

Abstract
Recent studies have revealed that tumor cells decrease their immunogenicity by epigenetically repressing the expression of highly immunogenic antigens to survive in immunocompetent hosts. We hypothesized that these epigenetically hidden “stealth” antigens should be favorable targets for cancer immunotherapy due to their high immunogenicity. To identify these stealth antigens, we treated human lung cell line A549 with DNA methyltransferase inhibitor 5‐aza‐2′‐deoxycytidine (5Aza) and its prodrug guadecitabine for 3 d in vitro and screened it using cDNA microarray analysis. We found that the gene encoding sperm equatorial segment protein 1 (SPESP1) was re‐expressed in cell lines including solid tumors and leukemias treated with 5Aza, although SPESP1 was not detected in untreated tumor cell lines. Using normal human tissue cDNA panels, we demonstrated that SPESP1 was not detected in normal human tissue except for testis and placenta. Moreover, we found using immunohistochemistry SPESP1 re‐expression in xenografts in BALB/c‐nu/nu mice that received 5Aza treatment. To assess the antigenicity of SPESP1, we stimulated human CD4+ T‐cells with a SPESP1‐derived peptide designed using a computer algorithm. After repetitive stimulation, SPESP1‐specific helper T‐cells were obtained; these cells produced interferon‐γ against HLA‐matched tumor cell lines treated with 5Aza. We also detected SPESP1 expression in freshly collected tumor cells derived from patients with acute myeloid leukemia or lung cancer. In conclusion, SPESP1 can be classified as a stealth antigen, a molecule encoded by a gene that is epigenetically silenced in tumor cells but serves as a highly immunogenic antigen suitable for cancer immunotherapy.
Keywords: cancer immunoediting, cancer immunotherapy, DNA methylation, stealth antigens, tumor immunoescape
We identified stealth antigen SPESP1. SPESP1 is encoded by a gene silenced in tumors epigenetically, but re‐expressed in tumors exposed to a DNA methyltransferase inhibitor. SPESP1 is a highly immunogenic because a SPESP1‐derived peptide efficiently activated CD4+ T‐cells. This suggests that SPESP1 is a favorable target for cancer immunotherapy.

Abbreviations
- 5Aza
5‐aza‐2′‐deoxycytidine
- CT
cancer/testis
- HLA
human leukocyte antigen
- ICI
immune checkpoint inhibitor
- IFN
interferon
- IHC
immunohistochemistry
- SPESP1
sperm equatorial segment protein 1
1. INTRODUCTION
The marked efficacy of ICIs in patients with cancer has proven that the immune system can control tumor progression through activation of tumor‐reactive T‐cells in the host. 1 , 2 In addition, ICI therapy has revealed that the immunosuppressive tumor microenvironment allows tumor cells to grow even in immunocompetent hosts, in which tumor‐reactive T‐cells circulate systemically. These findings indicate that relief from immunosuppressive conditions in T‐cells is necessary for effective induction of tumor‐specific immune responses leading to tumor eradication.
CT antigens are known to constitute a family of highly immunogenic tumor antigens; CT antigens are widely expressed in various tumors, but their expression is restricted to testis and/or placenta in normal tissues. 3 Therefore, CT antigens are considered good candidate targets for cancer immunotherapy. Indeed, immune responses against CT antigens have been observed in cancer patients; for instance, NY‐ESO‐1 was identified as a spontaneous immune target in a variety of cancer patients, 4 , 5 and KK‐LC‐1 was identified as a predominant target in a patient with cervical carcinoma who experienced complete cancer regression after adoptive transfer of tumor‐infiltrating lymphocytes. 6 Given their high immunogenicity and restricted expression in normal somatic tissues, some T‐cell epitope peptides have been identified from various CT antigens and subsequently used in clinical trials for cancer immunotherapy; however, such peptides have led to the desired outcomes in only a minority of patients. 7 , 8 , 9 , 10
One of the difficulties in cancer treatment is that tumor cells acquire resistance to treatment to which cells have been subjected. Although chemotherapy and radiotherapy can control tumor growth at first exposure, tumor cells eventually develop resistance to these treatments, resulting in tumor progression. 11 , 12 , 13 In addition, several reports have indicated that immunotherapy, including ICI and chimeric antigen receptor‐T‐cell therapy, also leads to treatment‐resistant tumors through a variety of mechanisms, including loss of surface expression of MHC class I, loss‐of‐function mutations in genes encoding IFN‐γ pathway‐related proteins, and loss of expression of neoantigens and targeted antigens. 14 , 15 , 16 One mechanism of antigen loss in tumor cells was revealed using a transgenic mouse model in which expression of a highly immunogenic antigen was silenced by methylation of the promoter region of a gene encoding an antigen; expression of the antigen was restored by treatment with a DNA methyltransferase inhibitor. 17 In addition, expression of highly immunogenic neoantigens is transcriptionally repressed in recurrent tumors of patients with glioma. 18 These observations suggested that usefully immunogenic antigens for cancer immunotherapy are likely to be found in DNA‐demethylated tumors.
