

Abstract
The prevalence of neuroendocrine prostate cancer (NEPC) arising from adenocarcinoma (AC) upon potent androgen receptor (AR) pathway inhibition is increasing. Deeper understanding of NEPC biology and development of novel therapeutic agents are needed. However, research is hindered by the paucity of research models, especially cell lines developed from NEPC patients. We established a novel NEPC cell line, KUCaP13, from tissue of a patient initially diagnosed with AC which later recurred as NEPC. The cell line has been maintained permanently in vitro under regular cell culture conditions and is amenable to gene engineering with lentivirus. KUCaP13 cells lack the expression of AR and overexpress NEPC‐associated genes, including SOX2, EZH2, AURKA, PEG10, POU3F2, ENO2, and FOXA2. Importantly, the cell line maintains the homozygous deletion of CHD1, which was confirmed in the primary AC of the index patient. Loss of heterozygosity of TP53 and PTEN, and an allelic loss of RB1 with a transcriptomic signature compatible with Rb pathway aberration were revealed. Knockdown of PEG10 using shRNA significantly suppressed growth in vivo. Introduction of luciferase allowed serial monitoring of cells implanted orthotopically or in the renal subcapsule. Although H3K27me was reduced by EZH2 inhibition, reversion to AC was not observed. KUCaP13 is the first patient‐derived, treatment‐related NEPC cell line with triple loss of tumor suppressors critical for NEPC development through lineage plasticity. It could be valuable in research to deepen the understanding of NEPC.
Keywords: cell line, cultured tumor cells, neuroendocrine tumor, prostate cancer, xenograft
In this study, we report the establishment of a novel patient‐derived xenograft model of t‐NEPC and the successful establishment of the KUCaP13 cell line. The cell line can be maintained permanently in vitro under regular cell culture conditions and is amenable to gene engineering. The cell line should be a valuable tool for research to identify novel therapeutic targets of NEPC and to develop effective therapeutic agents.

1. INTRODUCTION
Prostate cancer is an androgen‐driven disease and standard treatment for advanced cases is androgen deprivation. However, tumors eventually gain resistance and regrow as castration‐resistant prostate cancer (CRPC). Although the majority of CRPC are still dependent on the androgen receptor (AR) pathway, tumor cells can sometimes escape AR pathway inhibition by completely changing their phenotype to become non–AR‐expressing cells. These cells are typically characterized by their resemblance to small cell carcinoma of the lung and are called treatment‐related neuroendocrine prostate cancer (t‐NEPC). 1 It has long been debated whether t‐NEPC arises from minor neuroendocrine cells pre‐existing around prostatic ducts, or by transdifferentiation of adenocarcinoma cells. 2 , 3 Recent genomic studies showed that t‐NEPC shares major driver genomic alterations with adenocarcinoma, such as TMPRSS‐ERG gene fusion. 4 Experimental models have shown that adenocarcinoma can indeed transdifferentiate to NEPC by lineage plasticity. 5 , 6 , 7
Clinically, t‐NEPC is very aggressive, does not express prostate‐specific antigen (PSA), often metastasizes to the liver, and displays lytic bone metastasis. Based on treatment of small cell carcinoma of the lung, platinum‐based chemotherapy is administered. However, its efficacy is limited. 8 The incidence of t‐NEPC has been rapidly increasing recently due to the widespread use of potent AR axis–targeting agents such as abiraterone and enzalutamide. The development of novel therapeutic agents to treat t‐NEPC is urgently needed but is hindered by the lack of pertinent in vitro cell models of t‐NEPCs.
In this study, we report the establishment of a novel patient‐derived xenograft (PDX) model of t‐NEPC and the successful establishment of the KUCaP13 cell line. The cell line can be maintained permanently in vitro under regular cell culture conditions and is amenable to gene engineering. The cell line should be a valuable tool for research to identify novel therapeutic targets of NEPC and to develop effective therapeutic agents.
2. MATERIALS AND METHODS
2.1. Patient and tissue sample
The tissue sample for the establishment of PDX was obtained after penectomy of a 60‐year‐old Japanese male patient with t‐NEPC penile metastasis. Before the operation, the patient provided written informed consent to use the surgical specimen as a bioresource for future research purposes, signing the informed consent form approved by the ethics committee of Kyoto University Hospital (approval number G52).
2.2. Patient‐derived xenograft
A portion of the harvested penile tumor tissue was immediately submerged in ice‐cold normal saline solution, minced into 20‐30 mm3 bits, and transplanted into subcutaneous tissue of anesthetized C.B‐17/IcrCrj SCID mice (Charles River Japan). Tumor sizes were calculated as previously described. 9 All animal experiments were performed in accordance with the Guidelines for Animal Experiments of Kyoto University and approved by the Animal Research Committee at Kyoto University Graduate School of Medicine.
2.3. Establishment of cell line
Harvested xenograft tissues were washed in phosphate‐buffered saline (PBS), minced with scissors, transferred to microtubes, and centrifuged at 400 g for 5 minutes at room temperature. Supernatants were discarded, and samples were dissociated by gentle shaking for 30 minutes at 37°C in 1 mg/mL collagenase type I solution, followed by 0.25% trypsin. Trypsin was inactivated by addition of four volumes of growth medium consisting of RPMI1640, 10% fetal bovine serum, 12.5 mmol/L HEPES, penicillin, and streptomycin. Cultures were filtered through 100‐µm cell strainers and centrifuged, and the supernatant was discarded. Cell pellet was resuspended in Pharm Lyse (BD), incubated at room temperature for 15 minutes, and centrifuged. The supernatant was removed, and the cell pellet resuspended in growth medium was seeded into dishes. The cells were incubated at 37°C in an atmosphere of 5% CO2. Half the medium was removed and replaced with fresh medium two to three times a week. Cancer cells did not attach to the dishes but formed spherical colonies. Colonies containing living cells settled to the bottom without attachment to the culture dish, while dead cells tended to float. Thus, periodically replacing the half of the supernatant medium was efficient in maintaining the rate of living cells. Dilution passages were performed when the cell concentration exceeded 106 cells/mL. The colonies grown in excess were mechanically dissociated by pipetting. Cells were suspended in CP‐1 (Kyokuto Pharmaceutical Industrial) according to the manufacturer's protocol, frozen overnight in Bicell (Nihon Freezer) at −80°C, and stored in liquid nitrogen.
Detailed methods of genome sequencing, protein analysis, and gene engineering can be found in Supplementary Methods (Document S1).
