2 H, CH2), 0.90 ppm (t, J=7.4 Hz, 3 H, CH3); 13C{1H} NMR (50 MHz, [D6]DMSO): δ=163.7 (CO), 162.9 (CO), 150.8 (arom. C‐N), 134.2 (arom. CH), 130.6 (arom. CH), 129.4 (arom. CH), 128.5 (arom. CH), 124.2 (arom. C), 121.8 (arom. C), 120.1 (arom. C), 107.6 (arom. C), 103.8 (arom. CH), 58.8 (CH2‐OH), 45.5 (N‐CH2), 21.0 (CH2), 11.4 ppm (CH3); ESI‐MS: 320.2 [M+Na]+.
6‐((2‐Iodoethyl)amino)‐2‐propyl‐1 H ‐benzo[de]isoquinoline‐1,3(2 H )‐dione (6): In a flame‐dried Schlenk flask under N2 atmosphere, triphenylphosphine (329.7 mg, 1.26 mmol, 1.25 equiv) and imidazole (171.1 mg, 2.51 mmol, 2.5 equiv) were dissolved in dry THF (25 mL). I2 (331.79 mg, 1.31 mmol, 1.3 equiv) was added and the reaction was stirred at room temperature for 15 min, leading to a brown solution. Afterwards, the starting material 5 (300 mg, 1.01 mmol, 1 equiv) was added, the reaction mixture was heated to 65 °C and left to stir for 3 h. After cooling of the reaction mixture, EtOAc (25 mL) was added and the mixture was extracted twice with 10 % Na2S2O4 (35 mL) in water and twice with brine (35 mL). The organic layer was dried over Na2SO4, filtered, and evaporated to dryness under reduced pressure. The crude product was purified by flash column chromatography over silica (EtOAc/n‐hexane 0:1→1:1) yielding the product as a yellow solid. Yield: 270 mg (0.661 mmol, 65 %). Analytical data: R f (n‐hexane/EtOAc 8:2): 0.10; 1H NMR (200 MHz, chloroform‐d): δ=8.58 (dd, J=7.3, 1.0 Hz, 1 H, arom. CH), 8.45 (d, J=8.4 Hz, 1 H, arom. CH), 8.14 (dd, J=8.5, 1.1 Hz, 1 H, arom. CH), 7.64 (dd, J=8.5, 7.3 Hz, 1 H, arom. CH), 6.72 (d, J=8.4 Hz, 1 H, arom. CH), 5.61 (d, J=5.9 Hz, 1 H, NH), 4.11 (m, 2 H, N‐CH2), 3.81 (q, J=6.2 Hz, 2 H, NH‐CH2), 3.51 (t, J=6.4 Hz, 2 H, I‐CH2), 1.75 (m, 2 H, CH2), 1.00 ppm (t, J=7.4 Hz, 3 H, CH3); 13C{1H} NMR (50 MHz, chloroform‐d): δ=164.7 (CO), 164.2 (CO), 148.1 (arom. C‐N), 134.2 (arom. CH), 131.4 (arom. CH), 129.9 (arom. CH), 125.9 (arom. CH), 125.3 (arom. C), 123.5 (arom. C), 120.7 (arom. C), 111.8 (arom. C), 104.7 (arom. CH), 45.4 (NH‐CH2), 41.9 (N‐CH2), 21.6 (CH2), 11.7 (CH3), 3.7 ppm (I‐CH2); ESI‐MS: 304.1 [M−I+Na]+, 408.8 [M+H]+, 430.7 [M+Na]+.
1‐(2‐((1,3‐Dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)amino)ethyl)‐3‐methyl‐1 H ‐imidazol‐3‐ium iodide (7): In a flame‐dried Schlenk flask under N2 atmosphere, the starting material 6 (250 mg, 0.612 mmol, 1 equiv) was dissolved in dry THF (25 mL). 1‐Methylimidazole (73 μL, 75.42 mg, 0.918 mmol, 1.5 equiv) was added, the reaction mixture was heated to 65 °C and left to stir for 72 h, during which a yellow solid precipitated. The reaction mixture was allowed to cool to room temperature and the solution was concentrated under reduced pressure. The leftover reaction mixture was centrifuged for 10 min at 10 000 rpm to collect the yellow solid, which was washed twice with THF and dried under reduced pressure to yield the pure product as a yellow solid. Yield: 210 mg (0.428 mmol, 69 %). Analytical data: 1H NMR (200 MHz, [D6]DMSO): δ=9.15 (s, 1 H, N‐CH‐N), 8.63 (dd, J=8.6, 1.1 Hz, 1 H, arom. CH), 8.51 (dd, J=7.3, 0.9 Hz, 1 H, arom. CH), 8.34 (d, J=8.5 Hz, 1 H, arom. CH), 7.79 (m, 4 H, 2× arom. CH, 2× N‐CH=CH‐N), 6.96 (d, J=8.6 Hz, 1 H, NH), 4.56 (t, J=5.6 Hz, 2 H, CH2‐N), 3.96 (m, 7 H, CH2‐N, CH2‐NH, N‐CH3), 1.66 (p, J=7.4 Hz, 2 H, CH2), 0.95 ppm (t, J=7.4 Hz, 3 H, CH3); 13C{1H} NMR (75 MHz, [D6]DMSO): δ=163.7 (CO), 163.0 (CO), 149.9 (arom. C‐N), 136.9 (arom. CH), 130.8 (arom. CH), 129.3 (arom. CH), 128.5 (arom. CH), 124.7 (arom. C), 123.6 (arom. C), 122.9 (N‐CH=CH‐N), 122.0 (arom. C), 120.4 (arom. C), 108.9 (arom. C), 104.2 (arom. CH), 47.5 (NH‐CH2), 42.6 (N‐CH2), 35.8 (N‐CH3), 21.0 (CH2), 11.4 ppm (CH3); ESI‐MS: 363.1 [M−I−]+; HR ESI‐MS: calcd: 363.1816 [M−I−]+; found: 363.1783 [M−I−]+.