In the present study, we propose the existence of a new type of tumor antigen, which we called “stealth antigens,” that are highly immunogenic but whose expression is silenced by epigenetic modification of the encoding gene in tumor cells. Specifically, we examined whether such stealth antigens are suitable targets for cancer immunotherapy. We identified sperm equatorial segment protein 1 (SPESP1) as a stealth antigen using our criteria, and demonstrated that this protein and the corresponding transcript showed little or no expression in untreated tumor cell lines, but are expressed in cell lines following treatment with DNA methyltransferase inhibitor 5‐aza‐2′‐deoxycytidine (5Aza). SPESP1 transcription was detected not only in various cell lines but also in freshly isolated cancer cells from clinical samples subjected to treatment with 5Aza; and expression of SPESP1 was restricted to testis and placenta in normal tissues. Moreover, 5Aza‐treated tumor cells were recognized by SPESP1‐specific human CD4+ helper T‐cells, suggesting that SPESP1 is a highly immunogenic stealth antigen. Collectively, our findings may provide a new therapeutic target for cancer immunotherapy with improved efficacy.
2. MATERIALS AND METHODS
2.1. Cell lines and mice
Human tumor cell lines A549 (lung adeno carcinoma), HSC4 (HLA‐DR1/4; tongue squamous cell carcinoma), Lu65 (HLA‐DR4/15; lung large cell carcinoma cell line), and EBC1 (lung squamous cell carcinoma) were obtained from the RIKEN BioResource Center (Tsukuba, Ibaraki, Japan). HT29 (colorectal adenocarcinoma) and WiDr (colorectal adenocarcinoma) were purchased from the American Type Culture Collection (ATCC). L‐cells (mouse fibroblasts expressing transfected genes encoding HLA class II molecules) were obtained from Dr. R. Karr (Karr Pharma) and Dr. T. Sasazuki (Kyushu University). AML cell lines NOMO‐1, and KG‐1, and CML cell line MEG‐01 were acquired from the Japanese Collection of Research Bioresources Cell Bank. CML cell lines KT‐1 and KU812 were the kind gifts of Dr. Yasukawa (Ehime University, Ehime, Japan). Cell lines were maintained in RPMI‐1640 medium (Nacalai Tesque) or DMEM (Nacalai Tesque) supplemented with 10% fetal bovine serum (Biowest, #S1650), penicillin (100 U/mL), streptomycin (100 μg/mL), and anti‐mycoplasma drug MC‐210 (KAC Co., Ltd.; 0.5 μg/mL) as suppliers recommended. No authentication assays were performed for any of the cell lines. BALB/c‐nu/nu mice (female, 8‐10 wk old at the study start) were purchased from Charles River Laboratories Japan, and maintained and handled in accordance with protocols approved by the Asahikawa Medical University Institutional Animal Care and Use Committee.
To assess the expression of genes encoding stealth antigens in tumor cell lines treated with DNA methyltransferase inhibitors, tumor cell lines were treated with a DNA methyltransferase inhibitor, 5Aza (Sigma‐Aldrich) or guadecitabine SGI‐110 (AdooQ Bioscience), at 10 μmol/L for 3 d. Cells were then collected and total RNA was purified using an RNeasy Mini Kit (Qiagen) in accordance with the manufacturer's instructions. Total RNA was reverse‐transcribed to cDNA using a PrimeScript 1st strand cDNA Synthesis Kit (TaKaRa Bio) in accordance with the manufacturer's protocol. Microarray analysis was performed using a 3D‐gene array (Toray); probing and processing were performed as suggested by the manufacturer. For assessment of SPESP1 expression in normal human tissues, we used Human MTC panels I and II (Clontech). These samples were amplified using a LightCycler 480 Probes Master (Roche Diagnostics) and TaqMan probes purchased from Applied Biosystems as follows: SPESP1 (Hs00377364_m1) and Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH, Hs02758991_g1); Gapdh, a housekeeping gene, was used as an internal control. mRNA expression was measured by quantitative real‐time PCR (qPCR) using the LightCycler 480 System and Software (Roche Diagnostics); relative expression of each mRNA was calculated using the ΔΔCt method.
2.2. Assessing re‐expression of SPESP1 by 5Aza treatment of xenograft tumor tissues from immunodeficient mice
WiDr and EBC1 cell lines were injected intradermally into BALB/c‐nu/nu mice. Tumor‐bearing mice were injected intraperitoneally with 5Aza (1.6 mg/kg), formulated in PBS, on days 5, 10, 15, and 20. PBS was injected as a control. On day 25, mice were anesthetized in a chamber by isoflurane narcosis (>5%) and then euthanized by cervical dislocation. Tumor tissues were collected and homogenized using a BioMasher II disposable homogenizer (Nippi. Inc) for preparation of RNA. For immunohistochemistry (IHC), tumor tissues were fixed in 4% paraformaldehyde, and 2‐μm‐thick sections were generated from formalin‐fixed, paraffin‐embedded tissue blocks. Rabbit anti‐SPESP1 polyclonal antibody (1:1000; HPA051040, ATLAS) was used for detection of SPESP1. Images were acquired and analyzed using a BZ‐X700 fluorescence microscope with BZ‐H3 software (KEYENCE).
2.3. Evaluating re‐expression of SPESP1 by 5Aza treatment in freshly collected AML and lung cancer cells
Fresh AML and lung cancer samples were obtained from bone marrow of a patient newly diagnosed with AML and from pleural fluid of a patient with lung cancer, respectively. Each sample was incubated in the presence or absence of 10 μmol/L 5Aza. At 3 d later, samples were collected for RNA preparation and gene expression analysis for SPESP1. The institutional ethics committee approved this study, and written informed consent was obtained from all patients who provided samples.
