

Abstract
The crosstalk between angiogenesis and immunity within the tumor microenvironment (TME) is critical for tumor prognosis. While pro-angiogenic and immunosuppressive TME promote tumor growth, anti-angiogenic and immune stimulatory TME inhibit tumor progression. Therefore, there is a great interest in achieving vascular normalization to improve drug delivery and enhance anti-tumor immunity. However, anti-vascular endothelial growth factor (VEGF) mechanisms to normalize tumor vessels have offered limited therapeutic efficacies for cancer patients. Herein, we report that Myct1, a direct target of ETV2, was nearly exclusively expressed in endothelial cells. In preclinical mouse tumor models, Myct1 deficiency reduced angiogenesis, enhanced high endothelial venule formation, and promoted an anti-tumor immune environment, leading to restricted tumor progression in Myct1 knockout mice. Analysis of The Cancer Genome Atlas (TCGA) datasets revealed a significant (p<0.05) correlation between MYCT1 expression, angiogenesis, and anti-tumor immunity in human cancers, as suggested by the decreased FOXP3 expression and increased anti-tumor macrophages in patients with low MYCT1 expression. Mechanistically, MYCT1 interacted with tight junction protein Zona Occludens 1 and regulated Rho GTPase-mediated actin cytoskeleton dynamics, thereby promoting endothelial motility in the angiogenic environment. Myct1-deficient endothelial cells facilitated trans-endothelial migration of cytotoxic T lymphocytes and polarization of M1 macrophages. Myct1 targeting combined with anti-PD1 treatment significantly (p<0.05) increased complete tumor regression and long-term survival in anti-PD1-responsive and -refractory tumor models in mice. Our data collectively support a critical role for Myct1 in controlling tumor angiogenesis and reprogramming tumor immunity. Myct1-targeted vascular control, in combination with immunotherapy, may become an exciting therapeutic strategy.
One-sentence summary:
Myct1 inhibition controls tumor angiogenesis, remodels tumor immunity, and improves immunotherapy outcomes in mouse tumor models.
Introduction
Tumor vessels are irregular, tortuous, and leaky. These malfunctioning vessels lead to a hypoxic environment where tumors thrive and eventually metastasize to secondary sites. Following the initial report on the vasculature-dependent nature of solid tumor progression 50 years ago (1), the notion of preventing new vessel formation as a way to treat cancers generated substantial interest. Extensive research has identified several critical pro-angiogenic factors, including vascular endothelial growth factors (VEGFs) and fibroblast growth factors, as potential targets for anti-angiogenic therapies (2, 3). However, despite impressive potential shown in preclinical studies, anti-angiogenic strategies like anti-VEGF treatment generated only modest clinical outcomes in cancer patients, possibly due to temporary vascular normalization, angiogenic adaptive responses, and/or acquired resistance by the tumor (4, 5). Pro-angiogenic factors like VEGFs are also required for physiological vascular maintenance, contributing to the related toxicities and poor clinical outcomes of anti-VEGF therapies (6–8). As such, new targets are required to develop anti-angiogenic approaches that may empower cancer management.
Immunotherapies such as immune checkpoint blockers and adoptive immune cell transfer have revolutionized the field of oncotherapy by enabling the regression and long-term control of previously incurable and aggressive tumors. However, they fail to produce clinical benefits in 50%−80% of treated patients (9), suggesting that further studies focusing on the functional crosstalk between different immunotherapeutic approaches and the constituents of the tumor microenvironment are needed. Accumulating data show that the abnormal nature of the tumor vessels also profoundly influences the outcome of different immunotherapeutic strategies (10, 11). Moreover, several recent preclinical studies have shown that a combination of immune checkpoint blockade with anti-angiogenic treatment could result in improved outcomes (12, 13). Better understanding of the crosstalk between the angiogenic determinants and immunotherapies may lead to more effective clinical strategies.
In this study, we identified Myct1 as a direct target of ETS transcription factor ETV2, a critical factor for vascular development, regeneration, and tumor angiogenesis (14–18). We found that although Myct1 is dispensable for vascular development and homeostasis, it is crucial for tumor progression through the regulation of tumor angiogenesis and tumor immunity. Remarkably, Myct1 inhibition in combination with anti-PD1 led to dramatic tumor regression. These findings establish a critical role for Myct1 in modulating the tumor vasculature and remodeling of immune constituents of the tumor microenvironment.
Results
Myct1 is a direct target of ETV2 and is an angiogenic regulator gene.
We first analyzed the expression profiles of more than 8,000 human cancer patients (from thirteen different cancer types) from The Cancer Genome Atlas (TCGA) database. We generated a list of the top 20 potential angiogenesis regulatory genes (Table 1 and table S1), whose expression correlates with “seed genes” that are well characterized in angiogenesis and endothelial cell (EC) biology (See Materials and Methods for details; table S2) (19–21). Comparing this list with the ETS transcription factor ETV2 transcriptional target genes (15), we found Flt1, Myct1, Ptprb, and S1pr1 as top potential angiogenesis regulatory genes downstream of ETV2. Unlike the other genes, Myct1 has never been implicated in angiogenesis; hence, we identified Myct1 as a potential angiogenic gene. Consistent with this idea, we found that Myct1 expression was mostly restricted to ECs (fig. S1A). Next, we performed a gene set variation analysis with TCGA-derived patient datasets using a signature of genes upregulated during angiogenesis to generate an “Angiogenic score” for every patient (table S3) (22). We found that MYCT1 expression was significantly (p<0.05) correlated with the “Angiogenic score” in all cancers analyzed (Fig. 1A and fig. S2A). Moreover, we found increased Myct1 expression in the tumor-ECs compared to the non-tumor-ECs in mouse models (Fig. 1B). In line with this, our analysis of a recently published single-cell RNA sequencing dataset (GSE110501) (23) on the heterogeneous mouse tumor stromal population revealed that Myct1 expression is observed exclusively in the tumor-ECs (fig. S3A–C). Additionally, in the same dataset, compared to the EC population from a healthy heart, the tumor-EC population had a higher number of cells that expressed Myct1 (fig. S3D). Together, these observations suggest that Myct1 plays a role in tumor angiogenesis/vasculature in both human cancers and mouse tumors.
Table 1.
Top 20 regulators of angiogenesis identified by their correlated expression with seed genes
| Rank | Gene Symbol | Full name | PCC |
|---|---|---|---|
| 1 | GPR4 | G Protein-Coupled Receptor 4 | 0.92 |
| 2 | LDB2 | LIM Domain Binding 2 | 0.91 |
| 3 | CXorf36 | Chromosome X open reading frame 36 | 0.91 |
| 4 | ADGRL4 | Adhesion G Protein-Coupled Receptor L4 | 0.91 |
| 5 | FLT1 | Fms Related Tyrosine Kinase 1 | 0.9 |
| 6 | CLEC14A | C-Type Lectin Domain Containing 14A | 0.9 |
| 7 | ECSCR | Endothelial Cell Surface Expressed Chemotaxis and Apoptosis Regulator | 0.9 |
| 8 | RP11–389C8.2 | RP11–389C8.2 | 0.9 |
| 9 | ESAM | Endothelial Cell Adhesion Molecule | 0.9 |
| 10 | MYCT1 | Myc Target 1 | 0.9 |
| 11 | CD93 | Cluster of Differentiation 93 | 0.9 |
| 12 | PLVAP | Plasmalemma Vesicle Associated Protein | 0.89 |
| 13 | S1PR1 | Sphingosine-1-Phosphate Receptor 1 | 0.89 |
| 14 | PTPRB | Protein Tyrosine Phosphatase Receptor Type B | 0.89 |
| 15 | ARHGEF15 | Rho Guanine Nucleotide Exchange Factor 15 | 0.89 |
| 16 | GIMAP8 | GTPase, IMAP Family Member 8 | 0.89 |
| 17 | PCDH12 | Protocadherin 12 | 0.88 |
| 18 | EXOC3L2 | Exocyst complex component 3-like 2 | 0.88 |
| 19 | ZNF366 | Zinc Finger Protein 366 | 0.88 |
| 20 | GIMAP6 | GTPase, IMAP Family Member 6 | 0.88 |
See also Supplementary Table 1. PCC = Pearson correlation coefficient
Figure 1: ETV2 direct target gene Myct1 is a regulator of angiogenesis but is not required for vascular development and homeostasis.
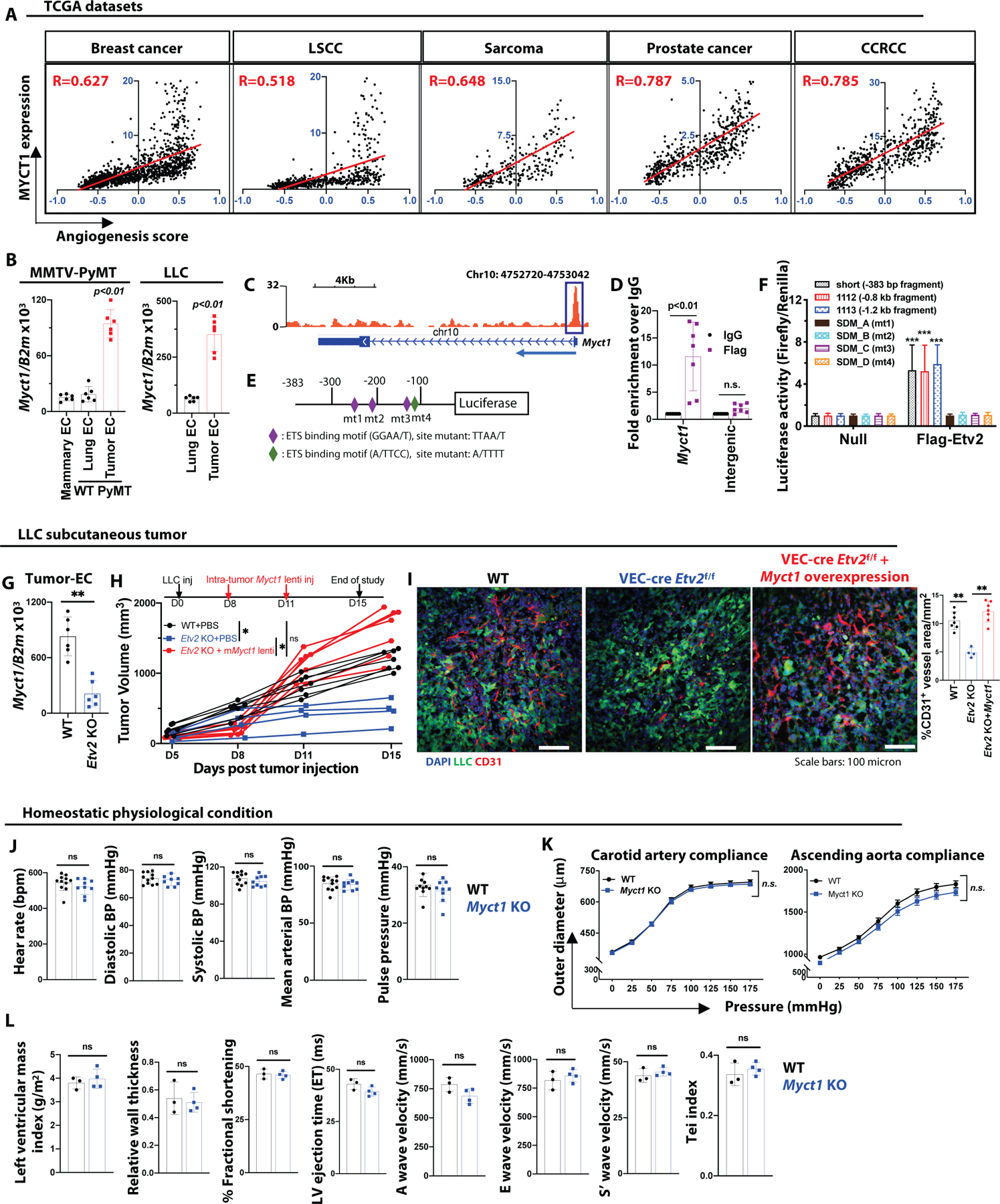
(A) Correlation between the ‘angiogenic score’ and MYCT1 expression in TCGA-derived breast cancer (TCGA-BRCA), LSCC (TCGA-LUSC), sarcoma (TCGA-SARC), prostate cancer (TCGA-PRAD), and CCRCC (TCGA-KIRC) patient datasets (Pearson’s χ2 test; R values are indicated with respective datasets). (B) Analysis of Myct1 expression in CD31+CD45− ECs isolated from the mammary glands of healthy wild-type (WT) mice, and the lungs and mammary tumors of WT MMTV-PyMT mice at 21 weeks of age (left) and lungs and tumors of the tumor-bearing WT mice at 14-days post LLC tumor transplantation (right). n=6 mice per group. Data is presented as mean with standard deviation (SD) from one of two biological replicates. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test (left) and two-tailed Student’s t-test (right). (C) A genomic snapshot illustrating the ETV2 binding peak to the Myct1 promoter region. (D) ChIP-qPCR analysis of ETV2 binding to the Myct1 promoter region. Data is presented as mean with SD. Statistical significance was analyzed by Two-way ANOVA followed by Tukey’s multiple comparison test. mt=mutant. (E and F) A graphical representation of the luciferase construct design (E) and analysis of the normalized reporter activity (F) for ETV2 binding motif on Myct1. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; ***p<0.001, compared to the respective values in the absence of ETV2 (null). (G) Analysis of Myct1 expression in CD31+CD45− ECs isolated from the tumors of WT and VEC-Cre;Etv2f/f (Etv2 KO)mice at 15-days post LLC tumor transplantation. n=6 mice per group. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test. (H and I) Tumor growth (H) and representative images with quantification for CD31+ vessel density (I) in PBS (control) and Myct1 lentiviral overexpression construct (intra-tumor) treated WT and Etv2 KO mice. n=7 mice per group. Scale bars, 100μm. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; *p<0.05, at the end of the study. (J and K) Analysis of vital cardiovascular parameters (J) and pressure-diameter measurements (K) in the Myct1 KO and WT mice. n=9 (Myct1 KO) and 11 (WT) mice. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test (J) and One-way ANOVA with Tukey’s multiple comparison test (K); n.s.=not significant. (L) Analysis of clinically important cardiac functional parameters measured by Doppler echocardiogram of Myct1 KO and WT mice. n=3 (WT) and 4 (Myct1 KO) mice. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; n.s.=not significant.
