

Abstract
The metabolism in tumors is reprogrammed to meet its energetic and substrate demands. However, this metabolic reprogramming creates metabolic vulnerabilities, providing new opportunities for cancer therapy. Metabolic vulnerability as a therapeutic target in esophageal squamous cell carcinoma (ESCC) has not been adequately clarified. Here, we identified pyruvate dehydrogenase (PDH) component X (PDHX) as a metabolically essential gene for the cell growth of ESCC. PDHX expression was required for the maintenance of PDH activity and the production of ATP, and its knockdown inhibited the proliferation of cancer stem cells (CSCs) and in vivo tumor growth. PDHX was concurrently upregulated with the CD44 gene, a marker of CSCs, by co‐amplification at 11p13 in ESCC tumors and these genes coordinately functioned in cancer stemness. Furthermore, CPI‐613, a PDH inhibitor, inhibited the proliferation of CSCs in vitro and the growth of ESCC xenograft tumors in vivo. Thus, our study provides new insights related to the development of novel therapeutic strategies for ESCC by targeting the PDH complex‐associated metabolic vulnerability.
Keywords: cancer stemness, CPI‐613, metabolic vulnerability, pyruvate dehydrogenase
Inoue et al. found that pyruvate dehydrogenase (PDH) component X expression is necessary for PDH activity and is involved in the cancer stemness of esophageal squamous cell carcinoma (ESCC) cells, thereby being metabolically essential for the tumor growth of ESCC. This study provides new insights related to the development of novel therapeutic strategies for ESCC by targeting the PDH complex‐associated metabolic vulnerability.

Abbreviations
- CSCs
cancer stem cells
- ESCC
esophageal squamous cell carcinoma
- PDH
pyruvate dehydrogenase
- PDHX
PDH component X
- TCA
tricarboxylic acid
1. INTRODUCTION
Metabolic reprogramming is a major hallmark of cancer and is characterized by the alteration of glycolysis, glutaminolysis, lipid metabolism, amino acid metabolism, mitochondrial metabolism, nucleic acid biogenesis, pentose phosphate pathway, autophagy‐lysosomal degradation pathway, and other biosynthetic and bioenergetic pathways. 1 , 2 These cancer metabolisms provide tumor cells not only with essential energy, but also with the materials required to support large‐scale biosynthesis for their rapid proliferation, survival, stemness, metastasis, and resistance to anticancer drugs. 3 As cancer is a genetic disease caused primarily by genetic and epigenetic alterations, metabolic reprogramming is often triggered by gene aberrations, including copy number aberrations (gene amplification and deletion) and somatic mutations of metabolism‐related genes. 2 , 3 , 4 Thus, the metabolism is often reprogrammed to meet the energetic and substrate demands of the tumor; however, this reprogramming often generates metabolic vulnerabilities in the tumor, providing new opportunities for cancer therapy. 2 , 3 , 4 Therefore, the identification of metabolically essential genes for cancer cells leads the development of novel cancer therapies targeting the specific metabolic vulnerability.
Esophageal cancer is the eighth most common cancer and the sixth most common cause of cancer‐related death worldwide. 5 There are two major histological types of esophageal cancer: squamous cell carcinoma and adenocarcinoma. 5 In Asia, the most common type is ESCC, which is associated with environmental factors such as chronic smoking and alcohol consumption. 6 , 7 Although our understanding of the pathogenesis has improved and there have been advances in therapeutic strategies, the 5‐year relative survival rate of patients with ESCC with distant metastasis remains low at only 4.3% due to rapid progression, local recurrence, and distant metastasis. 8 Recent studies demonstrated that immunotherapy can improve the survival of patients with locally advanced and metastatic ESCC. 9 However, novel therapeutic strategies are needed for advanced ESCC and the molecular pathogenesis of this disease remains unclear. We previously identified many genes related to the pathogenesis of ESCC by high‐throughput screening of copy number aberrations and epigenomic alterations, including DNA methylation. 10 , 11 , 12 , 13 , 14 , 15 , 16 Among them, genes expected to be druggable targets were found. In particular, small molecule compounds for the inhibitor of cIAP1/2, which we identified as a target of 11q22‐23 amplification in ESCC, have been developed. 17 However, metabolic vulnerability as a therapeutic target in ESCC has not been adequately understood.
Cancer cells utilize aerobic glycolysis, known as the Warburg effect, to generate energy for their survival and proliferation due to mitochondrial dysfunction resulting from the mutations of genes coding enzymes associated with the TCA cycle. 2 , 18 , 19 However, although there is interest in the glycolytic phenotype of many cancers, recent studies have demonstrated that cancer cells require fully functional mitochondria to support their proliferation and survival. 2 , 18 , 19 The connecting link between glycolysis and the TCA cycle is the PDH complex, which catalyzes pyruvate, a metabolite from glucose, to acetyl‐CoA in the mitochondria. Thus, the PDH complex is a rate‐limiting enzyme that connects glycolysis to the TCA cycle, and consists of E1 (PDH composed of catalytic α [PDHA] and regulatory β [PDHB] subunits), E2 (dihydrolipoyl transacetylase [DLAT]), E3 (dihydrolipoyl dehydrogenase [DLD]), and PDHX as an E3‐binding protein. 18 , 19 , 20 The inhibition of PDH activity induces aerobic glycolysis as the Warburg effect by catalyzing pyruvate to lactate in cancer cells, promoting the tumor suppressor function of the PDH complex. 19 , 20 , 21 , 22 , 23 In contrast, inhibition of PDH activity by depleting PDHB was reported to resemble the Warburg effect, inhibiting breast cancer growth. 19 , 24 Therefore, the role of the PDH complex in cancer is controversial and may be dependent on the cellular context, including the pathological condition of cancer cells. 19 In this study, using siRNA‐based screening for genes related to several metabolic pathways, we identified PDHX as a metabolically essential gene for the growth of ESCC cells. Our study provides new insights related to the development of novel therapeutic strategies against ESCC by targeting PDHX as a key molecule in its metabolic vulnerability.