2.4. Addressing antigenicity of SPESP1 in vitro
The complete culture medium for all procedures consisted of AIM‐V medium (Invitrogen, Carlsbad, CA) supplemented with 3% human male AB serum (Innovative Research). All blood materials were acquired after informed consent was appropriately obtained. SPESP1‐derived peptide SPESP131‐49 (QNLNHYIQVLENLVRSVPS) was selected from the entire amino acid sequence of SPESP1 using computer‐based algorithms developed by Southwood et al 19 ; the peptide then was commercially synthesized (GenScript). The procedure for the induction of peptide‐specific CD4+ helper T‐cell lymphocytes (HTLs) has been described in detail previously. 20 PBMCs were acquired from healthy humans using Ficoll‐Conray centrifugation. Next, CD14+ monocytes were purified from PBMCs using CD14 MACS microbeads (Miltenyi Biotec) and differentiated into dendritic cells (DCs) after 7 d of culture with granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) (50 ng/mL) and interleukin (IL)‐4 (1000 IU/mL) at 37°C in an incubator with 5% CO2. SPESP1 peptide‐pulsed DCs (3 μg/mL for 3 h at room temperature) were cocultured with autologous CD4+ T‐cells (isolated using CD4 MACS microbeads; Miltenyi Biotech) in 96‐well flat‐bottomed culture plates. At 7 d after peptide stimulation, CD4+ T‐cells were restimulated in individual microcultures with SPESP1 peptide‐pulsed γ‐irradiated autologous PBMCs (3 μg/mL); 2 d later, recombinant human IL‐2 (10 IU/mL) was added. After 2 cycles of restimulation, T‐cells exhibited a significant response, indicated by cytokine release in response to SPESP1 peptide‐pulsed irradiated autologous PBMCs. T‐cells were expanded in 24‐well plates by weekly restimulation with cognate peptides and irradiated autologous PBMCs (3 μg/mL). For evaluating peptide‐specific reactions, HTLs (1‐1.5 × 105) were cocultured with autologous PBMCs (1.5 × 105), HLA‐DR‐expressing L‐cells (3 × 104), tumor cell lines (3 × 104), or DCs (5 × 103). Tumor cell lines were pretreated with 500 U/mL IFN‐γ and 10 μM of 5Aza for 72 h to upregulate the expression of HLA‐DR and SPESP1, respectively; all reagents then were removed before further assays. To determine antigen specificity and HLA‐DR restriction, anti‐HLA‐DR monoclonal antibody (mAb) L243 (IgG2a, prepared from supernatants of hybridoma HB‐55, which was obtained from the ATCC) and anti‐HLA class I mAb W6/32 (IgG2a; ATCC) were added to the culture at 10 μg/mL during incubation. Culture supernatants were collected for evaluation of IFN‐γ and granzyme B production using ELISA kits (BD Pharmingen and MABTECH, respectively).
2.5. Statistical analysis
Data were analyzed by unpaired Student t test, one‐way ANOVA with the Holm post hoc test, or unpaired t test. All tests were run as two‐tailed analyses. P‐values < .05 were considered statistically significant.
3. RESULTS
3.1. Identification of a re‐expressed tumor antigen SPESP1 in 5Aza‐treated tumor cell lines
Based on cancer immunoediting theory, tumor cells (including tumor cell lines) derived from established tumors are supposed to lack antigenic molecules, excluding mutation‐associated antigens and neoantigens acquired in the late phase of tumor development. 21 One mechanism whereby tumor cells hide these antigenic molecules, as reported by DuPage et al, consists of epigenetic silencing of genes encoding highly antigenic molecules; methylation of the promoter region results in decreased expression of the corresponding gene. 17 Therefore, we hypothesized that exposure to a DNA methyltransferase inhibitor would reveal hidden antigenic molecules in tumor cells by allowing re‐expression of genes encoding highly antigenic molecules; such molecules are expected to be favorable targets for antitumor immunity. To test this hypothesis, we cultured the A549 human lung cancer cell line in the presence or absence of a DNA methyltransferase inhibitor (5‐aza‐2‐deoxycytidine; 5Aza) for 3 d and screened 25 000 genes in both cultures using cDNA microarray analysis (Figure 1A). Based on our above criteria, we defined sperm equatorial segment protein‐1 (SPESP1), a gene known to be uniquely expressed in human sperm, 22 , 23 as a gene that is re‐expressed following 5Aza treatment; we confirmed the re‐expression of SPESP1 in 5Aza‐treated A549 cells using qPCR (Figure 1B). SPESP1 expression was not detected in any normal human tissues, with the exception of testis and placenta, as assessed using multiple tissue cDNA panels (Figure 1C). We obtained similar results from interrogation of the Human Protein Atlas database (https://www.proteinatlas.org/search/SPESP1; data not shown). These results suggested that SPESP1 may be a candidate stealth tumor antigen, although this protein also belongs to the CT antigen family. We next assessed whether SPESP1 is generally re‐expressed in various human tumor cell lines following 5Aza treatment. Specifically, we treated HT29, WiDr, HSC4, EBC1, and Lu65 cell lines with 5Aza (10 μmol/L) for 3 d, and then used qPCR to assess expression levels of SPESP1. We found that SPESP1 was re‐expressed in all 5 of the tested tumor cell lines following exposure to 5Aza (Figure 1D). DNA methyltransferase inhibitors are used clinically in patients with hematologic malignancies including myelodysplastic syndromes and leukemia. 24 We therefore tested if SPESP1 also was re‐expressed in human AML (NOMO‐1 and KG‐1) and CML (MEG‐01, KT‐1, and KU812) cell lines when those lines were treated with 5Aza. Consistent with our findings in solid tumor cell lines, AML and CML cell lines exhibited SPESP1 re‐expression following exposure to either of the DNA methyltransferase inhibitors (Figure 1E). Because its prodrug guadecitabine SGI‐110 is also evaluated in clinical settings, 25 we also addressed whether it re‐expresses SPESP1 in the tumor cell lines as well as 5Aza. As well as 5Aza, SGI‐110 induced re‐expression of SPESP1 in the solid tumor and leukemia cell lines (Figure 1F,G). Exposure to a chemotherapy drug, gemcitabine did not induce re‐expression of SPESP1 in the tumor cell lines tested (data not shown). These results suggested that SPESP1 may be a general target antigen for patients with cancer.