We identified one potential ETV2 binding site in the Myct1 promoter region by analyzing previously published ETV2 ChIP-Seq data (Fig. 1C) (15). This binding was validated by ChIP-qPCR (Fig. 1D) (14). Only ETV2 and no other ETS transcription factors, such as FLI1 and ERG, activated different luciferase constructs made with varying sizes of the Myct1 promoter region, all containing the ETS binding motif. Mutations of the ETS binding motifs reduced luciferase activity, implying that these sites were critical for ETV2 binding (Fig. 1E, 1F, and S4A). In vitro overexpression of Myct1 rescued the tube-like structure formation, sprouting, and migration defects observed in the Etv2 deficient ECs (fig. S4B–E). Finally, Myct1 was downregulated in the tumor-ECs of the Etv2 conditional knockout (KO) mice (Fig. 1G) and intra-tumoral lentiviral Myct1 expression rescued impaired tumor growth and angiogenesis observed in Etv2 deficiency (17) (Fig. 1H and 1I). In this experiment, intra-tumoral lentiviral Myct1 injection resulted in enforced expression of Myct1 in ECs, as well as in tumor cells and hematopoietic cells (fig. S4F). To assess whether the observed phenotypic rescue is from non-endothelial enforced Myct1 expression, we generated Myct1 overexpressing Lewis lung carcinoma (LLC) tumor cells and found that there is no growth advantage for these Myct1 overexpressing tumor cells in the wild-type (WT) mice (fig. S4G and S4H). Additionally, tumor growth patterns in our bone-marrow chimeric mice suggest that Myct1 expression in hematopoietic cells does not contribute to tumor growth (as described below; see Fig. 2G–J). Together, these results suggested that Myct1 was a direct target gene of ETV2 that regulates the angiogenic functionalities of ECs.
Figure 2: Endothelial Myct1 is required for tumor growth and angiogenesis in mice.
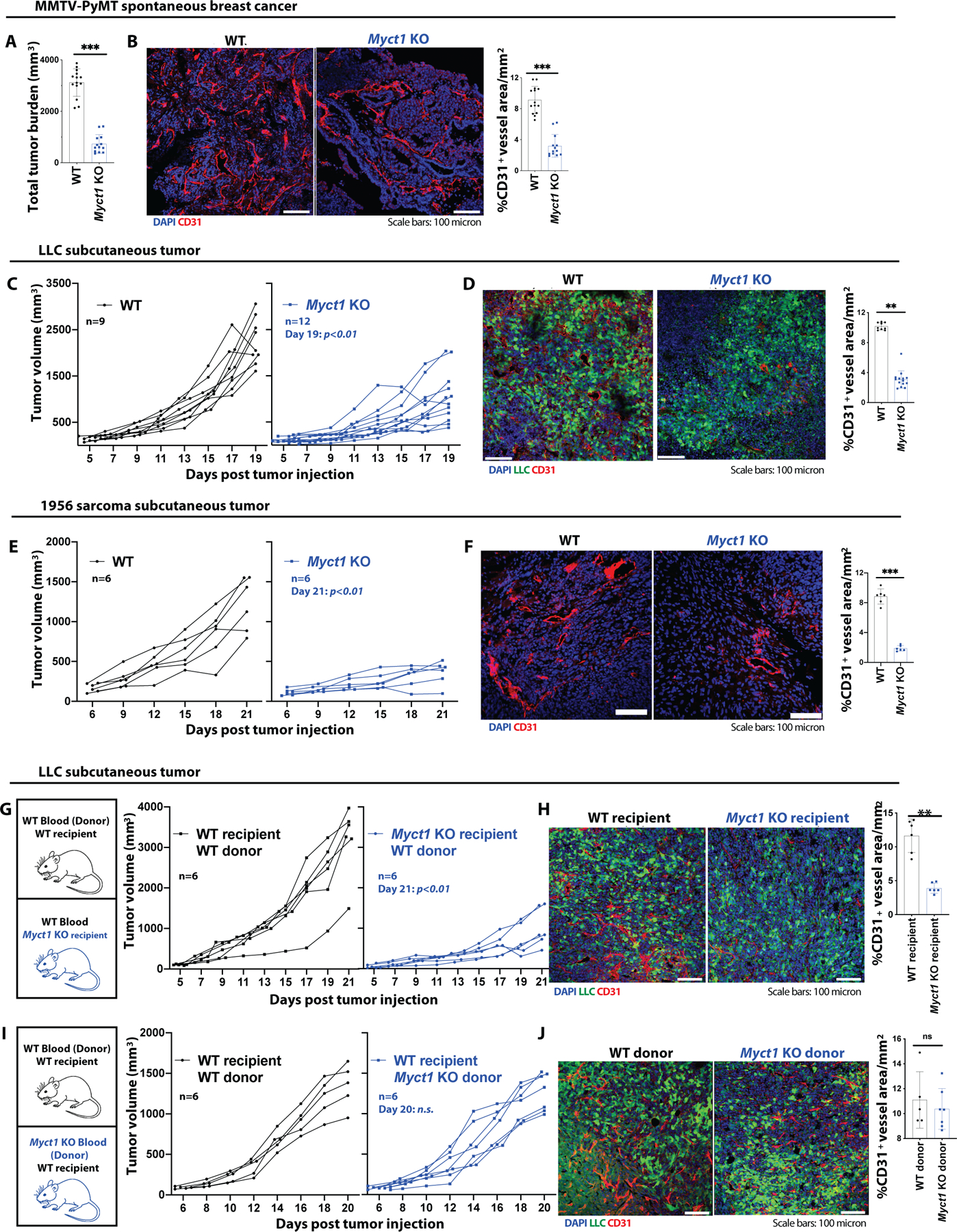
(A and B) Analysis of total tumor burden (A) and representative images with quantification of CD31+ vessel density (B) in WT and Myct1 KO MMTV-PyMT mice at 21 weeks of age. n=14 mice per group. Scale bars, 100μm. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; ***p<0.001. (C, D) Analysis of LLC tumor growth (C) and representative images with quantification for CD31+ vessel density (D) in WT and Myct1 KO mice. Scale bars, 100μm. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01, at the end of the study. (E and F) Analysis of 1956-sarcoma tumor growth (E) and representative images with quantification for CD31+ vessel density (F) in WT and Myct1 KO mice. Scale bars, 100μm. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.001, at the end of the study. (G-J) Analysis of tumor growths (G and I) and representative images with quantification for CD31+ vessel density (H and J) in stromal Myct1 KO (G and H) and hematopoietic Myct1 KO mice (I and J). Scale bars, 100μm. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; n.s.=not significant and **p<0.01, at the end of the study.
Myct1 is dispensable for vascular development and homeostasis in mice and zebrafish.
We generated Myct1−/− (Myct1 KO) mice by utilizing the CRISPR/CAS9 technology. Briefly, we designed gRNA to target the first exon of the Myct1 gene and injected it together with Cas9 mRNA into fertilized eggs (Details in the Materials and Method section; fig. S4I–K). By crossing the candidate knockout founder to wild-type mice, we generated heterozygous (Myct1+/−) offspring and obtained Myct1 KO mice from brother-sister matings. Myct1 expression was undetected in the ECs isolated from the lungs of the KO animals (fig. S4L), confirming the efficient deletion of the gene. Myct1 KO mice appeared normal and exhibited histologically regular vasculature in different organ beds. Vital cardiovascular parameters (Fig. 1J), compliance profiles of the ascending aorta and carotid artery (Fig. 1K), and clinically relevant cardiac functions (Fig. 1L) were similar between the Myct1 KO and littermate control mice. Similarly, myct1 zebrafish morphants showed minimal defects in vascular development (fig. S4M). Together, these observations suggested that Myct1 was dispensable for vascular development, maintenance, and homeostatic functions.
Myct1 is required for efficient tumor growth and tumor vasculature in multiple mouse models.
Since high Myct1 expression is observed in tumor ECs, we determined whether Myct1 deficiency has any impact on tumor growth by employing five different (one transgenic, three subcutaneous transplantation, and one orthotopic transplantation) mouse models of cancer. For the transgenic model, we developed MMTV-PyMT; Myct1−/− mice as a spontaneous breast cancer model and tracked the development and progression of the tumor in the mammary gland (fig. S5A). For transplantation models, we subcutaneously transplanted Lewis lung carcinoma (LLC-GFP) (17), B16F10 melanoma, and 1956 sarcoma tumor cells (24) and orthotopically transplanted PyMT-BO1 mammary tumor cells (25) to the mammary fat pad as described previously (17, 26). Compared to WT mice, Myct1 KO mice showed retarded tumor growth in all five of the tumor models; the growth restricted tumors had reduced counts of tumor vessels, which had better pericyte coverages (Fig. 2A–2F and fig. S5B–S5F). Moreover, Myct1 KO mice exhibited subsided intra-tumoral hypoxia (fig. S6A), improved vascular perfusion (fig. S6B), and reduced vascular leakage (fig. S6C). These data demonstrated that although not essential for vascular development and maintenance, Myct1 deficiency led to reduced tumor growth and normalization of tumor vessels.
Endothelial Myct1 is critical for tumor growth in mice.
While Myct1 expression was mostly restricted to the ECs, Myct1 was also expressed in hematopoietic stem and progenitor cells (fig. S1A. See also fig. S3A–C) (27–29). To evaluate whether the observed growth restrictive phenotype is dependent on EC-specific Myct1, we generated two distinct bone marrow chimeras: ‘WT hematopoietic/Myct1 KO background’ and ‘Myct1 KO hematopoietic/WT background’ (fig. S7A) mice. ‘WT hematopoietic/Myct1 KO background’ chimeric mice exhibited impaired tumor growth and vascular network formation, a phenotype similar to Myct1 KO mice (Fig. 2G and 2H). However, ‘Myct1 KO hematopoietic/WT background’ chimeric mice exhibited tumor growth comparable to WT mice (Fig. 2I and 2J). Moreover, endothelial-specific Myct1 deletion in mice (Cdh5-cre Myct1f/f) recapitulated the global Myct1 KO phenotype of retarded tumor growth and reduced angiogenesis, together suggesting that hematopoietic Myct1 expression was dispensable for tumor progression (fig. S7B–D).
To investigate whether Myct1 plays any role(s) in tumor cells, we assessed Myct1 expression in the tumor cell lines utilized earlier in the present study and found that Myct1 expression was not detectable (fig. S8A). To investigate whether synthetic Myct1 expression in tumor cells contributes to growth, we genetically modified LLC tumor cells with either Myct1 shRNA or Myct1 overexpressing lentivirus and the PyMT-BO1 tumor cells with Myct1 siRNA. We found that either Myct1 siRNA or shRNA treatment or enforced expression of Myct1 in the tumor cells did not play any role in tumor growth kinetics (fig. S8B–E). Additionally, tumor explants from neither the WT nor the Myct1 KO MMTV-PyMT mice exhibited any growth defects in the WT recipient mice. However, explants from both types of mice showed tumor growth defects in the Myct1 KO recipient mice (fig. S8F). Together these data suggest that Myct1 function in the observed phenotype is tumor cell independent. We also modified a human fibroblast cell line (BJ-5ta) that does not express MYCT1 by treating with MYCT1 shRNA or enforcing the expression of MYCT1 with lentivirus. We found that neither of the modifications impacted the growth kinetics and angiogenic sprouting of the fibroblasts (fig. S8G–I). Collectively, these data suggested an endothelial-specific role of Myct1 in tumor growth and angiogenesis.
Myct1 is also required for vascular regeneration in mice.
To investigate whether Myct1 is required exclusively for tumor angiogenesis, we utilized a mouse hindlimb ischemia injury model as described previously (18). We found that following the injury, Myct1 KO mice had a lower blood perfusion recovery and neovascularization of the injured area compared to WT mice (fig. S8J and S8K), suggesting that the Myct1 requirement is not exclusive to tumor angiogenesis - other forms of neovascularization also require Myct1.
MYCT1 interacts with ZO1 and regulates EC motility through actin cytoskeleton in vitro.
Because very little is known about the cellular functions of Myct1, we first assessed the subcellular localization of MYCT1 in ECs. To this end, we generated mouse cardiac EC lines (MCEC) expressing either N-terminal HA-tagged or C-terminal FLAG-tagged mouse Myct1 (Fig. 3A). Flow cytometric analysis revealed that in the intact cells, whereas the anti-HA antibody recognized HA-MYCT1, the anti-FLAG antibody did not recognize MYCT1-FLAG. On the contrary, both the antibodies recognized HA-MYCT1 and MYCT1-FLAG in permeabilized cells (Fig. 3A). Immunofluorescence with anti-HA and anti-FLAG antibodies showed that MYCT1 is indeed present at the plasma membrane, as well as in the cytoplasm, where it was found colocalized mainly with GM130+ and GIANTIN+ Golgi apparatus (fig. S9A–C). Collectively, these results demonstrated that MYCT1 is a membrane-spanning protein with an extracellular N-terminal and an intracellular C-terminal, consistent with a previous structural prediction based on bioinformatic analysis of MYCT1 (30).
Figure 3: Membrane-localized MYCT1 regulates endothelial actin cytoskeleton dynamics in the angiogenic environment.