2. MATERIALS AND METHODS
2.1. Antibodies and reagents
The antibody against PDHX (10951‐1‐AP) was purchased from Proteintech, the antibody against β‐actin (A5441) was from Sigma‐Aldrich, the antibody against CD44v9 (RV3) was from Cosmo Bio, and the antibody against Ki‐67 was from DAKO (#M7240). CPI‐613 was purchased from Cayman Chemical.
2.2. Construction of the siRNA library and cell growth assay
The Cherry‐Pick siRNA library containing four pooled siRNAs for 224 metabolism‐related genes was synthesized by Horizon Discovery. Cells (1 × 104 cells/well) were plated in 96‐well plates and siRNA was transfected the next day at 10 nmol/L using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer's instructions. After 3 days, cells were stained with 0.1% crystal violet (CV) as described in previous papers. 25 , 26 The stained cells were lysed with a 2% SDS solution and the optical density (OD) was measured at 560 nm using a microplate reader. The OD values of cells relative to the control cells transfected with pooled siRNA for the negative control (siNC) were arbitrarily set to 100% to determine the percentage of viable cells.
2.3. In vivo tumor growth assay
All animal experiments were carried out according to the guidelines and approval by the Tokyo Medical and Dental University Animal Care and Use Committee as described in previous papers. 25 , 26 Six‐week‐old female BALB/c nude mice were purchased from Charles River Laboratories. Cells (1 × 107 cells in 100 µL of PBS with 50% Matrigel) were injected subcutaneously into the flank of nude mice. The tumor volume was calculated using the following formula: 4/3 × π × (1/2 × smaller diameter)2 × (larger diameter)/2. shVector cells and PDHX‐inhibited HSC‐40A cells (1 × 107 cells in 200 µL of PBS) were injected into the intraperitoneal cavity of nude mice. For treatment with CPI‐613, when the tumor volume reached 100‐150 mm3, CPI‐613 was administered by intraperitoneal (IP) injection at 25 mg/kg two times per week. D5W (5% dextrose in water) was used as a control vehicle.
2.4. Tumor sphere formation assay
Cells were seeded at a density of 103 cells/ml in 6‐well Ultra‐Low Attachment Plates in DMEM‐F12 1:1 media (Wako) containing 1× B‐27 supplement (Thermo Fisher Scientific), 4 µg/mL of insulin (I‐6634, Sigma‐Aldrich), 20 ng/mL of FGF2 (F0291, Sigma‐Aldrich), 10 ng/mL of EGF (E9644, Sigma‐Aldrich), and 5% Pen/Strep (Thermo Fisher Scientific). At 10 days, three random fields per well were digitally imaged using an EVOS cell imaging system (Thermo Fisher Scientific). The diameters of each sphere were measured on the long axis using Adobe Photoshop Elements 14 (Adobe). The average size of spheres from three wells was calculated and tumor spheres with a diameter >70 µm were counted.
2.5. Publicly available data for the gene expression and the copy number in human cancer
An expression dataset (GSE44021) for tumor and nontumor tissue in 113 paired samples of primary ESCC was downloaded from the Gene Expression Omnibus (GEO). The frequencies of genes highly expressed in tumor tissues relative to the corresponding nontumor tissue were calculated. The Cancer Genome Atlas (TCGA) data for the expression and copy number were downloaded from the “cBioportal” website (https://www.cbioportal.org). For correlation analysis in 92 ESCC cases, a log2 transformation was applied to the expression values and copy number values, and the Pearson scores for the correlation were computed. The top 20% (18 of 92 cases) of the cohort was considered “PDHX‐high” and the remaining was considered “PDHX‐low”.
2.6. Statistical analysis
Significance was assessed by the two‐tailed Student's t‐test using GraphPad Prism Version 7.0 (GraphPad Software, Inc.). The correlation between PDHX expression in primary ESCC samples and the clinicopathological variables was analyzed using the chi‐squared test. Differences with P values lower than .05 were considered to be significant.
Additional materials and methods are described in the Supporting Information.