FIGURE 1.
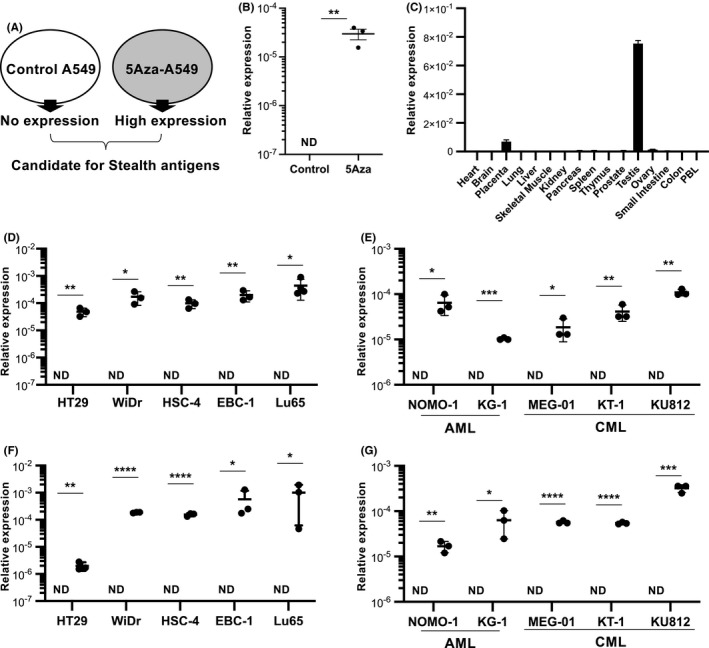
Identification of SPESP1 as a gene encoding a stealth antigen candidate. A, Schema of defining re‐expressed genes in a 5Aza‐treated tumor cell line. A tumor cell line was treated with or without 10 μmol/L 5Aza for 3 d, and total RNA was prepared for use as a probe against a cDNA microarray. B, Expression levels of SPESP1 in the A549 cell line treated with/without 5Aza was evaluated by qPCR. C, Expression levels of SPESP1 in normal human tissues were evaluated by qPCR. D, Solid tumor cell lines HT29, WiDr, HSC4, EBC1, and Lu65 and (E) AML cell lines NOMO‐1 and KG‐1 and CML cell lines MEG‐01, KT‐1, and KU812 were treated with/without 10 μmol/L 5Aza for 3 d. F, Solid tumor cell lines and (G) AML and CML cell lines were also treated with/without 10 μmol/L SGI‐110 for 3 d. Expression levels of SPESP1 were evaluated by qPCR. Statistical significance was determined using a two‐tailed unpaired t test. ND, not detected, *P < .05, **P < .01, ***P < .001, ****P < .0001. Error bar indicates SD. Experiments were performed with at least 3 biological replicates and are representative of at least 2 independent experiments
3.2. Re‐expression of SPESP1 following in vivo treatment with 5Aza
To address whether SPESP1 is re‐expressed by 5Aza in tumor tissues in vivo, we used a human cell line xenograft model. BALB/c‐nu/nu mice were transplanted intradermally with EBC1 or WiDr cell lines and then treated intraperitoneally with PBS or 5Aza (1.6 mg/kg) on days 5, 10, 15, and 20 after tumor implantation. At 5 d after the final PBS or 5Aza treatment, tumor tissue was collected from each animal and analyzed for SPESP1 transcript accumulation (Figure 2A). Gene expression of SPESP1 was detected in tumors of mice implanted with either EBC1 or WiDr and treated with 5Aza, but not in tumors of implanted mice treated with PBS (Figure 2B). Furthermore, SPESP1 protein expression was confirmed by IHC analysis in tumor tissues obtained from 5Aza‐treated mice (Figure 2C), demonstrating that both the SPESP1 gene and protein are re‐expressed in tumor cells in vivo following 5Aza treatment.
FIGURE 2.