3. RESULTS
3.1. Establishment of novel t‐NEPC PDX
NEPC tissues were harvested from the penile metastasis of a patient with localized prostatic adenocarcinoma who had metastatic recurrence to the lung and penis after 18 months of neoadjuvant hormone therapy with a luteinizing hormone–releasing hormone agonist and bicalutamide followed by radiation to the prostate (Figure 1A,B). At the time of recurrence, serum testosterone was at the castrate level, and PSA was undetectable. Both the penile tissue and subsequent prostate biopsy specimen showed a small cell carcinoma morphology (Figure 1C). The resected penile tumor tissues were implanted into the flanks of two SCID mice. One of the two grafts survived and was serially passaged. The growth of the tumor was very rapid and could not be suppressed by castration of the host mouse (Figure 1D). After three stable passages, this PDX line was named KUCaP13 as one of the series of PDXs established at our institution. 9 , 10 , 11
FIGURE 1.
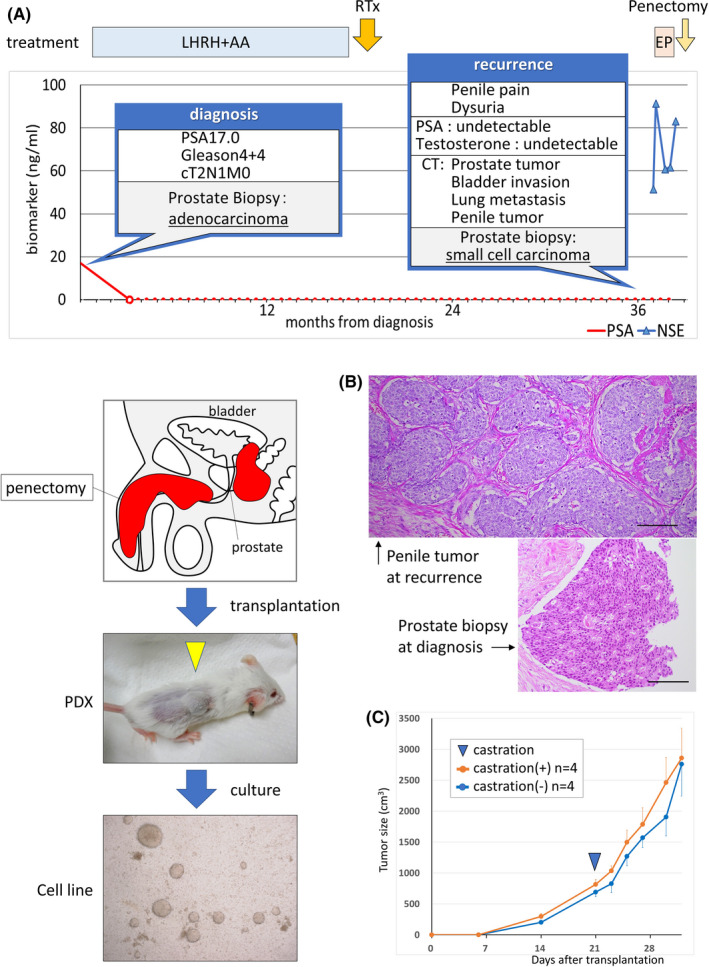
A, Clinical course of the index patient. The patient presented with adenocarcinoma. Prostate‐specific antigen (PSA) decreased to an undetectable level upon androgen deprivation. After whole pelvic radiotherapy with curative intent, the tumor recurred with penile metastasis without PSA elevation. The microscopic image of prostate biopsy as well as penile metastasis showed morphology of small cell carcinoma. AA, androgen receptor antagonist; EP, chemotherapy with etoposide and cisplatin; LHRH, luteinizing hormone–releasing hormone; RTx, whole pelvic radiotherapy. B, Schema of establishment of cell line derived from patient tumor. Cancer cells grew as floating culture and formed spherical colonies. C, Microscopic images of patient tumors. The prostate biopsy at the diagnosis showed cribriform pattern, compatible with prostate adenocarcinoma. By contrast, the tumor tissue at the recurrence showed sheet of uniform cells, frequent mitosis, and high nuclear/cytoplasmic ratio, compatible with small cell carcinoma. Scale bars: 100 µm. D, Growth of KUCaP13 PDX in vivo with/without castration. The average tumor volumes of four mice are plotted on the Y‐axis for each group with vertical bars showing standard error
3.2. Establishment of NEPC cell line
KUCaP13 PDX tumors were harvested, disseminated, and cultured in vitro. On day 5, fibroblasts derived from mice were scattered and attached to the bottom of the culture dish. On the other hand, cancer cells formed colonies and grew as floating cells even with plate centrifugation or the use of collagen‐coated dishes. One week after starting culture, several colonies of cells could be observed, which grew in size and number over time. The colonies eventually developed into spheres. Dissociation and passage at appropriate times were necessary to avoid central necrosis of the growing spheres. Because fibroblasts attached to the culture dish and cancer cells were maintained in floating culture, it was easy to dissociate the two types of cells, and the number of fibroblasts decreased over time with serial passaging. The KUCaP13 cell line was maintained in vitro for more than 2 years. Thus, we considered that an immortal cell line was established. We confirmed that the KUCaP13 cell line contained no reminiscent mouse‐derived cells by PCR (Figure 2A) using mouse‐ and human‐specific primers. Short tandem repeats‐PCR analysis (Promega) showed that KUCaP13 is a genuine new cell line without contamination by existing cell lines (Figure S1). Chromosome analysis of KUCaP13 cells 6 months after beginning in vitro culture showed chromosomal instability. The chromosome number of 20 cells in metaphase was 56‐78, and the modal number was 62‐64 (Figure 2B). Chromosome‐doubling cells, defined as cells with >100 chromosomes, were found in 12% (6/50) of the cells. A representative karyogram is presented in Figure 2C. The established cell line was also reimplanted into the subcutaneous tissue of SCID mice to confirm growth in vivo. The xenograft derived from the cell line (CDX) showed very rapid growth, similar to the original PDX.
FIGURE 2.