3. RESULTS
3.1. Identification of metabolically essential genes for cell growth in ESCC
To identify the metabolic vulnerability in the cell growth of ESCC, we constructed an siRNA library containing each of four pooled siRNAs for a total of 224 genes related to various metabolic pathways, including the TCA cycle and glycolysis, referring to the KEGG pathway (https://www.genome.jp/kegg/pathway.html) (Tables S1 and Table S2). We then transfected each of the four pooled siRNAs for 224 genes into two ESCC cell lines, KYSE850 and KYSE170 cells, and identified 43 genes causing a greater than 30% reduction in cell survival rate in both cell lines (Figure 1A and Table S3). Furthermore, the in silico analysis of the expression dataset (GSE44021), which was obtained by NCBI Gene Expression Omnibus (GEO), revealed that the expression of 13 of 43 genes was frequently upregulated in ESCC tumor tissues relative to the corresponding nontumor tissues in >30% of 113 ESCC cases (Figure 1B). They included genes related to the TCA cycle (PC, PDK1, PDHX), glycolysis (LDHB, HK2, HIF1A), amino acid metabolism (ASNS, SHMT2, PSAT1, DHFR, GLDC), nucleotide metabolism (PRPS1), and autophagy (RB1CC1), suggesting possible metabolic vulnerability in ESCC (Table S3). Among these 13 genes, we focused on the PDHX (PDHX) gene because its oncogenic significance is largely unknown. When the pooled siRNAs (named siPDHX‐1) and two individual siRNAs (siPDHX‐2 and ‐3) were transfected into KYSE850 and KYSE170 cells, respectively, we confirmed the inhibition of cell growth following effective knockdown of PDHX expression (Figure 1C). This growth inhibition was also demonstrated by shRNA‐mediated knockdown of PDHX expression (Figure 1D). Furthermore, tumor growth in xenograft mice was markedly suppressed in PDHX‐inhibited KYSE850 cells compared with the control cells (shVector) (Figure 1E). Tumors composed of PDHX‐inhibited KYSE850 cells had a significant reduction in the number of Ki67‐positive cells, a marker of cell proliferation, compared with tumors composed of control cells (shVector) (Figure 1F). Therefore, PDHX expression was suggested to be required for the growth of ESCC cells in vitro and in vivo.
FIGURE 1.

Inhibition of tumor growth by PDHX knockdown in ESCC cells. A, Identification of genes required for cell growth in ESCC cell lines. The Venn diagram shows the common 43 genes in KYSE850 and KYSE170 cells. B, Frequency of upregulation in the ESCC tumor tissue relative to the corresponding nontumor tissue for 43 genes. C and D, Effects of PDHX knockdown on cell growth. The cell growth rate is reported as the relative ratio compared with day 0. Error bars indicate the SD and are not visible. Data are presented as the mean ± SD. P values were calculated using the two‐sided Student's t test (*P < .0001). E, In vivo tumor growth assay. Scatter plot of tumor weight and tumor volume. Scale bars: 1 cm. Error bars indicate the SD. Data are presented as the mean ± SD. P values were calculated using the two‐sided Student's t‐test (*P < .05, **P < .01). F, Immunohistochemistry (IHC) analysis of Ki‐67 protein in the resected tumors. The percentage of Ki‐67‐positive cells is indicated. Scale bars: 50 μm. P value was calculated using the two‐tailed Student's t test (*P < .0001)
PDHX is a component of the PDH complex that catalyzes the conversion of pyruvate to acetyl‐CoA, thereby connecting glycolysis to the TCA cycle, where it can generate ATP as a form of energy in the mitochondria (Figure 2A). Inhibition of PDH activity suppresses this conversion, resulting in glycolysis via the conversion of pyruvate to lactate by lactate dehydrogenase (18,19,24). When PDHX expression was inhibited in KYSE850 and KYSE170 cells, PDH activity was reduced, followed by an increase in intracellular lactate and pyruvate (Figures 2B,C and Figure S1). Furthermore, the production of intracellular ATP markedly decreased in PDHX‐inhibited cells compared with the control cells (shVector) (Figure 2D). A decrease in ATP production was also observed after siRNA‐mediated knockdown of PDHX expression (Figure 2E). Taken together, PDHX plays a metabolically essential role in the growth of ESCC cells through the maintenance of PDH activity followed by ATP production via the TCA cycle.
FIGURE 2.

Reduction of TCA cycle activity by PDHX knockdown. A, Schema of the catalyzation of pyruvate to acetyl‐CoA via the PDH complex. B‐E, Relative changes in PDH activity (B), lactate production (C), and ATP production (D) are indicated in PDHX‐inhibited cells compared with control cells. Error bars indicate the SD. Data are presented as the mean ± SD. P values were calculated using the two‐sided Student's t test (*P < .005, **P < .0001)
3.2. Expression status of PDHX in primary ESCC samples
We next examined the expression levels of PDHX protein by immunohistochemical analysis in 70 primary ESCC samples. PDHX expression was assessed using the Histo‐score (H‐score) based on the staining intensity and percentage of positive cells. In normal esophagus tissue, PDHX is moderately expressed in differentiating cells, but not in the basal layer, as the main location of stem cells (H‐score = 100) (Figure 3A). When cases with an H‐score of >150 were defined as having “up‐regulation”, as demonstrated by the immunostaining of the ESCC tumor in case 1 (Figure 3A), PDHX expression was up‐regulated in 24 of 70 ESCC cases (34.3%) (Figure 3B). However, there was no significant association between clinical factors and PDHX expression status (Table S4). Of note, the distribution of PDHX staining was heterogeneous within the tumor and its expression was lower in the keratin pearl, a structure representing terminal differentiation of cancer cells, than that around undifferentiating cells (Figure 3C). A similar distribution of PDHX expression was observed in xenograft tumors of KYSE850 control cells (shVector) (Figure 3D). These observations suggested an association between cancer stemness and PDHX expression. CD44, especially the variant form (CD44v9), is a well‐known marker of CSCs in human cancers, including ESCC, 27 and is also known as an adhesion molecule localized on the cell surface of CSCs. 27 , 28 , 29 Immunofluorescence analysis revealed that PDHX was co‐expressed in CD44v9‐positive stem cells within the ESCC tumor tissue; however, this was not the case in CD44v9‐positive stem cells within the basal layer of adjacent noncancerous tissue (Figure 3E). Thus, PDHX expression may be closely associated with CSC properties in ESCC cells.
