

Abstract
Immunotherapy has revolutionized cancer treatment, however, not all tumor types and patients are completely responsive to this approach. Establishing predictive pre‐clinical models would allow for more accurate and practical immunotherapeutic drug development. Mouse models are extensively used as in vivo system for biomedical research. However, due to the significant differences between rodents and human, it is impossible to translate most of the findings from mouse models to human. Pharmacological development and advancing personalized medicine using patient‐derived xenografts relies on producing mouse models in which murine cells and genes are substituted with their human equivalent. Humanized mice (HM) provide a suitable platform to evaluate xenograft growth in the context of a human immune system. In this review, we discussed recent advances in the generation and application of HM models. We also reviewed new insights into the basic mechanisms, pre‐clinical evaluation of onco‐immunotherapies, current limitations in the application of these models as well as available improvement strategies. Finally, we pointed out some issues for future studies.
Keywords: human specificity, humanized mice, immunology, immunotherapy, patient‐derived xenografts
Establishing predictive pre‐clinical models leads toward more accurate and practical immunotherapeutic development. Humanized mice (HM) provide a suitable platform to discern human‐specific disease pathogenesis and evaluate an array of novel therapeutics. This review discusses recent progresses in the production and deployment of HM in the study of cancer immunotherapy.

1. INTRODUCTION
Cancer immunotherapy utilizes host immune system to eradicate tumor cells. Systematic pre‐clinical cancer immunotherapy is dependent on selecting, or preferably, developing appropriate animal models. Among various animal models, humanized mice (HM) have been extensively utilized for in vivo studies of human cancer immunology and immunotherapy. Different models are eligible for evaluating new anti‐cancer therapies namely patient‐derived xenografts (PDXs), humanized PDX and genetically engineered mice. 1
The pre‐requisite of a successful immunotherapy is a functional immune system of the patient since these methods recruit the host immune cells to combat growing tumors. This limits our ability to test the efficacy of these approaches in conventional experimental models. Early murine studies have evaluated the efficacy of anti‐CTLA4 immunotherapy targeting the CTLA‐4 receptor of a mouse model of fibrosarcoma and ovarian cancer. 2 , 3 Favorable results of this investigation promoted the production of human antibodies which was further tested in cynomolgous monkey as the only identified species with cross‐reactivity. 4 Nevertheless, testing human specific antibodies in cynomolgous monkeys had its pitfalls: complete cross‐reactivity could not be obtained and predictive data for human clinical usage was not provided. Therefore, deploying HM models would facilitate the evaluations on the interaction between immune system and tumors and, also, would yield more clinically reliable results. 5 , 6 HM are the best mouse model for cancer immunotherapy studies and are consisted of three elements: (a) immunodeficient host mice, (b) human immune cells and (c) human tumor cells. In current review, we explain recent advances in the “humanization” of mouse models which improve their application in the study of immunology and immunotherapy. Moreover, we discuss limitations of using these models and strategies that can remove these limitations. Finally, we explain how these improvements shape the future of employing HM models in cancer studies.
2. AN OVERVIEW ON IMMUNODEFICIENT MICE
The main challenge in the engraftment of human cancer cells in immune‐competent rodents is the xenogenic immune rejection. Table 1 shows the evolution of different immunodeficient mice, although modifications are needed. The major improvement in deploying scid mouse model was backcrossing of the scid mutation to non‐obese diabetic (NOD/Lt) strain background which associated with lower NK cell and myeloid function and, as a result, enhanced human engraftment of immune cells. 7 Moreover, backcrossing with NOD mice introduced a receptor that is highly homologous to a human equivalent called signal regulatory protein alpha (Sirpα). Murine macrophages (MQ) express Sirpα which is able to bind to counterpart CD47, a “don't eat me” signal protein, on human immune cells and subsequently inhibit phagocytosis. 8 Another remarkable milestone was the introduction of a mouse strain knocked‐out in the interleukin (IL)‐2 receptor common gamma chain (IL2Rγ) gene 9 that not only this mice would have functionally impaired adaptive immune system but, more importantly, disabled NK cell development. 10 The combination with NOD‐scid (NSG) 11 mice and RAG (NRG) 12 mice revolutionized human cell engraftment. Similar to NSG, NOG mice have NOD‐scid background with truncated IL2γc gene which enables binding but not signaling of cytokines. 13 Another improvement of human engraftment was achieved by interbreeding of NOG and BALB/c‐Rag2null which generated BRG mice. In addition, integration of NOD/Lt Sirpα polymorphism into BRG mice further refined human cell reconstitution. 14 Successful engraftment of human hematopoietic immune cells is achieved in NSG and NOG and provided a suitable animal models for initial immunologic studies of immunotherapy. 15 According to preliminary studies immune reconstitution is not yet optimal. In this review, we aimed to study novel approaches that improve hematopoietic reconstitution in the host mice for studies.
