

Abstract
Bardet-Biedl syndrome (BBS) is a ciliopathy characterized by retinitis pigmentosa, obesity, polydactyly, cognitive impairment and renal failure. Pathogenic variants in 24 genes account for the molecular basis of >80% of cases. Toward saturated discovery of the mutational basis of the disorder, we carefully explored our cohorts and identified a hominid-specific SINE-R/VNTR/Alu type F (SVA F) insertion in exon 13 of BBS1 in 8 families. In six families, the repeat insertion was found in trans with c.1169T>G, p.Met390Arg and in two families the insertion was found in addition to other recessive BBS loci. Whole genome sequencing, de novo assembly and SNP array analysis were performed to characterize the genomic event. This insertion is extremely rare in the general population (found in 8 alleles of 8 BBS cases but not in >10,800 control individuals from gnomAD-SV) and due to a founder effect. Its 2,435 bp sequence contains hallmarks of LINE1 mediated retrotransposition. Functional studies with patient-derived cell lines confirmed that the BBS1 SVA-F is deleterious as evidenced by a significant depletion of both mRNA and protein levels. Such findings highlight the importance of dedicated bioinformatics pipelines to identify all types of variation.
Keywords: Bardet-Biedl syndrome, BBS1, Mobile element insertion, SVA F, Founder effect
INTRODUCTION
Bardet-Biedl syndrome (BBS, MIM# 209900) is a rare autosomal ciliopathy associating retinitis pigmentosa, polydactyly, obesity, cognitive impairment and renal dysfunction. Its prevalence ranged from 1/160,000 live births 1 to <1/50,000 in consanguineous or isolated populations 2. Recent studies have delineated the antenatal and postnatal presentation of BBS patients 3,4 with pathogenic variants in 24 genes accounting for the molecular basis of a majority of cases 5. Among the known BBS genes, BBS1 contributes the highest fraction of BBS patients (~20%) 6 thanks to a founder variant (c.1169T>G, p.Met390Arg) 7. Among the unresolved cases in our cohort, several patients were heterozygous carriers of this variant suggesting two hypothesis: (1) a missed second primary driver allele in BBS1; or (2) participation of this variant as a second-site modifier in the presence of a different primary BBS driver locus 8,9. Notably, due to the high frequency of this variant in the general population (0.27% in non-Finnish Europeans; gnomAD) this variant could be detected coincidently.
In this study, we report the identification of a mobile element insertion of SVA F type in 8 families, a portion of which are in trans to the recurrent c.1169T>G in BBS1 (n=6), and a subset as secondary sites in addition to a known recessive BBS locus (n=2). Already described as a single case report 10, here we estimate the frequency of this variant in BBS (131 families) identifying this variant as the second most common pathogenic variant in BBS1. We also determined its full-length sequence using WGS and performed functional analysis to understand its consequences. In our cohorts, the insertion always occurs at the same genomic location and its recurrence in BBS populations is the result of a founder effect.
SUBJECTS AND METHODS
Subjects
Written informed consent was obtained from each participant and/or parent or legal guardian. The study protocols were approved by local Institutional Review Boards: “Comité de Protection des Personnes” (EST IV, N°DC-20142222), or the Lurie Children’s Hospital IRB (IRB 2019–3057 or IRB 2019–2950). Our research complies with the Declaration of Helsinki. All patients were diagnosed with BBS according to Beales’ clinical diagnostic criteria 11 (Supplementary Table 1). Patients (n=564) have been assembled into several cohorts (Supplementary Table 2).
Methods
All experimental procedures are reported as supporting information.
RESULTS
Identification of an SVA F insertion in BBS1
According to our tiered diagnostic paradigm, we screened the recurrent BBS variants (BBS1:p.Met390Arg and BBS10:p.Cys91Leufs*5) in cohort 1 (n=217) and cohort 2 (n=192) followed by targeted exome sequencing (TES) in cohort 1. We identified seven individuals who carry heterozygous p.Met390Arg changes without a second pathogenic BBS1 variant identified in trans. In one family (family I), we identified a novel heterozygous deletion encompassing exons 4 to 11 (Supplementary Figure 1). Among the six p.Met390Arg variation-bearing cases, we leveraged a high depth of coverage to identify another putative pathogenic variant in exon 13 of BBS1 with the deletion/insertion of 2 bases, c.1215_1216delinsT (p.(Ala406Glnfs*4)) in family A.II-3 (Figure 1B and supplementary Figure 2) and 2 others (B.II-1, C.II-1). Careful analysis of the genomic region revealed a more complicated event leading to incorrect variant calling (Figure 1C). Indeed, soft clipped reads part (82 bp) revealed the insertion of an SVA F retrotransposon with a possible truncation of 311 bases 5’ of the consensus sequence (Figure 1D).