FIGURE 3.

Immunostaining of the PDHX protein in primary ESCC tumors. A, Representative images of the normal esophagus and two cases (case 1 and case 2) of primary ESCC. Scale bars: 50 µm. B, The expression status of PDHX protein in primary ESCC (n = 70). Cases with an H‐score greater than 150, as indicated by red bars, were defined as ‘upregulation’ (24 of 70 cases, 34.3%). C and D, Distribution of PDHX staining within the tumor area containing terminal differentiation. Squares are enlarged in the right panels. Asterisk: areas containing the differentiating tumor cells. Scale bars: 100 µm. E, Immunofluorescence staining of PDHX (green) and CD44v9 (red). Nuclei were stained with DAPI (blue). White dashed lines indicate the margins of the tumor (left) or epithelium (Epi) (right) Scale bars: 50 µm
3.3. PDHX is involved in the proliferation of CSCs in ESCC by regulating CD44 expression
To evaluate the effects of PDHX knockdown on the proliferation of CSCs in ESCC cells, we performed a sphere formation assay using control cells (shVector) and PDHX‐inhibited cells (shPDHX). The number and size of the spheroids were markedly reduced in PDHX‐inhibited cells compared with control cells in KYSE850 and KYSE170 cells (Figure 4A). This inhibitory effect on sphere formation was confirmed by PDHX knockdown via another target sequence (Figure S2). Of note, the expression of both PDHX and CD44 genes was transcriptionally upregulated under the sphere condition compared with the two‐dimensional (2D) condition. Moreover, the level of CD44 expression markedly decreased in PDHX‐inhibited cells compared with that in control cells (shVector) under both culture conditions (Figure 4B). This decrease of CD44 expression was also observed after siRNA‐mediated knockdown of PDHX expression in ESCC cells (Figure S3). Furthermore, fluorescence‐activated cell sorting (FACS) and immunofluorescence analyses using specific antibodies against CD44v9 protein revealed that the population of CD44v9‐positive CSCs markedly decreased within the spheroids from PDHX‐inhibited cells compared with those from control cells (Figure 4C,D). Thus, PDHX expression is closely associated with the proliferation of CSCs in ESCC by positively regulating CD44 expression.
FIGURE 4.

Inhibition of CSC proliferation by PDHX knockdown. A, Tumor sphere formation assay using control (shVector) and PDHX‐inhibited (shPDHX) KYSE850 (left panel) and KYSE170 cells (right panel). Upper panels: representative images of tumor spheres. Scale bars: 200 µm. Lower panels: average size of spheres and the number of spheres larger than 70 µm are indicated as the mean ± SEM. Error bars indicate the SEM. P values were calculated using the two‐sided Student's t test (*P < .0001). B, Expression analysis of PDHX and CD44 mRNA under the sphere condition and two‐dimensional (2D) condition. P values were calculated using the two‐sided Student's t test (*P < .05). C, FACS analysis of CD44v9. Frequency of CD44v9‐positive CSCs in the sphere as the mean ± SD. Error bars indicate the SD. P values were calculated using the two‐sided Student's t test (*P < .0001). D, Immunofluorescence staining of CD44v9 (red) in the sphere. Nuclei were stained with DAPI (blue). Scale bars: 50 µm
3.4. Co‐amplification of PDHX and CD44 genes in human cancers, including ESCC tumors
Both PDHX and CD44 genes are located within 200 kb in the 11p13 region of the chromosome, which is well known for gene amplification in many types of human cancers, 30 , 31 , 32 including gastric (stomach) cancer (Figure S3). 30 Indeed, these two genes were involved in co‐amplification in 114 of 10, 967 cases (1.04%) of 32 cancer types in the TCGA PanCancer Atlas study, and their mutation frequencies were higher in gastric cancer (stomach adenocarcinoma) (17 of 440 cases, 3.9%), ESCC (3 of 92 cases, 3.3%), and head and neck squamous cell carcinoma (14 of 523 cases, 2.7%) (Figure 5A,B). Analysis of the copy number ratio of the genomic region within 15 Mb of 11p12‐p13 revealed that the PDHX and CD44 genes were localized within the smallest region of overlapping (SRO) in three ESCC cases with the 11p13 amplificon, strongly suggesting that these genes are targets of this amplicon (Figure 5C). In a correlation analysis of 92 ESCC cases, mRNA expression levels of PDHX and CD44 were positively correlated with their respective copy numbers, and the mRNA levels of both genes were significantly correlated (Figure 5D). We found that PDHX was co‐amplified with CD44 in YES3 cells, an ESCC cell line, using genomic‐PCR and FISH analyses, and its knockdown inhibited cell growth under both 2D and sphere culture conditions (Figure S4). In addition, our previous study demonstrated that the 11p13 region is frequently amplified in gastric cancer cell lines, including HSC‐40A cells, which were established from the ascites of a patient with gastric cancer, and the CD44 gene is a potential target of this 11p13 amplification (28). Moreover, the PDHX and CD44 genes were co‐amplified in HSC‐40A cells (Figure S5A,B), and PDHX knockdown inhibited in vitro cell growth and in vivo tumor formation in the peritoneal cavity after intraperitoneal injection into nude mice (Figure S5C,D). Thus, PDHX and CD44 are co‐activated by gene amplification and may coordinately function in cancer stemness in human cancers, including ESCC and gastric cancer.