Luminescent [M(COD)(NHC)Cl] complexes
General procedure: In a flame‐dried Schlenk flask under N2 atmosphere, 1.0 equiv of the imidazolium salt 4 and 0.5 equiv of the respective [M(COD)Cl]2 (M=Rh or Ir) were dissolved in dry CH2Cl2 (40 mL), which was previously degassed by three consecutive cycles of freeze–pump–thaw in the case of the Ir precursor to prevent its oxidation, per mmol of imidazolium salt. After addition of 3 equiv of DIPEA, the reaction mixture was heated at reflux overnight, leading to a color change from yellow to red. The reaction mixture was concentrated under reduced pressure and the crude product was purified by flash column chromatography over silica (EtOAc/n‐hexane 0:1→1:0), yielding the product as a dark‐red solid.
1,5‐Cyclooctadiene[1‐(1,3‐dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)‐3‐methyl‐1 H ‐imidazol‐3‐ide]iodorhodium(I) (8 a): The product was obtained as a dark‐red solid. Crystals were grown at −20 °C from a mixture of CH2Cl2 and hexane. Yield: 52 mg (0.079 mmol, 71 %). Analytical data: R f (n‐hexane/EtOAc, 2:8): 0.86; 1H NMR (400 MHz, chloroform‐d, measured at 243 K): δ=9.04 (d, J=7.8 Hz, 1 H, arom. CH), 8.71 (d, J=7.9 Hz, 1 H, arom. CH), 8.59 (d, J=7.2 Hz, 1 H, arom. CH), 7.91 (d, J=8.5 Hz, 1 H), 7.75 (t, J=7.9 Hz, 1 H, arom. CH), 7.23 (m, 2 H, N‐CH=CH‐N), 5.03 (dt, J=11.6, 6.5 Hz, 2 H, 2× COD CH), 4.09 (m, 6 H, 2× N‐CH2, N‐CH3), 3.46 (d, J=7.2 Hz, 1 H, COD CH), 2.36 (q, J=6.5 Hz, 1 H, COD CH), 2.20 (m, 1 H, COD CH2), 2.11 (d, J=8.4 Hz, 1 H, COD CH2), 1.64 (dtd, J=39.9, 13.2, 10.8, 7.0 Hz, 6 H, 3× COD CH2), 1.11 (m, 1 H, COD CH2), 0.98 (t, J=7.4 Hz, 3 H, CH3), 0.82 ppm (ddt, J=21.5, 14.1, 7.1 Hz, 1 H, COD CH2); 13C{1H} NMR (101 MHz, chloroform‐d, measured at 243 K): δ=184.9 (d, J=49.3 Hz, N‐C‐N), 163.5 (d, J=48.7 Hz, 2× CO), 140.93 (arom. C‐N), 131.6 (arom. CH), 130.6 (arom. CH), 128.2 (arom. CH), 128.1 (arom. CH), 127.8 (arom. CH), 127.1 (arom. C), 123.7 (N‐CH=CH‐N), 123.2 (arom. C), 123.0 (arom. C), 122.3 (arom. C), 96.6 (d, J=23.0 Hz, COD CH), 71.2 (COD CH), 53.3 (COD CH), 42.1 (N‐CH2), 39.1 (N‐CH3), 33.0 (COD CH2), 31.9 (COD CH2), 30.6 (COD CH2), 29.6 (COD CH2), 29.1 (COD CH2), 28.7 (COD CH2), 21.3 (CH2), 11.6 ppm (CH3); ESI‐MS: 529.8 [M−I]+; HR ESI‐MS: calcd: 530.1315 [M−I−]+; found: 530.1285 [M−I−]+.
1,5‐Cyclooctadiene[1‐(1,3‐dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)‐3‐methyl‐1 H ‐imidazol‐3‐ide]iodoiridium(I) (8 b): The product was obtained as a dark‐red solid. Yield: 50 mg (0.067 mmol, 66 %). Analytical data: R f (n‐hexane/EtOAc, 1:1): 0.39; 1H NMR (250 MHz, chloroform‐d): δ=8.69 (m, 3 H, 3× arom. CH), 7.78 (dd, J=8.5, 7.3 Hz, 2 H, 2× arom. CH), 7.18 (dd, J=13.4, 2.0 Hz, 2 H, N‐CH=CH‐N), 4.52 (s, 2 H, 2× COD CH), 4.19 (m, 6 H, N‐CH2, N‐CH3), 2.91 (d, J=30.2 Hz, 2 H, 2× COD CH), 1.91 (m, 6 H, 2× COD CH2, CH2), 1.42 (d, J=7.9 Hz, 2 H, COD CH2), 1.04 ppm (t, J=7.4 Hz, 5 H, COD CH2, CH3); 13C{1H} NMR (101 MHz, chloroform‐d, measured at 243 K): δ=182.2 (N‐C‐N), 164.1 (CO), 163.6 (CO), 140.8 (arom. C‐N), 131.7 (arom. CH), 130.9 (arom. CH), 128.9 (arom. CH), 128.8 (arom. CH), 128.4 (arom. C), 128.2 (arom. C), 126.8 (arom. C), 124.2 (arom. C), 123.1 (arom. C), 122.8 (d, J=8.5 Hz, N‐CH=CH‐N), 122.2 (arom. CH), 85.2 (d, J=28.6 Hz, COD CH), 51.8 (d, J=38.6 Hz, COD CH), 42.3 (N‐CH2), 38.2 (N‐CH3), 33.6 (COD CH2), 32.7 (COD CH2), 29.3 (d, J=26.3 Hz, COD CH2), 27.9 (COD CH2), 21.4 (CH2), 11.8 ppm (d, J=2.6 Hz, CH3); ESI‐MS: 619.8 [M−I]+; HR ESI‐MS: calcd: 620.1889 [M−I−]+; found: 620.1889 [M−I−]+.