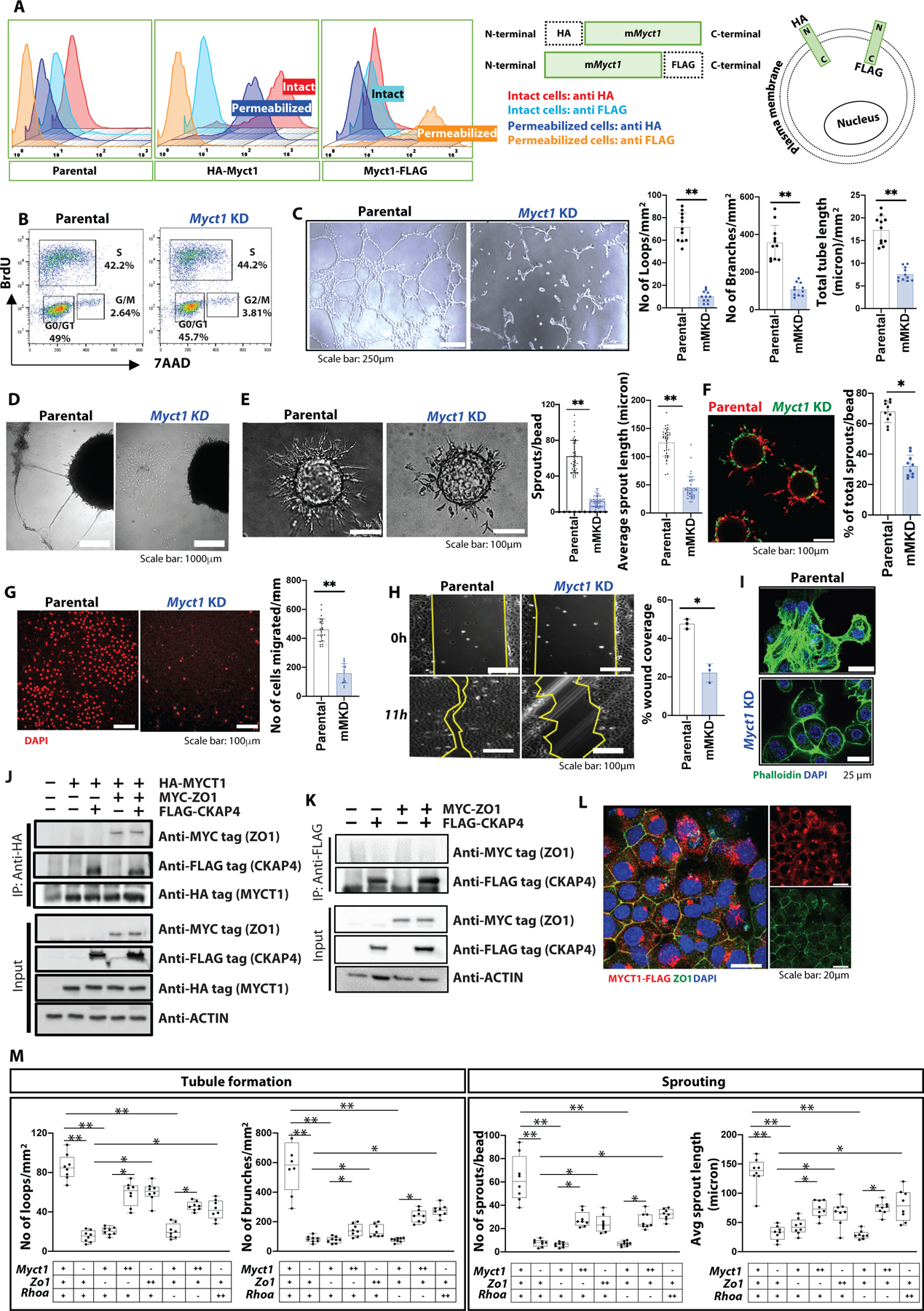
(A) (left) Flow cytometric analysis of HA- and FLAG-tagged MYCT1 overexpressing MCEC cells. (right) A graphical presentation of the HA- and FLAG-tagged MYCT1 protein situated at the plasma membrane. (B) A representative FACS chart of the BrdU proliferation assay with parental and Myct1 knockdown (KD) cells. (C) Representative images and quantifications from the Matrigel tube formation assay. n=12 independent observations per group. Scale bars, 250μm. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01. (D) Representative images from tumor-spheroid/EC co-culture assay. Scale bar, 1000μm. (E) Representative images and quantifications of sprouts from the fibrin gel sprouting assay. n≥32 independent observations per group. Scale bars, 100μm. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01. (F) Representative image and quantification of sprouts from the fibrin gel sprouting assay with 1:1 competitive seeding of parental (red) and Myct1 KD (green) MCEC cells on the beads. Scale bar, 100μm. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.05. (G) Representative images and quantifications of cell migration from the Boyden chamber assay. n≥12 independent observations per group. Scale bars, 100μm. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01. (H) Representative images and quantifications for recovery from the wound closure assay. n≥3 independent observations per group. Scale bars, 100μm. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.05. (I) Representative immunofluorescence images of actin filaments in a cultured monolayer of MCEC cells. Scale bars, 25μm. (J) Immunoprecipitation followed by western blot (IP-WB) analysis for MYCT1 in the total cell lysates of the parental and HA-tagged MYCT1 overexpressing (HA-MYCT1) MCEC cells transfected with either mock (empty vector control) or MYC-tagged ZO1, and FLAG-tagged CKAP4 expression plasmids. An anti-ACTIN antibody was used as a control. (K) IP-WB analysis for CKAP4 in the total cell lysates of the parental MCEC cells transfected with either mock (empty vector control), or MYC-tagged ZO1, or FLAG-tagged CKAP4 expression plasmids. An anti-ACTIN antibody was used as a control. (L) Representative immunofluorescence image of ZO1 (green) and MYCT1 (anti-FLAG, red) in a confluent monolayer of FLAG-tagged MYCT1 overexpressing MCEC cells. Scale bars, 20μm. (M) Quantifications from the Matrigel tube formation assay (left) and fibrin gel sprouting assay (right) with different combinations of Myct1, Zo1, and Rhoa KD and overexpressing MCEC cells. “+”, “++”, and “−” denotes parental, overexpression, and knockdown cells, respectively. n≥6 independent observations per group. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05, **p<0.01.
We next developed Myct1 knockdown (KD) MCEC cells (fig. S9D) and assessed EC functionalities. We did not observe any proliferative or maintenance disparity compared to the parental cells, as supported by the similar cell cycle distribution between parental and Myct1 KD cells (Fig. 3B and fig. S9E). However, Myct1 KD MCEC cells showed defects in tube-like structure formation in the Matrigel assay (Fig. 3C) and tumor spheroid/EC co-culture assay (Fig. 3D and fig. S9F). Myct1 KD MCEC cells also displayed defects in sprout formation on the fibrin gel matrix (Fig. 3E and 3F). Additionally, Myct1 KD MCEC cells lost their migratory characteristics, as shown in the Boyden chamber tumor-chemotaxis assay (Fig. 3G and fig. S9G) and wound-closure assay (Fig. 3H). Importantly, parental cells utilized in these studies did not show any difference compared to the sham shRNA or empty vector control transduced cells (fig. S9H and S9I). Together, these findings suggested that while Myct1 is not required for the steady-state maintenance of ECs, it is essential for ECs responses in the angiogenic environment.
Different phases of angiogenesis, such as sprouting, migration, and tube-like-structure formation, require EC alignment and directional movement in response to angiogenic cues (31). To understand why Myct1 KD ECs show defective migratory properties in the angiogenic environment, we assessed the downstream effectors of Myct1 in regulating EC motility. Specifically, we extracted RNA from sprouts formed in the fibrin gel matrix (see Fig. 3E) and performed a PCR array for genes that regulate cellular motility (fig. S9J). Genes that regulate actin cytoskeleton dynamics and cell adhesion turnover such as Capn2, Actin, Actinr2 and 3, Cfl1, Rdx, Myl12a, and Wasf1 were downregulated in the Myct1 KD MCEC sprouts (fig. S9J). Rhoa and Rhoc, which are required for the formation of actin stress fibers, cell retraction following protrusions, and overall movement (32), were also downregulated (fig. S9J), implying that Rho GTPase signaling is defective in Myct1 KD cells. Intriguingly, elements of RAC signaling, such as Rac1, Arhgef7, and Stat3, were upregulated in the Myct1 KD sprouts, suggesting an unbalanced hyperactivation of RAC signaling, which regulates the formation of leading-edge protrusion during cell movement (32). Consistent with this idea, Myct1 KD MCEC cells in both 2D culture (Fig. 3I) and scattered 3D tube structures (fig. S9K) formed virtually no stress fibers, which are critical for cellular contractility-relaxation, adhesion, and migration (33). Notably, Arhgdia, a Rho GDP dissociation inhibitor that keeps Rho GTPase proteins in their inactive forms (34), was highly upregulated in the Myct1 KD MCEC sprouts (fig. S9J), again implying a dysregulated Rho GTPase signaling. Congruent with this idea, Arhgdia knockdown moderately rescued the Myct1 KD MCEC phenotype (fig. S9L–N).
Next, we characterized MYCT1 binding partners by taking a target-directed approach. Using proteomics analysis, we identified ZO1 (Zona Occludens 1, also known as tight junction protein 1) and CKAP4 as potential candidates for binding MYCT1 (Supplementary Excel file 1). We confirmed ZO1 and CKAP4 as binding partners of MYCT1 by utilizing co-immunoprecipitation followed by western blot. Intriguingly, we found that, although ZO1 and CKAP4 do not bind directly to each other, MYCT1 binds both of them together in a complex (Fig. 3J and 3K). Immunofluorescence also supported the interaction between MYCT1 and ZO1 by co-localization (Fig. 3L). A similar binding pattern between MYCT1 and CKAP4 in HEK293T cells was also reported in a previous study (35). Since ZO1 plays a critical role in EC functions such as barrier formation, tension, and migration (36) and RHOA regulates endothelial tight junction maintenance and barrier formation in close association with ZO1 (37), we investigated whether there is a functional interplay between MYCT1, ZO1, and RHOA through a series of combined knockdown and overexpression of these genes in MCEC cells. We found that similar to Myct1 KD, downregulation of either Zo1 or Rhoa led to impaired sprouting and tube formation of ECs in the angiogenic environment. These angiogenic defects of Myct1 deficient ECs were partially rescued by Zo1 overexpression; likewise, Zo1 deficiency phenotypes were partially rescued by Myct1 overexpression (Fig. 3M and fig. S10A–C). We observed similar, although to a somewhat lesser extent, interplay between Myct1 and Rhoa as well (Fig. 3M and fig. S10A–C), suggesting that MYCT1-ZO1 complex works in close functional association with RHOA to control the actin cytoskeleton. Intriguingly, both the parental and Myct1 KD cells responded similarly to VEGF in a fibrin gel sprouting assay, suggesting that Myct1 might regulate ECs angiogenic functionalities independently of VEGF (fig. S10D). Collectively, these data suggested that MYCT1 regulates directional movement of ECs in the angiogenic environment through controlling actin cytoskeleton dynamics.
Endothelial MYCT1 function is evolutionarily conserved between human and mouse
Mouse MYCT1 protein shares an 85% sequence identity with the human MYCT1 protein (30, 35) (fig. S11A). To determine whether MYCT1 function is conserved between human and mouse, we knocked-down human MYCT1 in HUVEC (HUVEC hMKD) cells (fig. S11B). Similar to the Myct1 KD MCEC cells, HUVEC hMKD cells displayed severely impaired tube-like-structure generation, sprout formation, and migration capabilities (Fig. 4A–C). Importantly, enforced expression of mouse Myct1 almost completely rescued the human MYCT1 KD defects and vice-versa in vitro (Fig. 4A–F and fig. S11B). Moreover, both the mouse and human MYCT1 overexpression rescued the impaired tumor growth and reduced angiogenesis phenotype of the Myct1 KO mice in a subcutaneous tumor transplantation model (Fig. 4G, 4H, and fig. S11C). Again, although the lentiviral treatment induced Myct1 over expression in tumor cells and hematopoietic cells along with the ECs, as we described above (see fig. S8A–I), the observed rescue of the tumor phenotype in this experiment is most likely from endothelial Myct1. Together, these data suggest that the MYCT1 function in EC is conserved between human and mouse.
Figure 4: Endothelial MYCT1 function is evolutionarily conserved between human and mouse.
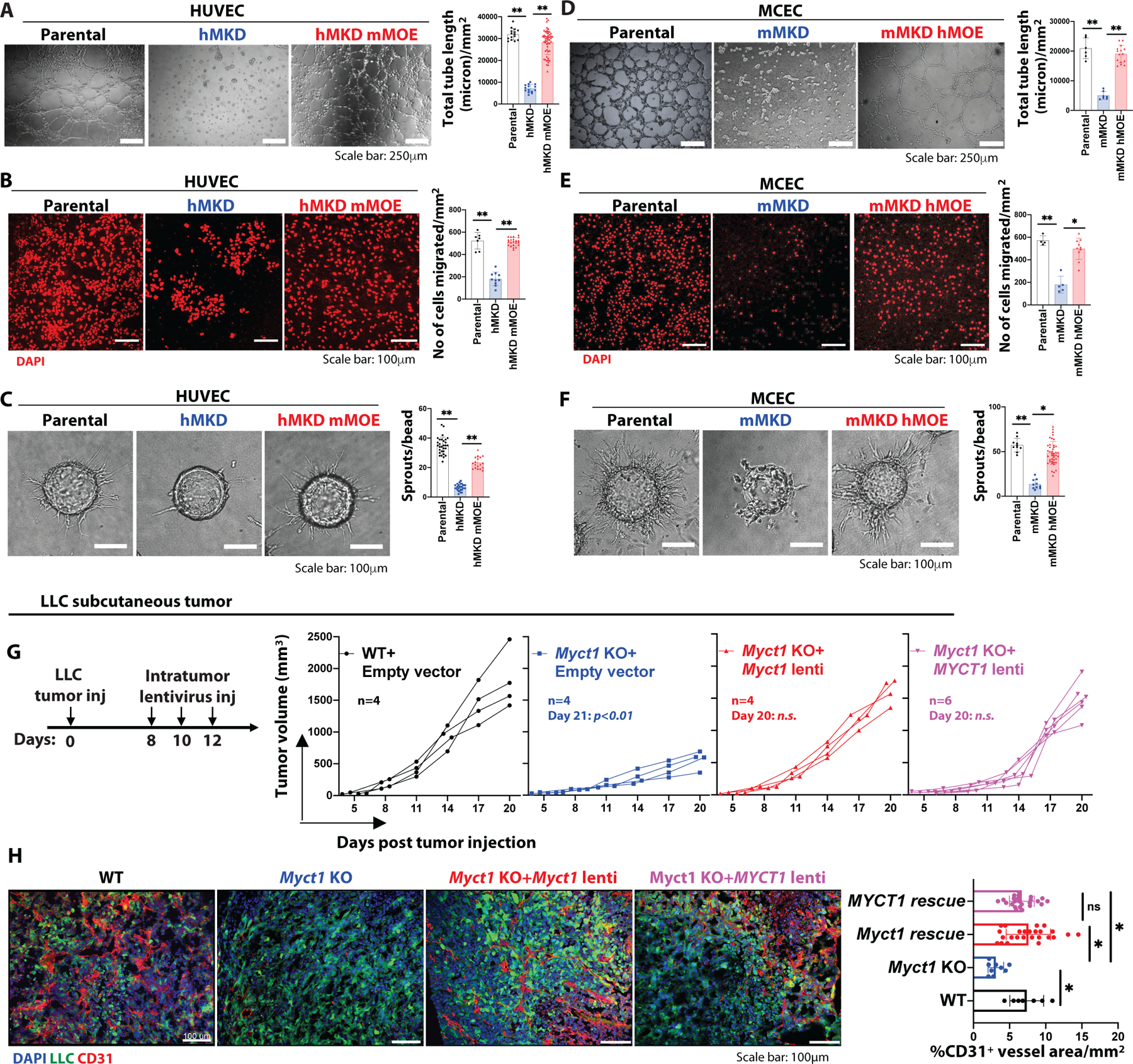
(A-C) Representative images and quantifications from the Matrigel tube formation assay (A), Boyden chamber migration assay (B), and fibrin gel bead sprouting angiogenesis assay (C) with parental, human MYCT1 KD (hMKD), and mouse Myct1 overexpressing human MYCT1 KD (hMKD mMOE) HUVEC cells. Scale bar, 250μm (A) and 100μm (B and C). n≥10 independent observations per group. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; **p<0.05. (D-F) Representative images and quantifications from the Matrigel tube formation assay (D), Boyden chamber migration assay (E), and fibrin gel sprouting assay (F) with parental, mouse Myct1 KD (mMKD), and human MYCT1 overexpressing mouse Myct1 KD (mMKD hMOE) MCEC cells. Scale bars, 250μm (D) and 100μm (E and F). n≥10 independent observations per group. Data is presented as mean with SD from one of three biological replicates. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05 and **p<0.01. (G and H) Analysis of tumor growth (G) and representative immunofluorescence images with quantification for CD31+ vessel density (H) in PBS (control) and either mouse Myct1 or human MYCT1 lentiviral overexpression constructs (intra-tumor) treated WT and Myct1 KO mice. Scale bars, 100μm. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; n.s.=not significant and *p<0.05, at the end of the study.