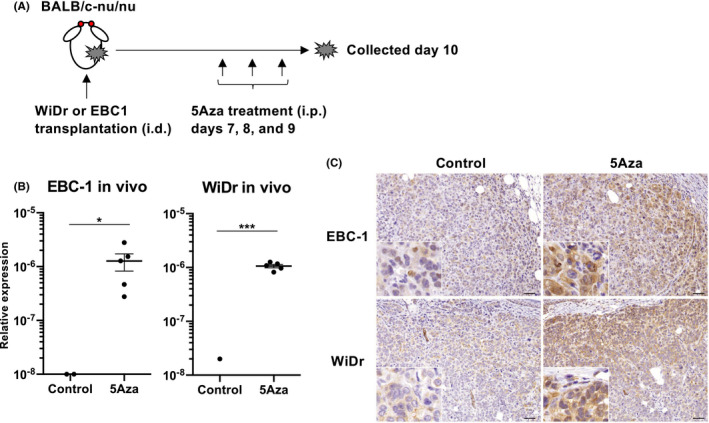
Re‐expression of SPESP1 in xeno‐transplanted tumor cell lines in vivo. A, Schema of 5Aza treatment in BALB/c‐nu/nu mice inoculated with human tumor cell lines WiDr and EBC1. Each tumor cell line was implanted intradermally into mice, which then were treated intraperitoneally with 5Aza (1.6 mg/kg) on days 7, 8, and 9 after implantation. Tumor tissue was collected for analysis of SPESP1 gene and protein expression on day 10 post‐implantation. B, Expression levels of SPESP1 in tumor tissues of WiDr and EBC1 in mice treated with/without 5Aza were evaluated by qPCR. Error bar indicates SD. C, Immunohistochemistry for SPESP1 was performed. Scale bars, 200 μm. Data are representative of at least 2 independent experiments
3.3. An immunogenic epitope in SPESP1 for activating CD4+ T‐helper cells
To validate the antigenicity of the SPESP1 protein, we used computer‐based algorithms to identify a SPESP131‐49 peptide (QNLNHYIQVLENLVRSVPS) as a candidate CD4+ T‐helper peptide. CD4+ T‐cells were purified from PBMCs of healthy donors and stimulated with SPESP131‐49 peptide in the presence of autologous DCs. We obtained 3 HTL lines specific for SPESP1 peptide and showed that these HTL lines produced IFN‐γ in response to SPESP131‐49 peptide‐pulsed autologous PBMCs in an HLA‐DR‐restricted manner (Figure 3A‐C). Furthermore, when these HTL lines were cocultured with mouse fibroblasts transfected with HLA‐DR allele gene (L‐cells), we found that one line (SP1) responded to L‐DR4, while other lines (SP2 and SP3) responded to L‐DR53 cells (Figure 3D‐F). To address whether the epitope recognized by SPESP1‐specific HTL lines was naturally presented to HLA class II on the surface of tumor cell lines treated with 5Aza, we cocultured SPESP1‐specific HTL lines with HLA‐DR‐matched tumor cell lines treated with or without 5Aza. As expected, DR4‐restricted HTL line SP1 enhanced the production of IFN‐γ and granzyme B when cocultured with 5Aza‐treated HLA‐DR4‐positive tumor cell lines (HT29, WiDr, and HSC4, but not EBC1), suggesting that SPESP1 does indeed serve as an immunologic target (Figure 3G,H).
FIGURE 3.
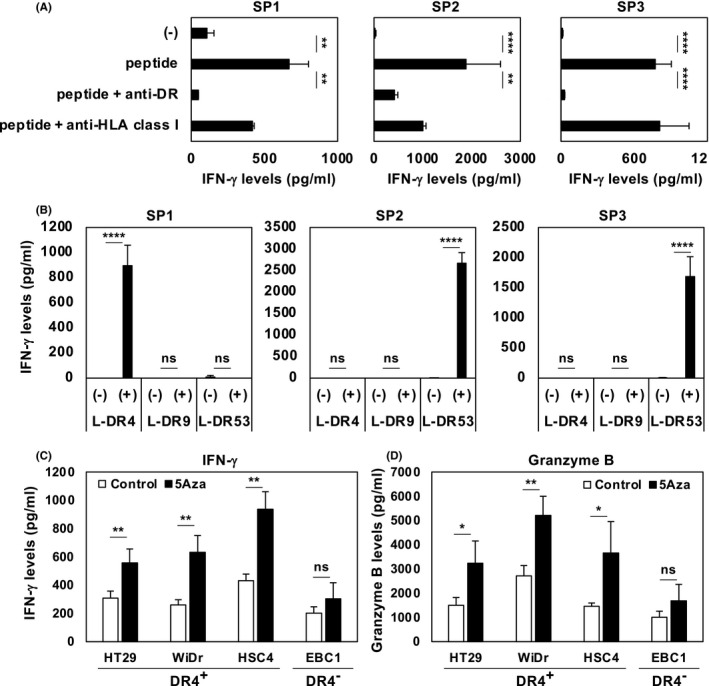
Assessing antigenicity of SPESP1. Helper T‐cell lymphocyte (HTL) responses against SPESP131‐49 peptide (QNLNHYIQVLENLVRSVPS) were assessed by evaluating levels of IFN‐γ or granzyme B produced by HTLs. A, HLA restrictions of 3 HTL lines specific for SPESP1 (SP1, SP2, and SP3) were evaluated using anti‐HLA‐DR mAb L243 and anti‐HLA class I mAb W6/32 (negative control). B, Each HTL line was cocultured with SPESP1 peptide‐pulsed L‐cells expressing the HLA‐DR4, DR9, or DR53 allele. C, D, HLA‐DR4‐restricted HTL line SP1 was cocultured with a DR4‐positive tumor cell line (HT29, WiDr, or HSC4) or with a DR4‐negative tumor cell line (EBC1) that had been pretreated with/without 5Aza (10 μmol/L) for 3 d. Supernatants were collected after 24 h and analyzed by ELISA for production of IFN‐γ (C) or granzyme B (D). Two independent experiments were performed with similar results. Statistical significance levels were determined with a two‐tailed unpaired t test. ns, not significant, *P < .05; **P < .01; ****P < .0001. Bars and error bars indicate mean and SD, respectively
3.4. Re‐expression of SPESP1 in freshly isolated human tumors
It was important to validate whether SPESP1 was re‐expressed in clinical samples exposed to 5Aza; this point would be critical to applying SPESP1‐targeting cancer immunotherapy in clinical settings in the future. Therefore, we treated freshly isolated tumor cells from patients with AML and lung cancer with 5Aza in vitro; as we showed above for tumor cell lines, SPESP1 was re‐expressed in fresh tumor cells following 5Aza exposure (Figure 4A,B). These results suggested that SPESP1 is a potential target stealth antigen suitable for cancer immunotherapy.