1,5‐Cyclooctadiene[1‐(2‐((1,3‐dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)amino)ethyl)‐3‐methyl‐1 H ‐imidazol‐3‐ide]iodorhodium(I) (9 a): The imidazolium salt 7 (50 mg, 0.102 mmol, 1 equiv) was dissolved in dry CH2Cl2 (6 mL) in a flame‐dried Schlenk flask under N2 atmosphere. To increase the solubility of the imidazolium salt, dry MeOH (approx. 4 mL) was added to the reaction mixture until the starting material was nearly fully dissolved. [Rh(COD)Cl]2 (25.14 mg, 0.051 mmol, 0.5 equiv) and DIPEA (52.02 μL, 39.54 mg, 0.306 mmol, 3 equiv) were added and the reaction mixture was left to reflux overnight. The reaction mixture was concentrated under reduced pressure and the crude product was purified by flash column chromatography over silica (EtOAc/n‐hexane 0:1→1:0), yielding the product as a dark‐yellow solid. Crystals were grown by slow evaporation of CDCl3. The product was stored in the dark. Yield: 21 mg (0.030 mmol, 29 %). Analytical data: 1H NMR (200 MHz, chloroform‐d): δ=8.43 (m, 3 H, arom. CH), 7.52 (dd, J=8.5, 7.3 Hz, 1 H, arom. CH), 6.81 (d, J=8.5 Hz, 1 H, arom. CH), 6.72 (d, J=1.9 Hz, 1 H, N‐CH=CH‐N), 6.62 (d, J=1.9 Hz, 1 H, N‐CH=CH‐N), 6.25 (t, J=5.9 Hz, 1 H, NH), 5.25 (m, 2 H, 2× COD CH), 4.90 (dtd, J=12.6, 8.6, 7.9, 5.2 Hz, 2 H, N‐CH2), 4.05 (m, 7 H, N‐CH2, N‐CH2, N‐CH3), 3.53 (m, 2 H, 2× COD CH), 2.30 (m, 4 H, 2× COD CH2), 1.76 (m, 6 H, 2× COD CH2, CH2), 0.98 ppm (m, 3 H, CH3); 13C{1H} NMR (50 MHz, chloroform‐d): δ=181.8 (d, J=48.4 Hz, N‐C‐N), 164.5 (d, J=25.9 Hz, 2× CO), 149.1 (arom. C‐N), 134.2 (arom. CH), 131.4 (arom. CH), 130.0 (arom. CH), 128.1 (arom. CH), 125.0 (arom. C), 122.6 (d, J=28.4 Hz, N‐CH=CH‐N), 120.7 (arom. C), 110.8 (arom. C), 103.9 (arom. CH), 97.0 (COD CH), 72.8 (d, J=14.3 Hz, COD CH), 71.0 (m, COD CH), 48.2 (N‐CH2), 41.9 (N‐CH2), 38.9 (N‐CH3), 33.1 (COD CH2), 31.9 (COD CH2), 30.3 (COD CH2), 29.1 (COD CH2), 21.6 (CH2), 11.7 ppm (CH3); ESI‐MS: 573.4 [M−I]+; HR ESI‐MS: calcd: 573.1737 [M−I−]+; found: 573.1731 [M−I−]+.
1,5‐Cyclooctadiene[1‐(2‐((1,3‐dioxo‐2‐propyl‐2,3‐dihydro‐1 H ‐benzo[de]isoquinolin‐6‐yl)amino)ethyl)‐3‐methyl‐1 H ‐imidazol‐3‐ide]iodoiridium(I) (9 b): In a flame‐dried Schlenk flask under N2 atmosphere, the imidazolium salt 7 (50 mg, 0.102 μmol, 1 equiv) was dissolved in dry MeCN (5 mL). The solution was degassed by three consecutive cycles of freeze–pump–thaw to prevent oxidation of the Ir precursor. After addition of freshly prepared Ag2O (11.81 mg, 0.051 mmol, 0.5 equiv), the solution was left to stir at room temperature for 1 h in the dark, during which disappearance of the black Ag2O could be seen, which was followed by addition of [Ir(COD)Cl]2 (42.81 mg, 0.051 mmol, 0.5 equiv) leading to a color change to bright yellow. The solution was then left to stir overnight at room temperature. Afterwards, the reaction mixture was filtered through Celite (CH2Cl2/MeOH 9:1) and the filtrate was concentrated under reduced pressure. Purification of the product was carried out by flash column chromatography over silica (EtOAc/n‐hexane 0:1→1:0), yielding a dark‐yellow oily residue, which was taken up in as little EtOAc as possible and precipitated from n‐hexane to yield the product as a dark‐yellow solid. The product was stored in the dark. Yield: 18 mg (0.023 mmol, 22 %). Analytical data: 1H NMR (400 MHz, chloroform‐d): δ=8.49 (m, 3 H, arom. CH), 7.58 (dd, J=8.4, 7.3 Hz, 1 H, arom. CH), 6.72 (m, 3 H, arom. CH, N‐CH=CH‐N), 6.51 (t, J=5.9 Hz, 1 H, NH), 5.04 (ddd, J=13.9, 6.0, 4.5 Hz, 1 H, 1× CH2), 4.67 (dtt, J=11.6, 8.9, 4.4 Hz, 2 H, 2× COD CH), 4.31 (ddd, J=13.8, 8.1, 4.3 Hz, 1 H, 1× CH2), 4.08 (m, 7 H, N‐CH3, N‐CH2, NH‐CH2), 3.04 (m, 2 H, 2× COD CH), 2.22 (m, 4 H, 2× COD CH2), 1.74 (m, 6 H, 2× COD CH2, CH2), 1.00 ppm (t, J=7.4 Hz, 3 H, N‐CH3); ESI‐MS: 662.9 [M−I]+; HR ESI‐MS: calcd: 663.2311 [M−I−]+; found: 663.2323 [M−I−]+.