Analysis of mouse tumor endothelial transcriptome reveals Myct1 regulation of angiogenesis and immune responses.
To better understand the tumor endothelial heterogeneity and the potential roles of Myct1 in tumor-ECs, we performed single-cell RNA-sequencing on sorted tumor-ECs from the PyMT-BO1 orthotopic tumor bearing WT and Cdh5-cre Myct1f/f mice using the 10X-genomics platform. Unsupervised hierarchical and Seurat cell-clustering analysis (38–41) (using a total of 965 ECs) revealed heterogeneity in the tumor endothelium. We identified distinct clusters of angiogenic, tip-like, stalk-like, proliferating, and transitionary ECs (Fig. 5A, 5B, and fig. S12A). Compared to WT tumor-ECs, Myct1 KO tumor-ECs had a vastly different transcriptional landscape, as evidenced by the reduced angiogenic and tip-like cell populations that are enriched for biological processes related to growth factor activities and invasive vascularization as per Gene Set Enrichment Analysis (Fig. 5C–H) (42). Myct1 KO tumor-ECs had increased stalk-like and transitionary cell populations that were enriched for biological processes related to leukocyte transendothelial migration, oxidative phosphorylation, and antigen presentation and processing (Fig. 5I–N), collectively reflecting on the Myct1 requirement for aggressive tumor angiogenesis and tumor immune modulation. We applied SCENIC (Single-cell rEgulatory Network Inference and Clustering) (43), which scans co-expression of transcription factors and putative target genes, and found that WT and Myct1 KO tumor-ECs were in transcriptionally distinct cellular states (Fig. 5O). While angiogenic transcriptional networks were driving the gene expressions in WT tumor-ECs, transcription factors for immune responses were among the prominent transcriptional drivers for gene expressions in Myct1 KO tumor-ECs (Fig. 5P).
Figure 5: Single cell RNA sequencing of tumor endothelium from WT and Cdh5-cre Myct1f/f mice signifies Myct1 functions in tumor angiogenesis.
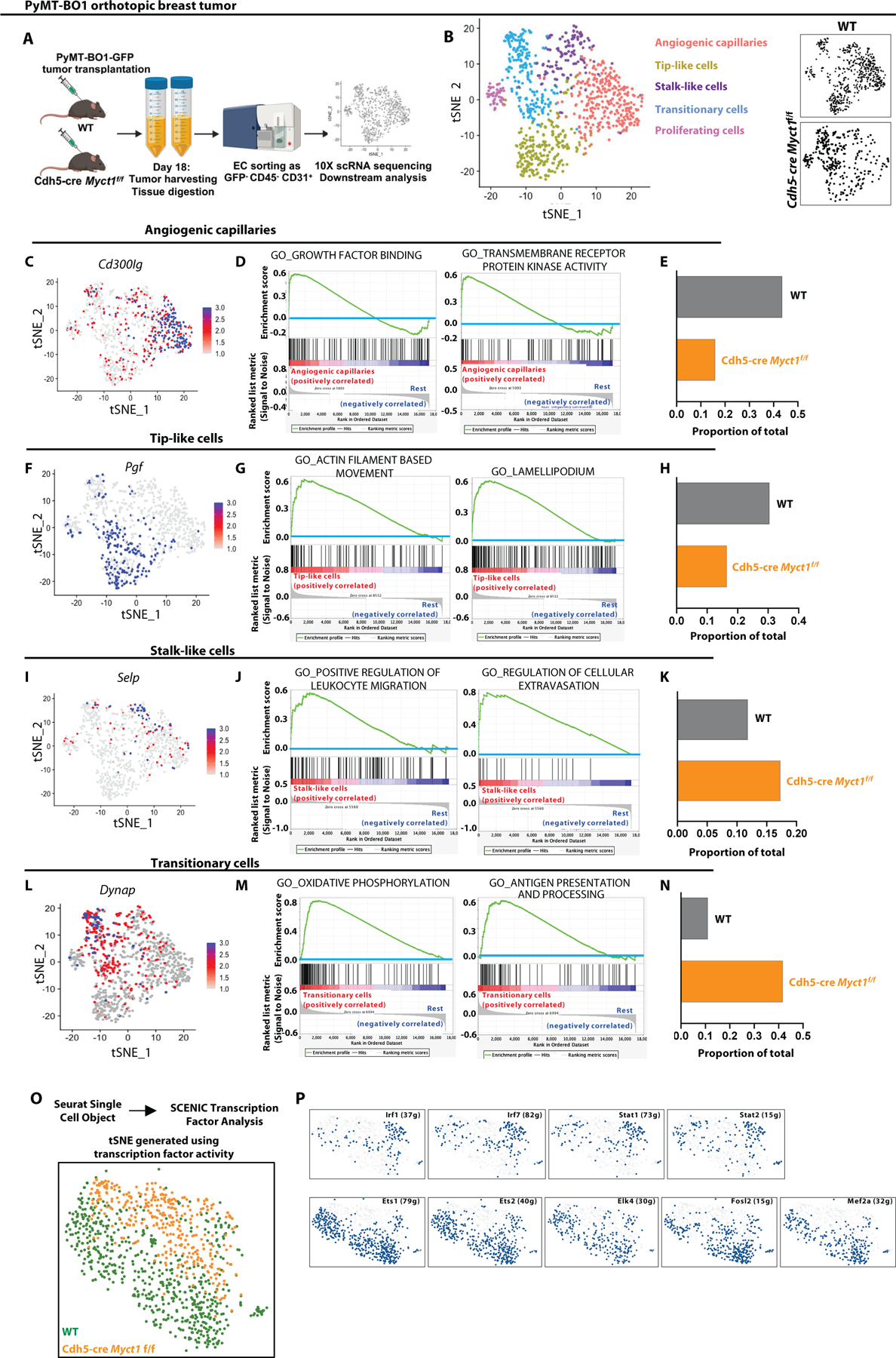
(A) A schematic representation of tumor endothelium sample collection and processing from WT and Cdh5-cre Myct1f/f mice (n = 5 per group) for single cell RNA sequencing. (B) (Left) tSNE projection color-coded for 995 endothelial cells sorted from tumor mass (PYMT-BO1 orthotopic transplantation tumor model). (Right) tSNE projection of endothelial cells grouped for different genotypes. (C-E) Feature plot showing Cd300lg expression pattern (C), gene set enrichment analysis depicting enriched molecular pathways (D), and normalized proportion of cells (E) in the angiogenic capillaries sub-cluster of the tumor endothelial cells from WT and Cdh5-cre Myct1f/f mice derived by analyzing the SCTtransform data (normalized). (F-H) Feature plot showing Pgf expression pattern (F), gene set enrichment analysis depicting enriched molecular pathways (G), and normalized proportion of cells (H) in the Tip-like cells sub-cluster of the tumor endothelial cells from WT and Cdh5-cre Myct1f/f mice derived by analyzing the SCTtransform data (normalized). (I-K) Feature plot showing Selp expression pattern (I), gene set enrichment analysis depicting enriched molecular pathways (J), and normalized proportion of cells (K) in the Stalk-like cells sub-cluster of the tumor endothelial cells from WT and Cdh5-cre Myct1f/f derived by analyzing the SCTtransform data (normalized). (L-N) Feature plot showing Dynap expression pattern (L), gene set enrichment analysis depicting enriched molecular pathways (M), and normalized proportion of cells (N) in the Transitionary cells sub-cluster of the tumor endothelial cells from WT and Cdh5-cre Myct1f/f derived by analyzing the SCTtransform data (normalized). (O and P) tSNE projection (O) and area under the curve (AUC) plots for differentially enriched transcription factor activity modules (P) in WT and Cdh5-cre Myct1f/f tumor endothelium generated by using SCENIC analysis.
Additionally, we observed similar patterns of transcriptional activities even in the WT tumor-ECs grouped as Myct1high and Myct1low expressing cells in one other tumor model. Briefly, we performed single-cell RNA-sequencing on sorted tumor-ECs from the LLC subcutaneous transplantation tumor model with only WT mice using the 10X-genomics platform. Unsupervised hierarchical cell-clustering analysis (using a total of 1977 ECs) revealed a similar heterogeneity and Myct1 expression pattern in the tumor-ECs (fig. S13A–D). Gene Set Variance Analysis for biological activities on the tumor-ECs based on high and low Myct1 expression revealed a similar enrichment pattern as the WT vs Myct1 KO tumor-ECs. As such, Myct1low ECs were enriched for biological processes related to leukocyte transendothelial migration, whereas Myct1high ECs were enriched for processes related to active vascularization (fig. S13E).
Myct1 deficiency leads to increased formation of high endothelial venules (HEV) in mouse tumors and an overall immunostimulatory tumor microenvironment.
While Myct1 KO mice exhibited reduced tumor vessel formation, we found that they developed more intra-tumoral high endothelial venules (HEV) compared to the WT mice (Fig. 6A and fig. S14A–C). HEVs are specialized vascular structures that mediate large scale lymphocyte extravasation in lymphoid organs and inflammatory sites. In solid tumors, HEVs preferentially facilitate infiltration of CD8+ cytotoxic T lymphocytes (CTL) into the tumor, and their presence is correlated with reduced tumor growth and favorable prognosis in cancer patients (44). The potential role(s) of Myct1 in leukocyte adhesion and endothelial transmigration (see Fig. 5I–K and fig. S13E) and HEV formation (Fig. 6A and fig. S14A–C) suggested a possible alteration of tumor immune environment in the Myct1 KO mice. Indeed, flow cytometric analysis revealed that tumors from Myct1 KO mice exhibited an increase of CTLs and a decrease of immunosuppressive regulatory T cells (Treg), as evident by the increased ‘CD8-to-Treg ratios’ in all the tumor models (Fig. 6B, 6C and fig. S14D, S14E). Likewise, tumors from Myct1 KO mice had an increased M1-macrophage population and a decreased M2-macrophage population; further manifested by the increased ‘M1-to-M2 ratios’ in all the tumor models (Fig. 6B, 6C). Similar to the Myct1 KO mice, Etv2 conditional KO mice exhibited normalization of tumor vasculature and an immunostimulatory tumor microenvironment, as evidenced by the increased ‘CD8-to-Treg’ and ‘M1-to-M2’ ratios (fig. S14F; also see Fig. 1H, 1I) (17). Intriguingly, this anti-tumor immune environment was reversed to a pro-tumor environment following enforced Myct1 expression (fig. S14F). Moreover, analysis of TCGA-derived cancer datasets using an immune-cell-deconvolution algorithm from CIBERSORT (45) revealed similar trends. Tumor samples with low MYCT1 expression showed decreased FOXP3 expression, a marker for regulatory T cells, and increased ‘M1-to-M2 ratio’ compared to the high expression group (Fig. 6D). Additionally, in some cancer types, tumor samples with low MYCT1 expression also exhibited higher frequencies of activated natural killer (NK) cells and activated dendritic cells (DC) (fig. S14G). Collectively, these observations suggested that Myct1 deficiency in tumor endothelial cells promotes an anti-tumor immune microenvironment.
Figure 6: Myct1 deficiency promotes an immunostimulatory tumor microenvironment.