FIGURE 5.
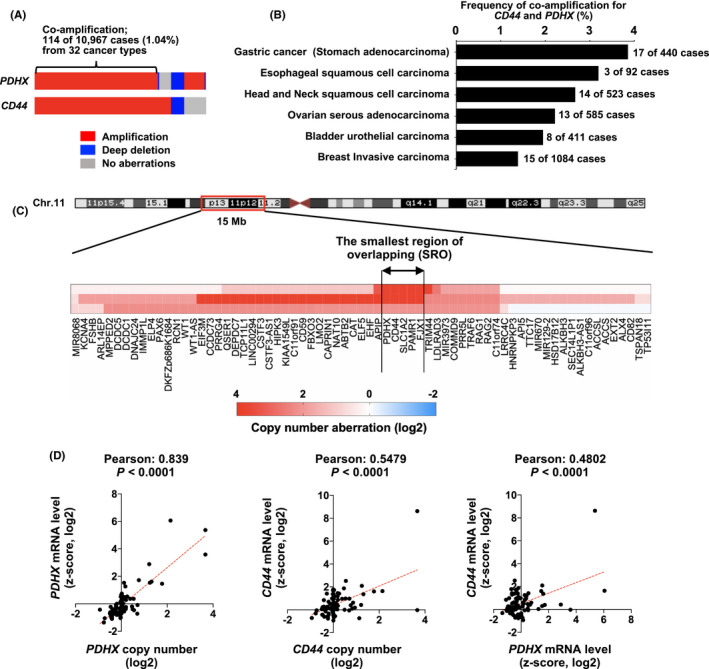
Co‐amplification of PDHX and CD44 genes in human cancers, including ESCC tumors. A, Frequency of amplification of PDHX and CD44 genes in 10 967 cases of 32 cancer types in the TCGA PanCancer Atlas study. B, Frequency of co‐amplification of PDHX and CD44 genes in various human cancers. C, Analysis of the copy number ratio for genomic region within 15 Mb at the 11p12‐p13 region in three ESCC cases having 11p13 amplification. The heat map shows the copy number ratio (Log2) for each gene within this genomic region. The SRO containing the PDHX and CD44 genes is indicated by the double‐headed arrows. D, The correlation between copy number and the corresponding mRNA expression of PDHX and CD44 in 92 ESCC cases
3.5. Antitumor effects of CPI‐613, a PDH inhibitor, in ESCC cells
The PDH complex consists of five major subunits, PDHA1, PDHB, DLAT, DLD, and PDHX. 20 , 21 Based on the mRNA expression in 92 ESCC cases, PDHA1, PDHB, DLAT, and DLD expression was significantly upregulated in the highest 20% of 92 ESCC tumors with high PDHX expression (PDHXhigh‐cases, n = 18) compared with the lowest 20% with low PDHX expression (PDHXlow‐cases, n = 18), suggesting that activation of PDH complex is associated with the expression of PDH‐related genes in ESCC tumors (Figure 6A). Furthermore, the expression of PDHA1, PDHB, DLAT, and DLD was transcriptionally upregulated and partially decreased in PDHX‐inhibited cells compared with that in control cells (shVector) under the sphere condition, not the 2D condition (Figure S6). As lipoic acid (lipoate) is covalently joined as a catalytic co‐factor for the inactivation of the PDH complex, a lipoate derivative, CPI‐613, can potently induce mitochondrial dysfunction via the inhibition of PDH activity and exhibits strong antitumor activity in numerous cancers. 33 , 34 We examined the antitumor effects of CPI‐613 on tumor growth of KYSE850 and KYSE170 cells highly expressing PDHX in vitro and in vivo. As shown in Figure 6B, CPI‐613 markedly reduced the number and size of the spheroids formed by both KYSE850 and KYSE170 cells. Furthermore, in vivo tumor growth was markedly inhibited by intraperitoneal administration of CPI‐613 compared with a vehicle in the KYSE850 xenograft model (Figure 6C). Thus, high expression of PDHX may be closely associated with the upregulation of other PDH‐related genes, and treatment with CPI‐613 is therapeutically effective for ESCC tumors highly expressing PDHX, especially ESCC stem cells.
FIGURE 6.