X‐ray crystallography
Single crystals suitable for X‐ray diffraction experiments were analyzed by using a Rigaku XtaLAB Supernova or a Rigaku XtaLAB Synergy (CuKα, λ=1.54184 Å) diffractometer. The crystals were handled in perfluorinated oil, mounted on fiber loops and cooled by a cold gas flow (N2) throughout the measurement. The obtained diffraction data was processed with the CrysAlisPro (Oxford Diffraction) software package. The structure solutions were computed by using SHELXT (intrinsic phasing) [73] and were refined against F 2 by using SHELXL. [74] The OLEX2[75] software package was used as a graphical user interface.
Photochemical characterization
The steady‐state absorption spectra were recorded with the commercially available spectrophotometers V‐770 (JASCO) and UV‐1800 (Shimadzu), while for stationary emission spectroscopy the spectrofluorometers FP‐8300 (JASCO) and Fluorolog‐3 (Horiba) were used. The time‐resolved emission spectroscopy was performed by using a combination of a streak camera (Hamamatsu C10 627‐11) with a monochromator (Princeton Instruments SP2150i). The samples were excited by a laser diode with a wavelength of 405 nm (PicoQuant LDH‐P‐C‐405B). The used rectangular quartz glass cuvettes (Starna) have internal dimensions of 10 mm×2 mm, holding a sample volume of 700 μL. The luminescence quantum yields were determined by measurements with an Ulbricht sphere (Hamamatsu C9920‐02 system equipped with a Spectralon integrating sphere). Its quantum yield accuracy is <10 % according to the manufacturer. All experiments were carried out in non‐degassed solvents, namely acetone (99.6 % for spectroscopy, Acros Organics), DMSO (Uvasol, for spectroscopy, Merck) and water (for spectroscopy, Acros Organics).
Cellular imaging
Cell culture: Work in cell culture was performed in sterile conditions by using a laminar flow bench. All necessary materials were either purchased sterile, autoclaved, or superficially disinfected with 70 % of ethanol in water before entering the device. The PT‐45 cell line was received from H. Kalthoff, Universitätsklinikum Eppendorf (Hamburg, Germany); HT‐29 cells were received from A. Schumacher, Max‐Delbrück Center (Berlin, Germany). The authenticity of the cells used in this work was confirmed by STR profiling and comparison to the STR data in the Expasy database (for HT‐29, https://web.expasy.org/cellosaurus). PT‐45 cells are not represented in public STR databases, therefore the stability of the profile was ensured by comparing it to the STR profile of the founder cell in our laboratory. STR analyses were performed as described by Dirks et al. [76] and analyzed with a CEQ8800 sequencer (Beckman Coulter).
All cell lines were cultured in DMEM (Dulbecco′s Modified Eagle Medium, high glucose) with addition of 10 % fetal calf serum, 1 % sodium pyruvate, 100 units mL−1 Penicillin, and 100 μg mL−1 Streptomycin. Cells were maintained in cell culture dishes with a diameter of 10 cm at 37 °C and 5–10 % CO2 in a humidified incubator. The culture medium was replaced regularly by fresh DMEM to keep the cells in optimal growth conditions. Reaching a confluency of 80 % or higher, the cells were passaged. Therefore, the cells were treated with 0.05 % Trypsin/EDTA in DMEM (2.5 mL) at 37 °C and 5–10 % CO2 for approximately 3 min, until detachment of the cells from the surface of the culture dish became visible under the microscope. In the following, fresh DMEM (3.5 mL) was added and the solution was transferred to a 50 mL Falcon tube and centrifuged at 1800 rpm for 2 min or 1500 rpm for 3 min. The remaining medium was removed and replaced with fresh DMEM (10 mL). Depending on the desired cell count, an aliquot of the cell suspension was diluted with the respective amount of DMEM to a total volume of 8–10 mL in a new culture dish with a diameter of 10 cm and the cells were grown under the conditions stated above.
Sample preparation for confocal microscopy
Preparation of acid‐cleaned cover slips: The cover slips were separated and placed in a 250 mL Schott bottle. 1 m HCl (250 mL) was added, the solution heated to 60 °C, and left overnight. When the solution had cooled to room temperature, the HCl was removed and the cover slips were first rinsed two times with ultrapure water (250 mL) and then sonicated two times in ultrapure water (250 mL) for 30 min. Afterwards, the cover slips were treated with increasing amounts of ethanol. During this procedure, the cover slips were successively sonicated for 30 min in 50 %, 70 %, and 95 % EtOH in ultrapure water. Afterwards, the solution was replaced by absolute EtOH, the bottle was sealed and the cover slips were stored.