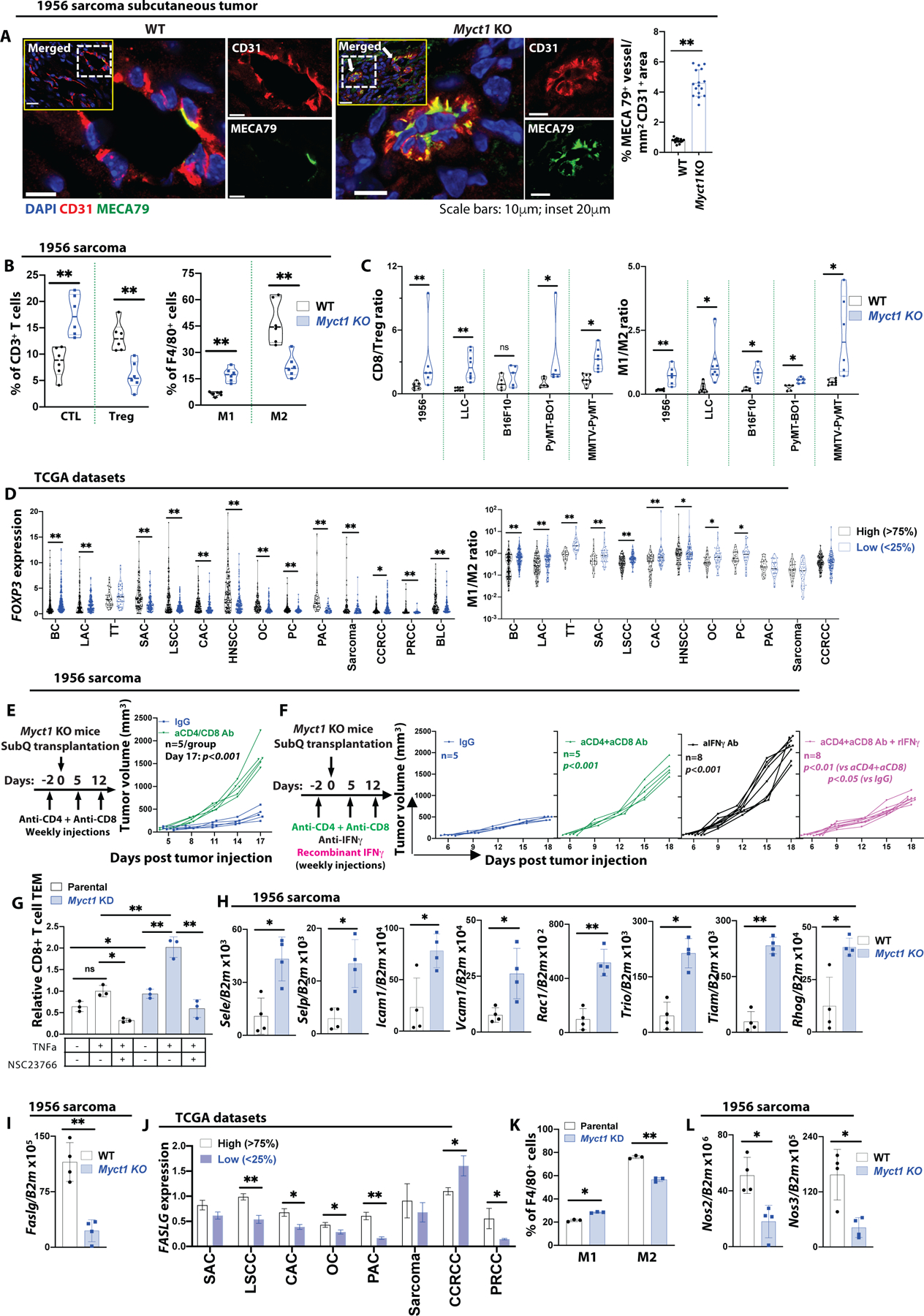
(A) Representative immunofluorescence images and quantification of MECA79+ high endothelial venules (HEVs) in 1956 sarcoma tumor. White dotted boxed area from the inset is presented as zoomed-in. Arrows in the inset indicate the MECA79 expressing vessels. Scale bars, 10μm and 20μm (inset). Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01. (B) CTL (CD8+ T cells) and Treg (CD4+FOXP3+ T cells) cells as a percentage of CD3+ T cells and M1 (iNOS+ macrophages) and M2 (CD206+ macrophage) populations as a percentage of F4/80+ cells. n=6 mice per group. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01. (C) Analysis of CD8/Treg and M1/M2 ratios as measures of immunosuppression in the tumor microenvironment of WT and Myct1 KO mice. n=6 mice per group. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; n.s.=not significant, *p<0.05, and **p<0.01. (D) (top) Analysis of FOXP3 expression in high vs. low MYCT1 expressing tumors in patients from the TCGA database. (bottom) Analysis of M1/M2 population ratios in high vs. low MYCT1 expressing tumors obtained by analyzing the TCGA patient datasets with the CIBERSORT algorithm. Datasets utilized were BC (TCGA-BRCA), LAC (TCGA-LUAD), TT (TCGA-TGCT), SAC (TCGA-STAD), LSCC (TCGA-LUSC), CAC (TCGA-COAD), HNSCC (TCGA-HNSC), OC (TCGA-OV), PC (TCGA-PRAD), PAC (TCGA-PDAC), Sarcoma (TCGA-SARC), CCRCC (TCGA-KIRC), PRCC (TCGA-KIRP), and BLC (TCGA-BLCA). Data is presented as scatter plot with the mean value. Statistical significance was analyzed by Mann-Whitney U test; *p<0.05 and **p<0.01 (E) Analysis of tumor growth in either IgG or combined anti-CD4 and anti-CD8 neutralizing antibody treated Myct1 KO mice. n=5 mice per group. Data is presented as mean with SD from one of two biological replicates. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; p<0.001, at the end of the study. (F) Analysis of tumor growth in either IgG, or anti-IFNγ neutralizing antibody, or combined anti-CD4 and anti-CD8 neutralizing antibody, or combined anti-CD4 and anti-CD8 neutralizing antibody with IFNγ cytokine treated Myct1 KO mice. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; ‘p-values’ indicated on the figure are calculated at the end of the study. (G) Transendothelial migration (TEM) of CD8+ T cells through parental and Myct1 KD MCEC cell barrier with TNFα and Rac1 inhibitor NSC23766 pre-treatment. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; n.s.=not significant, *p<0.05, and **p<0.01. (H and I) Analysis of mRNA expression of the indicated genes in CD31+CD45− ECs isolated from the 1956 sarcoma tumors. n=4 mice per group. Statistical significance was analyzed by Statistical significance was analyzed by two-tailed Student’s t-test; *p<0.05 and **p<0.01. (J) FASLG expression profile in high vs. low MYCT1 expressing tumors in SAC (TCGA-STAD), LSCC (TCGA-LUSC), CAC (TCGA-COAD), OC (TCGA-OV), PC (TCGA-PRAD), PAC (TCGA-PDAC), Sarcoma (TCGA-SARC), CCRCC (TCGA-KIRC), and PRCC (TCGA-KIRP) patients from the TCGA database. Data is presented as mean with standard error of mean. Statistical significance was analyzed by two-tailed Student’s t-test; *p<0.05 and **p<0.01. (K) Polarization of peripheral blood-derived monocytes to M1 or M2 phenotype with LPS+IFNγ or IL4 cytokine treatment, respectively, in a coculture assay with either parental or Myct1 KD MCEC cells, expressed as a percentage of F4/80+ macrophage population. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; *p<0.05 and **p<0.01. (L) Analysis of Nos2 and Nos3 mRNA expression in CD31+CD45− ECs isolated from the 1956 sarcoma tumor. n=4 mice per group. Data is presented as mean with SD. Statistical significance was analyzed by two-tailed Student’s t-test; **p<0.01.
Vascular normalization-mediated tumor growth restriction observed in Myct1 KO mice is dependent on adaptive immunity.
If, indeed, the increased CTLs in Myct1 KO tumor contributed to the tumor growth restriction, the ablation of CTLs should restore the tumor growth. To this end, we utilized neutralizing antibodies to deplete CD4+ and CD8+ T cell compartments in the tumor-bearing Myct1 KO mice and found that the tumor growth restriction was completely abrogated (Fig. 6E and fig. S14H), suggesting that the presence of the adaptive immunity was essential for the anti-tumor activity of Myct1-mediated tumor vessel normalization. This crosstalk between tumor vascular control and adaptive immunity was partly IFNγ-mediated. While IFNγ neutralizing antibody abrogated tumor growth restriction phenotype of Myct1 KO mice, mimicking the CD4+ and CD8+ T cell depletion, recombinant IFNγ treatment partially restored the phenotype in CD4+ and CD8+ T-cell-depleted Myct1 KO mice (Fig. 6F). Together, our data demonstrate that Myct1-mediated tumor vascular control actively shapes the tumor microenvironment through a close engagement with adaptive immunity. Notably, we also observed a small but statistically significant (p<0.05) increase in tumor vessels in the CD4+ and CD8+ T cell-depleted Myct1 KO mice (fig. S14I). This observation was similar in principle to a previous report that depletion of CD8+ T cells was associated with a dramatic increase in tumor microvascular frequency (46). However, the lack of Myct1 could be the reason why we observed only a modest increase of tumor vasculature even after T cell depletion.
Myct1-deficient ECs promote T cell trafficking and skew macrophage polarization in the angiogenic environment.
Since both the Myct1 KO and Myct1low tumor-ECs were enriched for leukocyte adhesion and transendothelial migration (Fig. 5I–K and fig. S13E), we evaluated whether Myct1-deficient ECs supported more T cell infiltration. By utilizing a modified T cell transendothelial migration assay as described previously (47), we found that CD8+ T cells transmigrated through the Myct1-deficient EC barrier more compared to the WT EC barrier (Fig. 6G). However, Treg cells did not display any difference in transmigration between the WT or Myct1 deficient ECs (fig. S14J). Mechanistically, Myct1 KO tumor-ECs showed increased expression of endothelial adhesion molecules E-and P-selectin, ICAM-1, and VCAM-1 (Fig. 6H and fig. S14K). Moreover, Myct1 KO tumor-ECs had increased expression of Rac1 and associated effector molecules such as Trio, Tiam, and Rhog (Fig. 6H and fig. S14K). This upregulation of the Rac1-mediated pathway is crucial as RAC1 inhibition by NSC23766 abrogated the increased T cell migration phenotype (Fig. 6G and fig. S14J). Additionally, tumor endothelium is known to express immune-suppressor molecules, such as PDL1, PDL2, Fas ligand, and TRAIL (48–51). We found that the expression of Fas ligand (Faslg), which can induce apoptosis of infiltrating CD8+ T cells by binding to the cognate FAS receptor (51), was downregulated in Myct1 KO tumor-ECs (Fig. 6I). We observed a similar trend in the tumor samples with low MYCT1 expression compared to the samples with high MYCT1 expression in some human cancer types (Fig. 6J). Finally, we investigated whether Myct1 expression in tumor endothelium had any direct impact on macrophage polarization. In the presence of respective M1 or M2 polarizing cytokines, Myct1-deficient ECs promoted more M1-like and less M2-like macrophage polarization from monocytes (Fig. 6K). Intriguingly, tumor-ECs in Myct1 KO mice exhibited reduced expression of Nos2 and Nos3 (Fig. 6L and fig. S14L), which have been shown to affect M1- and M2-macrophage polarization (52–54), suggesting a potential role for nitric oxide synthase (NOS) in the observed anti-tumor macrophage skewing. Together, this series of investigations provided a mechanistic explanation for the observed anti-tumor T-cell and macrophage remodeling phenotype in the Myct1 KO mice.
Targeting of Myct1 improves anti-PD1 immunotherapy outcomes in mice.
The success of the immune checkpoint blockade-mediated immunotherapeutic approach partially relies on the presence of CTLs in the tumor microenvironment. Since our data show that Myct1-deficient tumor endothelium promotes CTL infiltration, addition of anti-PD1 to prevent CTL exhaustion might provide a synergistic and superior treatment outcome. As such, we assessed the efficacy and usefulness of the combined anti-Myct1 and anti-PD1 treatment approaches in both anti-PD1-responsive and anti-PD1-refractory tumor models. First, we utilized an anti-PD1-responsive 1956 sarcoma subcutaneous transplantation tumor model that responds completely to an early-onset scheme of anti-PD1 treatment, but not to late-onset schemes (24). To this end, we validated different modalities of anti-PD1 therapy in this tumor model and found that a late-onset treatment starting from 9-days post-tumor transplantation did not result in total regression, but somewhat slowed tumor progression with eventual complete relapse of the tumor in WT mice (Fig. 7A). We treated the tumor-bearing Myct1 KO mice with this late-onset anti-PD1 scheme and observed dramatic tumor regression within 12 days of treatment initiation (Fig. 7B). This short-term regression led to complete tumor regression in all but one treated mouse (7 out of 8) (Fig. 7C). Supporting our observation that endothelial-specific Myct1 regulates the tumor growth and angiogenesis, Cdh5-cre Myct1f/f mice demonstrated a similar tumor regression with this late-onset anti-PD1 treatment scheme (fig. S15A). To determine whether a systemic anti-Myct1 approach confers similar anti-tumor activity, we utilized a Myct1 directed siRNA-peptide nanoparticle treatment approach in WT mice (17, 55) either alone or in combination with DC101 (a VEGF receptor 2 (VEGFR2) blocking antibody) and/or anti-PD1, following a similar late-onset treatment scheme (Fig. 7D). We found that combined anti-PD1 and anti-Myct1 treatment restricted tumor progression in all the treated mice (Fig. 7D), with a complete regression in 25% of the mice (2 out of 8) (Fig. 7E). In comparison, combined anti-PD1 and DC101 treatment also produced significant (p<0.05) short-term tumor growth restriction, though to a somewhat lesser extent (Fig. 7D), with an eventual relapse of tumor growth in all mice (Fig. 7E). Remarkably, anti-PD1 treatment with the dual blockade of Myct1 and VEGFR2 resulted in complete tumor regression in all of the treated mice (fig. 7E). Since combined Myct1 siRNA and anti-PD1 treatment resulted a complete tumor regression in 2 out of 8 mice (Fig. 7E), while anti-PD1 treatment in Myct1 KO mice led to complete tumor regression in 7 out of 8 mice (Fig. 7C), we assessed whether this was due to a sub-optimal Myct1 siRNA treatment. We utilized an extended anti-Myct1 treatment approach that started before the onset and continued after the termination of the anti-PD1 treatment. We found that this prolonged anti-Myct1 treatment resulted in the restriction of tumor progression for a longer period, with about 60% mice showing complete tumor regression (14 out of 24 mice) (Fig. 7F).
Figure 7: Myct1-targeted siRNA-peptide nanoparticle co-treatment improves anti-PD1 immunotherapy.