FIGURE 4.
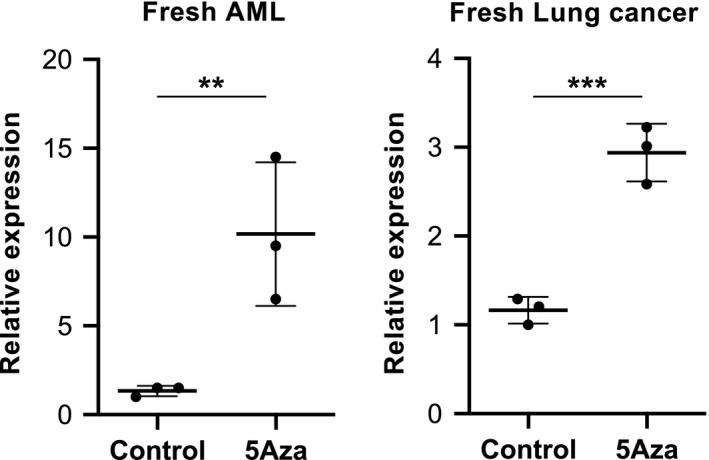
Re‐expression of SPESP1 in freshly isolated human tumors. Freshly isolated AML (A) and lung cancer cells (B) from bone marrow (BM) and pleural fluid of patients, respectively, were treated with control and 10 μmol/L 5Aza in vitro for 3 d. Expression levels of SPESP1 were evaluated by qPCR. Two independent experiments were performed with similar results. Statistical significance levels were determined with a two‐tailed unpaired t test, **P < .05; ***P < .01. Bars and error bars indicate mean and SD, respectively
4. DISCUSSION
In the present work we showed that SPESP1 is a highly immunogenic tumor antigen. We further demonstrated that the gene encoding this molecule is epigenetically silenced in tumor cell lines and fresh tumor cells but is re‐expressed following treatment with 5Aza. Together, these results validated the potential for development of cancer immunotherapy targeting stealth antigens such as SPESP1. However, multiple issues will need to be assessed before stealth antigen‐targeting cancer immunotherapy can be tested in a clinical study. Notably, we will need to evaluate the antitumor efficacy of stealth antigen‐targeting immunotherapy in in vivo mouse models. Mouse experiments will depend on 2 preceding steps. First is to identify mouse stealth antigens using mouse tumor cell lines and their syngeneic mouse T‐cell epitopes; second is to establish a mouse therapy model for evaluating the efficacy of mouse stealth antigen‐based cancer immunotherapy. Indeed, we have already identified several candidate mouse stealth antigens and are exploring epitope peptides of these antigens for use in establishing a mouse model of stealth antigen‐targeting cancer immunotherapy (data not shown). We also noted that ICI therapy presumably will be needed for implementation of a successful cancer vaccine therapy targeting stealth antigens, given that cancer vaccine therapy alone does not provide desirable effects in the absence of ICI therapy. 26 , 27
We have, in the current study, identified SPESP1 as re‐expressed tumor antigen in all tumor cells tested after treatment with 5Aza, suggesting that SPESP1 may constitute a new target for cancer immunotherapy. SPESP1 has been known to be rarely detected in some tumors including liver, carcinoid, skin, and lymphoma based on the Human Protein Atlas database (https://www.proteinatlas.org/search/SPESP1). However, the expression frequency of SPESP1 is too low to target for cancer immunotherapy without treatment with hypomethylating agents. Although this work proposes only SPESP1 as a human stealth antigen, we have identified other candidate stealth antigens in human and are evaluating their utility as targets of cancer immunotherapy (data not shown). These results, in both human and mouse, suggest that stealth antigens could be subcategorized as members of a new tumor antigen family, just as carcinoembryonic antigens have been. Given the costs associated with drug development, it will be important to know whether a given stealth antigen is shared among multiple tumor tissues or limited to individual tumors; the use of stealth antigens for personalized medicine would increase the cost of this modality. If suitable stealth antigens depend on the individual (for instance, due to various HLA types), we will have to define several stealth antigens and validate which is available as a shared antigen among multiple patients. We, in this study, identified the immunogenic SPESP1 peptide for activating HTLs because they, especially Th1 cells, play a critical role for promoting and maintaining acquired immune responses. Recently, Kreiter S et al demonstrated in several mouse tumor models that most tumor neoantigens were recognized by CD4+ T‐cells and vaccination with such helper epitopes promoted antitumor immune responses. 28 Because it was difficult for us to control tumor growth by targeting just a single antigen due to their heterogeneity, we have to activate immune responses against not only the target antigen but also endogenously released tumor antigens from dying tumor cells. From this viewpoint, activating Th1 cells would be needed in the tumor‐bearing host. This is why our strategy prefers to activate HTLs by vaccination with a helper epitope peptide. Although we have not assessed whether SPESP131‐49 peptide includes killer T‐cell epitopes, the SPESP1 peptide would have the potential to activate CD8+ killer T‐cells because it is a 19‐mer long peptide. This remains to be addressed.