Antitumor effects of CPI‐613 on ESCC cells. A, Comparison analysis of the expression of PDH‐related genes, PDHA1, PDHB, DLAT, and DLD, between cases with high expression and with low expression of PDHX. P values were calculated using the two‐sided Student's t test (*P < .005, **P < .01, ***P < .05). B, Tumor sphere formation assay using control cells (nontreatment, NT) and CPI‐613‐treated cells. Average size of spheres and the number of spheres larger than 70 µm are indicated as the mean ± SEM. Error bars indicate the SEM. P values were calculated by the two‐sided Student's t test (*P < .0001, **P < .0005, ***P < .005). C, In vivo tumor growth assay. Scatter plot of tumor weight and tumor volume are presented as the mean ± SD. Error bars indicate the SD. P values were calculated using the two‐sided Student's t test (*P < .001). D, A schema illustrating the key findings in this study. PDHX is closely associated with cancer stemness together with CD44v9 by activating the mitochondrial metabolism via PDH activity and may be a potential target for cancer therapy
4. DISCUSSION
The identification of genes that are metabolically essential for cancer cells is a useful approach for the development of cancer therapies targeting metabolic vulnerability. In this study, siRNA‐based screening of genes related to metabolic pathways identified PDHX as a metabolically essential gene in the cell growth of ESCC cells. PDHX expression is necessary for the maintenance of PDH activity and the production of ATP energy via the TCA cycle, and functions in the proliferation of CSCs along with in vivo tumor growth in ESCC. The PDHX gene was co‐upregulated with the CD44 gene, which is involved in cancer stemness, by amplification at the 11p13 region in ESCC tumors. CD44v9, a variant isoform of CD44, interacts with and stabilizes xCT, a subunit of the cystine‐glutamate transporter, which positively regulates the intracellular level of glutathione (GSH) for ROS scavenging, thereby affecting antioxidant levels in CSCs. 35 , 36 As increased PDH activity generates ROS in the mitochondria, CD44v9‐mediated antioxidants may protect CSCs from cytotoxicity via the accumulation of ROS. Therefore, the PDHX/CD44 axis coordinately functions in cancer stemness in ESCC and the co‐amplification of these two genes may promote the addiction to energy production via mitochondrial metabolism in CSCs. Furthermore, high expression of PDHX is closely associated with the upregulation of other PDH‐related genes, and pharmacological targeting of the PDH complex using CPI‐613 was therapeutically effective for ESCC tumors highly expressing PDHX. Thus, our study provides new insights related to the development of novel therapeutic strategies targeting the metabolic vulnerability of ESCC (Figure 6D).
A central connection between glycolysis and the mitochondrial TCA cycle is the PDH complex. 2 , 18 , 19 PDH activity is negatively regulated by the phosphorylation of PDHA, a component of the PDH complex, via pyruvate dehydrogenase kinase (PDK). 37 The PDK family of enzymes comprises four members (PDK1‐PDK4) and is frequently overexpressed in cancers, and inhibition of PDKs induces cancer cell death through increased PDH activity in the mitochondria and subsequent excessive ROS production. 37 , 38 , 39 As PDK inhibitors have been reported as potential therapeutic agents for cancer, 37 PDKs have been implicated in oncogenesis by promoting glycolysis as the Warburg effect via the reduction of PDH activity. 38 , 39 , 40 , 41 , 42 Consistent with these studies, our siRNA‐based screening demonstrated that knockdown of PDKs (PDK1‐4) inhibited the growth of KYSE850 and KYSE170 cells. However, PDHX knockdown, followed by the reduction of PDH activity, also inhibited the growth of ESCC cells. Thus, biased metabolic conditions, such as excess or reduced PDH activity, are toxic to cancer cells, suggesting the importance of the metabolic balance between glycolysis and the TCA cycle for cancer cell growth. The expression level of PDHX was increased in the spheroids of ESCC cells and in CD44v9‐positive cells within primary ESCC tumors. Therefore, activation of the mitochondrial metabolism via PDH activity is indispensable for CSC properties, suggesting the PDH complex to be a metabolically reasonable target for CSCs in the pathological context of ESCC.
The molecular mechanism underlying the transcriptional regulation of PDHX expression in the CD44v9‐positive cell population of ESCC remains unknown. Recently, Kumazoe et al. reported that PDHA1 expression was upregulated in CD44‐positive pancreatic CSCs via transcriptional factors, such as forkhead box O3 (FOXO3) and peroxisome proliferator‐activated receptor‐γ co‐activator‐1β (PGC‐1β), and suggested that the upregulation of PDHA1 is essential for CSC properties. 43 Our study suggests that PDHX expression is closely associated with the upregulation of other PDH‐related genes, including PDHA1. Thus, FOXO3 and PGC‐1β may be common transcriptional regulators of PDH‐related genes; however, this requires further investigation. In our present study, we were unable to define the mechanism by which PDHX positively regulates CD44 expression. The components of the PDH complex, including PDHX and PDHA1, were reported to localize in the nucleus in addition to the mitochondria, and the nuclear PDH complex controls gene expression by mediating histone acetylation. 44 , 45 Thus, the nuclear PDH complex may be associated with epigenetic regulation controlling the expression of CSC‐essential genes, including the CD44 gene.
Cancer cells require both aerobic glycolysis and mitochondria metabolism to generate energy. Indeed, combined treatment with 2‐deoxyglucose (2DG), which acts as an inhibitor of glucose metabolism, and metformin, a commonly prescribed drug for type 2 diabetes, which prevent oxidative phosphorylation in mitochondria, could synergistically induce cell death by reducing intracellular ATP levels in ESCC cells. 46 Furthermore, treatment with sulfasalazine, an inhibitor of xCT that functions as an antioxidant, effectively suppresses tumor growth, cancer stemness, and metastasis of ESCC cells. 47 , 48 , 49 Thus, the potential applications of metabolic inhibitors have been experimentally demonstrated in ESCC. For future clinical application, it will be necessary to identify specific metabolic vulnerability in ESCC tumors of individual patients.
The inhibition of mitochondrial metabolism as a new cancer therapeutic treatment has been evaluated in clinical trials. 50 Recent evidence suggests that defining the genetic status of enzymes related to the mitochondrial metabolism and mitochondrial activity using PET imaging can help stratify cancer patients who will benefit from treatment with inhibitors targeting the mitochondrial metabolism. 50 The lipoate derivative CPI‐613 is a first‐in‐class agent that targets the PDH complex in mitochondrial metabolism and is currently used in clinical trials for acute myeloid leukemia (AML), pancreatic cancer, myelodysplastic syndromes (MDS), and lymphoma. 51 , 52 , 53 PDHX was amplified in human cancers, including in ESCC and gastric cancer, and high expression of PDHX is closely associated with the upregulation of other PDH‐related genes. Thus, gene amplification and/or high expression of PDHX may be a biomarker to predict high PDH activity in the tumor and to identify tumors susceptible to CPI‐613. Further validation of this idea using a large cohort of tumor samples and cancer cell lines is required for the development of precision cancer medicine based on PDH activity in human cancers, including ESCC and gastric cancer.