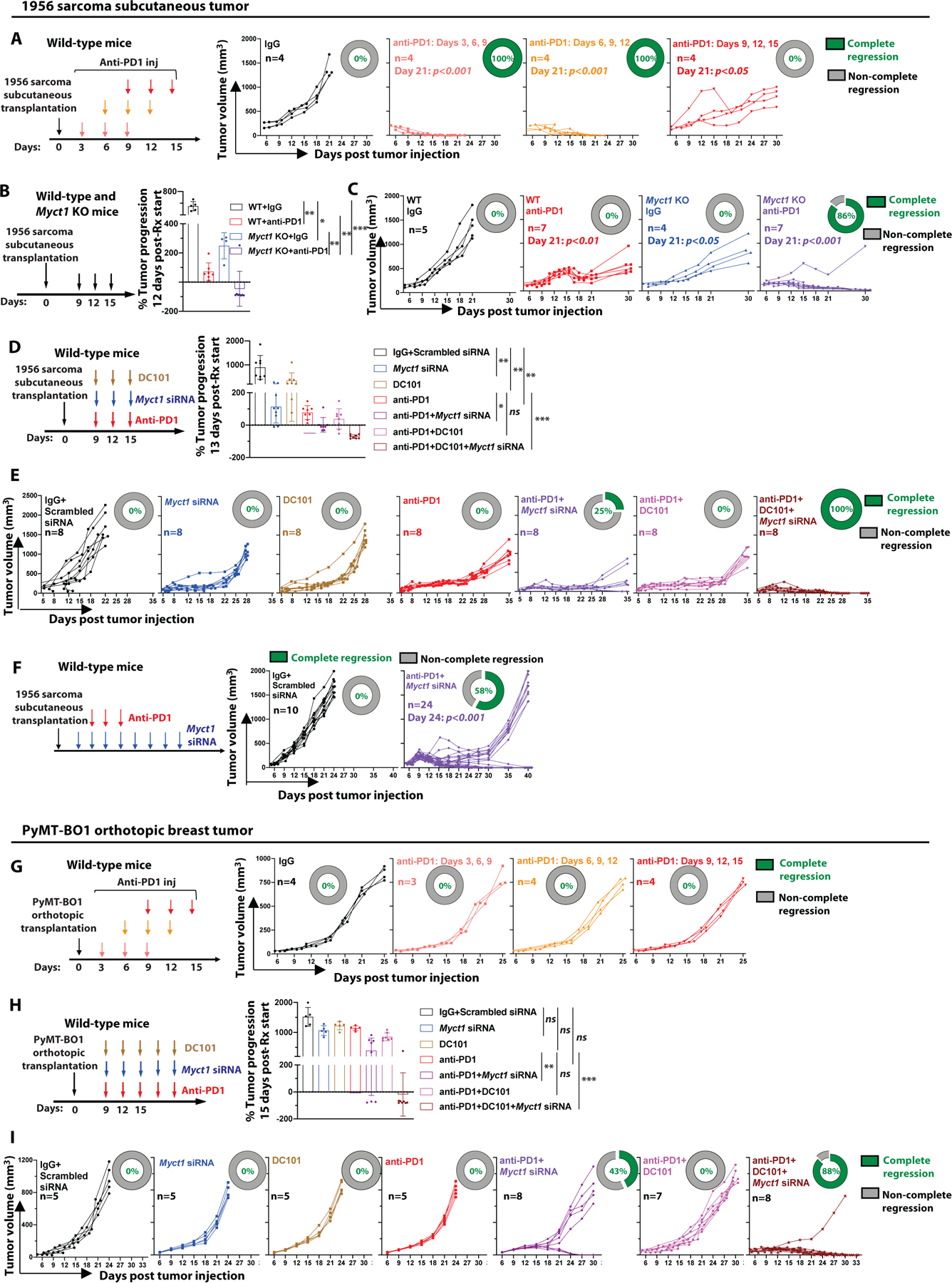
(A) Kinetics of tumor growth in 1956 sarcoma tumor bearing WT mice treated with anti-PD1 antibody in different indicated schemes. Donut charts display the percentage of subjects underwent complete regression (green) and non-complete regression (gray). Day-21 statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test. (B) Analysis of tumor progression in 1956 sarcoma tumor bearing WT and Myct1 KO mice at 12-days after anti-PD1 treatment initiation. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05, **p<0.01, and ***p<0.001. (C) Tumor growth in 1956 sarcoma tumor bearing WT and Myct1 KO mice treated with the anti-PD1 antibody. Donut charts display the percentage of subjects underwent complete regression (green) and non-complete regression (gray). Day-21 statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test. (D) Analysis of tumor progression in 1956 sarcoma tumor bearing WT mice at 13-days after treatment initiation as indicated. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05, **p<0.01, and ***p<0.001. (E) Tumor growth in 1956 sarcoma tumor bearing WT mice with different treatments as indicated. Donut charts display the percentage of subjects underwent complete regression (green) and non-complete regression (gray). (F) Tumor growth in 1956 tumor bearing WT mice treated with the anti-PD1 antibody in combination with an extended anti-Myct1 siRNA-peptide nanoparticle treatment. Day-21 statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test. (G) Kinetics of tumor growth in PyMT-BO1 tumor bearing WT mice treated with anti-PD1 antibody in different schemes. Donut charts display the percentage of subjects underwent complete regression (green) and non-complete regression (gray). (H) Tumor progression in PyMT-BO1 tumor bearing WT mice at 15-days after treatment initiation as indicated. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; n.s.=not significant, **p<0.01, and ***p<0.001. (I) Tumor growth in PyMT-BO1 tumor bearing WT mice with different treatments as indicated. Donut charts display the percentage of subjects underwent complete regression (green) and non-complete regression (gray).
Next, we assessed whether Myct1 targeting could sensitize anti-PD1-refractory tumors. Here, we utilized an orthotopic breast tumor model with PyMT-BO1 tumors that do not respond to anti-PD1 treatment (Fig. 7G). We treated tumor-bearing WT mice with Myct1-directed siRNA-peptide nanoparticles either alone or in combination with DC101 and/or anti-PD1. Although DC101 failed to induce any sensitivity to anti-PD1 treatment, combined anti-Myct1 and anti-PD1 treatment resulted in a substantial short-term tumor regression (Fig. 7H), with complete tumor regression in 43% of the treated mice (3 out of 7) (Fig. 7I). Intriguingly, similar to the 1956 sarcoma model, dual blockade of Myct1 and VEGFR2 with anti-PD1 treatment generated both the maximal short-term tumor restriction and long-term complete tumor regression (7 out of 8 mice) (Fig. 7H and 7I), suggesting that the collective blockade of both the VEGF and MYCT1 pathways might provide better and longer-lasting vascular control, resulting in improved outcomes with anti-PD1 immunotherapy in both sensitive and refractory tumor models.
Dormant tumor cells can reinitiate tumor growth after treatment (56–58). To address if dormant tumor cells still existed in these transplant tumor models after anti-Myct1 and anti-PD1 treatment, we treated the tumor-regressed mice (pooled from the experiments with 1956 sarcoma tumor model described in Fig. 7E and 7F) with monoclonal neutralizing antibodies against CD4, CD8, and IFNγ, three major components of adaptive immunity. None of the tumor-regressed mice developed any tumor mass after the alleviation of the immune surveillance for a period of over 90 days (fig. S15B), suggesting that anti-PD1 treatment combined with anti-Myct1 targeting, with or without other anti-angiogenics, not only brought complete tumor regression but also destroyed any potential dormant tumor cells residing in the equilibrium phase. Intriguingly, re-challenging both the tumor-regressed mice (pooled from the experiment with PyMT-BO1 tumor model described in Fig. 7H) and naïve control mice with the PyMT-BO1 tumor cells resulted in unrestrained tumor growth without any statistical difference (fig. S15C), suggesting that anti-Myct1 and anti-PD1 combination treatment did not induce long-lived immunological memory.
To further understand how Myct1-directed siRNA-peptide nanoparticle treatment improves the outcome of the anti-PD1 immunotherapy, we harvested and analyzed tumors seven days after treatment initiation in the anti-PD1 refractory PyMT-BO1 tumor model, at a time when the tumors just started to respond to combination treatment (fig. S16A). Flow cytometric analysis revealed that anti-Myct1 treatment increased CTLs and M1 macrophages, while reducing the number of Treg cells and M2 macrophages. Conversely, anti-PD1 treatment increased the percentage of Granzyme B+ CTLs, but did not alter the immune microenvironment. Notably, anti-Myct1 and anti-PD1 combined treatment increased both the influx of CTLs and the percentage of Granzyme B+ CTLs, suggesting that the combination treatment produces considerably superior tumor control due to the additive nature of the individual treatments on CTLs (Fig. 8A). Immunofluorescence observations supported the conclusion that anti-Myct1 treatment reduced angiogenesis, improved vascular normalization, and increased the infiltration of CD8+ T cells (Fig. 8B–E). Intriguingly, although the siRNA-peptide nanoparticle treatment downregulated Myct1 expression in both the tumor and non-tumor endothelium (fig. S16B), unlike in the tumor masses, there were no alterations in the immune constituents of the non-tumor tissues (fig. S16C). We observed a similar modification of immune constituents of the tumor masses in the Myct1 KO mice with the anti-PD1 treatment, supporting the notion of the additive nature of the treatments (fig. S16D and S16E). Downregulation of Myct1 by the nanoparticles led to the upregulation of several adhesion molecules, among which Selp and Icam1 reached statistical significance (p<0.05), only in the tumor endothelium but not in the non-tumor tissues (Fig. 8F and fig. S17A), corroborating the observations in the Myct1 KO mice (see Fig. 6H). Importantly, Myct1-directed siRNA-peptide nanoparticle treatment largely did not induce any non-specific immune responses in either the tumor-infiltrating hematopoietic cells or tumor cells (Fig. 8F, fig. S17B, and S17C). Collectively, these observations suggest that Myct1 targeting generates tumor-EC-focused changes that allows anti-PD1 treatment to mount more effective anti-tumor responses.
Figure 8: Anti-Myct1 works synergistically with anti-PD1 treatment to improve outcomes in mice.
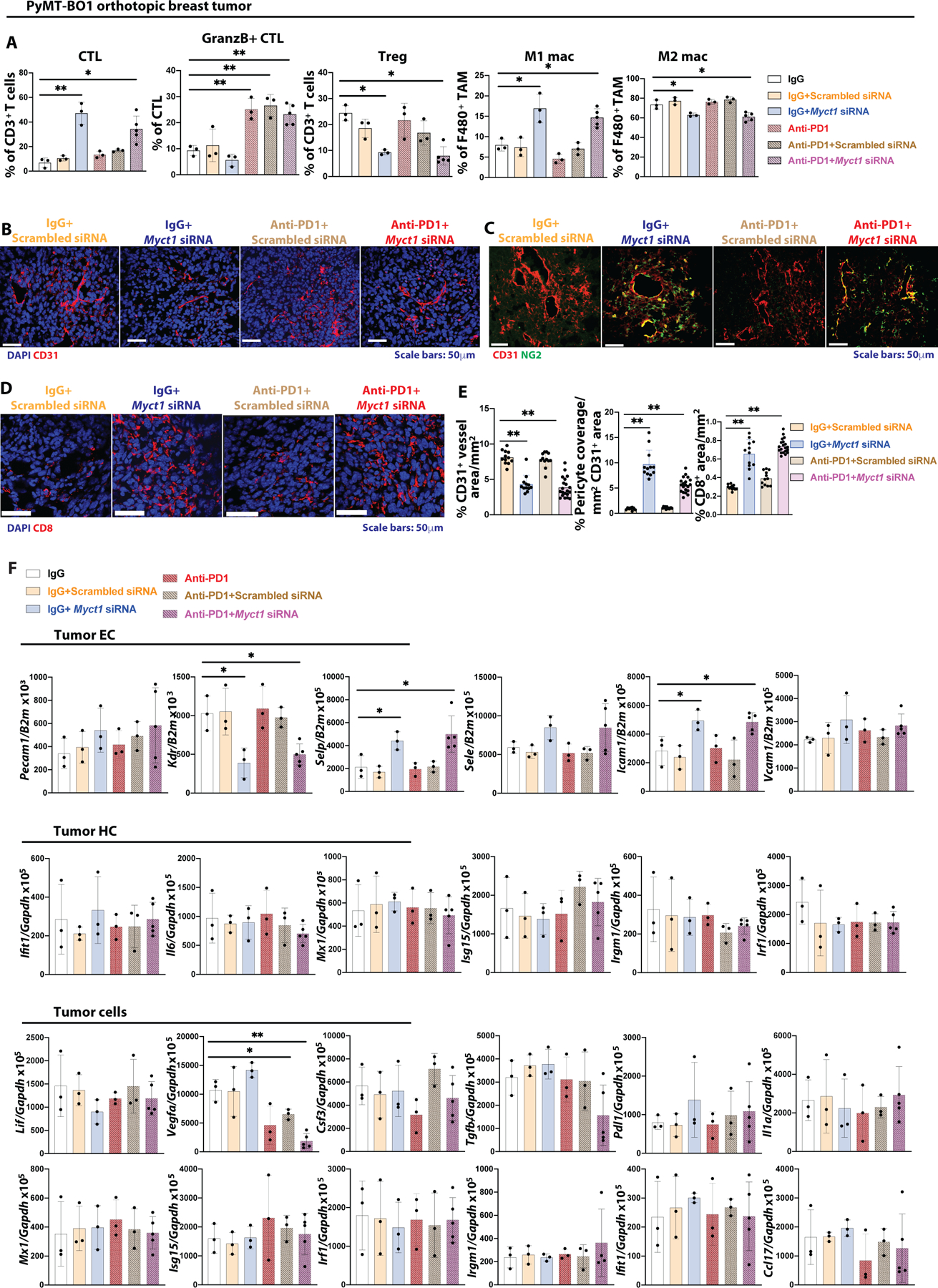
(A) Analysis of CTL (CD8+ T cells) and Treg (CD4+FOXP3+ T cells) cells as a percentage of CD3+ T cells, GranzB+ CTL as % of CTL cells, M1 (iNOS+ macrophages) and M2 (CD206+ macrophage) populations as a percentage of F4/80+ cells in the tumor of the PyMT-BO1 tumor bearing WT mice with different combination of treatments as indicated. n=3–6 mice per group. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05 and **p<0.01. (B-E) Representative images (B-D) and quantifications (E) for CD31+ vascular density (B), pericyte coverage (C), and CD8+ T cell infiltration in the tumor mass of PyMT-BO1 tumor bearing WT mice with different combination of treatments as indicated. n=3–6 mice per group. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; **p<0.01. (F) Analysis of the expression of the indicated genes in the tumor ECs, hematopoietic cells (HC), and tumor cells sorted from the PyMT-BO1 tumor bearing WT mice with different combination of treatments as indicated. n=3–6/group. Data is presented as mean with SD. Statistical significance was analyzed by One-way ANOVA with Tukey’s multiple comparison test; *p<0.05 and **p<0.01.