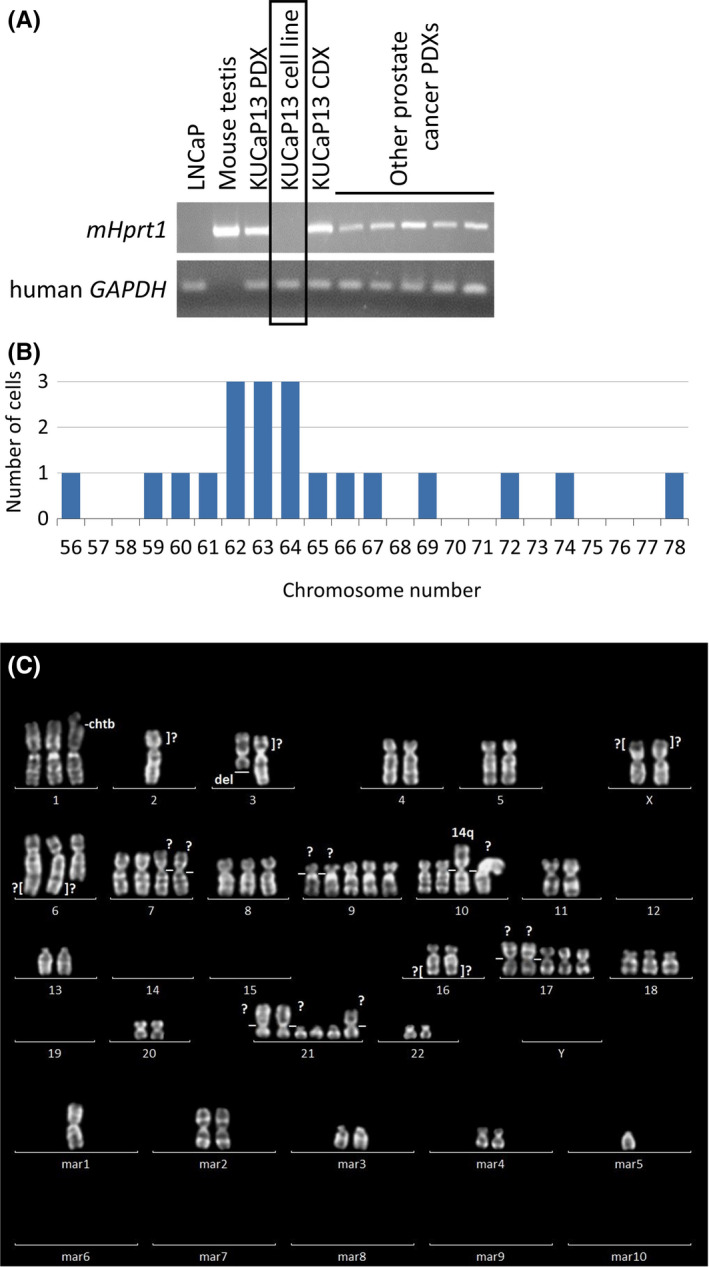
A, Confirmation of human origin of the KUCaP13 cell line. Cell lines and patient‐derived xenograft (PDX) series established in our laboratory were tested for mouse‐specific (mHprt1) and human‐specific (GAPDH) genes. All the PDXs include both mouse‐ and human‐originated genes. In contrast, KUCaP13 and LNCaP cell lines only express human‐originated gene. mHprt1: mouse hypoxanthine guanine phosphoribosyl transferase 1. B, Chromosome number of 20 KUCaP13 cells in metaphase. Modal number was 62‐64. 12% (6/50 cells) were tetraploid. C, A representative karyogram of KUCaP13
3.3. Genomic and transcriptomic characterization of KUCaP13
KUCaP13 PDX and KUCaP13 cell lines were characterized by multiple methods, including whole‐genome sequencing, whole‐exome sequencing (WES), RNA sequencing, Western blotting, and immunohistochemistry (IHC). Copy number alterations (CNAs) were evaluated using shallow whole‐genome sequencing (Figure 3A). The CNA profiles of PDX and cell lines were similar. Notable alterations were amplification of MYC at chromosome 8 and loss of PTEN, RB1, and TP53. The overall CNA pattern of the patient penile tumor tissue sample was also similar, albeit with changes of less magnitude due to contamination by peripheral nontumor cells. Moreover, WES revealed a nonsynonymous single‐nucleotide variant of PTEN (H61R) and frameshift deletion of TP53 (R77fs) with loss of heterozygosity (LOH). The distribution of the types of genomic aberrations identified by WES is shown in Figure S2. Catalogue Of Somatic Mutations In Cancer (COSMIC)‐annotated variants are listed in Table 1. Notably, a homozygous deletion of CHD1 in chromosome 5 was maintained from patient tissue to the cell line (Figure 3B). The homozygous deletion of CHD1 is one of the specific gene alterations in prostate adenocarcinoma, 12 and supports the origin of these models as prostate adenocarcinoma.
FIGURE 3.
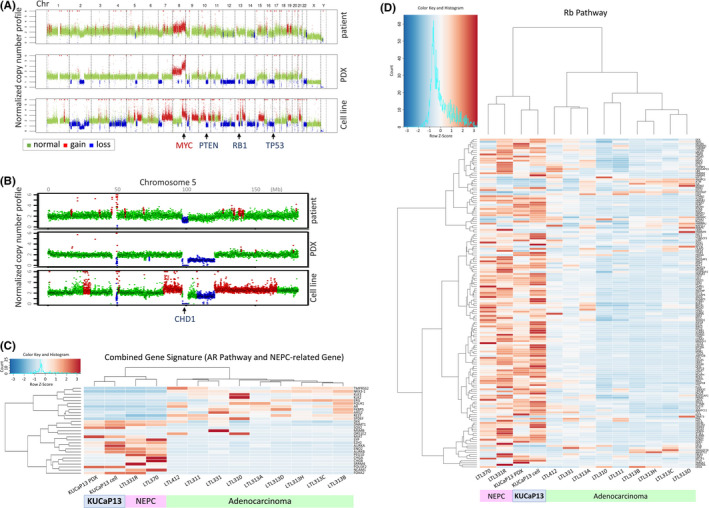
A, Copy number alterations assessed by shallow whole‐genome sequencing. DNA from patient penile tumor at recurrence, KUCaP13 patient‐derived xenograft (PDX), and cell line were compared. B, Loss of CHD1 locus at chromosome 5. C, Unsupervised clustering of KUCaP13 PDX and cell line with prostate cancer PDX series (adenocarcinoma and neuroendocrine prostate cancer [NEPC]) for androgen receptor (AR) pathway– and NEPC‐associated genes. D, Unsupervised clustering of KUCaP13 PDX and cell line with prostate cancer PDX series (adenocarcinoma and NEPC) for Rb pathway–associated genes. KUCaP13 clustered together with known NEPC PDXs
TABLE 1.
The list of Catalogue Of Somatic Mutations In Cancer (COSMIC)‐annotated variants identified in the KUCaP13 cell line by whole‐exome sequencing
| Chromosome | Start | End | Reference nucleotide | Alternative nucleotide | Gene | Variant type | COSMIC annotation | Total reads | Variant reads | Variant allele frequency |
|---|---|---|---|---|---|---|---|---|---|---|
| 1p34.2 | 43394663 | 43394663 | G | A | SLC2A1 | Synonymous SNV | ID=COSM1296455;OCCURENCE=1(urinary_tract) | 328 | 204 | 0.622 |
| 1q32.2 | 210415576 | 210415576 | A | G | SERTAD4 | Nonsynonymous SNV | ID=COSM1929444;OCCURENCE=1(liver) | 97 | 72 | 0.742 |
| 1q44 | 248801602 | 248801602 | ‐ | CA | OR2T35 | Stopgain | ID=COSM246232;OCCURENCE=1(prostate),1(large_intestine) | 203 | 194 | 0.956 |
| 3p21.2 | 52027854 | 52027859 | CCTTGG | ‐ | RPL29 | Nonframeshift deletion | ID=COSM3720236;OCCURENCE=1(haematopoietic_and_lymphoid_tissue) | 11 | 6 | 0.545 |
| 8q12.1 | 55542355 | 55542355 | C | A | RP1 | Nonsynonymous SNV | ID=COSM486530;OCCURENCE=1(kidney) | 123 | 78 | 0.634 |
| 10q23.31 | 89685287 | 89685287 | A | G | PTEN | Nonsynonymous SNV | ID=COSM5042;OCCURENCE=5(central_nervous_system),3(lung) | 58 | 58 | 1 |
| 12q24.31 | 122359397 | 122359397 | ‐ | GAGGAGGAGGAGAAA | WDR66 | Nonframeshift insertion | ID=COSM1359529;OCCURENCE=1(breast),13(large_intestine) | 39 | 24 | 0.615 |
| 12q24.31 | 125478381 | 125478381 | ‐ | CTG | BRI3BP | Nonframeshift insertion | ID=COSM3720593;OCCURENCE=1(haematopoietic_and_lymphoid_tissue) | 24 | 23 | 0.958 |
| 15q15.1 | 42246020 | 42246020 | C | T | EHD4 | Nonsynonymous SNV | ID=COSM187750;OCCURENCE=1(large_intestine) | 185 | 93 | 0.503 |
| 16p13.13 | 11850204 | 11850204 | G | A | ZC3H7A | Synonymous SNV | ID=COSM3817183;OCCURENCE=1(breast) | 69 | 39 | 0.565 |
| 17p13.1 | 7578222 | 7578223 | TC | ‐ | TP53 | Frameshift deletion | ID=COSM392317,COSM392319,COSM13120,COSM392318,COSM392316;OCCURENCE=2(lung),1(oesophagus),2(central_nervous_system),3(large_intestine),1(pancreas),1(breast),1(ovary) | 155 | 154 | 0.994 |
| 17q21.2 | 38975308 | 38975319 | GCCGCCGTGGCC | ‐ | KRT10 | Nonframeshift deletion | ID=COSM1323981;OCCURENCE=1(ovary) | 105 | 75 | 0.714 |
| 17q23.3 | 61571327 | 61571327 | G | A | ACE | Nonsynonymous SNV | ID=COSM3403103,COSM3403102,COSM3403101;OCCURENCE=1(central_nervous_system) | 266 | 166 | 0.624 |
| Xp21.3 | 27765400 | 27765411 | GAGGAGGAGGAG | ‐ | DCAF8L2 | Nonframeshift deletion | ID=COSM1625801;OCCURENCE=1(liver) | 61 | 36 | 0.59 |
| Xq13.1 | 69497289 | 69497289 | G | A | ARR3 | Synonymous SNV | ID=COSM194905;OCCURENCE=1(large_intestine) | 193 | 193 | 1 |
Only the variants with allele frequency of >0.5 are listed.
COSMIC, Catalogue Of Somatic Mutations In Cancer; SNV, single nucleotide variant.
We next assessed the transcriptome of the model by comparing it with a series of well‐characterized prostate cancer PDXs, including those developed from patients with both adenocarcinoma and NEPC. The PDX panel included LTL‐331 and LTL‐331R, which are a pair of adenocarcinomas and t‐NEPCs that developed upon castration of the host mouse. 13 , 14 Unsupervised clustering by AR pathway genes and NEPC‐related genes 14 showed a clear distinction between adenocarcinoma and NEPC, and both KUCaP13 PDX and cell lines were clustered with NEPC (Figure 3C). Interestingly, KUCaP13 PDX and cell line did not show significant elevation of typical “neuronal” genes such as SYP and CHGA. However, expression of key genes implicated in NEPC, including SOX2, EZH2, AURKA, PEG10, POU3F2, and FOXA2, were significantly elevated compared with those in adenocarcinoma. Moreover, although only one allele of RB1 was lost and LOH could not be confirmed by WES, clustering by the Rb pathway signature 15 showed that KUCaP13 models clustered together with other NEPC PDXs with known RB1 alterations, indicating significant aberration of the Rb pathway in KUCaP13 (Figure 3D).
3.4. Characterization of KUCaP13 models by analysis of protein expression
The morphology of patient tissues at recurrence and all KUCaP13 PDX tissues were small cell carcinoma by hematoxylin and eosin staining, while the patient prostate needle biopsy tissue at initial diagnosis was adenocarcinoma (Figure 4A). IHC showed that KUCaP13 tissues expressed NSE, PEG10, and SOX2, and did not express AR. This protein expression pattern was maintained from the patient's penile tumor tissue to CDX. Conversely, prostate biopsy tissue at diagnosis expressed AR but not NSE. PEG10 and SOX2 were positive in only a minority of cells at diagnosis. Remarkably, CHD1 expression was negative at diagnosis and remained negative in KUCaP13 PDX and cell line, corresponding to the loss of the gene at the genomic level. Protein expression analysis indicated that the KUCaP13 models represented features of NEPC derived from prostate adenocarcinoma, and that the protein expression pattern was maintained from patient penile tissue at recurrence to the cell line and CDX. The protein expression patterns were confirmed by Western blotting (Figure 4B).
FIGURE 4.
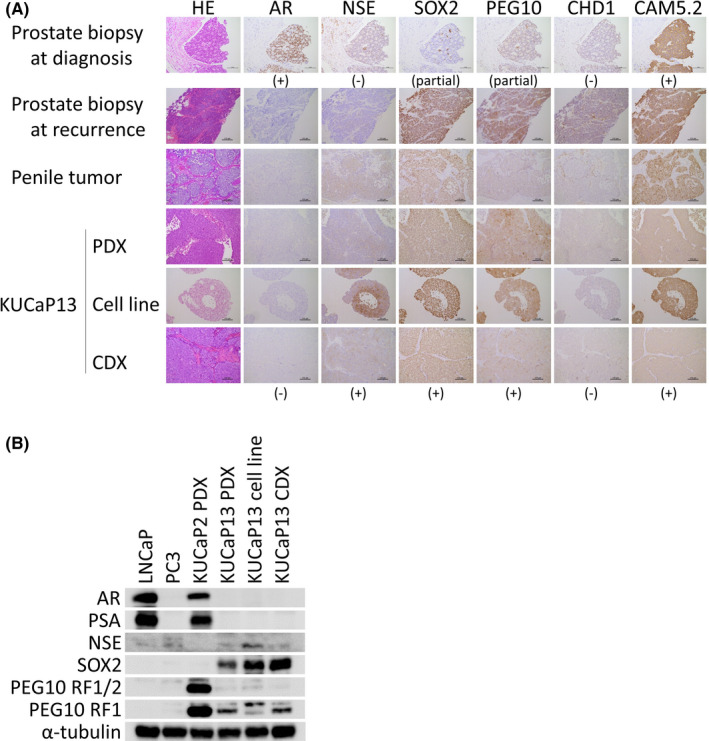
A, Immunohistochemistry of KUCaP13. Prostate biopsy at diagnosis showed typical adenocarcinoma with AR expression, while the patient tumors at recurrence as well as KUCaP13 PDX and cells (cell line and cell line–derived xenograft [CDX]) showed negative expression of androgen receptor (AR) and expression of NE‐associated genes (NSE, SOX2, PEG10). Note that CHD1 expression is already absent at initial diagnostic biopsy. The CAM5.2 cytokeratin was positive at diagnosis and remained positive throughout the transdifferentiation. B, Western blotting of AR pathway– and NE‐associated genes in KUCaP13. KUCaP2, a prostate adenocarcinoma PDX established at our laboratory, is included as a reference
3.5. Feasibility of gene engineering using KUCaP13
We tested the feasibility of gene engineering using two approaches to evaluate the utility of the KUCaP13 cell line as a research tool. First, we evaluated whether gene knockdown using shRNA is possible. We knocked down PEG10, a gene we have previously reported to be important in promoting the growth and invasion of NEPC. 14 PEG10 was successfully knocked down by lentiviral transduction using shRNA (Figure 5A,B). Interestingly, in contrast to our previous report using the PC3 and DU145 cell lines, 14 PEG10 knockdown did not suppress the growth of KUCaP13 in vitro (Figure 5C). However, in vivo growth was profoundly suppressed by PEG10 knockdown in KUCaP13 (Figure 5D,E). CDX tumors from KUCaP13 shPEG10 and KUCaP13 control mice could easily be regrown in vitro. Again, there was no difference in growth in vitro (Figure S3), confirming that PEG10 exerted its growth‐promoting effect only in vivo in the t‐NEPC–derived model.
FIGURE 5.
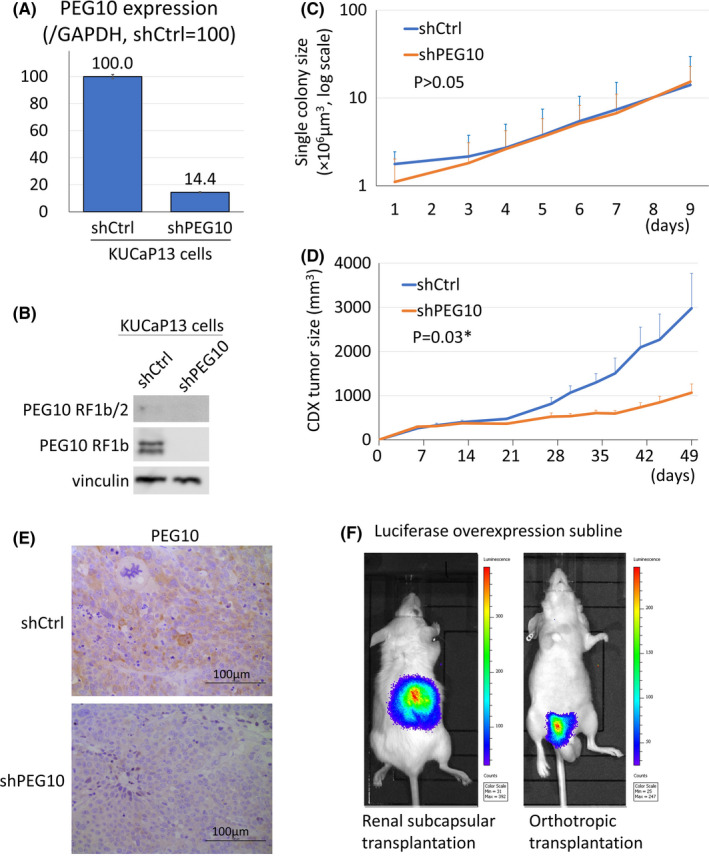
A, PEG10 mRNA expression upon knockdown in the KUCaP13 cell line. PEG10 expression was normalized to human glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). Gene expression is shown relative to shCtrl_KUCaP13. B, PEG10 protein expression upon knockdown in the KUCaP13 cell line. Both long (RF1b/2) and short (RF1b) isoforms of PEG10 are shown. C, In vitro growth assay upon PEG10 knockdown in the KUCaP13 cell line. Colony size was measured in septuplicate. The error bars show standard deviation. Y‐axis shows average colony size in logarithmic scale. Doubling time of shCtrl and shPEG10 KUCaP13 cells were similar (2.17 and 1.99 days, respectively, P > .05). D, In vivo growth of PEG10 knockdown KUCaP13. The average tumor size of four mice for each group is shown. Error bars show standard error. There was a significant difference in doubling time between shCtrl and shPEG10 KUCaP13 CDX (11.5 and 25.6 days, respectively, P = .03). E, PEG10 expression in CDX tumor tissue. PEG10 expression was decreased in shPEG10 KUCaP13 CDX compared with shCtrl KUCaP13 CDX. F, Detection of KUCaP13 transduced with luciferase in vivo. KUCaP13 implanted both in the renal subcapsule and orthotopically in the prostate is evaluable with luminescence
Next, to assess the feasibility of gene introduction into KUCaP13, firefly luciferase was successfully transduced using lentivirus. We confirmed that KUCaP13 could also grow in the subrenal capsule or orthotopically in the mouse prostate (Figure 5F). We also examined whether KUCaP13 could be used as a model of metastasis by intracardiac injection of the luciferase‐expressing KUCaP13. No metastasis was observed after 3 months (data not shown).
3.6. Utility of KUCaP13 as an in vitro preclinical model to test drugs
We next tested whether KUCaP13 could be used as an in vitro preclinical model to test drug efficacy in vitro. Equal numbers of colonies were dispensed into 96‐well plates. A dilution series of cisplatin was added to the wells. After a 48‐hour incubation, cell viability was analyzed using the CellTiter‐Glo 3D assay. We examined the cell viability in decaplicate samples for each dilution. A clear dose‐response curve with reasonable standard deviations was obtained (Figure 6A, Figure S4). Thus, we confirmed that KUCaP13 is a useful tool to evaluate the effectiveness of the candidate compound. Finally, given the controversy over whether neuroendocrine differentiation is reversible with EZH2 inhibitors, we tested the effect of the EZH2 inhibitor EPZ6438 (Tazemetostat) in KUCaP13. Although H3K27me significantly decreased with administration of EPZ6438 at concentrations ≥0.1 µmol/L, there was no change in the expression of AR or SOX2, and no difference in growth (Figure 6B,C). These findings suggested that EZH2 inhibition alone was not sufficient to reverse the lineage in this model.
FIGURE 6.
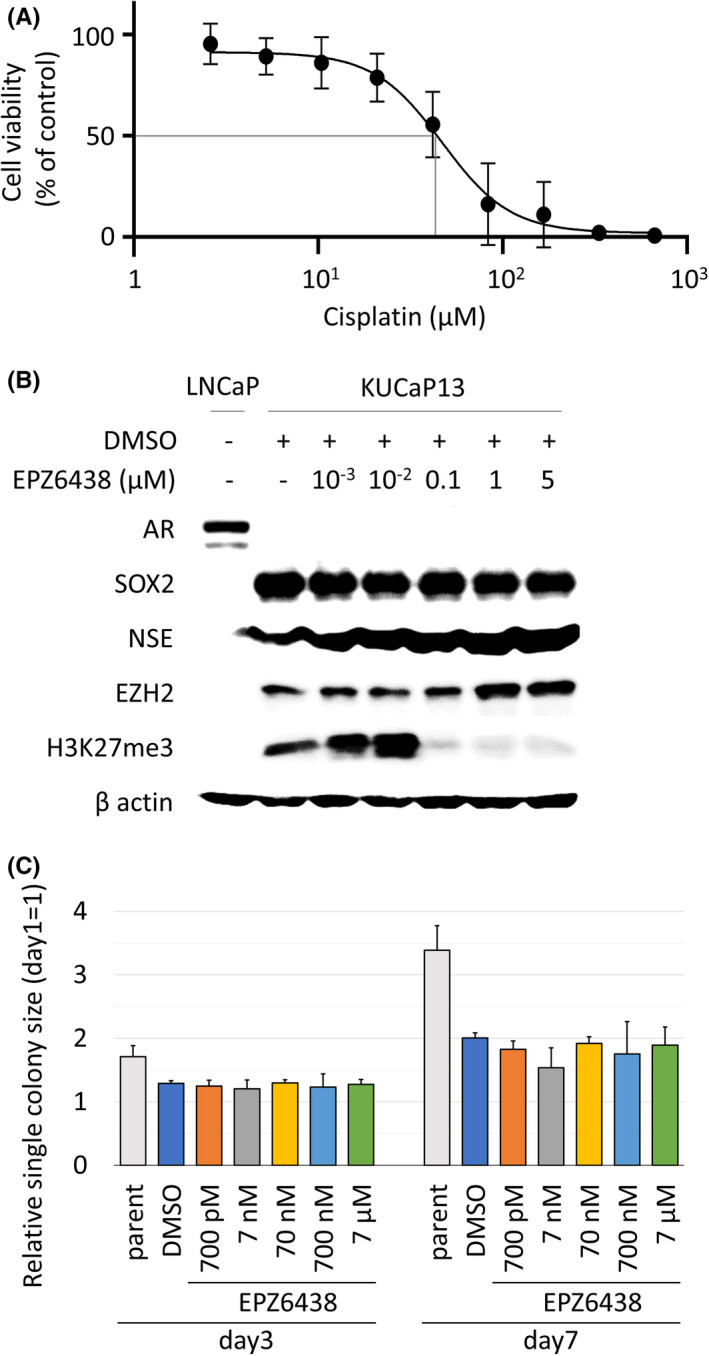
A, Dose‐response curve of KUCaP13 treated with cisplatin. IC50 was 45.4 µmol/L. B, Protein expression of the KUCaP13 cell line upon treatment with EZH inhibitor EPZ6438 (Tazemetostat). The target effect of EPZ6438 is evaluated by H3K27me3 expression. Even though EPZ6438 was active at ≥0.1 µmol/L, there was no change in androgen receptor (AR), SOX2, and NSE expression. DMSO, dimethyl sulfoxide. C, In vitro growth of the KUCaP13 cell line upon treatment with EZH2 inhibitor EPZ6438 (Tazemetostat). There was no dose‐dependent decline in growth (P > .05 by one‐way ANOVA test)
4. DISCUSSION
The biology of NEPC has been poorly understood until recently due to the rarity of the disease and lack of pertinent study models. However, recent advances in genomic sequencing have revealed some of the key features of NEPC. Genomic analysis of t‐NEPC tissues revealed that aberrations in the p53 and Rb pathways are critically important for the development of t‐NEPC. 16 , 17 , 18 Another pathway that may independently lead to t‐NEPC development is the amplification of MYCN and AURKA. 19 Subsequent in vivo and in vitro studies that reproduced these genetic aberrations in mice and cell lines suggest that upon potent AR pathway inhibition, adenocarcinoma transdifferentiates to completely change its phenotype and becomes t‐NEPC. 5 , 6 , 20 This process is considered lineage plasticity and may be driven by epigenetic factors that include SOX2 and EZH2, which are upregulated during the transdifferentiation process.
Researchers have long attempted to replicate the transdifferentiation process using cell lines. Many earlier studies focused on placing cellular stress on LNCaP adenocarcinoma models. LNCaP morphologically changes to a neuron‐like appearance with some expression of neuroendocrine (NE) markers when cultured in an androgen‐deprived medium. Other conditions induce similar changes in LNCaP. These include treatments with cyclic adenosine monophosphate, cytokines, and growth factors. As various stimuli can induce similar NE‐like phenotypes, it is possible that the NE phenotype is a default state for LNCaP cells under cellular stress. 21 However, a critical difference between clinical t‐NEPC and NE‐like LNCaP cells is that the latter are generally quiescent and nonproliferative. Therefore, it is impossible to identify a therapeutic target for t‐NEPC in these models. The most successful LNCaP model of t‐NEPC was established based on the results of recent genomic studies. LNCaP cells with double or triple knockdown of major tumor suppressors TP53, RB1, and PTEN were able to reproduce lineage plasticity and transdifferentiate to t‐NEPC with aggressive growth. 6 The important role of EZH2 and SOX2 was clarified by the model, and EZH2 is now a focus of new therapeutic targets for NEPC. 22
Despite these advances, cell models to study the biology of t‐NEPCs are still scarce. In addition to engineered models, models directly originating from clinical samples are valuable, as there is a large heterogeneity among t‐NEPCs that cannot be fully captured by gene engineered cell models. The only widely used cell line established from a patient with NEPC is NCI‐H660. NCI‐H660 was initially established from a lymph node metastasis at autopsy in a patient with extrapulmonary small cell lung cancer. The cell line was later found to be of prostatic origin due to the presence of TMPRSS‐ERG fusion. 23 However, NCI‐H660, whose primary tumor was not treated with androgen deprivation therapy for prostate cancer, is not a genuine t‐NEPC model. 24 In contrast, KUCaP13 was established from a patient tumor that transdifferentiated into t‐NEPC after androgen deprivation, and the clinical course of the index patient is well known. We confirmed the homozygous deletion of CHD1, a well‐known genomic signature specific to prostate cancer, in the patient tissue as well as in PDX and cell line of KUCaP13, confirming its origin as prostate adenocarcinoma. At the genomic level, KUCaP13 harbored PTEN (H61R) and TP53 (R77fs) pathogenic mutations with loss of the other allele for both genes, exhibiting LOH. Experimentally, the loss of CHD1 is synthetically lethal in PTEN‐deficient prostate cancer. 25 , 26 However, studies of human prostate cancers have shown cases of combined CHD1 and PTEN protein loss, suggesting that certain genomic backgrounds tolerate the dual loss of these genes. 27 As only a small archival formalin‐fixed tissue of prostate biopsy was available for the primary tumor in our study, we could not examine the presence of the PTEN mutation in the primary tumor. Therefore, it was not possible to determine whether the LOH of PTEN was already present in the primary adenocarcinoma, or if it occurred at a later stage, closer to the time of transdifferentiation to NEPC when the cell lineage was altered in KUCaP13. Although only monoallelic loss was present and LOH was not confirmed for RB1, transcriptomic analysis revealed a significant aberration in the Rb pathway genes and KUCaP13 clustered together with other NEPC PDXs with known Rb pathway aberrations. These results show that KUCaP13 is the first patient‐derived model of NEPC with dysfunctional p53, PTEN, and Rb1 pathways. A recent study reported a circulating tumor cell (CTC)‐derived eXplant and a cell line from a CRPC patient resistant to enzalutamide. Although the patient tumor showed adenocarcinoma, the established eXplant from CTC showed an AR‐null, neuroendocrine phenotype with TP53, PTEN, and RB1 loss, again highlighting the importance of the loss of these tumor suppressors in cells with the NEPC phenotype. 28
Although gene expression analysis demonstrated that KUCaP13 clustered together with other well‐characterized PDXs of t‐NEPC, KUCaP13 was unique in that it did not overexpress typical neuronal genes, such as CHGA and SYP. In addition, REST, one of the master regulators of neuronal gene expression, 29 was not altered either. However, KUCaP13 exhibited high expression of genes associated with lineage plasticity and aggressive growth, such as SOX2, EZH2, AURKA, PEG10, POU3F2 (encoding BRN2), 30 and FOXA2, and showed very rapid growth both in vitro and in vivo. Of the well‐known NEPC markers, ENO2 (encoding NSE) was also elevated. These expression patterns indicate that genetic programs that promote NE marker expression and aggressive growth diverge at some point during transdifferentiation, and each of these programs may contribute to the heterogeneity observed in clinical t‐NEPC.
Organoids are emerging as a valuable in vitro model derived from patient tissues to study the biology of t‐NEPC. In a recent report, a series of t‐NEPC organoids were developed and used to study the biological role of EZH2 in t‐NEPC. 31 EZH2 inhibition by shRNA or the inhibitor drug GSK503 suppressed EZH2 target genes and downregulated stem cell and neuronal programs. However, re‐expression of AR was not observed. Lack of re‐expression of AR upon EZH2 inhibition was compatible with our data using KUCaP13. However, it contrasts to an earlier report using LNCaP with TP53 and RB1 knockdown, 6 possibly due to an earlier more “plastic” disease state in the gene‐engineered LNCaP model. One of the advantages of the regular 2D cell culture over organoids is easier handling and lower maintenance costs. While multiple expensive growth factors are necessary for organoid culture, KUCaP13 can be maintained in regular growth medium. In addition, the results of chemical screening may be modulated in organoids by additional growth factors that are generally not required for regular cell culture. Therefore, a large‐scale, high‐throughput chemical screening is easier with KUCaP13 than with organoids.
We confirmed that gene knockdown using shRNA was possible using KUCaP13. PEG10 is a placental gene that was previously associated with the growth and invasion of NEPC. 14 In the original study, we used DU145 and PC3 cell lines as models of NEPC. PEG10 knockdown suppressed both in vitro and in vivo growth. In contrast, in the present study, PEG10 knockdown in KUCaP13 did not affect cell growth in vitro. However, the knockdown significantly suppressed in vivo tumor growth. This discrepancy in phenotype observed between different cell lines highlights the importance of using pertinent disease models when studying the role of a gene in a certain disease state, and emphasizes the value of KUCaP13 in studying the biology of t‐NEPC. We also attempted to knock down SOX2 by shRNA in KUCaP13 to evaluate its significance in t‐NEPC. However, SOX2 knockdown cells did not survive to propagate (data not shown), suggesting that SOX2 may be crucial for the survival of t‐NEPCs. Another interpretation is that because SOX2 is also associated with stemness, it is possible that SOX2 knockdown specifically affected the anchorage‐independent growth of KUCaP13 in vitro. Further studies are needed to elucidate the functional significance of SOX2 in t‐NEPC biology.
Similar to most small cell lung cancer cell lines and NCI‐H660, KUCaP13 grows as floating cells in culture dishes. Therefore, it is important to establish a method to measure tumor growth in vitro. We confirmed that measurement of colony size is a valid method to evaluate tumor growth. However, manual measurement of colony size is not suitable for high‐throughput screening. In regular cell viability assays, the measurement of spheroid‐forming cells is generally not possible. Therefore, we used the CellTiter‐Glo 3D®️ assay. This is a very sensitive assay that allows measurement of ATP in spheroid‐forming cells and organoids. We confirmed that the cytotoxicity of the drugs could be accurately measured using this assay in KUCaP13. The method can be applied in the future for chemical screening of compounds to target t‐NEPC.
Finally, we transduced KUCaP13 with luciferase, which allowed in vivo quantification of tumor growth (KUCaP13_luc). One of the disadvantages of PDX models is that the tumor volume must be measured manually. Therefore, it is difficult to precisely evaluate tumors implanted into the subrenal capsule until the tumor grows to a certain extent, and it is impossible to measure tumor growth serially in an orthotopic model. On the other hand, KUCaP13_luc grows both in the subrenal capsule and orthotopically in the prostate, and its growth can be monitored serially. As we can easily convert a PDX to a cell line and create a mouse model (CDX) in the KUCaP13 models, it is now possible to knock down or overexpress a gene in the model and study the function of the gene in a subrenal or orthotopic model, which may contribute to a broader understanding of t‐NEPC biology. We also evaluated whether KUCaP13_luc could be used as a metastasis model by intracardiac injection of the cells. However, no metastasis was observed.
5. CONCLUSIONS
We established a novel t‐NEPC cell line, KUCaP13, from a PDX and confirmed its origin as prostate adenocarcinoma. Genomic and transcriptomic studies confirmed its characteristics as NEPC with aberrations in the PTEN, p53, and Rb pathways. The feasibility of gene engineering using lentivirus and bidirectional conversion between in vivo and in vitro models was confirmed. KUCaP13 could be a valuable model to study the biology of t‐NEPC and to develop novel therapeutics, including its use in high‐throughput compound screening.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Figure S1‐S4
Document S1
ACKNOWLEDGMENTS
We thank Ms Junko Hirao and Ms Eriko Komaki for their technical assistance, and Dr Yukihiro Nagatani at Hikone Municipal Hospital, Shiga, Japan, for preparing archival tissue samples at the initial diagnosis of the index patient. The experiments with IVIS were conducted through the Joint Usage/Research Center Program of the Radiation Biology Center, Kyoto University. This study was supported by Grants‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science and by a research grant from the Japanese Foundation for Prostate Research.
Okasho K, Mizuno K, Fukui T, et al. Establishment and characterization of a novel treatment‐related neuroendocrine prostate cancer cell line KUCaP13. Cancer Sci. 2021;112:2781–2791. 10.1111/cas.14935
REFERENCES
- 1. Akamatsu S, Inoue T, Ogawa O, Gleave ME. Clinical and molecular features of treatment‐related neuroendocrine prostate cancer. Int J Urol. 2018;25(4):345‐351. 10.1111/iju.13526 [DOI] [PubMed] [Google Scholar]
- 2. Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. 10.3389/fonc.2014.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu Y, Ippolito JE, Garabedian EM, Humphrey PA, Gordon JI. Molecular characterization of a metastatic neuroendocrine cell cancer arising in the prostates of transgenic mice. J Biol Chem. 2002;277(46):44462‐44474. 10.1074/jbc.M205784200 [DOI] [PubMed] [Google Scholar]
- 4. Lotan TL, Gupta NS, Wang W, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24(6):820‐828. 10.1038/modpathol.2011.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53‐ and RB1‐deficient prostate cancer. Science. 2017;355(6320):84‐88. 10.1126/science.aah4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78‐83. 10.1126/science.aah4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zou M, Toivanen R, Mitrofanova A, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration‐resistant prostate cancer. Cancer Discov. 2017;7(7):736‐749. 10.1158/2159-8290.CD-16-1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aggarwal R, Huang J, Alumkal JJ, et al. Clinical and genomic characterization of treatment‐emergent small‐cell neuroendocrine prostate cancer: a multi‐institutional prospective study. J Clin Oncol. 2018;36(24):2492‐2503. 10.1200/JCO.2017.77.6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida T, Kinoshita H, Segawa T, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen‐dependent prostate cancer xenograft model derived from a bicalutamide‐treated patient. Cancer Res. 2005;65(21):9611‐9616. 10.1158/0008-5472.CAN-05-0817 [DOI] [PubMed] [Google Scholar]
- 10. Terada N, Shimizu Y, Kamba T, et al. Identification of EP4 as a potential target for the treatment of castration‐resistant prostate cancer using a novel xenograft model. Cancer Res. 2010;70(4):1606‐1615. 10.1158/0008-5472.CAN-09-2984 [DOI] [PubMed] [Google Scholar]
- 11. Yoshikawa T, Kobori G, Goto T, et al. An original patient‐derived xenograft of prostate cancer with cyst formation. Prostate. 2016;76(11):994‐1003. 10.1002/pros.23188 [DOI] [PubMed] [Google Scholar]
- 12. Augello MA, Liu D, Deonarine LD, et al. CHD1 loss alters AR binding at lineage‐specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell. 2019;35(4):603‐617.e8. 10.1016/j.ccell.2019.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin D, Wyatt AW, Xue H, et al. High fidelity patient‐derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272‐1283. 10.1158/0008-5472.CAN-13-2921-T [DOI] [PubMed] [Google Scholar]
- 14. Akamatsu S, Wyatt AW, Lin D, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12(6):922‐936. 10.1016/j.celrep.2015.07.012 [DOI] [PubMed] [Google Scholar]
- 15. Ertel A, Dean JL, Rui H, et al. RB‐pathway disruption in breast cancer: differential association with disease subtypes, disease‐specific prognosis and therapeutic response. Cell Cycle. 2010;9(20):4153‐4163. 10.4161/cc.9.20.13454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tan HL, Sood A, Rahimi HA, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20(4):890‐903. 10.1158/1078-0432.CCR-13-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conteduca V, Oromendia C, Eng KW, et al. Clinical features of neuroendocrine prostate cancer. Eur J Cancer. 2019;121:7‐18. 10.1016/j.ejca.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aparicio AM, Shen L, Tapia ELN, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22(6):1520‐1530. 10.1158/1078-0432.CCR-15-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beltran H, Rickman DS, Park K, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487‐495. 10.1158/2159-8290.CD-11-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dardenne E, Beltran H, Benelli M, et al. N‐Myc induces an EZH2‐mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30(4):563‐577. 10.1016/j.ccell.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang YH, Zhang YQ, Huang JT. Neuroendocrine cells of prostate cancer: biologic functions and molecular mechanisms. Asian J Androl. 2019;21(3):291‐295. 10.4103/aja.aja_128_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies A, Zoubeidi A, Selth LA. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat Cancer. 2020;27(2):R35‐R50. 10.1530/ERC-19-0420 [DOI] [PubMed] [Google Scholar]
- 23. Mertz KD, Setlur SR, Dhanasekaran SM, et al. Molecular characterization of TMPRSS2‐ERG gene fusion in the NCI‐H660 prostate cancer cell line: a new perspective for an old model. Neoplasia. 2007;9(3):200‐206. 10.1593/neo.07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carney DN, Gazdar AF, Bepler G, et al. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985;45(6):2913‐2923. [PubMed] [Google Scholar]
- 25. Zhao D, Lu X, Wang G, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN‐deficient cancer. Nature. 2017;542(7642):484‐488. 10.1038/nature21357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rahmy S, Cheng X, Wang M, et al. Organ‐specific regulation of CHD1 by acute PTEN and p53 loss in mice. Biochem Biophys Res Commun. 2020;525(3):614‐619. 10.1016/j.bbrc.2020.02.136 [DOI] [PubMed] [Google Scholar]
- 27. Boysen G, Rodrigues DN, Rescigno P, et al. SPOP‐mutated/CHD1‐deleted lethal prostate cancer and abiraterone sensitivity. Clin Cancer Res. 2018;24(22):5585‐5593. 10.1158/1078-0432.CCR-18-0937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faugeroux V, Pailler E, Oulhen M, et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell‐derived eXplant model. Nat Commun. 2020;11(1):1884. 10.1038/s41467-020-15426-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Coleman IM, Brown LG, et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration‐resistant prostate cancer. Clin Cancer Res. 2015;21(20):4698‐4708. 10.1158/1078-0432.CCR-15-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bishop JL, Thaper D, Vahid S, et al. The master neural transcription factor BRN2 is an androgen receptor‐suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017;7(1):54‐71. 10.1158/2159-8290.CD-15-1263 [DOI] [PubMed] [Google Scholar]
- 31. Puca L, Bareja R, Prandi D, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun. 2018;9(1):2404. 10.1038/s41467-018-04495-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S4
Document S1