Preparation of poly‐l‐lysine‐coated cover slips: All steps were performed with a laminar flow bench. The previously acid‐cleaned cover slips were used directly from the storage container and placed separately in a 6‐well plate. After the remaining EtOH had dried off, the cover slips were covered with 0.01 % poly‐l‐lysine solution (1 mL) each and shaken at room temperature for 30 min. The poly‐l‐lysine solution was removed and the cover slips were washed ten times for 5 min with ultrapure water (1 mL) each. After washing, the cover slips were placed in absolute EtOH (1 mL), the EtOH was removed, and the cover slips were left to dry.
Cell preparation: All steps were performed with a laminar flow bench. When the cells had reached a confluency of 80 % or higher, the cells were passaged as described. The cell pellet was resuspended in fresh DMEM (10 mL) and the cells were counted. Thereafter, 10 μL of the cell suspension was transferred to a Neubauer counting chamber, the cell count was averaged and extrapolated to 10 mL. To reach optimal conditions for microscopy, the cells were grown in a 6‐well plate equipped with a poly‐l‐lysine‐coated cover slip. Therefore, the desired amount of cell suspension was diluted with DMEM to a total volume of 2 mL per well to reach a cell number of 1.5×105 cells per m The cells were left to grow on the cover slips at 37 °C and 5–10 % CO2 in a humidified incubator overnight.
Treatment with LysoTracker Red: All steps were performed with a laminar flow bench. For co‐localization studies with LysoTracker Red, the cells were treated with LysoTracker Red before being treated with the respective fluorescent compound (9 a or 9 b). Therefore, the commercially available 1 mm stock solution of LysoTracker Red in DMSO was diluted with DMEM to a final concentration of 50 nm, then 2 mL of the mixture were added to the cells grown on poly‐l‐lysine‐coated cover slips and they were incubated for 30 min at 37 °C and 5–10 % CO2 in a humidified incubator. After incubation, the medium was removed, the cells were washed with PBS and treatment of the cells with the respective luminescent compound was performed as described.
Treatment with luminescent compounds 9 a, 9 b: All steps were performed with a laminar flow bench. To treat the cells with the respective luminescent probe, a 1 mm stock solution of the respective compound in DMSO was prepared and diluted with fresh DMEM to a final concentration of 2 μm and added to the prepared cells grown on poly‐l‐lysine‐coated cover slips. During this procedure, the medium in the wells was removed, and replaced by 2 mL of the 2 μm solution of the luminescent compound in DMEM. The cells were then incubated for 30 min at 37 °C and 5–10 % CO2 in a humidified incubator. After treatment, the medium was removed, the cells were washed with 1 mL of PBS, and aldehyde fixed.
Aldehyde fixation: Aldehyde fixation was performed in non‐sterile conditions. For the fixation of the cells treated with the respective luminescent probes, 1 mL of a solution of 4 % of paraformaldehyde in PBS was added to the cells grown on poly‐l‐lysine‐coated cover slips and incubated for 15 min at room temperature. In the following, the cells were washed three times for 5 min with PBS (1 mL) and treated with the respective luminescent probe or subjected to permeabilization.
Treatment with Alexa Fluor 555 Phalloidin: For the treatment of the aldehyde‐fixed cells with Alexa Fluor 555 Phalloidin, the cell membrane was permeabilized. The cells were incubated for 10 min with a solution of 0.2 % Triton‐X 100 in PBS and washed two times for 5 min with PBS in the following. The commercially available stock solution of Phalloidin Alexa Fluor 555 (8 μL) was diluted with 1 % BSA (392 μL) in PBS and the cover slips were incubated with the resulting solution (30 μL) for 15 min at room temperature in the dark. After incubation, the cover slips were washed three times for 5 min with PBS (1 mL) and once with ultrapure water (1 mL) and mounted on microscopy slides as described.
Treatment with Mito‐ID red: Treatment with Mito‐ID red was performed on aldehyde‐fixed cells. The commercially available detection reagent (1 μL) was diluted with ultrapure water (2499 μL) and the cover slips were incubated with the resulting solution (100 μL) for 30 min at room temperature in the dark. In the following, the cover slips were successively washed with PBS (1 mL) for 5 min and ultrapure water (1 mL) for 5 min and mounted on microscopy slides as described.
Treatment with ER‐ID red: Treatment with ER‐ID red was performed on aldehyde‐fixed cells. The commercially available detection reagent (1 μL) was diluted with ultrapure water (999 μL) and the cover slips were incubated with the resulting solution (100 μL) for 30 min at room temperature in the dark. In the following, the cover slips were washed for 5 min with PBS (1 mL) and mounted on microscopy slides as described in the following.
