

Abstract
Mature teratomas are usually benign tumors that rarely undergo malignant transformation. We report an advanced neuroblastoma arising in a mature teratoma of the ovary. Whole‐exome sequencing identified extensive copy‐neutral loss of heterozygosity (LOH) in both neuroblastoma and teratoma elements, suggesting that the neuroblastoma evolved from the teratoma. In addition, several truncating germline heterozygous variants in tumor suppressor genes, including RBL2 and FBXW12, became homozygous as a result of LOH. Collectively, we speculate that extensive LOH in teratoma cells may force heterozygous germline variants to become homozygous, which, in turn, may contribute to the development of neuroblastoma with the acquisition of additional chromosomal changes.
Keywords: cancer genome/genetics, copy‐neutral loss of heterozygosity, genomic analysis, malignant transformation, neuroblastoma, teratoma
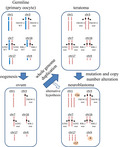
A model of malignant transformation from teratoma to neuroblastoma.
Abbreviations
- AYA
adolescent‐and‐young‐adult
- FFPE
formalin‐fixed paraffin‐embedded
- NSE
neuron‐specific enolase
- MIBG
123I‐metaiodobenzylguanidine
- VAFs
variant allele frequencies
- CN‐LOH
copy‐neutral LOH
- UPD
uniparental disomy
1. INTRODUCTION
A mature teratoma is a subtype of a germ cell tumor that occurs in both adults and children. Most cases of mature teratomas are benign, and malignant transformation rarely occurs. 1 Although carcinomas are common malignancies arising in mature teratomas, embryonal malignancies, including neuroblastoma, can also occur. 2 Previous studies have shown additional somatic mutations in genes such as TP53, PIK3CA, and CDKN2A during malignant transformation in squamous cell carcinoma and adenocarcinoma. 3 However, genetic changes that contribute to the malignant transformation of teratoma to neuroblastoma and their clonal relationship have not been previously reported.
Neuroblastoma is an embryonal malignant tumor originating from the sympathetic nervous system. It is the most common type of extracranial malignant tumor in children, and over 90% cases of neuroblastoma are found in children younger than 10 y of age. Neuroblastoma in adolescents and young adults (AYA) is rare, often showing an indolent course and poor prognosis. 4
Here, we report a 15‐y‐old girl with neuroblastoma arising in a mature cystic teratoma of the ovary who was successfully treated using a multidisciplinary treatment approach. We identified the underlying genetic changes within the elements of the mature cystic teratoma and neuroblastoma using whole‐exome sequencing and copy number analysis.
2. MATERIALS AND METHODS
2.1. Pathological diagnosis and DNA extraction
After surgical resection, samples were immediately fixed with formaldehyde. FFPE sections were prepared from different tumor regions, and pathological evaluation was performed. For genetic analysis, neuroblastoma‐enriched lesions and teratoma‐enriched lesions were pathologically reviewed using the respective FFPE sections to identify tissue regions for DNA extraction. At 8 mo after tumor resection, samples cut from the paraffin block, as well as peripheral blood, underwent DNA extraction using a GeneRead DNA FFPE Kit and DNeasy Blood & Tissue Kit (Qiagen), respectively, in accordance with the manufacturer's instructions.
2.2. Whole‐exome sequencing
DNA libraries were prepared using SureSelect Human All Exon V5 (Agilent Technologies) in accordance with the manufacturer's instructions. Enriched exome libraries were sequenced on an Illumina HiSeq 2500 instrument. Somatic variants were called using Genomon v.2.3.0 with the following parameters: (a) VAFs < 0.05 in normal samples, (b) Empirical Bayesian Mutation Calling P ≥ .0001, and (c) variants not only present in unidirectional reads. Mapping errors were removed by visual inspection using the Integrative Genomics Viewer tool. To narrow down the disease‐causing candidate germline variants, mutations with frequencies in the normal population <1% and VAFs in peripheral blood >0.2 were selected. Mapping errors were also removed by visual inspection on the Integrative Genomics Viewer browser (http://www.broadinstitute.org/igv/).
2.3. Copy number analysis
Allele‐specific copy numbers were calculated by allele frequencies and sequenced depths of single nucleotide polymorphisms using our in‐house CNACS pipeline, the scripts of which are available at https://github.com/papaemmelab/toil_cnacs.
2.4. Ethics approval and consent to the study
This study was reviewed and approved by the Institutional Review Board of St. Luke's International Hospital, Kyoto University, and the University of Tokyo. Written informed consent was obtained from the patient and her parents.
2.5. Data availability
Sequencing data were deposited at the European Genome‐phenome Archive (http://www.ebi.ac.uk/ega/) under the accession number EGAS00001005116.
3. RESULTS
3.1. Case presentation
A 15‐y‐old girl complained of lumbar pain and abdominal distention for 1 mo. Contrast‐enhanced MRI and computed tomography showed a bulky solid mass with heterogeneous enhancement, measuring 11 × 9 × 11 cm, in the left ovary (Figure S1A‐E). The tumor consisted of cystic and solid components, including calcification and minimal fat. Mediastinal and para‐aortic lymphadenopathies were also observed. Left salpingo‐oophorectomy, partial omentectomy, and pelvic and para‐aortic lymphadenectomy were performed, although para‐aortic lymph nodes were partially unresectable. Histopathological examination revealed that the tumor consisted of 10% teratoma elements and 90% neuroblastoma elements (Figure 1A‐C). The teratoma component contained tissues derived from all 3 germ layers, thereby characterized as a mature cystic teratoma. By contrast, the neuroblastoma component was mainly composed of poorly differentiated, dense, chromatin‐rich small round cells with a low mitosis‐karyorrhexis index. Immunohistochemistry of neuroblastoma elements showed cells positive for anti‐synaptophysin, anti‐chromogranin, and tyrosine hydroxylase. The DNA index was diploidy, and MYCN was not amplified. Evaluation of the para‐aortic lymph node showed similar findings to the neuroblastoma component. Bone marrow aspiration showed metastasis of neuroblastoma cells positive for NSE. Cytogenetic analysis showed 45,X,−X,+1,der(1;8)(q10;q10),del(9)(q?),add(11)(p15),der(14;22)(q10;q10),+17 in 4 of 20 cells analyzed. Both bone scintigraphy and MIBG scintigraphy showed no abnormal uptake of radioactive tracers. Serum NSE was elevated (up to 129 ng/mL), but urine catecholamine metabolites did not increase. On the basis of these findings, we diagnosed the patient with neuroblastoma arising in a mature teratoma of the left ovary with multiple metastases.
FIGURE 1.
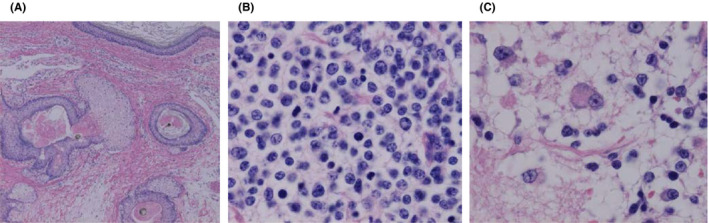
Histopathological evaluation of ovarian tumor. A‐C, H&E staining of the tumor. Neuroblastomatous tissue consisting of dense, solid proliferation of small round cells with increased chromatin. There are minor components with multilayered squamous epithelium with skin appendage‐like structures and choriocapillary epithelium
Mediastinal and para‐aortic lymphadenopathies progressed rapidly 1 wk after surgery, leading to a compressed ureter and hydronephrosis (Figure S2A,B). After confirming normal renal function, multiagent chemotherapy for neuroblastoma, consisting of cyclophosphamide (1200 mg/m2/d, day 1 for the 1st course; days 1‐2 for subsequent courses), vincristine (1.5 mg/m2/d, day 1), pirarubicin (40 mg/m2/d, day 3), and cisplatin (20 mg/m2/d, days 1‐5), was initiated. Chemotherapy successfully reduced the size of the metastatic lymph nodes, and serum NSE was reduced to 15.7 ng/mL 2 wk after the initiation of treatment. After 5 courses of chemotherapy, we administered high‐dose chemotherapy, consisting of busulfan (3.2 mg/kg/d, days −6 to −3) and melphalan (140 mg/m2/d, day −2), followed by autologous peripheral blood stem cell rescue. Neutrophil engraftment was achieved on day 12 after transplantation, and she did not develop transplant‐related complications. The patient underwent lymphadenectomy of residual para‐aortic lymph nodes on day 48 from transplantation, showing no viable cells. She received 19.8 Gy of local irradiation to the mediastinum and retroperitoneum, followed by 9 courses of 13‐cis‐retinoic acid. At 9 y after diagnosis, she is alive without any evidence of disease recurrence.
3.2. Genomic analysis of neuroblastoma‐ and teratoma‐enriched elements
To identify genetic abnormalities that may facilitate malignant transformation and to determine the clonal relationship between teratoma and neuroblastoma, we performed whole‐exome sequencing using DNA extracted from neuroblastoma‐enriched lesions and mature teratoma‐enriched lesions, as well as peripheral blood.
Table 1 shows the 14 somatic mutations and representative germline variants. Although 14 genes including ARHGEF1 and KCNJ10, whose expression changes were reported in neuroblastoma cell lines 5 , 6 were mutated, no mutations were found in neuroblastoma driver genes such as ALK and ATRX. In total 11 of the 14 somatic mutations found in the neuroblastoma‐enriched lesions were also found in the teratoma‐enriched lesions with a low allele frequencies of <4% (Table 1), a finding that was in agreement with the histopathological finding that the teratoma lesions contained ~1%‐5% of neuroblastoma components. Therefore, we concluded that no somatic mutations were identified in teratoma lesions.
TABLE 1.
Somatic mutations and representative germline single nucleotide variants in peripheral blood, neuroblastoma‐enriched lesions, and teratoma‐enriched lesions
| Type | Gene | Exonic function | Amino acid change | VAF (neuroblastoma‐enriched lesions) | VAF (teratoma‐enriched lesions) | VAF (peripheral blood) |
|---|---|---|---|---|---|---|
| Somatic | ANKRD31 | Nonsynonymous SNV | L604P | 0.42 | 0.01 | 0.00 |
| ARHGEF1 | Nonsynonymous SNV | T662M | 0.52 | 0.04 | 0.00 | |
| CNBP | Synonymous SNV | Q98Q | 0.36 | 0.00 | 0.00 | |
| CNGA3 | Nonsynonymous SNV | D199V | 0.60 | 0.06 | 0.00 | |
| DHX34 | Synonymous SNV | L138L | 0.37 | 0.03 | 0.00 | |
| GRAP2 | Nonsynonymous SNV | N192K | 0.41 | 0.04 | 0.00 | |
| INO80D | Frameshift deletion | 868_895del | 0.24 | 0.01 | 0.00 | |
| KCNJ10 | Nonsynonymous SNV | R26G | 0.07 | 0.00 | 0.00 | |
| KDM4E | Synonymous SNV | P327P | 0.45 | 0.00 | 0.00 | |
| KIAA1217 | Nonsynonymous SNV | A539S | 0.50 | 0.02 | 0.00 | |
| P2RY2 | Nonsynonymous SNV | S141F | 0.44 | 0.05 | 0.00 | |
| SH3TC1 | Nonsynonymous SNV | A282S | 0.45 | 0.01 | 0.00 | |
| SVIL | Nonsynonymous SNV | E1545K | 0.40 | 0.02 | 0.00 | |
| ZKSCAN3 | Nonsynonymous SNV | V27M | 0.46 | 0.04 | 0.00 | |
| Germline | RBL2 | Stop gain | K911X | 0.95 | 0.90 | 0.51 |
| FBXW12 | Frameshift deletion | N353fs | 1.00 | 0.70 | 0.50 | |
| EZH2 | Splice site | 1547‐2A>C | 0.99 | 0.68 | 0.40 |
Next, we then evaluated copy number alterations in neuroblastoma‐enriched lesions and teratoma‐enriched lesions, as well as peripheral blood by a sequencing‐based method. Although no copy number alterations were found in peripheral blood (Figure 2A, upper panel), teratoma‐enriched lesions showed genome‐wide CN‐LOH, which is also known as uniparental disomy (UPD) (Figure 2A, middle panel). 7 , 8 , 9 Intriguingly, the neuroblastoma‐enriched lesions also showed genome‐wide CN‐LOH, suggesting that the neuroblastoma cells were derived from the teratoma cells. The neuroblastoma‐enriched lesions also showed additional copy number alterations including +1q, +17, and −X, which are characteristic of neuroblastoma (Figure 2A, lower panel).
FIGURE 2.
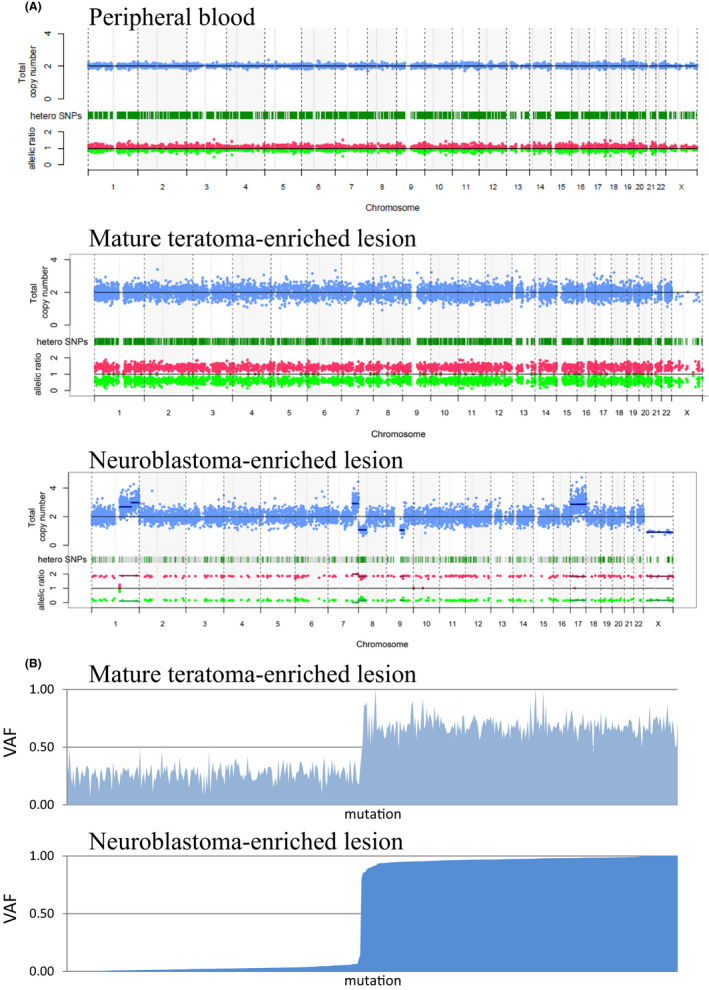
Genetic changes within the elements of the mature cystic teratoma and neuroblastoma by whole‐exome sequencing and copy number analysis. A, Copy number analyses of neuroblastoma and teratoma lesions. Copy number plot from peripheral blood (upper panel), teratoma‐enriched lesion (middle panel), and neuroblastoma‐enriched lesion (lower panel) by a sequencing‐based method. Note an allele‐specific copy number of almost 1:1 in peripheral blood compared with genome‐wide deviations from 1:1 in teratoma and neuroblastoma regions, demonstrating extensive LOH. B, Histograms of VAFs of 478 germline variants in a mature teratoma‐enriched lesion (upper panel) and in a neuroblastoma‐enriched lesion (lower panel). Mutations in each gene are shown in order sorted by the VAF value in the neuroblastoma lesion. Each mutated gene at the teratoma lesion is shown in the same order as at the neuroblastoma lesion
The extensive CN‐LOH in teratoma and neuroblastoma indicates that heterozygous germline variants would become homozygous in tumor cells, otherwise heterozygous germline variants would be lost and become wild‐type. Therefore, we next investigated whether the neuroblastoma harbored germline variants that became homozygous when CN‐LOH occurred. Of 478 rare germline variants detected in a peripheral blood sample, 248 heterozygous variants, including loss‐of‐function mutations in EZH2, FBXW12, and RBL2, showed VAFs of nearly 1.0 (0.97 ± 0.03) in neuroblastoma‐enriched lesions suggesting that these variants became homozygous, while the remaining 230 heterozygous variants showed VAFs of nearly 0.0 (0.02 ± 0.02) suggesting that these became wild‐type in neuroblastoma‐enriched lesions (Table 1 and Dataset S1). Conversely, VAFs of mature‐teratoma‐enriched lesions showed less contrast when relevant 248 and 230 variants were 0.69 ± 0.09 and 0.27 ± 0.07, respectively. Figure 2B shows histograms of VAFs of 478 germline variants in mature teratoma‐enriched lesions (Figure 2B, upper panel) and in neuroblastoma‐enriched lesions (Figure 2B, lower panel). Although the possibility remains that the discrepancy of VAFs was due to heterogeneity of teratoma component, we suspect that it is more likely to be caused by contamination of normal tissue in teratoma‐enriched lesions.
4. DISCUSSION
Neuroblastoma occurring in AYA tends to show an indolent course with less sensitivity to chemotherapy compared with neuroblastoma in infants and young children. Genetic changes in neuroblastoma of AYA have a lower frequency of MYCN amplification and higher frequency of ALK and ATRX mutation, which are in contrast with neuroblastoma in infants and young children, and suggests that the pathobiology of neuroblastoma may be different based on the age of onset. 4 , 10
To date, 18 female cases of ovarian neuroblastoma have been reported (Table S1), of which 13 arose from teratomas. Here, 6 of these 13 teratoma‐derived cases of neuroblastoma were advanced. Although the prognostic significance of a teratomatous component and treatment modalities were inconclusive because of the limited number of patients, local therapy without chemotherapy appears to be insufficient for patients with advanced disease. Despite the rapid progression of metastatic lymph nodes, our presented patient responded well to chemotherapy, and she remains in long‐term remission after multidisciplinary treatment, including high‐dose chemotherapy.
We performed whole‐exome sequencing and sequencing‐based copy number analysis of neuroblastoma‐enriched lesions and mature teratoma‐enriched lesions, and detected somatic mutations in 14 genes. However, no driver mutations were identified in the neuroblastoma‐enriched sample. Conversely, recurrent copy number alterations, including +1q, +17, and −X, were identified, suggesting that these copy number alterations contributed to tumor development.
Copy number analysis also showed that the neuroblastoma‐enriched sample and mature teratoma‐enriched sample shared extensive CN‐LOH. Genome‐wide LOH and low frequencies of somatic mutations in teratomas are consistent with previous reports that found most teratomas were in a CN‐LOH status because they arise from meiotic errors. 7 , 8 , 9 Furthermore, extensive CN‐LOH has not been reported in neuroblastoma, 11 suggesting that neuroblastoma developed from teratoma cells. However, it is also possible that common ancestry clones characterized with CN‐LOH generated both teratoma and neuroblastoma (Figure 3, alternative hypothesis). In addition, several heterozygous germline variants became homozygous because of extensive CN‐LOH. For instance, a heterozygous nonsense mutation in RBL2, which has a known role in neural crest cell differentiation and cancer suppression, 12 , 13 detected in peripheral blood became homozygous in neuroblastoma. Similar findings were also observed in the epigenetic regulator EZH2 14 and tumor suppressor gene FBXW12. 15 These findings suggested that genome‐wide LOH, which is a key pathognomonic change of a teratoma, may force heterozygous germline variants to become homozygous. Further investigation is mandatory to examine whether homozygous changes of tumor suppressor genes caused by genome‐wide LOH may induce teratoma to acquire additional mutations or copy number alterations, which result in malignant transformation.
FIGURE 3.
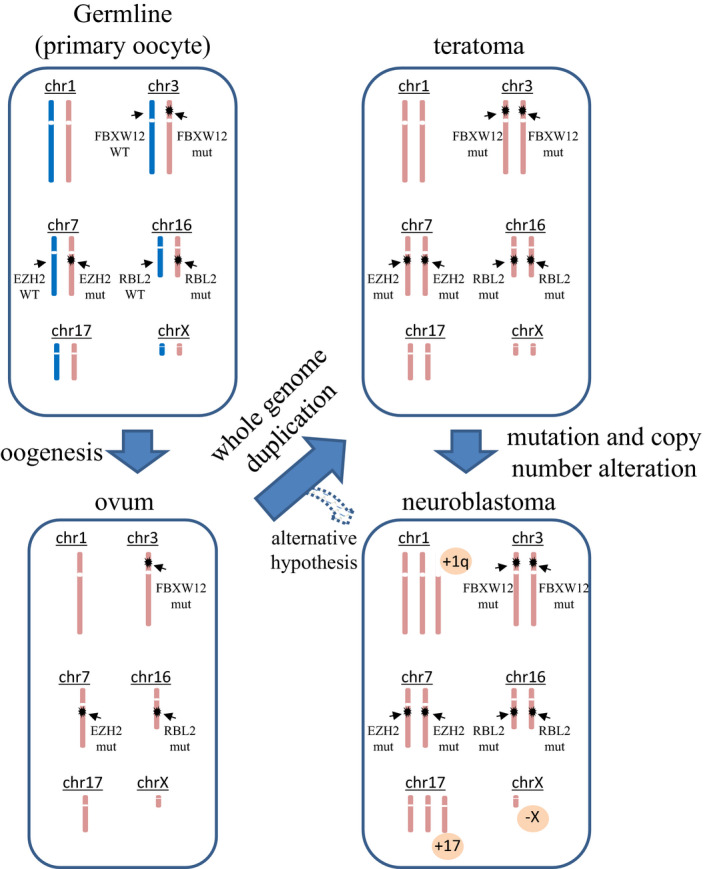
A model of malignant transformation from teratoma to neuroblastoma. A schematic diagram of tumorigenesis. During the process of oogenesis, germline cells (primary oocyte) become hemizygous. Errors in meiosis (whole genome duplication) lead to teratomas, in which heterozygous germline variants become homozygous. By acquiring additional mutations and/or copy number alterations, a part of the teratoma may develop neuroblastoma. Alternatively, common ancestry clones after whole genome duplication may independently develop to teratoma and neuroblastoma
We experienced an adolescent case of mature teratoma‐derived advanced neuroblastoma of the ovary. Considering the favorable outcome of our patient, teratoma‐derived malignancies may have different biological characteristics compared with de novo malignancies. To our best knowledge, this is the 1st case in which a clonal relationship between teratoma and neuroblastoma was genetically confirmed. Furthermore, our results may suggest a possible mechanism of malignant transformation of teratomas, in which multiple heterozygous germline variants become homozygous because of genome‐wide LOH, which is a hallmark cytogenetic abnormality of teratomas (Figure 3). Further studies to compare the genomic profiles of teratomas with and without malignant transformation are needed to elucidate tumorigenesis of somatic malignant neoplasms arising from teratomas.
DISCLOSURE
The authors have no conflicts of interest.
Supporting information
Supplementary Material
Dataset S1
ACKNOWLEDGMENTS
We would like to thank Dr. Yuichi Shiraishi and Dr. Satoru Miyano, The University of Tokyo, for data analysis with this study. This work was supported by the Japan Society for the Promotion of Science (JSPS) through Grants‐in‐Aid for Scientific Research (KAKENHI) (grant number JP26221308 and JP19H05656 to Seishi Ogawa) and also by the Japan Agency for Medical Research and Development (AMED), and the Project for Development of Innovative Research on Cancer Therapeutics (JP 20cm0106501h0005 to Seishi Ogawa).
Ono R, Ueno H, Yoshida K, et al. Clonal evidence for the development of neuroblastoma with extensive copy‐neutral loss of heterozygosity arising in a mature teratoma. Cancer Sci. 2021;112:2921–2927. 10.1111/cas.14931
REFERENCES
- 1. Rathore R, Sharma S, Agarwal S. Malignant transformation in mature cystic teratoma of the ovary: a retrospective study of eight cases and review of literature. Prz Menopauzalny Menopause Rev. 2018;17:63‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niwa Y, Yamamuro O, Kato N, Tsuzuki T. Two cases of primary ovarian neuroblastoma arising from mature cystic teratomas. Gynecol Oncol Case Rep. 2013;5:58‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cooke SL, Ennis D, Evers L, et al. The driver mutational landscape of ovarian squamous cell carcinomas arising in mature cystic teratoma. Clin Cancer Res. 2017;23:7633‐7640. [DOI] [PubMed] [Google Scholar]
- 4. Mossé YP, Deyell RJ, Berthold F, et al. Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer. 2014;61:627‐635. [DOI] [PubMed] [Google Scholar]
- 5. Raetz EA, Kim MKH, Moos P, et al. Identification of genes that are regulated transcriptionally by Myc in childhood tumors. Cancer. 2003;98:841‐853. [DOI] [PubMed] [Google Scholar]
- 6. Zschüntzsch J, Schütze S, Hülsmann S, Dibaj P, Neusch C. Heterologous expression of a glial Kir channel (KCNJ10) in a neuroblastoma spinal cord (NSC‐34) cell line. Physiol Res. 2013;62:95‐105. [DOI] [PubMed] [Google Scholar]
- 7. Kubota Y, Seki M, Kawai T, et al. Comprehensive genetic analysis of pediatric germ cell tumors identifies potential drug targets. Commun Biol. 2020;3:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heskett MB, Sanborn JZ, Boniface C, et al. Multiregion exome sequencing of ovarian immature teratomas reveals 2N near‐diploid genomes, paucity of somatic mutations, and extensive allelic imbalances shared across mature, immature, and disseminated components. Mod Pathol. 2020;33:1193‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shao L, Heider A, Rabah R. Single nucleotide polymorphism array and cytogenetic analyses of ovarian teratomas in children. Genes Chromosom Cancer. 2021;60(6):418‐425. [DOI] [PubMed] [Google Scholar]
- 10. Cheung NK, Zhang J, Lu C, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takita J, Hayashi Y, Kohno T, et al. Allelotype of neuroblastoma. Oncogene. 1995;11:1829‐1834. [PubMed] [Google Scholar]
- 12. Raschellà G, Tanno B, Bonetto F, Negroni A, Amendola R, Paggi MG. Retinoblastoma family proteins induce differentiation and regulate B‐myb expression in neuroblastoma cells. Med Pediatr Oncol. 2001;36:104‐107. [DOI] [PubMed] [Google Scholar]
- 13. Jori FP, Galderisi U, Piegari E, et al. RB2/p130 ectopic gene expression in neuroblastoma stem cells: evidence of cell‐fate restriction and induction of differentiation. Biochem J. 2001;360:569‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Takenobu H, Setyawati AN, et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene. 2018;37:2714‐2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Hackett CS, Zhang S, et al. The genetics of splicing in neuroblastoma. Cancer Discov. 2015;5:380‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Dataset S1
Data Availability Statement
Sequencing data were deposited at the European Genome‐phenome Archive (http://www.ebi.ac.uk/ega/) under the accession number EGAS00001005116.