TABLE 1.
Immunodeficient mouse strains for human immune system engraftment
| Mouse model | Strain/Characteristics | Life span | T | B | NK | DC | MQ | Com. | References |
|---|---|---|---|---|---|---|---|---|---|
| Nude | Spontaneous mutation of Foxn1 causing lack of thymic tissue | >18 mo | − | + | + | + | + | + | 117 |
| scid |
CB17‐Prkdcscid −/− Defect in DNA protein kinase, no functional rearrangement of antigen‐specific receptors |
<12 mo | − | − | + | + | + | + | 118 |
| NOD‐scid |
NOD.CB17‐Prkdc scid−/− Expression of the scid mutation on the NOD strain background |
<10 mo | − | − | FI | FI | FI | − | 119, 120, 121 |
| NSG |
NOD. Cg‐Prkdcscid IL2rgtm1Wjl/SzJ NOD‐scid combined with IL2rg−/− |
>18 mo | − | − | − | FI | FI | − | 11, 122 |
| NOG |
NOD cg‐PrkdcscidIl2rgtm1Sug Similar to NSG, with truncated IL2rg (enables binding but not signaling of cytokines) |
>18 mo | − | − | − | FI | FI | − | 39, 123 |
| NRG |
NOD. Cg‐ Rag1tm1Mom IL2rgtm1Wjl NOD, RAG1−/− and IL2rg−/− combined |
ND | − | − | − | FI | FI | − | 12, 15, 20 |
| BRG |
BALB/c Rag2null IL‐2Rgcnull interbreeding of NOG and BALB/c‐Rag2null |
ND | − | − | − | FI | FI | − | 14, 124 |
| BRGS |
BALB/c Rag2null IL‐2Rgcnull NOD.sirpa BRG mice with integration of the NOD/Lt Sirpa polymorphism |
ND | − | − | − | FI | FI | − | 125, 126 |
Abbreviations: Com., complement; DC, dentritic cell; FI, function impaired; Foxn1, forkhead box protein; IL2Rgc, interleukin‐2 receptor γ‐chain; MQ, macrophage; ND, not determine; NK, natural killer; NOD, non‐obese diabetic; NSG, NOD‐scid combination; Prkdc, protein kinase DNA activated, catalytic polypeptide; Rag, recombination activating gene; scid, severe combined immunodeficiency; SIRPa, signal regulatory protein a.
3. PATIENT‐DERIVED XENOGRAFTS AND CELL‐DERIVED XENOGRAFTS
Immunodeficient mice grafted with human cancer cells could be classified as PDXs and cell‐derived xenografts (CDXs) based on the type of samples or human cells used in transplantation. 16 CDXs are particularly useful in high through put screening assays and genetic modifications. Although CDXs come with some limitations like the selective proliferation of clonal cells. 17 In comparison with CDXs, PDX mouse models maintain more characteristics of their parental malignancy and thereby are stronger tools for investigating the effects of targeted therapy or chemotherapy. 18 In order to produce PDXs, fresh human tumor tissues are implemented into an immunodeficient mice in which the chance of rejection is lower. The size of tumor tissues is no larger than 2 mm3 and are implemented into the mice subcutaneously or orthotopically meaning at the same site of the tissue‐of‐origin. Normally the immunodeficient mice used for the generation of PDXs have combined T/B/NK cell deficiency and/or macrophage tolerance for human cells like NOD/SCID and NSG/NOG mice. 16 In addition, PDXs is especially beneficial for in vivo screening of targeted therapies using single‐mouse schedule. 19 Following such an approach not only decreases the number of mice and costs of evaluation but also allows for identifying the best treatment in a panel of PDXs and validating the efficacy of tested therapies in the selected target‐specific tumors. Thereby, these mouse models perfectly represent the original patient tumor which would serve as a more reliable platform to predict therapeutic outcomes. As an example, Dr Sidransky conducted a research on 237 patients with different tumor types using PDX mice and validated that these models are able to faithfully conserve the genetic profile of primary tumor. 20 While such pre‐clinical studies have yet to be developed for immunotherapies, investigating chemo‐/radio‐ and targeted therapies in HM are of particular interest.
3.1. Limitations of PDX and CDX models
One of the major drawbacks of both PDX and CDX mouse models for human oncogenesis is that the process of oncologic transformation from normal cells into malignant cells is missing. More importantly, generation of a PDX model is time consuming and can take as long as 6 (or more) months. Adding to this, certain tumor types are difficult to establish as PDX models such as prostate cancer which might be due to innumerable unknown factors in the development of prostate tumor. And as for tumors with genetic heterogenicity, if the genetic heterogeneity is not all represented in the dissected tumor that is passaged these tumors cannot always be recapitulated in serial passages.