Figure 1. Pedigree of BBS families and variant analysis.
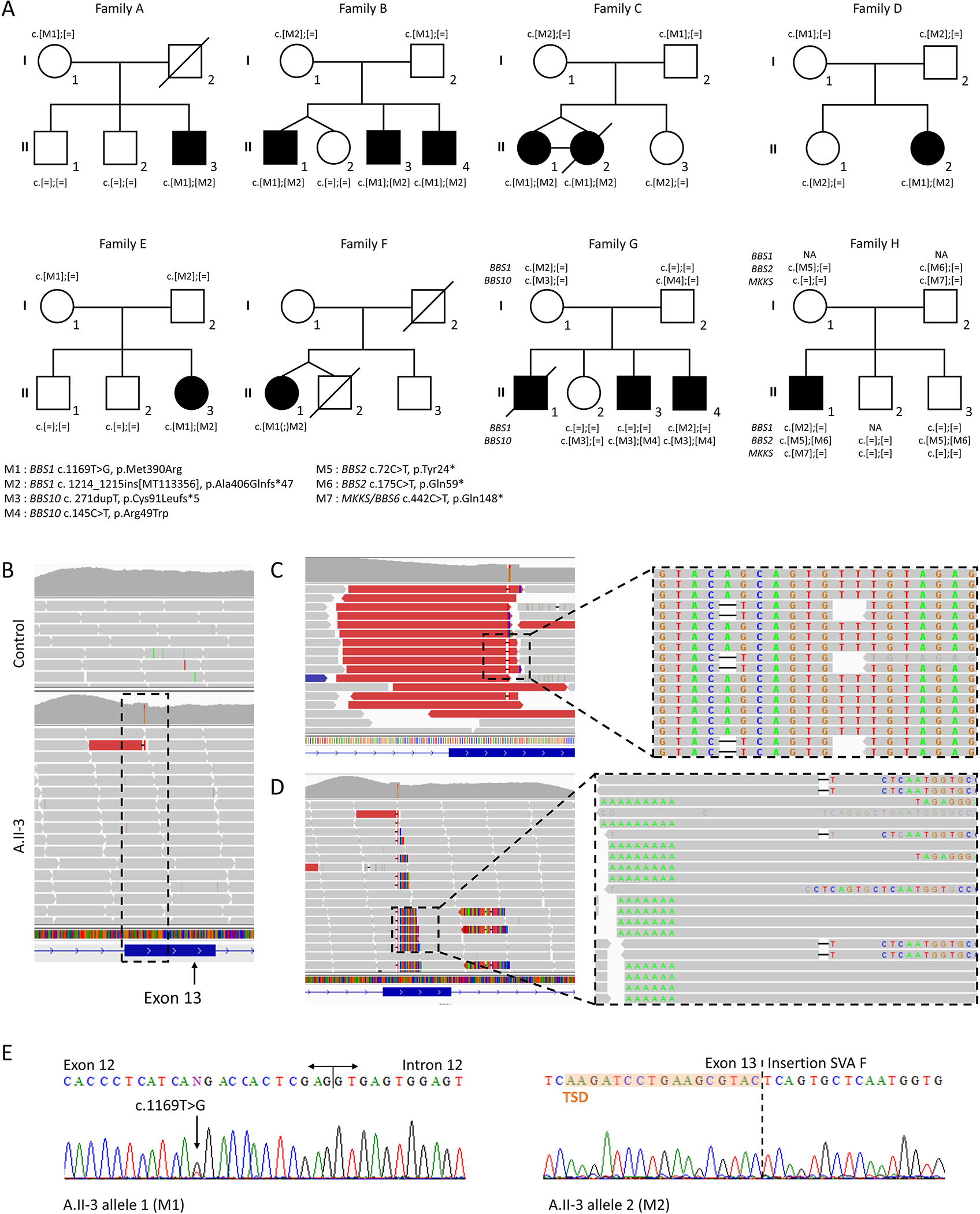
(A) 8 families carrying the SVA F insertion in exon 13 of BBS1.