Mounting of cover slips on microscopy slides: Cover slips were mounted on microscopy slides with ProLong Gold Anti Fade mountant. A small drop of the mountant free of air bubbles was transferred to the slide and covered with the cover slip. The mountant was left to harden in the dark at 4 °C for at least 72 h before the samples were imaged by confocal microscopy. The samples were stored under similar conditions. Confocal microscopy images were recorded with a Zeiss Axiovert 100 m confocal microscope (Dept. of Anatomy and Embryology at RUB) and a Leica TCS SP8 confocal laser scanning microscope (Chair of Biochemistry II at RUB).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We kindly acknowledge the help of Martin Gartmann, who recorded the NMR spectra of the complexes 8 a and 8 b, and of Dr. Dirk Wolters for measuring high‐resolution mass spectra. Support from Milena John during the revision of this work is also gratefully acknowledged. I.M.D. is very grateful to Britta Redeker and Dr. Abdelouahid Maghnouj, who introduced her to cell culture and cellular luminescence microscopy and helped to generate the results presented herein. Access to confocal microscopes in the Department of Anatomy and Embryology at the Ruhr‐Universität Bochum and at the Chair of Biochemistry II—Molecular Biochemistry is kindly acknowledged. S.W. and P.N. thank Prof. Dr. Martina Havenith for providing streak‐camera equipment. Open access funding enabled and organized by Projekt DEAL.
I. M. Daubit, S. Wortmann, D. Siegmund, S. Hahn, P. Nuernberger, N. Metzler-Nolte, Chem. Eur. J. 2021, 27, 6783.
References
- 1. Rosenberg B., VanCamp L., Cancer Res. 1970, 30, 1799–1802. [PubMed] [Google Scholar]
- 2. Rosenberg B., VanCamp L., Trosko J. E., Mansour V. H., Nature 1969, 222, 385–386. [DOI] [PubMed] [Google Scholar]
- 3. Rosenberg B., Van Camp L., Krigas T., Nature 1965, 205, 698–699. [DOI] [PubMed] [Google Scholar]
- 4. Giraldi T., Zassinovich G., Mestroni G., Chem. Biol. Interactions 1974, 9, 389–394. [DOI] [PubMed] [Google Scholar]
- 5. Giraldi T., Sava G., Mestroni G., Zassinovich G., Stolfa D., Chem. Biol. Interactions 1978, 22, 231–238. [DOI] [PubMed] [Google Scholar]
- 6. Liu Z., Sadler P. J., Acc. Chem. Res. 2014, 47, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma D.-L., Wang M., Mao Z., Yang C., Ng C.-T., Leung C.-H., Dalton Trans. 2016, 45, 2762–2771. [DOI] [PubMed] [Google Scholar]
- 8. Ma D.-L., Chan D. S.-H., Leung C.-H., Acc. Chem. Res. 2014, 47, 3614–3631. [DOI] [PubMed] [Google Scholar]
- 9. Leung C.-H., Zhong H.-J., Chan D. S.-H., Ma D.-L., Coord. Chem. Rev. 2013, 257, 1764–1776. [Google Scholar]
- 10. Geldmacher Y., Oleszak M., Sheldrick W. S., Inorg. Chim. Acta 2012, 393, 84–102. [Google Scholar]
- 11. Carrasco A. C., Rodriguez-Fanjul V., Habtemariam A., Pizarro A. M., J. Med. Chem. 2020, 63, 4005–4021. [DOI] [PubMed] [Google Scholar]
- 12. Conesa J. J., Carrasco A. C., Rodriguez-Fanjul V., Yang Y., Carrascosa J. L., Cloetens P., Pereiro E., Pizarro A. M., Angew. Chem. Int. Ed. 2020, 59, 1270–1278; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1286–1294. [Google Scholar]
- 13. Dömötör O., Enyedy E. A., J. Biol. Inorg. Chem. 2019, 24, 703–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Konkankit C. C., Marker S. C., Knopf K. M., Wilson J. J., Dalton Trans. 2018, 47, 9934–9974. [DOI] [PubMed] [Google Scholar]
- 15. Lord R. M., McGowan P. C., Chem. Lett. 2019, 48, 916–924. [Google Scholar]
- 16. Petrini A., Pettinari R., Marchetti F., Pettinari C., Therrien B., Galindo A. N., Scopelliti R., Riedel T., Dyson P. J., Inorg. Chem. 2017, 56, 13600–13612. [DOI] [PubMed] [Google Scholar]
- 17. Tabrizi L., Chiniforoshan H., Dalton Trans. 2017, 46, 2339–2349. [DOI] [PubMed] [Google Scholar]
- 18. Simpson P. V., Schmidt C., Ott I., Bruhn H., Schatzschneider U., Eur. J. Inorg. Chem. 2013, 5547–5554. [Google Scholar]
- 19. McConnell J. R., Rananaware D. P., Ramsey D. M., Buys K. N., Cole M. L., McAlpine S. R., Bioorg. Med. Chem. Lett. 2013, 23, 2527–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oehninger L., Spreckelmeyer S., Holenya P., Meier S. M., Can S., Alborzinia H., Schur J., Keppler B. K., Wölfl S., Ott I., J. Med. Chem. 2015, 58, 9591–9600. [DOI] [PubMed] [Google Scholar]
- 21. Oehninger L., Küster L. N., Schmidt C., Muñoz-Castro A., Prokop A., Ott I., Chem. Eur. J. 2013, 19, 17871–17880. [DOI] [PubMed] [Google Scholar]
- 22. Streciwilk W., Terenzi A., Lo Nardo F., Prochnow P., Bandow J. E., Keppler B. K., Ott I., Eur. J. Inorg. Chem. 2018, 3104–3112. [Google Scholar]
- 23. Streciwilk W., Terenzi A., Misgeld R., Frias C., Jones P. G., Prokop A., Keppler B. K., Ott I., ChemMedChem 2017, 12, 214–225. [DOI] [PubMed] [Google Scholar]
- 24. Streciwilk W., Terenzi A., Cheng X., Hager L., Dabiri Y., Prochnow P., Bandow J. E., Wölfl S., Keppler B. K., Ott I., Eur. J. Med. Chem. 2018, 156, 148–161. [DOI] [PubMed] [Google Scholar]
- 25. Fan R., Bian M., Hu L., Liu W., Eur. J. Med. Chem. 2019, 183, 111721–111736. [DOI] [PubMed] [Google Scholar]
- 26. Zhang J.-J., Muenzner J. K., Abu el Maaty M. A., Karge B., Schobert R., Wölfl S., Ott I., Dalton Trans. 2016, 45, 13161–13168. [DOI] [PubMed] [Google Scholar]
- 27. Gothe Y., Romero-Canelón I., Marzo T., Sadler P. J., Messori L., Metzler-Nolte N., Eur. J. Inorg. Chem. 2018, 2461–2470. [Google Scholar]
- 28. Gothe Y., Marzo T., Messori L., Metzler-Nolte N., Chem. Eur. J. 2016, 22, 12487–12494. [DOI] [PubMed] [Google Scholar]
- 29.Y. Gothe, Ph.D. thesis, Ruhr-Universität Bochum, Bochum, Germany, 2016.