To target stealth antigens such as SPESP1, the combinational use of a hypomethylating agent is required. Although hypomethylating agents are likely to cause myelosuppression, Griffiths EA et al demonstrated that vaccination in combination with 5Aza induced target antigen‐specific T‐cell responses in a phase I study. 29 Therefore, the undesirable impact of the hypomethylating agent on immune cells could be controlled.
Tumor immune escape is achieved by silencing not only genes encoding highly immunogenic tumor antigens but also by changes (through DNA methylation) in the expression of loci encoding other immune‐related molecules. For instance, Kitajima et al 30 observed that hyperactivation of DNMT1 (DNA (cytosine‐5)‐methyltransferase 1) and EZH2 (enhancer of zeste homolog 2) in KL cells suppressed the expression of STING (stimulator of interferon genes), a protein that induces expression of type I IFNs, which in turn activates the immune system's ability to recognize cyclic nucleic acids such as cGAMP (cyclic guanosine monophosphate‐adenosine monophosphate). 31 , 32 Therefore, loss of STING would favor tumor survival. In other work, Peng et al 33 reported that expression of chemokine‐encoding genes CXCL9 and CXCL10 was silenced by methylation of their promoter regions in tumor tissues. Frequent DNA methylation would require both DNMT1 activation and abundantly available S‐adenosyl‐methionine (SAM), which is a substrate for DNMT1. Because SAM is synthesized in the methionine cycle of one‐carbon metabolism, ectopic DNA methylation in tumor cells should be affected by cellular metabolism. Given reports of silencing of genes encoding highly immunogenic tumor antigens in tumor cells, 17 , 18 inflammation by antigen‐specific immune responses could trigger DNA methylation by boosting one‐carbon metabolism in tumor tissues. Therefore, studies clarifying the relationship between tumor‐specific immune responses and tumor metabolism are expected to be valuable for the development of new cancer immunotherapies.
In conclusion, we propose a new era of cancer immunology, in which the strategy of cancer immunotherapy is based on the relationship between immune escape and silencing of genes encoding highly immunogenic tumor antigens. In the present work, we identified immunogenic tumor antigen SPESP1, which is hidden in tumor cells but re‐expressed in tumor cells treated with a DNA methyltransferase inhibitor. SPESP1 may serve as a favorable target for cancer immunotherapy.
DISCLOSURE
This study was funded, in part, by Otsuka Pharmaceutical Co., Ltd.
ACKNOWLEDGMENTS
This work was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (H Kobayashi 16K15244 and T Ohkuri 20K07367) and Japan Agency for Medical Research and Development (AMED) (A Kosaka JP16lm0103005). The authors thank Mr. Hayakawa Toshiyuki and Ms. Hino Chihiro (at the Animal Laboratory for Medical Research, Center for Advanced Research and Education, Asahikawa Medical University) and Ms. Matsumoto Rie (at the Department of Pathology, Asahikawa Medical University) for devotedly maintaining the mice.
Kosaka A, Yajima Y, Hatayama M, et al. A stealth antigen SPESP1, which is epigenetically silenced in tumors, is a suitable target for cancer immunotherapy. Cancer Sci. 2021;112:2705–2713. 10.1111/cas.14973
Akemi Kosaka and Takayuki Ohkuri contributed equally to this work.
Funding information
Japan Society for the Promotion of Science (H Kobayashi 16K15244, and T Ohkuri 20K07367) and Japan Agency for Medical Research and Development AMED (A Kosaka JP16lm0103005).
Contributor Information
Hiroya Kobayashi, Email: hiroya@asahikawa-med.ac.jp.
Takayuki Ohkuri, Email: ohkurit@asahikawa-med.ac.jp.