DISCLOSURE
The authors declare no competing interests.
Supporting information
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Figures S1–S6
ACKNOWLEDGEMENTS
The authors thank Ayako Takahashi and Rumi Mori (Tokyo Medical and Dental University, Japan) for their technical assistance. This study was supported in part by Grants‐in‐Aid for Scientific Research (18K06954 to Jun Inoue; 16K14630 to Johji Inazawa), a Grant‐in‐Aid for Scientific Research on Innovative Areas “Conquering cancer through NEO‐dimensional systems understandings” (15H05908 to Johji Inazawa) from JSPS and MEXT, a research programme of the Project for Cancer Research and Therapeutic Evolution (P‐CREATE) and the Tailor‐Made Medical Treatment with the BioBank Japan Project (BBJ) from the Japan Agency for Medical Research and Development (AMED). This study was supported by Nanken‐Kyoten, TMDU.
Inoue J, Kishikawa M, Tsuda H, Nakajima Y, Asakage T, Inazawa J. Identification of PDHX as a metabolic target for esophageal squamous cell carcinoma. Cancer Sci. 2021;112:2792–2802. 10.1111/cas.14938
Jun Inoue and Masahiro Kishikawa contributed equally to this work.
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Mullen AR, DeBerardinis RJ. Genetically‐defined metabolic reprogramming in cancer. Trends Endocrinol Metab. 2012;23:552‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phan LM, Yeung SC, Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti‐cancer therapies. Cancer Biol Med. 2014;11:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma AK, Eils R, König R. Copy number alterations in enzyme‐coding and cancer‐causing genes reprogram tumor metabolism. Cancer Res. 2016;76:4058‐4067. [DOI] [PubMed] [Google Scholar]
- 5. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐386. [DOI] [PubMed] [Google Scholar]
- 6. Lin Y, Totsuka Y, He Y, et al. Epidemiology of esophageal cancer in Japan and China. J Epidemiol. 2013;23:233‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dotto GP, Rustgi AK. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell. 2016;29:622‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okuda M, Inoue J, Fujiwara N, Kawano T, Inazawa J. Subcloning and characterization of highly metastatic cells derived from human esophageal squamous cell carcinoma KYSE150 cells by in vivo selection. Oncotarget. 2017;8:34670‐34677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kojima T, Doi T. Immunotherapy for esophageal squamous cell carcinoma. Curr Oncol Rep. 2017;19:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li R, Li P, Xing W, Qiu H. Heterogeneous genomic aberrations in esophageal squamous cell carcinoma: a review. Am J Transl Res. 2020;12:1553‐1568. [PMC free article] [PubMed] [Google Scholar]
- 11. Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci. 2004;95:559‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nagata H, Kozaki K‐I, Muramatsu T, et al. Genome‐wide screening of DNA methylation associated with lymph node metastasis in esophageal squamous cell carcinoma. Oncotarget. 2017;8:37740‐37750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Komatsu S, Imoto I, Tsuda H, et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis. 2009;30:1139‐1146. [DOI] [PubMed] [Google Scholar]
- 14. Haruki S, Imoto I, Kozaki K‐I, et al. Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis. 2010;31:1027‐1036. [DOI] [PubMed] [Google Scholar]
- 15. Tanaka K, Imoto I, Inoue J, et al. Frequent methylation‐associated silencing of a candidate tumor‐suppressor, CRABP1, in esophageal squamous‐cell carcinoma. Oncogene. 2007;26:6456‐6468. [DOI] [PubMed] [Google Scholar]
- 16. Sonoda I, Imoto I, Inoue J, et al. Frequent silencing of lowdensity lipoprotein receptor‐related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004;64:3741‐3747. [DOI] [PubMed] [Google Scholar]
- 17. Cai Q, Sun H, Peng Y, et al. A potent and orally active antagonist (SM‐406/AT‐406) of multiple inhibitor of apoptosis proteins (IAPs) in clinical development for cancer treatment. J Med Chem. 2011;54:2714‐2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun W, Liu Q, Leng J, Zheng Y, Li J. The role of pyruvate dehydrogenase complex in cardiovascular diseases. Life Sci. 2015;121:97‐103. [DOI] [PubMed] [Google Scholar]
- 19. Cai Z, Li C‐F, Han F, et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol Cell. 2020;80(2):263‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou ZH, McCarthy DB, O'Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. 2001;98:14802‐14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF‐1‐mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177‐185. [DOI] [PubMed] [Google Scholar]
- 22. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF‐1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187‐197. [DOI] [PubMed] [Google Scholar]
- 23. Hitosugi T, Fan J, Chung T‐W, et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;44:864‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yonashiro R, Eguchi K, Wake M, Takeda N, Nakayama K. Pyruvate dehydrogenase PDH‐E1β controls tumor progression by altering the metabolic status of cancer cells. Cancer Res. 2018;78:1592‐1603. [DOI] [PubMed] [Google Scholar]
- 25. Gokita K, Inoue J, Ishihara H, Kojima K, Inazawa J. Therapeutic potential of LNP‐mediated delivery of miR‐634 for cancer therapy. Mol Ther Nucleic Acids. 2020;19:330‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inoue J, Fujiwara K, Hamamoto H, Kobayashi K, Inazawa J. Improving the efficacy of EGFR inhibitors by topical treatment of cutaneous squamous cell carcinoma with miR‐634 ointment. Mol Ther Oncolytics. 2020;19:194‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taniguchi D, Saeki H, Nakashima Y, et al. CD44v9 is associated with epithelial‐mesenchymal transition and poor outcomes in esophageal squamous cell carcinoma. Cancer Med. 2018;7:6258‐6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(‐) and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. [DOI] [PubMed] [Google Scholar]
- 29. Yae T, Tsuchihashi K, Ishimoto T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. [DOI] [PubMed] [Google Scholar]
- 30. Fukuda Y, Kurihara N, Imoto I, et al. CD44 is a potential target of amplification within the 11p13 amplicon detected in gastric cancer cell lines. Genes Chromosomes Cancer. 2000;29:315‐324. [DOI] [PubMed] [Google Scholar]
- 31. Starczynowski DT, Lockwood WW, Deléhouzée S, et al. TRAF6 is an amplified oncogene bridging the RAS and NF‐κB pathways in human lung cancer. J Clin Invest. 2011;121:4095‐4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ong C‐A, Shannon NB, Ross‐Innes CS, et al. Amplification of TRIM44: pairing a prognostic target with potential therapeutic strategy. J Natl Cancer Inst. 2014;106:dju050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stuart SD, Schauble A, Gupta S, et al. A strategically designed small molecule attacks alpha‐ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zachar Z, Marecek J, Maturo C, et al. Non‐redox‐active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med. 2011;89:1137‐1148. [DOI] [PubMed] [Google Scholar]
- 35. Woolbright BL, Rajendran G, Harris RA, Taylor JA 3rd. Metabolic flexibility in cancer: targeting the pyruvate dehydrogenase kinase: pyruvate dehydrogenase axis. Mol Cancer Ther. 2019;18:1673‐1681. [DOI] [PubMed] [Google Scholar]
- 36. Olson KA, Schell JC, Rutter J. Pyruvate and metabolic flexibility: illuminating a path toward selective cancer therapies. Trends Biochem Sci. 2016;41:219‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonnet S, Archer SL, Allalunis‐Turner J, et al. A mitochondria‐K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37‐51. [DOI] [PubMed] [Google Scholar]
- 38. Kaplon J, Zheng L, Meissl K, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene‐induced senescence. Nature. 2013;498:109‐112. [DOI] [PubMed] [Google Scholar]
- 39. Fujiwara S, Kawano Y, Yuki H, et al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013;108:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hitosugi T, Fan J, Chung TW, et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;44:864‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jagust P, de Luxán‐Delgado B, Parejo‐Alonso B, Sancho P. Metabolism‐based therapeutic strategies targeting cancer stem cells. Front Pharmacol. 2019;10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghosh P, Vidal C, Dey S, Zhang L. Mitochondria targeting as an effective strategy for cancer therapy. Int J Mol Sci. 2020;21:3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumazoe M, Takai M, Hiroi S, et al. The FOXO3/PGC‐1β signaling axis is essential for cancer stem cell properties of pancreatic ductal adenocarcinoma. J Biol Chem. 2017;292:10813‐10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eguchi K, Nakayama K. Prolonged hypoxia decreases nuclear pyruvate dehydrogenase complex and regulates the gene expression. Biochem Biophys Res Commun. 2019;520:128‐135. [DOI] [PubMed] [Google Scholar]
- 45. Chen J, Guccini I, Di Mitri D, et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet. 2018;50:219‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shafaee A, Pirayesh Islamian J, Zarei D, et al. Induction of apoptosis by a combination of 2‐deoxyglucose and metformin in esophageal squamous cell carcinoma by targeting cancer cell metabolism. Iran J Med Sci. 2019;44:99‐107. [PMC free article] [PubMed] [Google Scholar]
- 47. Chen RS, Song YM, Zhou ZY, et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin‐1/beta‐catenin pathway. Oncogene. 2009;28:599‐609. [DOI] [PubMed] [Google Scholar]
- 48. Kagami T, Yamade M, Suzuki T, et al. High expression level of CD44v8‐10 in cancer stem‐like cells is associated with poor prognosis in esophageal squamous cell carcinoma patients treated with chemoradiotherapy. Oncotarget. 2018;9:34876‐34888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li B, Li YY, Tsao SW, Cheung AL. Targeting NF‐kappaB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol Cancer Ther. 2009;8:2635‐2644. [DOI] [PubMed] [Google Scholar]
- 50. Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32:341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alistar A, Morris BB, Desnoyer R, et al. Safety and tolerability of the first‐in‐class agent CPI‐613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single‐centre, open‐label, dose‐escalation, phase 1 trial. Lancet Oncol. 2017;18:770‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pardee TS, Lee K, Luddy J, et al. A phase I study of the first‐in‐class antimitochondrial metabolism agent, CPI‐613, in patients with advanced hematologic malignancies. Clin Cancer Res. 2014;20:5255‐5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bellio C, DiGloria C, Spriggs DR, Foster R, Growdon WB, Rueda BR. The metabolic inhibitor CPI‐613 negates treatment enrichment of ovarian cancer stem cells. Cancers. 2019;11:1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1
Table S2
Table S3
Table S4
Figures S1–S6