- 30. Gothe Y., Marzo T., Messori L., Metzler-Nolte N., Chem. Commun. 2015, 51, 3151–3153. [DOI] [PubMed] [Google Scholar]
- 31. Sánchez-Mora A., Valdés H., Ramírez-Apan M. T., Nieto-Camacho A., Hernández-Ortega S., Canseco-González D., Morales-Morales D., Inorg. Chim. Acta 2019, 496, 119061. [Google Scholar]
- 32. Fernández-Moreira V., Thorp-Greenwood F. L., Coogan M. P., Chem. Commun. 2010, 46, 186–202. [DOI] [PubMed] [Google Scholar]
- 33. Coogan M. P., Pope S. J. A., The Chemistry of Molecular Imaging, 1st Ed.(Eds.: N. Long ,, Wong W.-T.), John Wiley and Sons Inc., New York, 2015, pp. 275–298. [Google Scholar]
- 34. Lanoë P.-H., Chan J., Gontard G., Monti F., Armaroli N., Barbieri A., Amouri H., Eur. J. Inorg. Chem. 2016, 1631–1634. [Google Scholar]
- 35. Lanoë P. H., Chan J., Groue A., Gontard G., Jutand A., Rager M. N., Armaroli N., Monti F., Barbieri A., Amouri H., Dalton Trans. 2018, 47, 3440–3451. [DOI] [PubMed] [Google Scholar]
- 36. Nelson D. J., Nolan S. P., Falivene L., Poater A., Cavallo L., Wu L., Salvador A., Dorta R., Hoyos M., Guest D., Navarro O., Dierick S., Markó I. E., Wagers P. O., Shelton K. L., Panzner M. J., Tessier C. A., Youngs W. J., Lazreg F., Cazin C. S. J., Wurm T., Asiri A. M., Hashmi A. S. K., Poyatos M., Guisado-Barrios G., Peris E., Mavila S., Lemcoff N. G., Schwartsburd L., Whittlesey M. K., Haynes M. T. I., Jackson E. P., Montgomery J., Bellemin-Laponnaz S., Dagorne S., Murphy L. J., Robertson K. N., Masuda J. D., Clyburne J. A. C., Slaughter L. M., in N-Heterocyclic Carbenes—Effective Tools for Organometallic Synthesis, (Ed.: Nolan S. P.), Wiley-VCH, Weinheim, 2014. [Google Scholar]
- 37. Hopkinson M. N., Richter C., Schedler M., Glorius F., Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- 38. Kos P., Plenio H., Chem. Eur. J. 2015, 21, 1088–1095. [DOI] [PubMed] [Google Scholar]
- 39. Kos P., Plenio H., Angew. Chem. Int. Ed. 2015, 54, 13293–13296; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13491–13494. [Google Scholar]
- 40. Halter O., Plenio H., Eur. J. Inorg. Chem. 2018, 2935–2943. [Google Scholar]
- 41. Banerjee S., Veale E. B., Phelan C. M., Murphy S. A., Tocci G. M., Gillespie L. J., Frimannsson D. O., Kelly J. M., Gunnlaugsson T., Chem. Soc. Rev. 2013, 42, 1601–1618. [DOI] [PubMed] [Google Scholar]
- 42. Llombart M., Poveda A., Forner E., Fernández-Martos C., Gaspar C., Muñoz M., Olmos T., Ruiz A., Soriano V., Benavides A., Martin M., Schlick E., Guillem V., Invest. New Drugs 1992, 10, 177–181. [DOI] [PubMed] [Google Scholar]
- 43. Freeman C. L., Swords R., Giles F. J., Expert Rev. Hematol. 2012, 5, 17–26. [DOI] [PubMed] [Google Scholar]
- 44. Groves L. M., Williams C. F., Hayes A. J., Ward B. D., Isaacs M. D., Symonds N. O., Lloyd D., Horton P. N., Coles S. J., Pope S. J. A., Dalton Trans. 2019, 48, 1599–1612. [DOI] [PubMed] [Google Scholar]
- 45. Langdon-Jones E. E., Symonds N. O., Yates S. E., Hayes A. J., Lloyd D., Williams R., Coles S. J., Horton P. N., Pope S. J., Inorg. Chem. 2014, 53, 3788–3797. [DOI] [PubMed] [Google Scholar]
- 46. Chu G. M., Fernández I., Guerrero-Martínez A., Ramírez de Arellano C., Sierra M. A., Eur. J. Inorg. Chem. 2016, 844–852. [DOI] [PubMed] [Google Scholar]
- 47. Wang D.-H., Gong Z., Sun R., Zhao D.-Z., New J. Chem. 2015, 39, 5991–5996. [Google Scholar]
- 48. Gutiérrez-Blanco A., Peris E., Poyatos M., Organometallics 2018, 37, 4070–4076. [Google Scholar]
- 49. Chianese A. R., Li X., Janzen M. C., Faller J. W., Crabtree R. H., Organometallics 2003, 22, 1663–1667. [Google Scholar]
- 50. Frey G. D., Herdtweck E., Herrmann W. A., J. Organomet. Chem. 2006, 691, 2465–2478. [Google Scholar]