REFERENCES
- 1. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous‐Cell Non‐Small‐Cell Lung Cancer. N Engl J Med. 2015;373:123‐135. 10.1056/NEJMoa1504627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus Ipilimumab versus sunitinib in advanced renal‐cell carcinoma. N Engl J Med. 2018;378:1277‐1290. 10.1056/NEJMoa1712126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643‐1647. 10.1126/science.1840703 [DOI] [PubMed] [Google Scholar]
- 4. Jäger E, Chen YT, Drijfhout JW, et al. Simultaneous humoral and cellular immune response against cancer‐testis antigen NY‐ESO‐1: definition of human histocompatibility leukocyte antigen (HLA)‐A2‐binding peptide epitopes. J Exp Med. 1998;187:265‐270. 10.1084/jem.187.2.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas R, Al‐Khadairi G, Roelands J, et al. NY‐ESO‐1 Based Immunotherapy of Cancer: Current Perspectives. Front Immunol. 2018;9:947. 10.3389/fimmu.2018.00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stevanović S, Pasetto A, Helman SR, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356:200‐205. 10.1126/science.aak9510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kobayashi H, Song Y, Hoon DS, Appella E, Celis E. Tumor‐reactive T helper lymphocytes recognize a promiscuous MAGE‐A3 epitope presented by various major histocompatibility complex class II alleles. Cancer Res. 2001;61:4773‐4778. [PubMed] [Google Scholar]
- 8. Ohkuri T, Wakita D, Chamoto K, Togashi Y, Kitamura H, Nishimura T. Identification of novel helper epitopes of MAGE‐A4 tumour antigen: useful tool for the propagation of Th1 cells. Br J Cancer. 2009;100:1135‐1143. 10.1038/sj.bjc.6604966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi N, Ohkuri T, Homma S, et al. First clinical trial of cancer vaccine therapy with artificially synthesized helper/ killer‐hybrid epitope long peptide of MAGE‐A4 cancer antigen. Cancer Sci. 2012;103:150‐153. 10.1111/j.1349-7006.2011.02106.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cebon JS, Gore M, Thompson JF, et al. Results of a randomized, double‐blind phase II clinical trial of NY‐ESO‐1 vaccine with ISCOMATRIX adjuvant versus ISCOMATRIX alone in participants with high‐risk resected melanoma. J Immunother Cancer. 2020;8(1):e000410. 10.1136/jitc-2019-000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin Z, Fan Z, Zhang X, Wan J, Liu T. Cellular plasticity and drug resistance in sarcoma. Life Sci. 2020;263:118589. 10.1016/j.lfs.2020.118589 [DOI] [PubMed] [Google Scholar]
- 12. Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, et al. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist Updat. 2020;53:100728. 10.1016/j.drup.2020.100728 [DOI] [PubMed] [Google Scholar]
- 13. Archer M, Dogra N, Kyprianou N. Inflammation as a driver of prostate cancer metastasis and therapeutic resistance. Cancers (Basel). 2020;12(10):2984. 10.3390/cancers12102984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zaretsky JM, Garcia‐Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med. 2016;375:819‐829. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov. 2017;7:264‐276. 10.1158/2159-8290.Cd-16-0828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour‐specific antigens underlies cancer immunoediting. Nature. 2012;482:405‐409. 10.1038/nature10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nejo T, Matsushita H, Karasaki T, et al. Reduced Neoantigen Expression Revealed by Longitudinal Multiomics as a Possible Immune Evasion Mechanism in Glioma. Cancer Immunol Res. 2019;7:1148‐1161. 10.1158/2326-6066.Cir-18-0599 [DOI] [PubMed] [Google Scholar]
- 19. Southwood S, Sidney J, Kondo A, et al. Several common HLA‐DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363‐3373. [PubMed] [Google Scholar]
- 20. Kobayashi H, Wood M, Song Y, Appella E, Celis E. Defining promiscuous MHC class II helper T‐cell epitopes for the HER2/neu tumor antigen. Cancer Res. 2000;60:5228‐5236. [PubMed] [Google Scholar]
- 21. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 22. Wolkowicz MJ, Shetty J, Westbrook A, et al. Equatorial segment protein defines a discrete acrosomal subcompartment persisting throughout acrosomal biogenesis. Biol Reprod. 2003;69:735‐745. 10.1095/biolreprod.103.016675 [DOI] [PubMed] [Google Scholar]
- 23. Fujihara Y, Murakami M, Inoue N, et al. Sperm equatorial segment protein 1, SPESP1, is required for fully fertile sperm in mouse. J Cell Sci. 2010;123:1531‐1536. 10.1242/jcs.067363 [DOI] [PubMed] [Google Scholar]
- 24. Wijermans P, Lübbert M, Verhoef G, et al. Low‐dose 5‐aza‐2'‐deoxycytidine, a DNA hypomethylating agent, for the treatment of high‐risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956‐962. 10.1200/jco.2000.18.5.956 [DOI] [PubMed] [Google Scholar]
- 25. Coral S, Parisi G, Nicolay HJ, et al. Immunomodulatory activity of SGI‐110, a 5‐aza‐2'‐deoxycytidine‐containing demethylating dinucleotide. Cancer Immunol Immunother. 2013;62:605‐614. 10.1007/s00262-012-1365-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishihara M, Tono Y, Miyahara Y, et al. First‐in‐human phase I clinical trial of the NY‐ESO‐1 protein cancer vaccine with NOD2 and TLR9 stimulants in patients with NY‐ESO‐1‐expressing refractory solid tumors. Cancer Immunol Immunother. 2020;69:663‐675. 10.1007/s00262-020-02483-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vansteenkiste JF, Cho BC, Vanakesa T, et al. Efficacy of the MAGE‐A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE‐A3‐positive non‐small‐cell lung cancer (MAGRIT): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2016;17:822‐835. 10.1016/s1470-2045(16)00099-1 [DOI] [PubMed] [Google Scholar]
- 28. Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692‐696. 10.1038/nature14426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Griffiths EA, Srivastava P, Matsuzaki J, et al. NY‐ESO‐1 vaccination in combination with decitabine induces antigen‐specific T‐lymphocyte responses in patients with myelodysplastic syndrome. Clin Cancer Res. 2018;24:1019‐1029. 10.1158/1078-0432.Ccr-17-1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kitajima S, Ivanova E, Guo S, et al. Suppression of STING Associated with LKB1 Loss in KRAS‐Driven Lung Cancer. Cancer Discov. 2019;9:34‐45. 10.1158/2159-8290.Cd-18-0689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohkuri T, Ghosh A, Kosaka A, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2:1199‐1208. 10.1158/2326-6066.Cir-14-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohkuri T, Kosaka A, Ishibashi K, et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti‐tumor immunity at a mouse tumor site. Cancer Immunol Immunother. 2017;66:705‐716. 10.1007/s00262-017-1975-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249‐253. 10.1038/nature15520 [DOI] [PMC free article] [PubMed] [Google Scholar]