(B) Next generation sequencing data displayed from the Integrative Genomics Viewer 23 surrounding the BBS1 locus from A.II-3 and one control individual.
(C) Magnified view of the incorrectly called deletion insertion with the sorting and coloring of the reads according to the “insert size”. The sequence reveals all reads ending at the exact same position with a “AG” deletion and a “T” insertion (c.1215_1216delinsT).
(D) Region of interest with the “Show soft-clipped bases” turned off revealing multiple reads with aberrant alignments on their right side and corresponding to an SVA F insertion. The spacing between the reads on the left (including the 3’ end of the SVA F with the poly(A) tail) and the reads on the right (including the 5’ end of the SVA F) corresponds to the TSD.
(E) Segregation analysis using Sanger sequencing for M1 (left) and M2 (right).
To screen additional patients not yet available by TES (cohort 2; 213 cases from 192 families), or an independent group with no pre-selection bias for BBS variations other than exclusion of BBS1 recessive cases (cohort 3; 134 cases from 126 families), we designed a duplex PCR including the SVA insertion point (Supplementary Figure 3). This led to the identification of 5 additional cases (family D to H) including 3 in trans to the p.Met390Arg and 2 as third or fourth allele (Figure 1A). Biallelic status of the variants and the exact same insertion point was confirmed for 7 individuals (Figure 1E).
To estimate the overall incidence of the SVA F as a contributor of the BBS1 mutational burden, we explored all our BBS1 positive cases (e.g. biallelic pathogenic variants). Data from 131 unrelated cases of European ancestry corresponding to 262 pathogenic alleles in total (cohort 4) could be retrieved and account for 44 different pathogenic variations. Among those, the c.1169T>G represents 72.1% (189 alleles) while the second most frequent variant is the SVA F with a frequency of 2.3% (6 alleles).
Determination of the full length SVA F sequence
To understand the consequences of this insertion, we determined its full-length sequence. Given the repeated sequence of the SVA F, alignment of short reads onto the human genome generate artifacts (multimapped reads, split reads, discordant read pairs) that can be rescued using a dedicated bioinformatics pipeline. The initial TES aligned reads allowed us to identify 99 bp from the SVA F (Figure 2B). Assembled reads from WGS data (A.II-3) output a 387 bp contig composed of BBS1 (38 bp before the breakpoint in exon 13), a TSD (17 bp) and 330 bp that matched the SVA F consensus sequence. In conclusion, WGS data enabled accurate characterization of an additional 250 bp SVA F sequence (Figure 2). Based on this and the consensus SVA F sequence 12, we walked on the genome and determined an inserted sequence of 2,435 bp (Figure 2B and Supplementary Figure 4 and 5). With the exception of a missing 311 bases (Figure 2A) in the 5’ part, hallmarks of L1 mediated retrotransposition could be identified (Figure 2). The variant can now be described as c.1214_1215ins[MT113356], p.(Ala406Glnfs*47).
Figure 2. Schematic of the inserted SVA F element in exon 13 of BBS1.
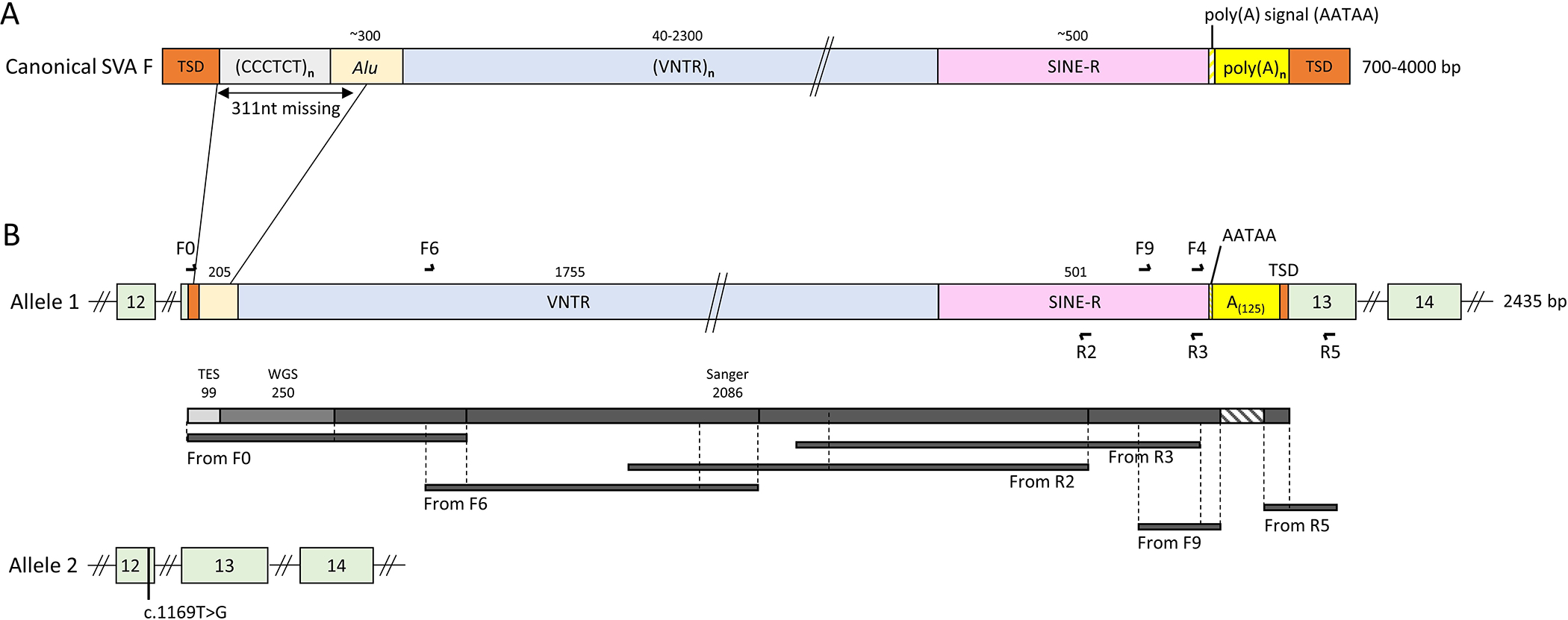
(A) Canonical SVA F structure composed of 6 parts: (1) hexameric CCCTCT repeats (2) Alu-like domain, (3) a Variable Number Tandem Repeats (VNTR) domain, (4) a Short Interspersed Nucleotide Elements (SINE-R) domain, (5) a poly(A) tail and (6) the target site duplication (TSD) at both extremities. Numbers above the scheme indicate the size of each element.
(B) Schematic of both BBS1 alleles in A.II-3 with primer positioning and amplification products highlighting each contribution to the resolution of the full-length sequence.
Functional impact of the SVA F insertion on BBS1
Predicted to create a premature stop codon, a reduced or absent mRNA expression due to nonsense-mediated decay potentially leading to a shorter protein (452 vs 593 AA) is likely to happen. Using primary cells from individual A.II-3, we demonstrated a significantly reduced BBS1 expression corresponding a priori to the loss of the SVA F allele (Figure 3A). However, a transcript carrying the SVA F is expressed (Supplementary Figure 6), albeit likely minor compared to the p.Met390Arg allele. Western blot analysis on protein extracted from A.II-3 cells revealed a single band corresponding to the normal BBS1 protein (Figure 3B) demonstrating the absence of a putative BBS1 polypeptide carrying the SVA F.
Figure 3. Quantification of BBS1 mRNA and protein levels in A.II-3.
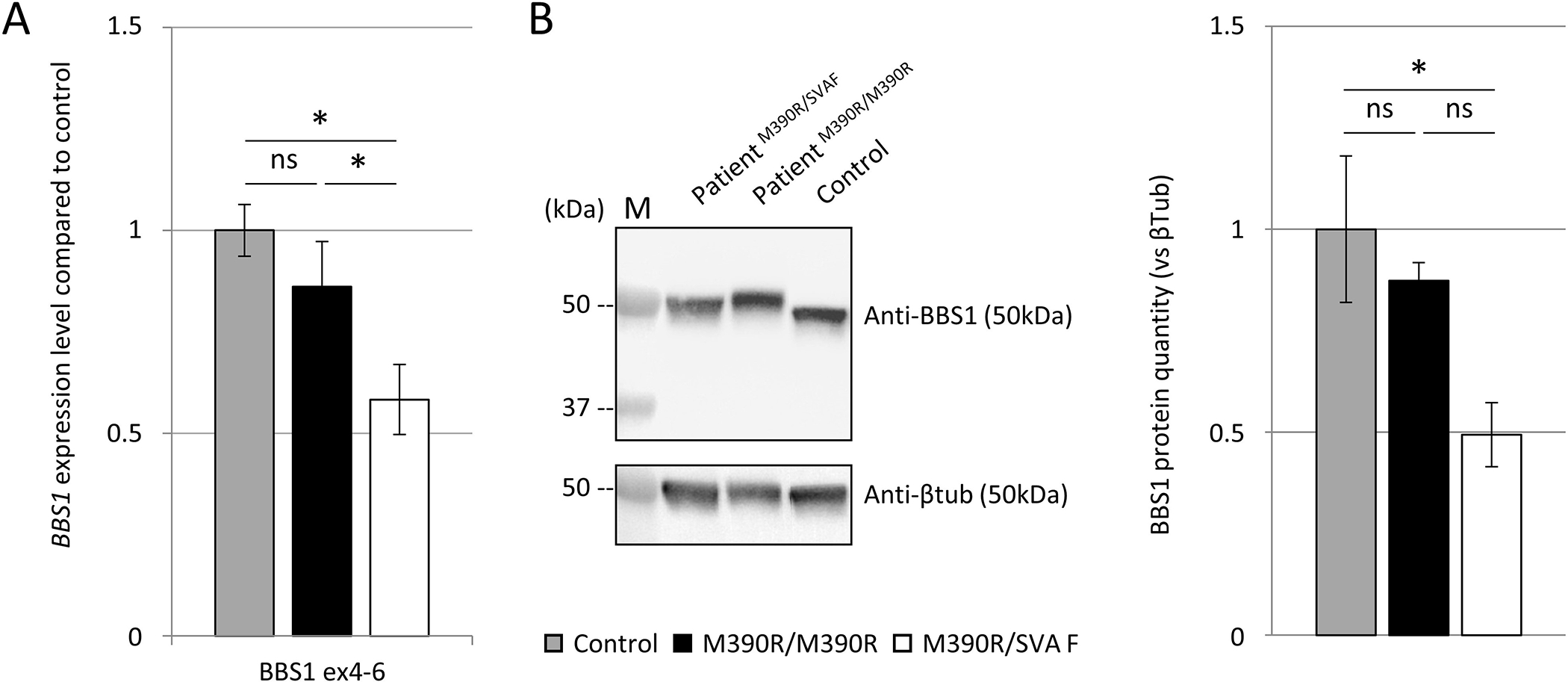
(A) Reduced BBS1 mRNA expression by RT-qPCR (A.II-3) compared to control or homozygous p.Met390Arg patient. The major fraction of BBS1 expression likely corresponds to the p.Met390Arg allele. Normalization was performed using both GAPDH and HPRT as reference genes. Mean of both results are shown. Controls are in gray (n=3), homozygous p.Met390Arg in black (n=2), and patient A.II-3 (p.Met390Arg and SVA F) in white (n=2). Bars show the mean of 2 independent experiments +/− SEM (n=2, t-test *: p<0.05, ns: not significant).
(B) Western blot analysis of BBS1 protein level. No truncated version of BBS1 could be detected. Half reduced BBS1 protein level in A.II-3 compared to control. Bars show mean of 3 independent experiments +/− SEM (n=3, t-test *: p<0.05, ns: not significant). M: molecular weight ladder (kDa).
Founder effect or recurrent mechanism?
Given the multiple carriers of this insertion in our cohorts (8 families), we wondered whether this MEI occurred as an independent (recurrent) mechanism or as the same mutational event derived from common ancestry (founder effect). First, no occurrence could be observed either from 1299 healthy individuals of European ancestry or from the 14,891 gnomAD SV controls 13. Second, genotyping of 4 different families with SVA F alleles, along with 3 patients carrying the c.1169T>G change in the homozygous state (Supplementary Table 5) revealed the common c.1169T>G haplotype (~1.614 Mb) as well an SVA F allele of a much smaller size (~56.6kb) (Supplementary Table 5). PCA analysis confirmed the geographical distribution of the patients (Supplementary Figure 7). Haplotypes shared by the four SVA F carriers were compared to find the position where recombination events are likely to have happened. The estimation of the most recent common ancestor age was 74 generations 95% CI [33;190]. If we assume that one generation lasts 25 years, this will indicate an age of 1850 years 95% CI [825;4750]. Overall, this MEI is an ultra-rare event that arose from a common ancestor on one of the most common haplotypes at this locus and more recently than the Met390Arg haplotype.
DISCUSSION
Although the diagnostic yield in BBS is relatively high, (60–100%), a substantial proportion of cases remain unresolved 3,5. This could be related to the attributes of the cohort (sample size and geographic origin), the clinical inclusion criteria (strict or more inclusive) or the genetic test used (TES, WES, WGS). In addition to the first BBS case with MEI reported by our group 14 and the recent SVA F description 10, this is the largest description of such cases in BBS patients. Although rarely described, a number of MEI cases (>100) have been reported in rare diseases 15 or in cancer 16. The majority are Alu insertions 17 and only a few SVA (n=16). Recently, large studies have evaluated the impact of MEI in patients between 0.04% to 0.15% of the cases 18,19.
BBS1 is often the target of structural variations 20. Interestingly, the BBS1 locus is GC rich and has a high density of L1 and Alu insertions that might explain the SVA F insertion (Supplementary methods). Given the nature of the SVA F sequence, which contains highly repeated elements, GC rich content and a poly(A) tail, complete amplification was not easily performed. We thus used the breakpoints identified by the TES and WGS to walk across the patient’s genome and determine a 2,435bp insertion in exon 13 of BBS1. This inserted sequence is identical to the one already described 10. Unlike most of the SVA elements in the human genome (63%) 21, this inserted SVA F is incomplete lacking 311 bases in 5’ and possibly as almost all SVA it is inactive 17. Notably, beside short read standard WGS we also performed linked-read sequencing (10x genomics) to see whether this technology could overcome any of the technical challenges encountered through WGS and analysis (Supplementary Figure 8). Consistent with other reports, we did not find any significant advantage and it is now discontinued 22. Interestingly sequencing of the insertion point revealed the exact same sequence for all patients and the genotyping of some individuals revealed a common shared haplotype. All families carrying the SVA F are of European ancestry: 4 originated from the western part of France (family A, C, E and F), one from the northern part of France (family B) and one from Denmark and Sweden (family D). These observations were highly suggestive of a single founder genomic event that we confirmed and estimated its age close to 1850 years.
In conclusion, we identified the recurrence of a specific SVA F element in exon 13 of BBS1 in multiple BBS families highlighting the importance of detecting such genetic events even by TES. We determined the full length sequence of the inserted SVA F, show its pathogenicity and demonstrated the founder effect. This rare allele is the second most frequent cause of BBS1 after the c.1169T>G variant. We have also developed a duplex PCR, offering a simple and inexpensive way to detect this variant prior to any other large genetic screen. Accordingly we have modified our BBS diagnostic strategy (Supplementary Figure 10).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients and their family members for their participation, and the French Bardet-Biedl association for their support. Part of TES was performed by the Plateforme GenomEast (IGBMC, Illkirch-Graffenstaden, France) and for this, we thank Serge Vicaire, Murielle Philipps and Bernard Jost. We would like to thank Amélie Piton for scientific discussions and acknowledge Pierre Lindenbaum and every member of the FranceGenRef consortium.
FUNDING INFORMATION
This work has been published under the framework of the IdEX Unistra and benefits from a funding from the state managed by the French National Research Agency as part of the investments for the future program (JM). This work was supported by US National Institutes of Health grants HD042601 (NK), GM121317 (NK), DK072301 (NK and EED). NK is a Distinguished Valerie and George D. Kennedy Professor. WGS were supported by Fondation APLM and CREGEMES. CD is supported by an FRM grant (ECO20170637509).
Footnotes
CONFLICT OF INTEREST STATEMENT
NK is a significant shareholder in Rescindo Therapeutics. The other authors declare no conflict of interest.
AVAILABILITY OF DATA AND MATERIAL
Data generated or analyzed during this study are included in the published article and the corresponding supplementary data. The raw sequencing data generated in the course of this study are not publicly available due to the protocol and the corresponding consents used that did not include such information. The variants are available in ClinVar (SCV001427246, SCV001427247) and the SVA F sequence in GenBank (MT113356).
REFERENCES
- 1.Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26(7):1039–1056. doi: 10.1007/s00467-010-1731-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gouronc A, Zilliox V, Jacquemont M-L, et al. High prevalence of Bardet-Biedl syndrome in La Réunion Island is due to a founder variant in ARL6/BBS3. Clinical Genetics. 2020;n/a(n/a). doi: 10.1111/cge.13768 [DOI] [PubMed] [Google Scholar]
- 3.Mary L, Chennen K, Stoetzel C, et al. Bardet-Biedl syndrome: Antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clinical Genetics. 2019;95(3):384–397. doi: 10.1111/cge.13500 [DOI] [PubMed] [Google Scholar]
- 4.Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl Syndrome uncovers differences among causative genes. Human Mutation. 2019;0(ja). doi: 10.1002/humu.23862 [DOI] [PubMed] [Google Scholar]
- 5.Shamseldin HE, Shaheen R, Ewida N, et al. The morbid genome of ciliopathies: an update. Genetics in Medicine. Published online February 14, 2020. doi: 10.1038/s41436-020-0761-1 [DOI] [PubMed] [Google Scholar]
- 6.Khan SA, Muhammad N, Khan MA, Kamal A, Rehman ZU, Khan S. Genetics of Human Bardet-Biedl Syndrome, an Updates. Clin Genet. Published online January 14, 2016. doi: 10.1111/cge.12737 [DOI] [PubMed] [Google Scholar]
- 7.Mykytyn K, Nishimura DY, Searby CC, et al. Evaluation of Complex Inheritance Involving the Most Common Bardet-Biedl Syndrome Locus (BBS1). The American Journal of Human Genetics. 2003;72(2):429–437. doi: 10.1086/346172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beales PL, Badano JL, Ross AJ, et al. Genetic Interaction of BBS1 Mutations with Alleles at Other BBS Loci Can Result in Non-Mendelian Bardet-Biedl Syndrome. The American Journal of Human Genetics. 2003;72(5):1187–1199. doi: 10.1086/375178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kousi M, Söylemez O, Ozanturk A, et al. Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nature Genetics. Published online October 12, 2020. doi: 10.1038/s41588-020-0707-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavares E, Tang CY, Vig A, et al. Retrotransposon insertion as a novel mutational event in Bardet-Biedl syndrome. Molecular Genetics & Genomic Medicine. 2019;7(2):e00521. doi: 10.1002/mgg3.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–446. [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon Y-J, Choi Y, Eo J, et al. Structure and Expression Analyses of SVA Elements in Relation to Functional Genes. Genomics & Informatics. 2013;11(3):142. doi: 10.5808/GI.2013.11.3.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins RL, Brand H, Karczewski KJ, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–451. doi: 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redin C, Le Gras S, Mhamdi O, et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alström syndromes. J Med Genet. 2012;49(8):502–512. doi: 10.1136/jmedgenet-2012-100875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taşkesen M, Collin GB, Evsikov AV, et al. Novel Alu retrotransposon insertion leading to Alström syndrome. Hum Genet. 2012;131(3):407–413. doi: 10.1007/s00439-011-1083-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crivelli L, Bubien V, Jones N, et al. Insertion of Alu elements at a PTEN hotspot in Cowden syndrome. Eur J Hum Genet. 2017;25(9):1087–1091. doi: 10.1038/ejhg.2017.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hancks DC, Kazazian HH. Active human retrotransposons: variation and disease. Current Opinion in Genetics & Development. 2012;22(3):191–203. doi: 10.1016/j.gde.2012.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner EJ, Prigmore E, Gallone G, et al. Contribution of retrotransposition to developmental disorders. Nature Communications. 2019;10(1):4630. doi: 10.1038/s41467-019-12520-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torene RI, Galens K, Liu S, et al. Mobile element insertion detection in 89,874 clinical exomes. Genetics in Medicine. Published online January 22, 2020. doi: 10.1038/s41436-020-0749-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindstrand A, Frangakis S, Carvalho CMB, et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. The American Journal of Human Genetics. 2016;99(2):318–336. doi: 10.1016/j.ajhg.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Xing J, Grover D, et al. SVA Elements: A Hominid-specific Retroposon Family. Journal of Molecular Biology. 2005;354(4):994–1007. doi: 10.1016/j.jmb.2005.09.085 [DOI] [PubMed] [Google Scholar]
- 22.Uguen K, Jubin C, Duffourd Y, et al. Genome sequencing in cytogenetics: Comparison of short-read and linked-read approaches for germline structural variant detection and characterization. Mol Genet Genomic Med. 2020;8(3):e1114–e1114. doi: 10.1002/mgg3.1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013;14(2):178–192. doi: 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.