- 51. Doyle M. J., Lapper M. F., J. Chem. Soc. Chem. Commun. 1974, 197, 679–680. [Google Scholar]
- 52. Jamil M. S. S., Alkaabi S., Brisdon A. K., Dalton Trans. 2019, 48, 9317–9327. [DOI] [PubMed] [Google Scholar]
- 53. Izawa H., Nishino S., Sumita M., Akamatsu M., Morihashi K., Ifuku S., Morimoto M., Saimoto H., Chem. Commun. 2015, 51, 8596–8599. [DOI] [PubMed] [Google Scholar]
- 54. Waern J. B., Desmarets C., Chamoreau L.-M., Amouri H., Barbieri A., Sabatini C., Ventura B., Barigelletti F., Inorg. Chem. 2007, 46, 3340–3348. [DOI] [PubMed] [Google Scholar]
- 55. Spaenig F., Olivier J. H., Prusakova V., Retailleau P., Ziessel R., Castellano F. N., Inorg. Chem. 2011, 50, 10859–10871. [DOI] [PubMed] [Google Scholar]
- 56. Jayabharathi J., Thanikachalam V., Saravanan K., Venkatesh Perumal M., Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2012, 91, 158–165. [DOI] [PubMed] [Google Scholar]
- 57. Cao H., Sun H., Yin Y., Wen X., Shan G., Su Z., Zhong R., Xie W., Li P., Zhu D., J. Mater. Chem. C 2014, 2, 2150–2159. [Google Scholar]
- 58. Bokarev S. I., Hollmann D., Pazidis A., Neubauer A., Radnik J., Kuhn O., Lochbrunner S., Junge H., Beller M., Bruckner A., Phys. Chem. Chem. Phys. 2014, 16, 4789–4796. [DOI] [PubMed] [Google Scholar]
- 59. Schallenberg D., Neubauer A., Erdmann E., Tanzler M., Villinger A., Lochbrunner S., Seidel W. W., Inorg. Chem. 2014, 53, 8859–8873. [DOI] [PubMed] [Google Scholar]
- 60. Tschierlei S., Neubauer A., Rockstroh N., Karnahl M., Schwarzbach P., Junge H., Beller M., Lochbrunner S., Phys. Chem. Chem. Phys. 2016, 18, 10682–10687. [DOI] [PubMed] [Google Scholar]
- 61. Stonelake T. M., Phillips K. A., Otaif H. Y., Edwardson Z. C., Horton P. N., Coles S. J., Beames J. M., Pope S. J. A., Inorg. Chem. 2020, 59, 2266–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Veale E. B., Tocci G. M., Pfeffer F. M., Kruger P. E., Gunnlaugsson T., Org. Biomol. Chem. 2009, 7, 3447–3454. [DOI] [PubMed] [Google Scholar]
- 63. Kucheryavy P., Khatmullin R., Mirzakulova E., Zhou D., Glusac K. D., J. Phys. Chem. A 2011, 115, 11606–11614. [DOI] [PubMed] [Google Scholar]
- 64. Nordlund T. M., in Topics in Fluorescence Spectroscopy: Techniques (Ed.: Lakowicz J. R.), Springer US, Boston, MA, 1999, pp. 183–260. [Google Scholar]
- 65. Lemmetyinen H., Tkachenko N. V., Valeur B., Hotta J.-i., Ameloot M., Ernsting N. P., Gustavsson T., Boens N., Pure Appl. Chem. 2014, 86, 1969–1998. [Google Scholar]
- 66. van Stokkum I. H. M., Larsen D. S., van Grondelle R., Biochim. Biophys. Acta—Bioenergetics 2004, 1657, 82–104. [DOI] [PubMed] [Google Scholar]
- 67. Laptenok S. P., Nuernberger P., Lukacs A., Vos M. H., Methods Mol. Biol. 2014, 1076, 321–336. [DOI] [PubMed] [Google Scholar]
- 68. Alexiou M. S., Tychopoulos V., Ghorbanian S., Tyman J. H. P., Brown R. G., Brittain P. I., J. Chem. Soc. Perkin Trans. 2 1990, 837–842. [Google Scholar]
- 69. Cunningham R. M., DeRose V. J., ACS Chem. Biol. 2017, 12, 2737–2745. [DOI] [PubMed] [Google Scholar]
- 70. Tian J., Liu R., Qu Q., Oncol. Lett. 2017, 13, 1437–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mandic A., Hansson J., Linder S., Shoshan M. C., J. Biol. Chem. 2003, 278, 9100–9106. [DOI] [PubMed] [Google Scholar]
- 72. King A. P., Marker S. C., Swanda R. V., Woods J. J., Qian S. B., Wilson J. J., Chem. Eur. J. 2019, 25, 9206–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dirks W. G., Drexler H. G., in Basic Cell Culture Protocols, (Eds.: Helgason C. D., Miller C. L.), Humana Press, Totowa, NJ, 2013, pp. 27–38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary