

Abstract
Ten‐eleven translocation 1 (TET1) is an essential methylcytosine dioxygenase of the DNA demethylation pathway. Despite its dysregulation being known to occur in human cancer, the role of TET1 remains poorly understood. In this study, we report that TET1 promotes cell growth in human liver cancer. The transcriptome analysis of 68 clinical liver samples revealed a subgroup of TET1‐upregulated hepatocellular carcinoma (HCC), demonstrating hepatoblast‐like gene expression signatures. We performed comprehensive cytosine methylation and hydroxymethylation (5‐hmC) profiling and found that 5‐hmC was aberrantly deposited preferentially in active enhancers. TET1 knockdown in hepatoma cell lines decreased hmC deposition with cell growth suppression. HMGA2 was highly expressed in a TET1high subgroup of HCC, associated with the hyperhydroxymethylation of its intronic region, marked as histone H3K4–monomethylated, where the H3K27‐acetylated active enhancer chromatin state induced interactions with its promoter. Collectively, our findings point to a novel type of epigenetic dysregulation, methylcytosine dioxygenase TET1, which promotes cell proliferation via the ectopic enhancer of its oncogenic targets, HMGA2, in hepatoblast‐like HCC.
Keywords: enhancer, epigenome profiling, hepatocellular carcinoma, hydroxymethylation, TET1
Ten‐eleven translocation 1 (TET1), a methylcytosine dioxygenase of the DNA demethylation pathway, is overexpressed in the subgroup of hepatocellular carcinoma with the hepatoblast‐like expression pattern. Comprehensive epigenome profilings revealed the ectopic enhancer activation of HMGA2 through cytosine hydroxymethylation, H3K27 acetylation, H3K4 monomethylation, and promoter‐enhancer interaction in a TET1‐dependent manner.
1. INTRODUCTION
Epigenomic aberration is one of the hallmarks of cancer. 1 , 2 , 3 The global hypomethylation and promoter hypermethylation of cancer‐related genes are frequently observed in human cancer cells. Genome‐wide alterations of histone modifications also result in the disruption of gene expression networks. 4 , 5 Recently, comprehensive analyses using genomic technologies have revealed altered epigenetic regulators, including DNA modifying enzymes, 4 histone modifiers, 5 and chromatin remodelers. 6 , 7 The ectopic activation of epigenetic regulators can promote oncogenic phenotypes and play essential roles during carcinogenesis. Therefore, it is thought to be a potential target for anticancer therapeutics. 8 , 9
The ten‐eleven translocation (TET) family of proteins, TET1, TET2, and TET3, encode 5‐methylcytosine (5‐mC) dioxygenases that convert 5‐mC to 5‐hydroxymethylcytosine (5‐hmC). 10 , 11 The enzymatic role of TET proteins of the mammalian DNA demethylation pathway suggests the involvement of epigenetic dysregulation in various human cancers. 12 , 13 , 14 , 15 , 16 Recent years have seen an increase in research conducted on TET1, especially with regard to its role in stem cell maintenance. 7 , 17 , 18 , 19 , 20 , 21 However, the role of aberrantly expressed TET1 in human cancers remains poorly understood. 22
In this study, we focused on the upregulation of TET1 in a subgroup of hepatocellular carcinoma (HCC). A functional assay using liver cancer cell lines revealed that the overexpression of TET1 promotes hepatoma cell proliferation. We performed a genome‐wide mapping of 5‐mC and 5‐hmC for TET1‐upregulated HCC and found that 5‐hmC was significantly enriched in active enhancer regions. The methylation status of this regulatory region changes dynamically during hepatic differentiation. An oncogenic effector gene, HMGA2, which is epigenetically upregulated by ectopic TET1 expression, was identified.
2. MATERIALS AND METHODS
2.1. Cell lines
The liver cancer cell lines HepG2 and Huh7 were obtained from the Cell Resource Center for Biomedical Research at Tohoku University and the Japanese Collection of Research Bioresources (Osaka, Japan), respectively. The cells were cultured in Dulbecco's modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin.
2.2. Clinical samples
HCC patients undergoing hepatectomy in the Hepato‐Biliary‐Pancreatic Surgery Division, Department of Surgery, Graduate School of Medicine, University of Tokyo were included in this study after providing informed consent. The surgical specimens were immediately cut into small pieces after resection, snap frozen in liquid nitrogen, and stored at ‒80°C.
2.3. RT‐qPCR
Total RNAs from cell lines and clinical tissue samples were isolated using TRIzol reagent (Invitrogen, #15596018) according to the manufacturer's instructions. One microgram of RNA was used for the generation of double‐stranded cDNA with the SuperScript Double‐Stranded cDNA Synthesis Kit (Invitrogen, #1197010) and analyzed by real‐time PCR using SYBR Green. The expression levels of the examined genes were normalized to β‐actin expression. The PCR conditions for each cycle were as follows: denaturation at 95°C for 15 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 20 seconds. The sequences of primers for TET1, HMGA2, and β‐actin are listed in Table S1.
2.4. Gene expression microarray
The genome‐wide analysis of the mRNA expression levels using U133 plus 2.0 human expression array (Affymetrix) was performed as described previously. 4 Briefly, 1 µg of RNA was used for the generation of double‐stranded cDNA with the SuperScript Double‐Stranded cDNA Synthesis Kit (Invitrogen) according to the manufacturer's protocol.
2.5. Immuno‐dot blot assay
The genomic DNA of HepG2 or Huh7 cells was isolated by phenol/chloroform treatment and precipitated with 70% ethanol. The genomic DNA of the clinical tissue samples was isolated using the QIAamp DNA Mini kit (QIAGEN, #51304). All genomic DNA was denatured at 95°C for 5 minutes in the 5‐hmC assay. The DNA samples were spotted onto a Hybond‐N+ nylon membrane (GE Healthcare, #RPN1210B). The membranes were then cross‐linked with UV at 0.18 J/cm2 and blocked with 5% nonfat milk in Tris‐buffered saline containing 0.1% Tween‐20 (TBS‐T), followed by incubation with the antibodies against 5‐mC (1:2000, Diagenode, #MAb‐081‐100) and 5‐hmC (1:2000, Active Motif, #39769) at 4°C overnight. The membranes were then washed and probed with an appropriate HRP‐conjugated secondary antibody (anti‐mouse IgG, GE Healthcare, #NA9310V, or anti‐rabbit IgG, GE Healthcare, NA9340V) and detected with the ECL Plus chemi‐luminescense assay kit (Thermo Scientific, #1896426 A and B). To measure the relative amount of each sample, the same blot was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2). Dot blot intensity was quantified by Multi Gauge software (version 3.0).
2.6. Knockdown assay
For the knockdown of endogenous TET1 or HMGA2, cells were transfected with 20 nM of specific small interfering RNAs (siRNAs) (Invitrogen) using Lipofectamine 2000 (Life Technologies, #11668‐019). The sense sequences of the TET1 and HMGA2 siRNAs are listed in Table S2. Stealth RNAi™ siRNA negative control med GC duplex 2 (Invitrogen, #12935‐112) was used as the control siRNA.
2.7. Western blotting analysis
HepG2 or Huh7 cells were washed twice with ice‐cold PBS and ruptured with RIPA buffer containing protease inhibitor (Roche, #1697498). Cell lysates were resolved by SDS‐polyacrylamide gel electrophoresis (PAGE) and then transferred onto PVDF membranes. The membranes were blocked for 1 hour with 5% non‐fat milk in TBS‐T and incubated with anti‐HaloTag antibody (Promega, #G928A), anti‐HMGA2 antibody (Active Motif, #61041), anti‐V5 antibody (Invitrogen, #1305726), anti‐nucleoporin antibody (BD Biosciences, #610498), or anti‐β‐actin antibody (Sigma, #A5441) at 4°C overnight. Membranes were washed for 20 minutes with TBS‐T, probed with appropriate HRP‐conjugated secondary antibodies, and detected with the ECL Plus chemi‐luminescence assay kit (GE Healthcare).
2.8. Growth assay
The control and transfected liver cancer cells were seeded at a density of 3 or 4 × 103 cells/well in 96‐well plates, respectively. Viable cells were counted using a cell counting kit‐8 (Dojindo, #CK04) at 1, 3, 5, and 7 days after transfection.
2.9. Overexpression assay
For the overexpression of TET1, the cells were transfected with HaloTag‐hTET1 (Promega), which contains full‐length human TET1. HaloTag control vector (Promega, #G6591) was used as a negative control. For the overexpression of HMGA2, cells were infected with pLenti6.3/V5‐hHMGA2, which contains full‐length human HMGA2. Full‐length of hHMGA2 was amplified by PCR and subcloned into a pLenti6.3/V5‐DEST lentiviral vector (Invitrogen). To package the lentivirus, 293T cells were cotransfected with the packaging plasmid, psPAX2 and pMD2.G, and a pLenti6.3/V5‐DEST plasmid containing the hHMGA or LacZ. pLenti6.3/V5‐LacZ was used as a negative control. All constructs were verified by Sanger sequencing.
2.10. Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation using anti‐H3K4 trimethylation (H3K4me3) rabbit polyclonal antibody (Active Motif, #39159) and mouse monoclonal antibodies against anti‐H3K4 monomethylation (H3K4me1) and anti‐H3K27 acetylation (H3K27ac) (kindly gifted by Dr Hiroshi Kimura, Osaka University 23 ) was performed as previously reported. 24 Briefly, HepG2 and Huh7 cells were cross‐linked with 1% formaldehyde for 10 minutes at room temperature, and the cross‐linked cell lysates underwent ultrasonic fragmentation and were incubated with antibodies bound to protein A‐ and G‐sepharose beads (GE Healthcare, #17127901 and #17061801) for H3K4me3 and magnet beads (Invitrogen, #112.02D) for H3K4me1 and H3K27ac at 4°C overnight. The beads were washed and eluted with elution buffer (0.5% SDS, 25 mM Tris‐HCl, 5 mM EDTA). The eluates were treated with 1.5 μg of pronase at 42°C for 2 hours and then incubated at 65°C overnight to reverse the cross‐links. The immunoprecipitated DNA was purified by the QIAquick PCR Purification Kit (QIAGEN, #28106).
2.11. Methylated and hydroxymethylated DNA immunoprecipitation (MeDIP and hmeDIP)
The genomic DNA of HepG2 or Huh7 cells was isolated by phenol/chloroform treatment and precipitated with 70% ethanol. The genomic DNA of clinical liver samples was isolated using the QIAamp DNA Mini kit (QIAGEN) according to the manufacturer's instructions. The genomic DNA was quantitated using the Qubit dsDNA HS kit (Invitrogen, #Q32854). Briefly, 20 μg (hmeDIP) or 2 μg (MeDIP) of genomic DNA from the cell lines and 5 μg of genomic DNA from the clinical samples were sonicated and denatured at 95°C for 5 minutes. Fragmented DNA was incubated with 4 μL of anti‐5‐mC antibody (Diagenode) or 4 μL anti‐5‐hmC antibody (Active Motif) at 4°C for 3 hours and bound to protein A‐ and G‐sepharose beads at 4°C for 1 hour. The beads were washed twice by wash buffer 1 (20 mM Tris‐HCl, 2 mM EDTA, 150 mM NaCl, 1% Triton‐X) and 2 (20 mM Tris‐HCl, 2 mM EDTA, 300 mM NaCl, 0.1% SDS, 1% Triton‐X) before eluting with the elution buffer (25 mM Tris‐HCl, 10 mM EDTA, 0.5% SDS, 100 mM NaCl). The eluates were treated with 100 μg of proteinase K at 55°C for 1 hour. The immunoprecipitated DNA was purified by phenol/chloroform treatment and precipitated with LiCl, glycogen, and 70% ethanol.
2.12. ChIP‐, MeDIP‐, and hmeDIP‐sequencing (ChIP‐, MeDIP‐ and hmeDIP‐seq)
Sequencing was conducted on an Illumina Genome Analyzer GAIIX using Cluster Generation (version 2 and 4) chemistries, as well as Sequencing by Synthesis Kits (version 3 and 4). Data collection was performed using Sequencing Control Software (version 2.5 and 2.6). Real‐Time Analysis (RTA) 1.5‐1.8 was used for base calling. Genomic mapping of short reads was performed using the sequence_pair mode of ELAND in the Illumina CASAVA pipeline (version 1.5‐1.8) (Table S3). Distribution of immunoprecipitated or enriched fragments was analyzed using model‐based analysis for ChIP‐seq (MACS). 25 Data were visualized using Integrative Genomics Viewer (IGV) (version 2.3.32).
2.13. Multiplex targeted sequencing of bisulfite (BS)‐treated amplicons
BS treatment of genomic DNA (200‐250 ng) was performed by the EZ DNA Methylation‐Gold Kit (ZYMO Research, #D5005) following the manufacturer's protocols. BS‐converted DNA was amplified with 16 primer sets for the HMGA2 enhancer region (Table S4) using KAPA HiFi HotStart Uracil + Ready Mix (KAPA Biosystems, #KK2801), and ligated with Illumina TruSeq adapters. The libraries of amplicons were generated by the Illumina TruSeq Nano DNA LT Sample Prep Kit (#15041757 and #15041759) according to the manufacturer's instructions, and quantified by the Agilent 2100 Bioanalyzer using High Sensitivity DNA chip. Multiplexed libraries were sequenced on the Illumina MiSeq, as 150‐bp paired‐end reads, following the manufacturer's recommendations. Sequenced reads were mapped to the top or bottom strand of the hg19 reference genome by Bismark 0.5.4 (Bowtie 0.12.2) using the default parameters. The uniquely mapped reads were processed to estimate the percentage of methylation. The methylation rate for each cytosine of the CG sites was estimated by dividing the number of C by the total number of C or T (read depth above 50).
2.14. Chromatin conformation capture (3C) assay
A total of 1.8 million of HepG2 and Huh7 cells were transfected with siCTL or siTET1. Preparation of 3C templates were following the previous report (Rao et al 26 ) except for skipping the step of DNA end‐repair and shearing. Briefly, 48 hours after transfection, cells were crosslinked by 1% formaldehyde for 10 minutes and quenched by 0.2 M glycine for 5 minutes. Cells were lysed in lysis buffer (10 mM Tris‐HCl pH8.0, 10 mM NaCl, 0.2% Igepal CA630) containing 1 × cOmplete, EDTA‐free (Roche #11873580001) on ice for 15 minutes; then, chromatins were digested with EcoRI (NEB #R0101) at 37°C overnight. DNA ends were ligated with T4 DNA Ligase (NEB #M0202) at 4 hours, followed by protein digestion with Proteinase K (NEB #P8107) and crosslink reversal. DNAs were purified by phenol‐chloroform extraction and ethanol precipitation; then, qPCR was performed. HepG2 and Huh7 cells with siCTL or siTET1 KD were used for 3C with the TaqMan 3C Chromosome Conformation Kits (Life Technologies, #4466151) according to the manufacturer's protocol. The sequences of the constant primer (forward) and the test primer (reverse) are listed in Table S5. A TaqMan probe was designed based on the negative strand DNA sequence located 23 bp upstream of the first EcoRI enzymatic digestion site. TaqMan quantitative real‐time PCR was performed with SsoAdvancedTM Universal Probes Supermix (Bio‐Rad, #172‐5280) according to the manufacturer's protocol, using the following cycling conditions: denaturation at 95°C for 10 seconds and annealing and extension at 60°C for 60 seconds. The PCR products were purified using a QIAGEN quick gel purification kit (QIAGEN, #28106), and the sequence of each chimeric DNA was verified by Sanger sequencing (Figure S6). To normalize primer efficiency, control PCR templates were generated by digestion and random ligation of bacterial artificial chromosomes (BAC) containing HMGA2 (Life Technologies, clone RP11‐462A13). A total of 10 μg of BAC clone was digested with EcoRI (Toyobo, #ECO‐153) and then ligated with T4 DNA ligase (NEB, #M0202S). The paired primers/probes designed for 3C‐qPCR assay were tested on the random ligation product, which contained all possible chimeric DNA ligation products in equal molar concentrations.
3. RESULTS
3.1. Clinicopathological features of TET1‐upregulated HCC
To elucidate the dysregulation of TET1 in the context of human liver cancer, we first examined its mRNA expression levels among clinical liver tissues using an expression microarray (U133 plus 2.0, Affymetrix). As shown in Figure 1A, TET1 was upregulated in 14 of 53 HCCs (26.9%) compared with noncancerous liver tissues. In general, TET1 is strongly expressed in human embryonic stem cells (hESC). In contrast, the expression level of TET1 in somatic tissues, including the adult liver, was lower, which is consistent with the results of murine somatic tissues. 17 Fetal liver and colon tissues showed relatively high levels of TET1 expression, as well as TET1‐upregulated HCCs. As for TET2 and TET3, there was no significant difference between HCCs and noncancerous livers (Figure S1). Interestingly, the aberrant expression patterns of TET1 mRNA were cancer type–dependent. Large‐scale transcriptome analyses (n = 4175, total) of The Cancer Genome Atlas (TCGA) research network are shown in Figure 1B. As previously reported by Hsu et al, 13 TET1 was relatively downregulated in breast cancers because the epithelium in normal breast maintains high levels of expression. However, a significant upregulation of TET1 was observed in lung cancers and head and neck squamous cell carcinomas, as well as HCCs.
FIGURE 1.
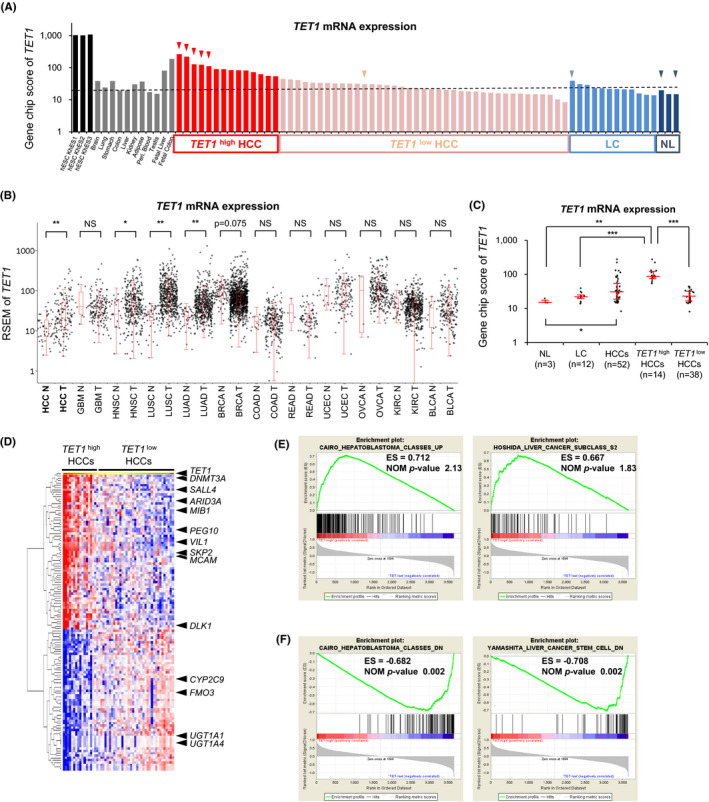
TET1 is upregulated in hepatoblast‐like HCC. A, TET1 expression among hESCs, somatic tissues, and HCCs in expression microarray (U133 plus 2.0, Affymetrix). TET1: gene name, CXXC6; probe name, 228906_at. HCC, hepatocellular carcinoma; hESC, human embryonic stem cell; LC, liver cirrhosis (corresponding noncancerous liver); NL, normal liver. B, TET1 expression among 12 types of cancer in RNA‐seq data of TCGA project. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; GBM, glioblastoma multiform; HCC, hepatocellular carcinoma; HNSC, head, and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; N, nontumor; OVCA, ovarian serous cystadenocarcinoma; READ, rectal adenocarcinoma; T, tumor; TCGA, The Cancer Genome Atlas; UCEC, uterine corpus endometrial carcinoma. The black dot represents the TET1 expression level (RSEM) of each case. Red line represents the median ± quartile. C, TET1 expression among clinical liver samples in expression microarray. Each dot represents the GeneChip score of TET1 (228906_at). Red line represents the median ± quartile. P‐values were measured using the Mann‐Whitney U‐test in (B) and (C). *P < .05; **P < .01; ***P < .001. D, Heatmap of the expression profiles using the probes which are highly correlated or inversely correlated with TET1 expression among clinical liver samples. E and F, Enrichment plot showing the enrichment score for the gene sets upregulated © and downregulated (F) in TET1 high HCCs
To clarify the relationship between the expression level of TET1 and the clinicopathological behaviors of HCCs, we investigated the clinical information of TET1‐upregulated cases (TET1 high HCCs; greater than twofold of noncancerous liver tissues, n = 14) and the other cases (TET1 low HCCs, n = 38) (Table 1). As shown in Table 1, TET1 high HCCs were characterized by higher serum alpha‐fetoprotein (AFP, P < .001), younger age (P = .003), and poorer pathological differentiation (P = .014) than TET1 low HCCs. No significant differences were observed with regard to gender, etiology, tumor size, or serum des‐gamma‐carboxy prothrombin (DCP) (Table 1 and Figure S2).
TABLE 1.
Clinicopathological characteristics of hepatocellular carcinoma (HCC) cases associated with TET1 expression
| Characteristic |
TET1 high HCCs (14/52) |
TET1 low HCCs (38/52) |
P‐value |
|---|---|---|---|
| Age, y | 61 (32, 76) a | 68 (48, 80) a | **.003 b |
| Sex | |||
| Male | 12 (86%) | 28 (74%) | **.344 c |
| Female | 2 (14%) | 10 (26%) | |
| Etiology | |||
| HBV | 5 (36%) | 12 (32%) | .779 c |
| HCV | 9 (64%) | 26 (68%) | |
| Tumor size, mm | 45 (20, 100) a | 30 (11, 120) a | **.056 b |
| Differentiation | |||
| Well | 0 (0%) | 9 (24%) | **.014 c |
| Mod | 10 (71%) | 25 (68%) | |
| Por | 4 (29%) | 3 (8%) | |
| AFP, ng/mL | 5770 (4, 145 972) a | 13 (3, 9700) a | ***<.001 b |
| DCP, mAU | 106 (12, 5354) a | 37 (10, 20 955) a | .358 b |
Abbreviations: AFP, alpha‐fetoprotein; DCP, des‐gamma‐carboxy prothrombin; HBV, Hepatitis B virus; HCV, Hepatitis C virus.
Median (minimum, max).
P‐value was measured by the Mann‐Whitney U test.
P‐value was measured by χ 2 test or Fisher's exact test.
P < .01.
P < .001.
To further evaluate the expression profiles of TET1 high HCCs (Figure 1C), we performed hierarchical clustering analysis using probes whose expression patterns among 53 HCCs were highly correlated (correlation coefficient ρ > .75) or inversely correlated (ρ < ‒0.5) with the expression levels of TET1. Epigenetic regulators, such as DNMT3A and ARID3A, and two imprinted genes, PEG10 and DLK1, were highly coexpressed with TET1 (Figure 1D). The oncofetal gene SALL4 27 was also upregulated in TET1 high HCCs. On the contrary, genes specific for matured hepatocytes (CYP2C9, FMO3, UGT1A1, and UGT1A4) were downregulated. Gene Set Enrichment Analysis (GSEA) indicated that TET1 high HCCs have hepatoblast‐like gene expression signatures. Cairo's gene sets, 28 representing overexpression in human hepatoblastoma (HBL), were found to overlap the most with the upregulated gene sets for TET1 high HCCs (Figure 1E). Hoshida's gene sets 29 for the G2 subclass, which is featured as hepatoblast‐like HCC, were highly ranked with a significant nominal P‐value (P = .015). The downregulated genes for TET1 high HCCs were well correlated with Cairo's gene sets downregulated in HBLs and with Yamashita's gene sets 30 downregulated in EpCAM‐positive liver cancer stem cells (Figure 1F). These results indicate that TET1 high HCCs may be derived from premature hepatic progenitor cells with high levels of TET1 expression.
3.2. Genome‐wide mapping of 5‐hmC in clinical HCC samples reveals that 5‐hmC is enriched at the transcriptional regulatory regions
The prevalence of TET1 overexpression in HCCs raises an intriguing possibility that dioxygenase TET1 may cause the aberrant hydroxymethylation of cytosine, which leads to malignant phenotypes via epigenetic disruption. To test this hypothesis, we first verified the mRNA levels of the TET family genes (Figure 2A, Figure S3A) and examined the total content of 5‐hmC and 5‐mC by immuno‐dot blot assay (Figure 2B,C). Consistent with the GSEA result of TET1 high HCCs, TET1 was highly expressed in the liver cancer cell line, HepG2 (Figure 2A), while 5‐hmC might be globally increased in TET1 high HCC patients (Figure 2B). Conversely, the total content of 5‐mC was decreased (Figure 2C).
FIGURE 2.
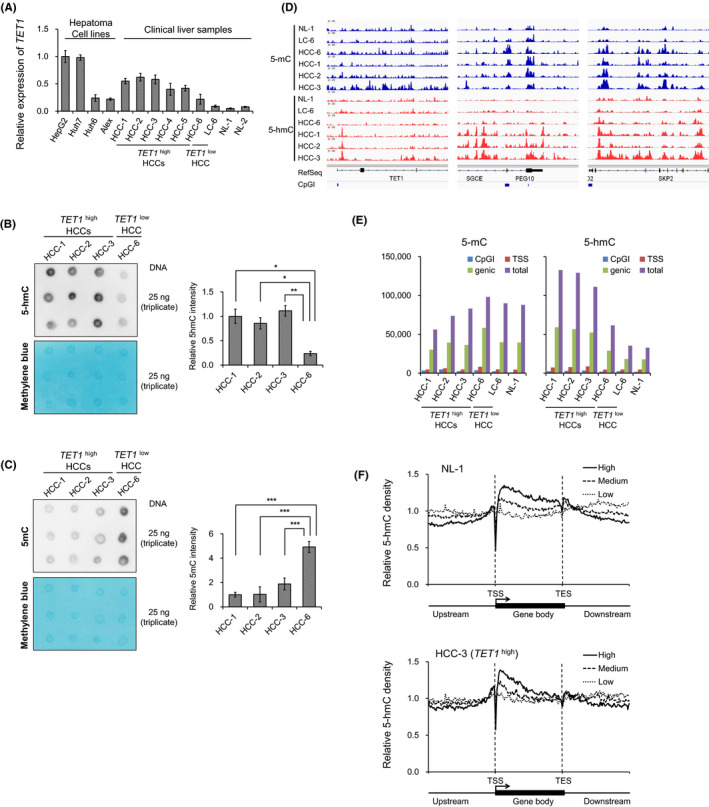
5‐mC and 5‐hmC profiles of clinical liver tissues. A, Relative TET1 mRNA expression of hepatoma cells and clinical liver samples by RT‐qPCR. B and C, Global 5‐hmC (B) and 5‐mC (C) levels in hepatocellular carcinomas (HCCs) quantified by immuno‐dot blot assay. Left, dot blot images of triplicate experiments. The methylene blue staining is used as a loading control for total genomic DNA. Right, quantified dot blot intensity by Multi Gauge (version 3.0) software compared with HCC‐1. Data are shown as the mean ± SD of triplicate experiments. P‐values are measured using Student's t‐test in (B) and (C). *P < .05; **P < .01; ***P < .001. D, Genome‐wide profiling of 5‐mC (upper) and 5‐hmC (lower) for clinical liver tissues (NL‐1 as a normal liver; LC‐6 as a noncancerous liver; HCC‐6 as a TET1low HCC; HCC‐1, ‐2, ‐3 as TET1high HCCs). Signal intensities are visualized using Integrated Genomics Viewer (IGV). E, The number of 5‐mC‐enriched regions (left) and 5‐hmC‐enriched regions (right) identified by MeDIP‐seq and hmeDIP‐seq, respectively. F, 5‐hmC distributions relative to human RefSeq gene position by hmeDIP‐seq according to the expression levels in clinical liver samples. TSS, transcription start site; TES, transcription end site. Expression level: high, GeneChip score >250; low, GeneChip score <25; medium, GeneChip score 25‐250
To dissect the genome‐wide distribution of 5‐hmC and 5‐mC in HCC, we performed a comprehensive profiling using a hmeDIP and MeDIP approach coupled with massively parallel sequencing (hmeDIP‐ and MeDIP‐seq), respectively (Figure 2D). At the TET1 gene locus, a prominent 5‐hmC peak downstream from the transcription start site (TSS) was observed in TET1 high HCCs, which is consistent with reports that human TET1 binds directly with its own promoter. 15 Imprinted gene PEG10 and cell cycle–related gene SKP2 were also aberrantly hydroxymethylated in a TET1 high HCC–specific manner. As shown in Figure 2E, the total numbers of 5‐hmC–marked regions were increased in TET1 high HCCs. Overall, 5‐mC and 5‐hmC were depleted at TSS regions but enriched on the gene‐body regions in clinical liver samples (Figure 2F). Consistent with previous findings in murine ESCs, 18 , 31 , 32 5‐hmC was preferentially enriched downstream of TSS (Figure 2F, Figure S3D). 5‐mC was widely distributed in the gene‐body region along the 3′ ends (Figure S3C). With regard to the gene expression statuses, 5‐hmC was more enriched downstream from the TSSs of highly expressed genes in clinical liver samples (Figure 2F). However, 5‐mC enrichment showed no difference according to the gene expression levels (Figure S3C). Taken together, 5‐hmC was enriched at the transcriptional regulatory region in TET1 high HCC.
3.3. TET1 knockdown inhibits proliferation of hepatoma cell lines
To evaluate the role of TET1 in liver cancer, we introduced siRNAs targeting TET1 into HepG2 and Huh7 cells (Figure 3A). These two cell lines showed high expression level of TET1 among the Broad Institute Cancer Cell Line Encyclopedia (Figure S4). The transient knockdown of TET1 resulted in a significant reduction of cell growth, indicating that TET1 promotes cell proliferation in liver cancer cells (Figure 3B). Immuno‐dot blot assay showed a total decrease of 5‐hmC after 48 hours of TET1 suppression (Figure 3C). Subsequently, we assessed the effect of exogenous overexpression via the HaloTag vector containing the full‐length human TET1 (Halo‐TET1). The induction efficiency of Halo‐TET1 and its enzymatic activity for methylcytosine oxidation were confirmed (Figure 3D,E). The overexpression of Halo‐TET1 rescued siRNA‐mediated growth inhibition in HepG2 cells (Figure 3F). Taken together, our data demonstrate that TET1 enhances cell proliferation in liver cancer cells, suggesting that TET1 functions as an oncogenic regulator in human HCC.
FIGURE 3.
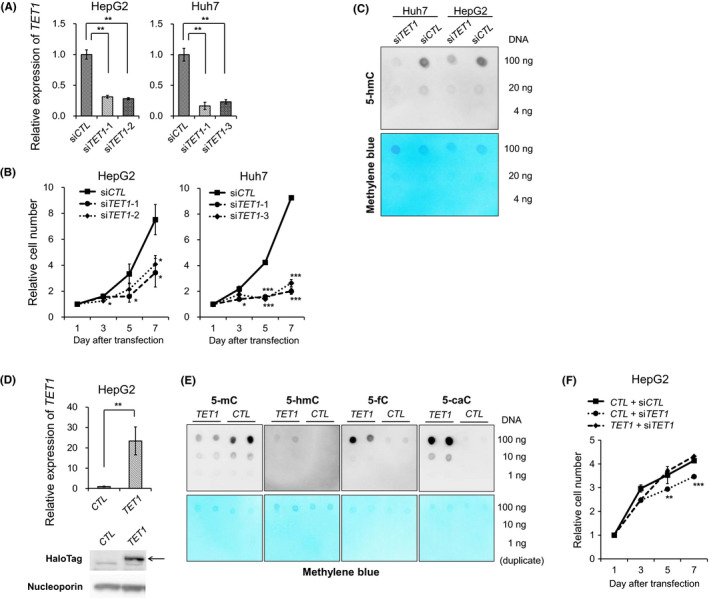
TET1 knockdown inhibits proliferation of hepatoma cell lines. A, Relative TET1 mRNA expression of hepatoma cells (HepG2 and Huh7) treated with control siRNA (siCTL) and TET1 siRNA (siTET1‐1, ‐2, and ‐3) by RT‐qPCR. B, The tumor proliferation curves of HepG2 and Huh7 cells treated with siCTL and siTET1 transfection. C, Global 5‐hmC levels in HepG2 and Huh7 cells treated with siCTL and siTET1 quantitated by immuno‐dot blot assay. The methylene blue staining was used as a loading control for total genomic DNA. D, Forced overexpression of full‐length TET1 in HepG2 using HaloTag vector. CTL, untreated; TET1, treated with the Halo‐TET1 vector. Upper, relative TET1 mRNA expressions by RT‐qPCR compared with cells with CTL. Lower, TET1 expression by Western blotting analysis using anti‐HaloTag antibody and anti‐Nucleoporin antibody. Arrow indicates the Halo‐TET1 proteins. E, Dot blot assays of methylcytosine oxidation using HEK293FT untreated (CTL) or treated with HaloTag vectors (TET1). Data are shown as duplicate experiments. The methylene blue staining is used as a loading control for total genomic DNA. F, The tumor proliferation curves of HepG2 and Huh7 cells treated with siCTL, siTET1, CTL, and TET1. Data in (A), (B), (D), and (F) are shown as the mean ± SD from triplicate experiments. P‐values were measured using Student's t‐test in (A), (B), (D), and (F). *P < .05; **P < .01; ***P < .001
3.4. TET1 is involved in transcriptional regulation via cytosine demethylation at active promoters and enhancers
To elucidate the impact of TET1 depletion on transcriptional regulation, we profiled the change of cytosine modifications along with the epigenomic status of the liver cancer cell lines. Compared with the histone modification statuses of HepG2 cells defined in the Encyclopedia of DNA Elements (ENCODE) project, 33 5‐hmC was preferentially enriched in enhancers (28.8%) and transcribed regions (27.1%), while 5‐mC was distributed in heterochromatin (40.2%) and transcribed regions (35.5%) (Figure 4A). In active promoter regions, 5‐mC and 5‐hmC showed a bimodal modification pattern that spanned 2‐3 kb with TSS at its center (Figure 4B,C, Figure S5). Although 5‐mC showed no apparent changes, we found a remarkable 5‐hmC decrease by TET1 knockdown in active promoters and enhancers (Figure 4D‐G). At active enhancer regions, 5‐hmC mainly located at the center of enhancers, but 5‐mC was depleted. These enhancer 5‐hmC signals were decreased by TET1 knockdown in HepG2 and Huh7 cells. Overall, the transcriptional activity was more affected by the level of cytosine hydroxymethylation in enhancers than in promoters. The changes of 5‐hmC accumulation around active promoters did not affect gene expression (Figure 4H). On the other hand, 5‐hmC reduction at active enhancers in TET1 knockdown cells was significantly associated with decreased gene expression (Figure 4I). These results indicate that TET1‐mediated enhancer hydroxymethylation may exert an impact on transcriptional regulation.
FIGURE 4.
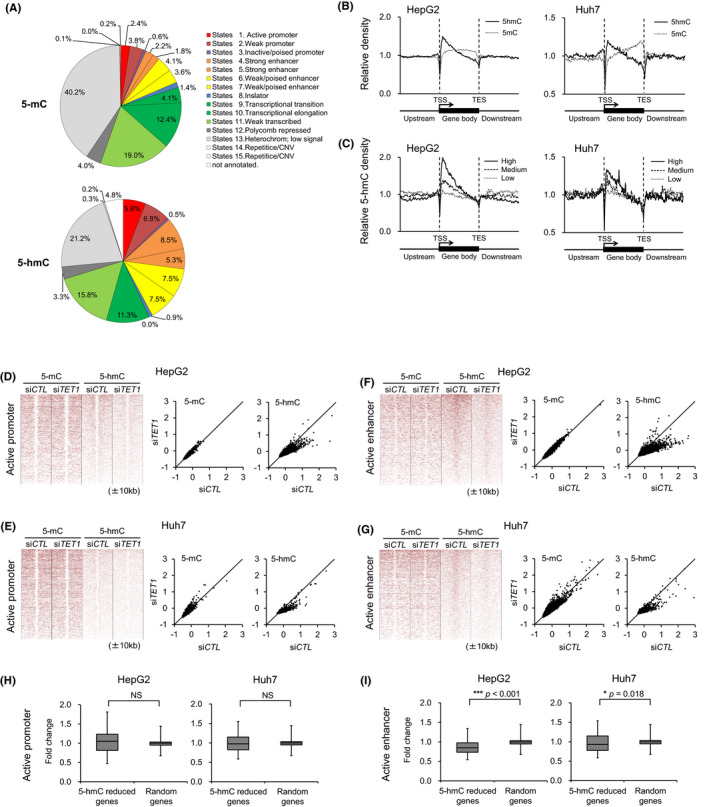
Genome‐wide distributions of 5‐hmC and the effect of TET1‐knockdown at the promoters and enhancers. A, Chromatin status of 5‐mC–enriched regions (upper) and 5‐hmC–enriched regions (lower) in HepG2 and Huh7 cells. B, 5‐hmC and 5‐mC distributions of HepG2 and Huh7 cells relative to human RefSeq gene position, respectively. C, 5‐hmC distributions relative to human RefSeq gene position by hmeDIP‐seq according to the expression levels in HepG2 and Huh7. Expression level: high, GeneChip score >250; low, GeneChip score <25; medium, GeneChip score 25‐250. D‐G, Heat map representation around active promoters and enhancers (±10 kb) with enriched 5‐mC and 5‐hmC in HepG2 (D, F) and Huh7 (E, G) cells with siCTL and siTET1. Active promoters (D, E) are classified by genomic elements (H3K4me3 and H3K27ac positive). Active enhancers (F, G) are classified by genomic elements (H3K4me3 negative, and H3K27ac and H3K4me1 positive). The heat map is rank‐ordered by 5‐hmC levels of siCTL. Z‐values of 5‐mC or 5‐hmC enrichments around active promoters (D, E) and active enhancers (F, G) (±5 kb) in HepG2 and Huh7 cells with siCTL and siTET1 are shown. Z‐value indicates normalized deviations based on a normal distribution. H, Boxplot of the expression changes in HepG2 and Huh7 cells with siCTL and siTET1 by expression microarray (U133 plus 2.0) at the active promoter of 5‐hmC reduced genes (HepG2, 578 probes; Huh7, 517 probes) and randomly selected genes (each 1000 probe). 5‐hmC–reduced genes show the genes whose 5‐hmC sum score of hmeDIP‐seq around active promoter (±200 bp) is reduced to one‐fifth by TET1 knockdown. I, Boxplot of the expression changes in HepG2 and Huh7 cells with siCTL and siTET1 by expression microarray (U133 plus 2.0) at active enhancer of 5‐hmC–reduced genes (HepG2, 94 probes; Huh7, 118 probes) and randomly selected genes (each 1000 probe). 5‐hmC–reduced genes show the genes whose 5‐hmC sum score of hmeDIP‐seq around active enhancer (±200 bp) is reduced to one‐fifth by TET1 knockdown. P‐values were measured using the Mann‐Whitney U‐test in (H) and (I). *P < .05; ***P < .001
3.5. Oncogenic target HMGA2 is dysregulated by TET1
The proliferative action of TET1 in liver cancer cells led us to consider that oncogenic target genes may be dysregulated by the aberrant deposition of 5‐hmC. To explore such downstream targets, we studied the gene expression changes of TET1 knockdown cells (Figure 5A). As a result, 177 and 320 genes were found to be downregulated (fold change < 0.5) by the transient knockdown of TET1 in HepG2 and Huh7 cells, respectively. Among these, 50 genes were identified as common targets according to the expression ratio of TET1 high HCCs over TET1 low HCCs. Although there was no deposition of 5‐hmC at the locus of the top‐ranked gene, DKK1, several hmC peaks were observed at the HMGA2 gene locus (Figure 5B).
FIGURE 5.
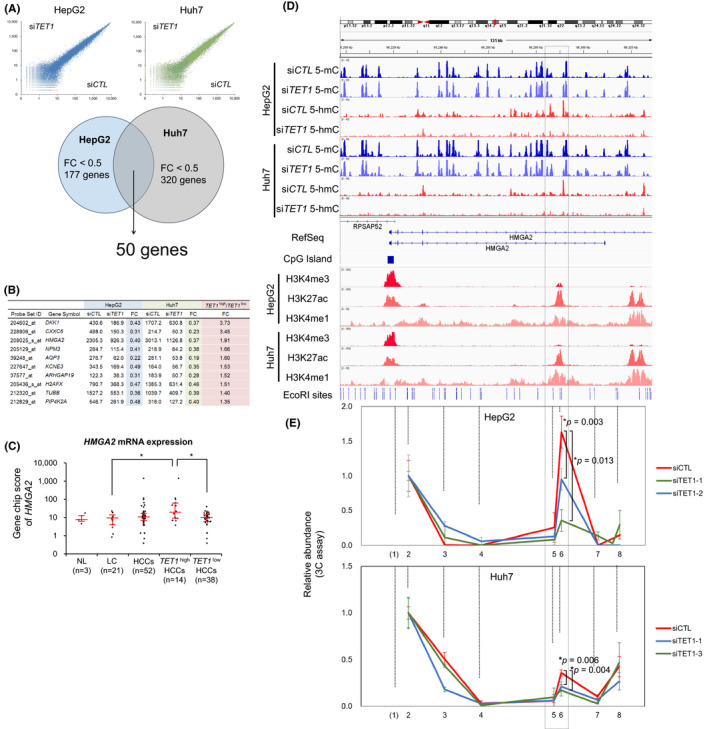
TET1 activates HMGA2 through enhancer hydroxymethylation. A, Expression changes by TET1 knockdown. Scatter plots (upper) demonstrate the GeneChip score of HepG2 (blue) and Huh7 (green). Venn diagram (lower) indicates the overlap of TET1‐upregulated genes. B, Representative genes upregulated by TET1. C, HMGA2 expression level among human clinical liver samples in expression microarray (U133 plus 2.0, Affymetrix). Each dot represents the GeneChip score of HMGA2 (208025_s_at). Red line represents the median ± quartile. D, The epigenetic statuses of the HMGA2 locus showing cytosine methylation (blue, upper), hydroxymethylation (red, upper), and histone modification (lower) of HepG2 and Huh7 cells treated with siCTL and siTET1 on the Integrative Genomics Viewer (IGV). The dotted‐line box intragenic enhancer regions identified by ChIP‐sequencing analysis of liver cancer cells. E, 3C‐qPCR analysis of long‐distance interactions at the enhancer of HMGA2, in which 5‐hmC was reduced by TET1 knockdown in HepG2 and Huh7 cells. Data in (C) are shown as the median ± quartile, and data in (E) are shown as the mean ± SD from triplicate experiments. P‐values were measured using the Mann‐Whitney U‐test in (C) and Student's t‐test in (E). *P < .05
The expression level of HMGA2 in TET1 high HCCs was significantly higher than in TET1 low HCCs and in noncancerous liver tissues (Figure 5C). In the intronic region of HMGA2, we found the hyperhydroxymethylated region, which was diminished after TET1 knockdown in both HepG2 and Huh7 cells (dotted‐line box in Figure 5D). In TET1 high HCCs, unlike TET1 low HCC and nontumor samples, the intronic 5‐hmC peak of HMGA2 was observed in a similar way (Figure S6). ChIP‐seq analyses of the hepatoma cell lines revealed that this region was marked with H3K27 acetylation and H3K4 monomethylation, known as active enhancer histone marks (Figure 5D, lower). Slight signals of H3K4 trimethylation were also implicated in the spatial interaction of this enhancer region to the promoter in vivo. We performed a 3C assay 34 and observed a specific interaction between the HMGA2 promoter and this hydroxymethylated enhancer (Figure 5E, Figure S7). This promoter‐enhancer interaction was validated by the Circularized Chromosome Conformation Capture (4C)‐like view of the Hi‐C data of HepG2 (Figure S8). Interestingly, this interaction was decreased by TET1 knockdown in both cell lines. These results suggest that TET1‐mediated hydroxymethylation plays an important role in the aberrant transcriptional activation of HMGA2.
To quantify the methylation levels of each CpG at the HMGA2 enhancer regions, we performed multiplex targeted sequencing of BS‐treated amplicons in the clinical liver samples. As shown in Figure S9, these CpGs were unmethylated in hESCs but were methylated in noncancerous liver tissues. In HCC tissues and liver cancer cell lines, this enhancer region was locally demethylated at the center of the H3K27ac peak. Compared with the epigenome profile in Roadmap Epigenomics data, the HMGA2 locus was fully covered by the H3K27 trimethylation of the normal liver tissue (E066). However, these polycomb repressive complex marks disappeared in HepG2 cells (E118) and in H1 hESC (E003), and an active enhancer mark appeared upon NANOG and SOX2 binding (Figure S10). These data suggest that the epigenetic status of the intronic HMGA2 enhancer is tightly regulated in a spatial and temporal manner.
Lastly, we analyzed the cellular effect of HMGA2 knockdown to determine its role in liver cancer. Knockdown efficiency for suppressing HMGA2 was verified by RT‐qPCR and Western blotting (Figure 6A). As expected, HMGA2 knockdown had an inhibitory effect on cell proliferation in HepG2 and Huh7 cells (Figure 6B). Next, we induced HMGA2 overexpression by infecting pLenti6.3/V5 vector (Figure 6C). Cell proliferation assays showed that the overexpression of full‐length HMGA2 partially cancelled the growth inhibition of TET1 knockdown in these cell lines (Figure 6D). Collectively, these results suggest that HMGA2 is a major oncogenic target of TET1 via epigenetic mechanisms, an enhancer hydroxymethylation in HCC (Figure 6E).
FIGURE 6.
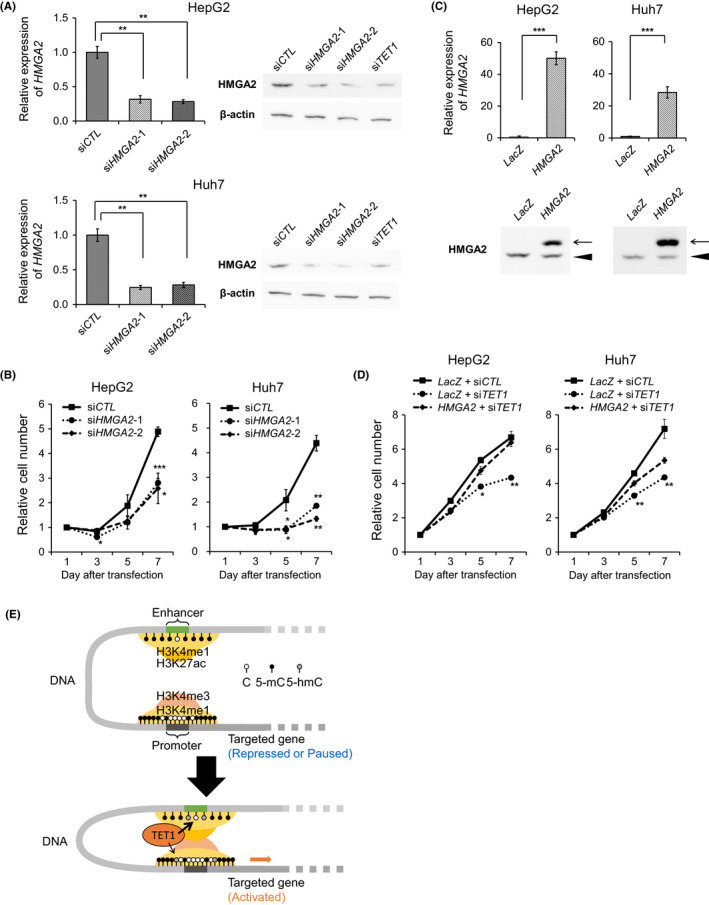
TET1‐targeted gene, HMGA2, enhances hepatoma cell proliferation. A, HMGA2 expression of HepG2 and Huh7 cells with siCTL, HMGA2 siRNA (siHMGA2‐1 and ‐2), and siTET1. Left, relative HMGA2 mRNA expression levels by RT‐qPCR compared with cells with siCTL. Right, HMGA2 expression by Western blotting analysis using anti‐HMGA2 antibody and anti‐β‐actin antibody. B, The tumor proliferation curves of HepG2 and Huh7 cells with siCTL and siHMGA2 by WST‐8. C, HMGA2 expression of HepG2 and Huh7 cells with control (LacZ) and HMGA2 overexpressed (HMGA2) by pLenti6.3/V5 vectors. Upper, relative HMGA2 mRNA expressions by RT‐qPCR compared with cells with LacZ. Lower, HMGA2 expression by Western blotting analysis using anti‐HMGA2 antibody. Arrow and arrowhead indicate the overexpressed HMGA2 proteins and endogenous HMGA2 proteins, respectively. D, The tumor proliferation curves of HepG2 and Huh7 cells with siCTL, siTET1, LacZ, and HMGA2 by WST‐8. Data are shown as the mean ± SD from triplicate experiments in (B) and (D). P‐values were measured using Student's t‐test. *P < .05; **P < .01; ***P < .001. E, Transcriptional regulation of oncogenic target HMGA2 through enhancer cytosine hydroxymethylation, histone modification, and chromatin interaction
4. DISCUSSION
In this study, TET1 was demonstrated to play an oncogenic role in liver cancer cells. TET1 upregulation was first revealed in a subgroup of HCCs presenting a hepatoblast‐like gene expression pattern. Contrary to the reported finding of TET1 in breast and prostate cancer, 13 we found that TET1 promotes hepatoma cell proliferation. To elucidate the oncogenic effectors regulated by TET1, we performed a genome‐wide analysis of cytosine methylation and hydroxymethylation for TET1‐upregulated HCCs. TET1 overexpression led to global hyperhydroxymethylation, preferentially in active enhancer regions. Among them, we identified the specific enhancer hydroxymethylation in a putative oncogene, HMGA2.
So far, TET1 has been described as a tumor suppressor in human cancers most likely due to its decreased expression levels and 5‐hmC depletion in most cancer tissues. 13 , 14 , 22 , 35 , 36 , 37 However, the expression level of TET1 in the previous reports was only verified by a limited number of cases or proven by semiquantitative antibody‐based experiments, such as Western blot or immunohistochemical assay. Thus, we re‐evaluated the TET1 mRNA expression levels using thousands of cancer transcriptome data. As shown in Figure 1B, the expression pattern of TET1 mRNA is cancer type–dependent. Generally, TET1 expression levels are extremely high in hESCs compared with differentiated somatic cells. 38 Another TET family protein, TET2, showed the opposite pattern: higher expression in terminally differentiated somatic cells than in hESCs (Figure S1). Several studies have demonstrated that TET2 plays an essential role in hematopoietic differentiation. 39 , 40 In addition, loss‐of‐function mutations of TET2 have been reported in myeloid and lymphoid malignancies. 9 , 39 In the context of solid cancer, there is a report of TET1 functions on tumorigenesis. Suppressive function for cell invasion was reported in prostate and breast cancers, 13 consistent with the result of TET1 downregulation in these types of cancer. In the present study, TET1 was upregulated in HCC and promoted cell proliferation of liver cancer cell lines, suggesting an oncogenic role in HCC development. Similarly, the potential oncogenic role of TET1 has been previously reported in several solid cancers, such as glioma, 41 mixed‐lineage leukemia–rearranged leukemia, 16 triple‐negative breast cancer, 42 and lung cancer. 43 These findings indicate that TET1 can function either as an oncogene or as a tumor suppressor depending on the cellular context.
Next, we explored 5‐mC and 5‐hmC mapping in clinical liver tissues and liver cancer cell lines. Although TET1 regulates 5‐hmC levels in transcriptional regulatory regions for murine ESCs, 19 , 32 , 44 the significance of 5‐hmC reduction at promoters or enhancers for gene regulation is poorly understood. 19 , 32 , 35 , 45 , 46 , 47 , 48 Overall, 5‐hmC is depleted at TSS regions but enriched at enhancer and transcribed regions. With regard to the gene expression, 5‐hmC is preferentially enriched in the downstream regions of TSS, especially for highly expressed genes. TET1 knockdown in hepatoma cells also shows the involvement of the cytosine demethylation at active promoters and enhancers. Notably, 5‐hmC reduction at active enhancers is associated with a decrease in gene expression. Although the global content of 5‐hmC is greatly decreased in tumor cells in vivo and in vitro, 12 , 13 , 14 whether the decreased content of 5‐hmC is caused by the downregulation or inactivation of TET proteins requires further study. Pfeifer et al also pointed out that a decreased content of 5‐hmC is not always associated with the activity of TET family proteins or Isocitrate dehydrogenase family proteins, 49 which play a role in the DNA demethylation pathway. As 5‐hmC is not maintained by DNMT1 during DNA replication, 50 the frequency of cell divisions has a great impact on the 5‐hmC levels, as well as on the enzymatic activity. Therefore, rather than the global amount of 5‐hmC and 5‐mC, their local distribution is thought to be much more important for epigenomic dysregulation via the aberrant expression of TET1.
Finally, we identified the HMGA2 gene as one of the important targets of TET1‐mediated enhancer hydroxymethylation during hepatocarcinogenesis. Although the oncogenic action of HMGA2 in HCC has been studied previously, 51 , 52 , 53 it is not well understood how HMGA2 is upregulated. Interestingly, the active enhancer mark, H3K27 acetylation, of this intronic region is observed only in a limited number of cell lines, such as H1ESC and HepG2, in the ENCODE database. Similarly, another ENCODE database (University of Washington) on DNase I hypersensitivity sites of 193 cell lines indicates that the open chromatin regions in HMGA2 locus highly depend on cell type.
Taken together, these results provide an important insight into the context‐dependent role of TET1 and 5‐hmC in cancer biology. GSEA analysis indicates that TET1 high HCCs show a hepatoblast‐like gene expression pattern. This subgroup of HCC is characterized by the overexpression of oncofetal genes 29 , 54 and a clinically worse prognosis. 27 Therefore, targeting the ectopic expression of TET1 would provide a translational impact to overcome these aggressive types of liver cancer.
DISCLOSURE
The authors have no competing interest to declare.
Supporting information
App S1
ACKNOWLEDGEMENTS
We thank Kaori Shiina, Hiroko Meguro, and Saori Kawanabe for their technical assistance in the experiments, and Ryota Yamanaka for his assistance with data processing. This work was supported by a Grant‐in‐Aid for Scientific Research (S) (24221011) (HA) and a Grant‐in‐Aid for Young Scientists (B) (25860521) (GN) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and the Program of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO), Japan.
Shirai K, Nagae G, Seki M, et al. TET1 upregulation drives cancer cell growth through aberrant enhancer hydroxymethylation of HMGA2 in hepatocellular carcinoma. Cancer Sci. 2021;112:2855–2869. 10.1111/cas.14897
REFERENCES
- 1. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415‐428. [DOI] [PubMed] [Google Scholar]
- 2. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baylin SB, Jones PA. A decade of exploring the cancer epigenome ‐ biological and translational implications. Nat Rev Cancer. 2011;11:726‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Esteller M. Cancer epigenomics: DNA methylomes and histone‐modification maps. Nat Rev Genet. 2007;8:286‐298. [DOI] [PubMed] [Google Scholar]
- 5. Chi P, Allis CD, Wang GG. Covalent histone modifications–miswritten, misinterpreted and mis‐erased in human cancers. Nat Rev Cancer. 2010;10:457‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481‐492. [DOI] [PubMed] [Google Scholar]
- 7. Koh KP, Yabuuchi A, Rao S, et al. Tet1 and Tet2 regulate 5‐hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37‐50. [DOI] [PubMed] [Google Scholar]
- 9. Shih AH, Abdel‐Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599‐612. [DOI] [PubMed] [Google Scholar]
- 10. Kriaucionis S, Heintz N. The nuclear DNA base 5‐hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kudo Y, Tateishi K, Yamamoto K, et al. Loss of 5‐hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103:670‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsu CH, Peng KL, Kang ML, et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012;2:568‐579. [DOI] [PubMed] [Google Scholar]
- 14. Liu C, Liu L, Chen X, et al. Decrease of 5‐hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS ONE. 2013;8:e62828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun M, Song CX, Huang H, et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc Natl Acad Sci USA. 2013;110:9920‐9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang H, Jiang X, Li Z, et al. TET1 plays an essential oncogenic role in MLL‐rearranged leukemia. Proc Natl Acad Sci USA. 2013;110:11994‐11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES‐cell self‐renewal and inner cell mass specification. Nature. 2010;466:1129‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams K, Christensen J, Pedersen MT, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y, Wu F, Tan L, et al. Genome‐wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim R, Sheaffer KL, Choi I, Won KJ, Kaestner KH. Epigenetic regulation of intestinal stem cells by Tet1‐mediated DNA hydroxymethylation. Genes Dev. 2016;30:2433‐2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khoueiry R, Sohni A, Thienpont B, et al. Lineage‐specific functions of TET1 in the postimplantation mouse embryo. Nat Genet. 2017;49:1061‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeschke J, Collignon E, Fuks F. Portraits of TET‐mediated DNA hydroxymethylation in cancer. Curr Opin Genet Dev. 2016;36:16‐26. [DOI] [PubMed] [Google Scholar]
- 23. Kimura H, Hayashi‐Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33:61‐73. [DOI] [PubMed] [Google Scholar]
- 24. Kaneshiro K, Tsutsumi S, Tsuji S, Shirahige K, Aburatani H. An integrated map of p53‐binding sites and histone modification in the human ENCODE regions. Genomics. 2007;89:178‐188. [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y, Liu T, Meyer CA, et al. Model‐based analysis of ChIP‐Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yong KJ, Chai L, Tenen TG. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med. 2013;368:2266‐2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cairo S, Armengol C, Reynies A, et al. Hepatic stem‐like phenotype and interplay of Wnt/beta‐catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell. 2008;14:471‐484. [DOI] [PubMed] [Google Scholar]
- 29. Hoshida Y, Nijman SMB, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385‐7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt‐beta‐catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67:10831‐10839. [DOI] [PubMed] [Google Scholar]
- 31. Pastor WA, Pape UJ, Huang Y, et al. Genome‐wide mapping of 5‐hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu H, D'Alessio AC, Ito S, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306‐1311. [DOI] [PubMed] [Google Scholar]
- 35. Lian CG, Xu Y, Ceol C, et al. Loss of 5‐hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang H, Liu Y, Bai F, et al. Tumor development is associated with decrease of TET gene expression and 5‐methylcytosine hydroxylation. Oncogene. 2013;32:663‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cimmino L, Dawlaty MM, Ndiaye‐Lobry D, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015;16:653‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li W, Liu M. Distribution of 5‐hydroxymethylcytosine in different human tissues. J Nucleic Acids. 2011;2011:870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quivoron C, Couronne L, Valle VD, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25‐38. [DOI] [PubMed] [Google Scholar]
- 40. Moran‐Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self‐renewal and myeloid transformation. Cancer Cell. 2011;20:11‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takai H, Masuda K, Sato T, et al. 5‐Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP‐methylosome complex. Cell Rep. 2014;9:48‐60. [DOI] [PubMed] [Google Scholar]
- 42. Good CR, Panjarian S, Kelly AD, et al. TET1‐mediated hypomethylation activates oncogenic signaling in triple‐negative breast cancer. Cancer Res. 2018;78:4126‐4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Filipczak PT, Leng S, Tellez CS, et al. p53‐suppressed oncogene TET1 prevents cellular aging in lung cancer. Cancer Res. 2019;79:1758‐1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamaguchi S, Hong K, Liu R, et al. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012;492:443‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5‐hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398‐402. [DOI] [PubMed] [Google Scholar]
- 46. Cimmino L, Abdel‐Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9:193‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu P, Xiao S, Xin X, et al. Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Res. 2013;23:352‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tan L, Xiong L, Xu W, et al. Genome‐wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP‐seq method. Nucleic Acids Res. 2013;41:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jin SG, Jiang Y, Qiu R, et al. 5‐Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71:7360‐7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Inoue A, Zhang Y. Replication‐dependent loss of 5‐hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899‐910. [DOI] [PubMed] [Google Scholar]
- 52. Morishita A, Zaidi MR, Mitoro A, et al. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013;73:4289‐4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu L, Wang Z, Lu R, Jiang W. Expression of high mobility group A2 is associated with poor survival in hepatocellular carcinoma. Pathol Oncol Res. 2012;18:983‐987. [DOI] [PubMed] [Google Scholar]
- 54. Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779‐6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
App S1