Discussion
Myct1 was first identified as a direct target gene of c-MYC in myeloid cells, as well as in laryngeal squamous cell carcinoma cells, in the context of c-MYC overexpression (59, 60). However, analysis of published scRNA sequencing datasets from different mouse organs revealed that Myct1 expression is mostly restricted to ECs and hematopoietic stem and progenitor cells. We have demonstrated that Myct1 is highly expressed in tumor ECs and is a regulator of angiogenesis. We found that Myct1 is not required for blood and vascular development. Instead, Myct1 seems to regulate acute angiogenic demand in pathological conditions such as cancer. Notably, data from human patients with varying MYCT1 expression across multiple different cancer types also suggest that MYCT1 is positively correlated with the angiogenic status of the cancers. Particularly, we show that MYCT1 regulates EC motility by interacting with ZO1 and CKAP4 and modulating Rho GTPases and actin cytoskeleton. It will be necessary to further delineate the crosstalk among MYCT1, ZO1, Rho GTPases, and CKAP4 in regulating the actin cytoskeleton and EC motility.
Myct1 deficient tumor vasculature is characterized by having more HEVs, facilitating CTLs infiltration, and promoting anti-tumor macrophage polarization. Tumor vessels inhibit CTLs activation and promote apoptosis of the infiltrating immune cells in part by upregulating immunosuppressive molecules, PDL1, PDL2, Fas ligand, and TRAIL (48–51). We found that Myct1 deficient ECs downregulate the expression of Fas ligand. We also found a similar trend with human cancers with reduced MYCT1 expression. Our data suggest that Myct1 deficient endothelium promotes an immunostimulatory microenvironment by enhancing CTLs infiltration and presumably by preventing CTLs apoptosis. Moreover, it has been reported that inducible-NOS (NOS2) inhibits the M1-macrophage population, whereas endothelial-NOS (NOS3) promotes the M2-macrophage polarization (52–54). Notably, the expressions of Nos2 and Nos3 were decreased in Myct1 deficient ECs, suggesting that enhanced anti-tumor macrophage polarization in Myct1 deficient tumor microenvironments may, in part, through the regulation of NO production of ECs. These findings collectively provide mechanistic insights into Myct1 deficient tumor vessels contributing to an immunostimulatory microenvironment.
Vascular normalization has been implicated to influence local tumor immune environment (61, 62). However, the molecular targets like VEGF and associated pathways also impact the immune cells in an endothelial cell-independent manner (63–66), raising a possibility of synergistic and/or independent effect rather than a sole consequence of vascular normalization on the tumor immune components. Myct1 targeting in endothelial cells leading to an anti-tumor microenvironment provides more definitive evidence for endothelial regulation of tumor immunity in mice. This data is consistent with the human cancer datasets that show the increased presence of immunostimulatory components in tumors with lower MYCT1 expression. Our finding supports the emerging notion that combined vascular and immune control would provide a synergistic anti-tumor activity (3, 48, 67–69). Indeed, our data demonstrate that targeting Myct1 improves the response to anti-PD1 therapy in treatment-responsive and -refractory tumors. Importantly, we observed that combined Myct1 and VEGF targeting with anti-PD1 treatment produced a superior tumor control, suggesting a potential synergy between Myct1 and VEGF pathways.
Our study’s limitations include that our data on Myct1 expression in tumor endothelium is confined to RNA, not protein. Additionally, our data on immune modulation are limited to CTL infiltration and M1 polarization. Global changes in tumor immunity by endothelial-specific Myct1 deficiency need to be further elucidated to better understand the crosstalk between angiogenesis and tumor immunity. Lastly, while siRNA-nanoparticle-mediated Myct1 targeting has provided proof of concept, it would be valuable to develop and validate an antibody-mediated MYCT1 blocking approach to realize the therapeutic potential of MYCT1 inhibition.
In summary, we have identified Myct1 as a regulator of tumor angiogenesis. Myct1-deficient ECs display suboptimal angiogenesis, facilitate HEV formation, enhance robust CTL infiltration, and promote inflammatory M1 macrophage polarization. Anti-PD1 antibody treatment in the context of Myct1 inhibition augments complete tumor elimination in mouse models. We propose that combined Myct1-inhibition and immunotherapy might become a treatment regimen for cancer patients.
Materials and Methods
Study design
The goal of the study was to investigate the role of Myct1 as a regulator of tumor angiogenesis and anti-tumor immunity and to characterize the mechanism of this regulation. The Cancer Genome Atlas (TCGA) datasets for thirteen different cancer types were analyzed to identify Myct1 as an angiogenic gene downstream of Etv2 (14, 19, 20, 22, 70). Myct1 requirement for tumor growth, angiogenesis, and antitumor immunity was assessed by using different preclinical mouse models of cancer (18, 26, 57, 71–76). Potential correlation between endothelial Myct1 expression and the immune output in the human tumor microenvironment was evaluated by analyzing cancer datasets using the CIBERSORT deconvolution algorithm (45). Using various in vitro assays, the role(s) of Myct1 in endothelial cells and the underlying mechanisms for regulating anti-tumor immunity were rigorously investigated (17, 77–79). Lastly, using both the knockout mice and siRNA-peptide nanoparticle mediated approach (17), the efficacy of Myct1 inhibition for treating tumors in combination with anti-PD1 immunotherapy was evaluated. For animal studies, the minimum number of subjects used in any experiments was 5/group to attain a statistical significance of p<0.05 with a power of at least 80%, considering the mean differences between experimental groups would be >20% and the pooled standard deviation would be ~20%. For in-vitro experiments, the anticipated mean difference was even more, and hence the sample size was kept at 3 or more/group. Age- and sex-matched mice were randomly assigned into groups in all experiments except experiments utilizing both the genetic and orthotopic breast tumor models, where only female mice were used. The investigators were not aware of the group allocation until the treatment, data collection, and data analysis were done. All experimental data were reliably reproduced in two or more individual biological replicates unless indicated otherwise. No data were excluded from analysis. Sample sizes, biological replicates, and statistical methods are provided in the corresponding figure legends. Primary data are reported in supplementary data files S1 and S2.
Mice
Myct1−/− (Myct1 KO) and Cdh5-Cre;Myct1f/f mice were generated at Washington University in St. Louis and Emory University, respectively. MMTV-PyMT mice were a gift from Mikala Egeblad, Cold Spring Harbor Laboratory, and crossed with Myct1 KO mice to generate Myct1 KO in the presence of MMTV-PyMT transgene (MMTV-PyMT Myct1−/−) at Washington University in St. Louis. The details of the mice generation can be found in the Supplementary Materials and Methods section. Animal husbandry, generation, handling, and experimentation were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Washington University School of Medicine in St. Louis.
Mouse tumor models
MMTV-PyMT transgenic mice were utilized to generate a spontaneous model of breast cancer, where MMTV-LTR drives the expression of mouse mammary gland-specific polyomavirus middle T-antigen. For tumor transplantation studies, LLC-GFP, B16F10 melanoma, and 1956 sarcoma tumor cells were injected subcutaneously to the back of the mice. PyMT-BO1 tumor cells were injected orthotopically to the mammary fat pad of the female mice. The details regarding the transplantation procedures, tumor growth monitoring, different treatment schemes, and downstream flow cytometry, immunofluorescence, and RNA expression analysis of the tumor masses can be found in the Supplementary Materials and Methods section.
Cell lines
Mouse cardiac endothelial cells (MCEC) and human umbilical vein endothelial cells (HUVEC) were purchased from CELLutions Biosystems Inc. (Cat: CLU510) and ATCC (Cat: ATCC CRL-1730), respectively. Overexpressing and shRNA lentiviral particles were utilized to generate different genetically modified overexpressing and knockdown stable cell lines. Further details about the cell culture conditions, lentiviral clones, production of lentiviral particles, antibiotic selection, and different treatment schemes for in vitro experiments are provided in the Supplementary Materials and Methods section.
Single-cell RNA sequencing
Endothelial cells sorted by flow cytometry from the tumor masses were loaded on a Chromium Single Cell Instrument (10X Genomics) to generate single-cell GEMs. Single-cell RNA-seq libraries were prepared using version 2 Chromium Single-cell 3′ Library, Gel Bead & Mutiplex Kit (10X Genomics). Sequencing was performed on Illumina NextSeq2500 and mapped to the mouse genome (build mm10) using CellRanger software (10x Genomics, version 2.1.1). Sequencing data is available as GSE157879 and GSE146819. Further details about the sequencing data quality control, normalization, integration, and downstream analysis can be found in the Supplementary Materials and Methods section.
Statistical Analysis
GraphPad Prism 8 software was used for performing statistical analysis and generating graphs/plots. Data are presented as mean with standard deviation for all the measurements. Statistical significance was determined by two-tailed unpaired Student’s t-test (for two groups) and One-way ANOVA with Tukey’s multiple comparison test (for more than two groups). Non-parametric tests were used for non-log transformed gene expression data from the TCGA database. p<0.05 was considered statistically significant.
Supplementary Material
Materials and Methods
Figure S1: Single-cell RNA sequencing in heterogenous cell populations from different mouse organs shows an almost endothelial-exclusive pattern of Myct1 expression.
Figure S2: Analysis of TCGA-derived cancer patient datasets shows correlation between the ‘angiogenic scores’ and MYCT1 expression.
Figure S3: Single cell RNA sequencing of heterogenous mouse tumor stromal compartments shows exclusive pattern of Myct1 expression in the endothelial cells.
Figure S4: Etv2 downstream target Myct1 is a regulator of angiogenesis; however, it is not required for vascular development and homeostasis in mice and zebrafish.
Figure S5: Endothelial Myct1 expression is essential for tumor growth and angiogenesis in mice.
Figure S6: Myct1-deficient mouse tumor vessels display improved vascular functions.
Figure S7: Endothelial Myct1 expression is relevant for tumor growth and angiogenesis in mice.
Figure S8: Endothelial Myct1 is critical for tumor growth and is required for vascular regeneration in vitro and in vivo.
Figure S9: MYCT1 regulates the actin cytoskeleton dynamics of endothelial cells in the angiogenic environment in vitro.
Figure S10: MYCT1 functionally interacts with ZO1 and RHOA to regulate the capillary characteristics of endothelial cells in the angiogenic environment in vitro.
Figure S11: MYCT1 is evolutionarily conserved between human and mouse.
Figure S12: Single cell RNA sequencing displays that tumor endothelial cells express classical endothelial marker genes.
Figure S13: UMAP projection of tumor endothelial cells following single cell RNA sequencing identifies different functionally distinct sub-populations in WT mice.
Figure S14: Myct1 deficiency leads to an anti-tumor immune environment both in humans and mice.
Figure S15: Endothelial Myct1 deficiency recapitulates global Myct1 KO phenotype in combination with anti-PD1 treatment in tumor-bearing mice.
Figure S16: Combined Myct1 and PD1 targeting does not alter immune constituents of the non-tumor tissues in tumor-bearing mice.
Figure S17: Combined Myct1 and PD1 targeting does not induce off-target and non-specific immune activation.
Table S1. Top 100 regulators of angiogenesis identified by their expression correlation with seed genes.
Table S2: List of genes (termed as “seed genes”) to identify angiogenic regulatory genes.
Table S3: List of genes used to generate the “angiogenic score” for patient datasets across different cancer types
Table S4: Sequences of primers used in the study.
Table S5: LC/MS proteomics screening for MYCT1 binding partners.
Data file S1: Subject-level data for main figures.
Data file S2: Subject-level data for supplementary figures.
Acknowledgements
We thank Mikala Egeblad at Cold Spring Harbor Laboratory for the generous gift of the MMTV-PyMT mice. We want to thank our colleagues at Washington University, Kory Lavine for MCEC cells, Andrew Yoo for BJ-5ta cells, Katherine Weilbaecher for PyMT-BO1-GFP-Luc cells, Luis Batista for pCDH-(LB12-FLAG-TERT)-EF1-NEO plasmid. We thank Robert D. Schreiber at Washington University for 1956 sarcoma cells and helpful suggestions with experimental design and data interpretation. We also want to thank Alexander S Krupnick at the University of Virginia for providing LLC-GFP cells. We thank Mike White and the Genome Engineering and iPSC Center (GEIC) at Washington University in St. Louis for the generation of Myct1 KO mice using CRISPR/CAS9 technology. We also thank Washington University Center for Cellular Imaging (WUCCI) and Pathology FACS core for providing access to the light microscopes and FACS facility, respectively. We thank Attila Kovacs and Carla J. Weinheimer of the Mouse Cardiovascular Phenotyping Core at Washington University School of Medicine for the echocardiogram analysis.
Funding:
This work was supported by the NIH grants R01HL149954 (to K.C.), R01HL55337 (to K.C), R01HL119291 (to C.P), and K08HL135400 (to C.M.H), Children’s Heart Research and Outcomes Center and Children’s Healthcare of Atlanta 00060337 (to C.P.) and Mallinckrodt Challenge Grant (to K.C. and D.H.F.).
Footnotes
Competing interests: Samuel A. Wickline has Equity in Trasir Therapeutics, Inc. Ashraf Ul Kabir and Kyunghee Choi have a patent pending relating to this work (Application Number: 63/093,595; Title: COMPOSITIONS AND METHODS FOR MODULATING MYCT1). All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are available in the main text or the supplementary materials. RNA sequencing data is available as GSE157879 and GSE146819. Global Myct1 knockout mice and Cdh5-cre Myct1 knockout mice can be provided to academic researchers via an MTA upon request to Kyunghee Choi and Changwon Park, respectively.
This is the author’s version of the work. It is posted here by permission of the AAAS for personal use, not for redistribution. The definitive version was published in Science Translational Medicine 2021 Mar 3;13(583):eabb6731. doi: 10.1126/scitranslmed.abb6731.
References
- 1.Folkman J, Tumor angiogenesis: therapeutic implications. N. Engl. J. Med 285, 1182–1186 (1971). [DOI] [PubMed] [Google Scholar]
- 2.Chung AS, Ferrara N, Developmental and Pathological Angiogenesis. Annual Review of Cell and Developmental Biology 27, 563–584 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA, Hallmarks of Cancer: The Next Generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Ye W, The Complexity of Translating Anti-angiogenesis Therapy from Basic Science to the Clinic. Developmental Cell 37, 114–125 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Bergers G, Hanahan D, Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrara N, Adamis AP, Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 15, 385–403 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML, Autocrine VEGF Signaling Is Required for Vascular Homeostasis. Cell 130, 691–703 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Zhang Y, Cao Z, Ji H, Yang X, Iwamoto H, Wahlberg E, Lanne T, Sun B, Cao Y, Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proc Natl Acad Sci U S A 110, 12018–12023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dammeijer F, Lau SP, van Eijck CHJ, van der Burg SH, Aerts J, Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev 36, 5–15 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Chauhan VP, Chen IX, Tong R, Ng MR, Martin JD, Naxerova K, Wu MW, Huang P, Boucher Y, Kohane DS, Langer R, Jain RK, Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc Natl Acad Sci U S A 116, 10674–10680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, Huang P, Lindeman N, Langer R, Jain RK, Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci U S A 116, 4558–4566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, Kienast Y, Mueller HJ, Ooi CH, Laoui D, De Palma M, Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med 9, (2017). [DOI] [PubMed] [Google Scholar]
- 13.Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, Feyen K, Tawney J, Hanahan D, Michael IP, Bergers G, Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, Janknecht R, Lim D-S, Choi K, ER71 Acts Downstream of BMP, Notch, and Wnt Signaling in Blood and Vessel Progenitor Specification. Cell Stem Cell 2, 497–507 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, Li D, Yu YYL, Kang I, Cha M-J, Kim JY, Park C, Watson DK, Wang T, Choi K, Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO reports 16, 654–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu CX, Lee TJ, Sakurai N, Krchma K, Liu F, Li D, Wang T, Choi K, ETV2/ER71 regulates hematopoietic regeneration by promoting hematopoietic stem cell proliferation. J Exp Med 214, 1643–1653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabir AU, Lee TJ, Pan H, Berry JC, Krchma K, Wu J, Liu F, Kang HK, Hinman K, Yang L, Hamilton S, Zhou Q, Veis DJ, Mecham RP, Wickline SA, Miller MJ, Choi K, Requisite endothelial reactivation and effective siRNA nanoparticle targeting of Etv2/Er71 in tumor angiogenesis. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park C, Lee T-J, Bhang SH, Liu F, Nakamura R, Oladipupo SS, Pitha-Rowe I, Capoccia B, Choi HS, Kim TM, Urao N, Ushio-Fukai M, Lee D, Miyoshi H, Kim B-S, Lim D-S, Apte RS, Ornitz DM, Choi K, Injury-Mediated Vascular Regeneration Requires Endothelial ER71/ETV2. Arteriosclerosis, Thrombosis, and Vascular Biology 36, 86–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masiero M, Simoes FC, Han HD, Snell C, Peterkin T, Bridges E, Mangala LS, Wu SY, Pradeep S, Li D, Han C, Dalton H, Lopez-Berestein G, Tuynman JB, Mortensen N, Li JL, Patient R, Sood AK, Banham AH, Harris AL, Buffa FM, A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24, 229–241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Costa NM, Cina D, Shrestha R, Bell RH, Lin YY, Asghari H, Monjaras-Avila CU, Kollmannsberger C, Hach F, Chavez-Munoz CI, So AI, Identification of gene signature for treatment response to guide precision oncology in clear-cell renal cell carcinoma. Sci Rep 10, 2026 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hänzelmann S, Castelo R, Guinney J, GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Q, Eichten A, Parveen A, Adler C, Huang Y, Wang W, Ding Y, Adler A, Nevins T, Ni M, Wei Y, Thurston G, Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res 78, 2370–2382 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Vesely M, Tumor Antigens Revealed by Exome Seqeuncing Drive Editing of Tumor Immunogenicity. All Theses and Dissertations (ETDs), (2013). [Google Scholar]
- 25.Su X, Esser AK, Amend SR, Xiang J, Xu Y, Ross MH, Fox GC, Kobayashi T, Steri V, Roomp K, Fontana F, Hurchla MA, Knolhoff BL, Meyer MA, Morgan EA, Tomasson JC, Novack JS, Zou W, Faccio R, Novack DV, Robinson SD, Teitelbaum SL, DeNardo DG, Schneider JG, Weilbaecher KN, Antagonizing integrin beta3 increases immune suppression in cancer. Cancer Res 76, 3484–3495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang GL, Zhang Y, Cao KX, Wang XM, Orthotopic Injection of Breast Cancer Cells into the Mice Mammary Fat Pad. J Vis Exp, (2019). [DOI] [PubMed] [Google Scholar]
- 27.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F, Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Heng TS, Painter MW, The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Schaum N, Karkanias J, Neff NF, May AP, Quake SR, Wyss-Coray T, Darmanis S, Batson J, Botvinnik O, Chen MB, Chen S, Green F, Jones R, Maynard A, Penland L, Pisco AO, Sit RV, Stanley GM, Webber JT, Zanini F, Baghel AS, Bakerman I, Bansal I, Berdnik D, Bilen B, Brownfield D, Cain C, Cho M, Cirolia G, Conley SD, Demers A, Demir K, de Morree A, Divita T, du Bois H, Dulgeroff LBT, Ebadi H, Espinoza FH, Fish M, Gan Q, George BM, Gillich A, Genetiano G, Gu X, Gulati GS, Hang Y, Hosseinzadeh S, Huang A, Iram T, Isobe T, Ives F, Kao KS, Karnam G, Kershner AM, Kiss BM, Kong W, Kumar ME, Lam J, Lee DP, Lee SE, Li G, Li Q, Liu L, Lo A, Lu WJ, Manjunath A, May KL, May OL, McKay M, Metzger RJ, Mignardi M, Min D, Nabhan AN, Ng KM, Noh J, Patkar R, Peng WC, Puccinelli R, Rulifson EJ, Sikandar SS, Sinha R, Szade K, Tan W, Tato C, Tellez K, Travaglini KJ, Tropini C, Waldburger L, van Weele LJ, Wosczyna MN, Xiang J, Xue S, Youngyunpipatkul J, Zardeneta ME, Zhang F, Zhou L, oungyunpipatkul JY, Castro P, Croote D, DeRisi JL, Kuo CS, Lehallier B, Nguyen PK, Tan SY, Wang BM, Yousef H, Beachy PA, Chan CKF, Huang KC, Weinberg K, Wu SM, Barres BA, Clarke MF, Kim SK, Krasnow MA, Nusse R, Rando TA, Sonnenburg J, Weissman IL, Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris: The Tabula Muris Consortium. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang WD, Chen HX, Wang YX, Chen ZP, Shan ZJ, Xu G, Bioinformatic analysis of c-Myc target from laryngeal cancer cell gene of laryngeal cancer. Journal of cancer research and therapeutics 12, 58–61 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Eilken HM, Adams RH, Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22, 617–625 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR, Cell migration: integrating signals from front to back. Science 302, 1704–1709 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Hall A, Rho GTPases and the actin cytoskeleton. Science 279, 509–514 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Mata R, Boulter E, Burridge K, The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 12, 493–504 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Gui J, Yin X, Pan Q, Liu X, Chu L, Transmembrane domain is crucial to the subcellular localization and function of Myc target 1. Journal of cellular and molecular medicine 20, 471–481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS, ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol 208, 821–838 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Citi S, Guerrera D, Spadaro D, Shah J, Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases 5, 1–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotech 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gribov A, Sill M, Luck S, Rucker F, Dohner K, Bullinger L, Benner A, Unwin A, SEURAT: visual analytics for the integrated analysis of microarray data. BMC Med Genomics 3, 21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S, SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, Rochaix P, Girard JP, Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 71, 5678–5687 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA, Robust enumeration of cell subsets from tissue expression profiles. Nature Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian L, Goldstein A, Wang H, Lo HC, Kim IS, Welte T, Sheng K, Dobrolecki LE, Zhang X, Putluri N, Phung TL, Mani SA, Stossi F, Sreekumar A, Mancini MA, Decker WK, Zong C, Lewis MT, Zhang XH-F, Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544, 250–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding Z, Xiong K, Issekutz TB, Regulation of chemokine-induced transendothelial migration of T lymphocytes by endothelial activation: differential effects on naive and memory T cells. Journal of Leukocyte Biology 67, 825–833 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Motz GT, Coukos G, The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nature Reviews Immunology 11, 702–711 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ, Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol 33, 3117–3126 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Sata M, Walsh K, TNFalpha regulation of Fas ligand expression on the vascular endothelium modulates leukocyte extravasation. Nat Med 4, 415–420 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G, Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 20, 607–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee WJ, Tateya S, Cheng AM, Rizzo-DeLeon N, Wang NF, Handa P, Wilson CL, Clowes AW, Sweet IR, Bomsztyk K, Schwartz MW, Kim F, M2 Macrophage Polarization Mediates Anti-inflammatory Effects of Endothelial Nitric Oxide Signaling. Diabetes 64, 2836–2846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu G, Zhang R, Geng S, Peng L, Jayaraman P, Chen C, Xu F, Yang J, Li Q, Zheng H, Shen K, Wang J, Liu X, Wang W, Zheng Z, Qi C-F, Si C, He JC, Liu K, Lira SA, Sikora AG, Li L, Xiong H, Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nature Communications 6, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lavin B, Gómez M, Pello OM, Castejon B, Piedras MJ, Saura M, Zaragoza C, Nitric oxide prevents aortic neointimal hyperplasia by controlling macrophage polarization. Arterioscler Thromb Vasc Biol 34, 1739–1746 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Hou KK, Pan H, Lanza GM, Wickline SA, Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials 34, 3110–3119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD, Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3, 991–998 (2002). [DOI] [PubMed] [Google Scholar]
- 57.Dunn GP, Old LJ, Schreiber RD, The three Es of cancer immunoediting. Annu Rev Immunol 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD, Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Yin X, Grove L, Rogulski K, Prochownik EV, Myc target in myeloid cells-1, a novel c-Myc target, recapitulates multiple c-Myc phenotypes. J Biol Chem 277, 19998–20010 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Qiu G, Xu Z, Huang D, Gong L, Li C, Sun X, Sun K, [Cloning and characterization of MTLC, a novel gene in 6q25]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics 20, 94–97 (2003). [PubMed] [Google Scholar]
- 61.Martin JD, Seano G, Jain RK, Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol 81, 505–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC, Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A 109, 17561–17566 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J, VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 73, 539–549 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, Zinzindohoue F, Yagita H, Tartour E, Taieb J, Terme M, VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 212, 139–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lai YS, Wahyuningtyas R, Aui SP, Chang KT, Autocrine VEGF signalling on M2 macrophages regulates PD-L1 expression for immunomodulation of T cells. Journal of cellular and molecular medicine 23, 1257–1267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shitara K, Nishikawa H, Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci 1417, 104–115 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Munn LL, Jain RK, Vascular regulation of antitumor immunity. Science 365, 544–545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johansson A, Hamzah J, Ganss R, More than a scaffold: Stromal modulation of tumor immunity. Biochim Biophys Acta 1865, 3–13 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK, Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 15, 325–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Z, Kang B, Li C, Chen T, Zhang Z, GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–w560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown CR, Reiner SL, Bone-marrow chimeras reveal hemopoietic and nonhemopoietic control of resistance to experimental Lyme arthritis. J Immunol 165, 1446–1452 (2000). [DOI] [PubMed] [Google Scholar]
- 72.Halabi CM, Broekelmann TJ, Lin M, Lee VS, Chu ML, Mecham RP, Fibulin-4 is essential for maintaining arterial wall integrity in conduit but not muscular arteries. Sci Adv 3, e1602532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin SW, Beis D, Mitchell T, Chen JN, Stainier DY, Cellular and molecular analyses of vascular tube and lumen formation in zebrafish. Development 132, 5199–5209 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Eckfeldt CE, Mendenhall EM, Flynn CM, Wang TF, Pickart MA, Grindle SM, Ekker SC, Verfaillie CM, Functional analysis of human hematopoietic stem cell gene expression using zebrafish. PLoS Biol 3, e254 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guy CT, Cardiff RD, Muller WJ, Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12, 954–961 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, Schreiber RD, Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res 5, 106–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Timmins NE, Nielsen LK, Generation of multicellular tumor spheroids by the hanging-drop method. Methods Mol Med 140, 141–151 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Nakatsu MN, Hughes CC, An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol 443, 65–82 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Steinritz D, Schmidt A, Balszuweit F, Thiermann H, Ibrahim M, Bolck B, Bloch W, Assessment of Endothelial Cell Migration After Exposure to Toxic Chemicals. J Vis Exp, e52768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Figure S1: Single-cell RNA sequencing in heterogenous cell populations from different mouse organs shows an almost endothelial-exclusive pattern of Myct1 expression.
Figure S2: Analysis of TCGA-derived cancer patient datasets shows correlation between the ‘angiogenic scores’ and MYCT1 expression.
Figure S3: Single cell RNA sequencing of heterogenous mouse tumor stromal compartments shows exclusive pattern of Myct1 expression in the endothelial cells.
Figure S4: Etv2 downstream target Myct1 is a regulator of angiogenesis; however, it is not required for vascular development and homeostasis in mice and zebrafish.
Figure S5: Endothelial Myct1 expression is essential for tumor growth and angiogenesis in mice.
Figure S6: Myct1-deficient mouse tumor vessels display improved vascular functions.
Figure S7: Endothelial Myct1 expression is relevant for tumor growth and angiogenesis in mice.
Figure S8: Endothelial Myct1 is critical for tumor growth and is required for vascular regeneration in vitro and in vivo.
Figure S9: MYCT1 regulates the actin cytoskeleton dynamics of endothelial cells in the angiogenic environment in vitro.
Figure S10: MYCT1 functionally interacts with ZO1 and RHOA to regulate the capillary characteristics of endothelial cells in the angiogenic environment in vitro.
Figure S11: MYCT1 is evolutionarily conserved between human and mouse.
Figure S12: Single cell RNA sequencing displays that tumor endothelial cells express classical endothelial marker genes.
Figure S13: UMAP projection of tumor endothelial cells following single cell RNA sequencing identifies different functionally distinct sub-populations in WT mice.
Figure S14: Myct1 deficiency leads to an anti-tumor immune environment both in humans and mice.
Figure S15: Endothelial Myct1 deficiency recapitulates global Myct1 KO phenotype in combination with anti-PD1 treatment in tumor-bearing mice.
Figure S16: Combined Myct1 and PD1 targeting does not alter immune constituents of the non-tumor tissues in tumor-bearing mice.
Figure S17: Combined Myct1 and PD1 targeting does not induce off-target and non-specific immune activation.
Table S1. Top 100 regulators of angiogenesis identified by their expression correlation with seed genes.
Table S2: List of genes (termed as “seed genes”) to identify angiogenic regulatory genes.
Table S3: List of genes used to generate the “angiogenic score” for patient datasets across different cancer types
Table S4: Sequences of primers used in the study.
Table S5: LC/MS proteomics screening for MYCT1 binding partners.
Data file S1: Subject-level data for main figures.
Data file S2: Subject-level data for supplementary figures.