4. HUMANIZATION FOR XENOGRAFT
By far, several types of HM have been employed in cancer research. Basically, mice are considered humanized after being engineered to express certain human proteins that are relevant to tumor growth. 21 Needless to say, the ultimate goal of humanization is to develop mice that are fully competent to human immune system and are able to mount proper anti‐cancer immune responses. Thereby, allowing for more accurate interpretations of therapeutic interventions. This objective requires implementing malignant and immune cells, ideally from the same donor, into an environment customized as fully compatible between graft and the host; which ensures that neither rejection of human cells by mouse immune cells nor human immune cell toxicity on the host would occur. Upon successful implementation, human leukocyte precursors are ultimately engrafted and receive full trophic support within the host. As mentioned earlier, only immunodeficient mice are suitable for this purpose. Three widely used mouse strains are (a) NSG mice which are characterized by complement deficiency (preventing lysis of human cells by mouse complement) and loss‐of‐function mutation of Sirpα (decreasing the phagocytosis of human CD47+ cells by mouse macrophages, (b) scid phenotype lacking of T lymphocytes and B lymphocytes as a result of mutations in the Prkdc (protein kinase, DNA activated, catalytic polypeptide) gene and (c) strains with mutation in IL‐2 receptor common γ chain (IL2rg) featuring profound NK cell deficiency 11 , 19 (Table 1).
There are three types of HM developed by two sources of human immune cells: PBMC and human CD34+. (a) Hu‐PBL (peripheral blood lymphocytes), (b) Hu‐CD34+ (also called Hu‐SCR) and (c) BLT mice (bone marrow–liver–thymus) (Figure 1). Each of these HM has their own advantages and limitations. In the following, the process of generating these HM is discussed. Table 2 compares the different features of HM models.
FIGURE 1.
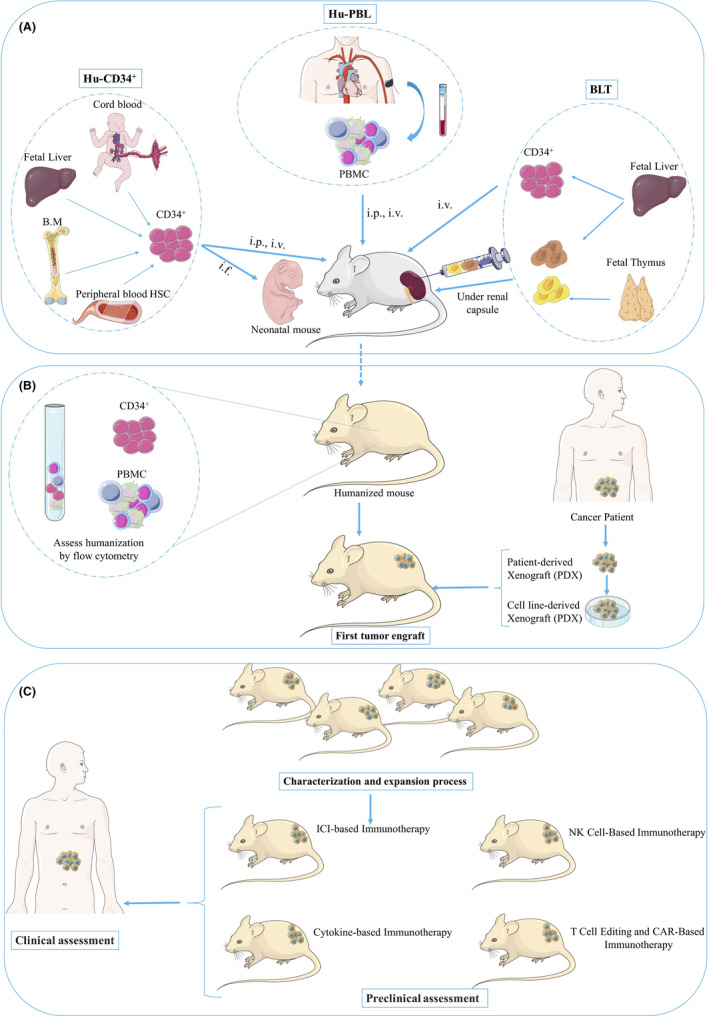
The major steps in the production of humanized mice. A, Demonstrates the humanization process of immunodeficient mice. Hu‐PBL: intravenous (iv) or intraperitoneal (ip) injection of peripheral blood mononuclear cells to an adult immunodeficient mouse. Hu‐ CD34+: IV, IP or intra‐femoral (if) injection of human CD34+ HSCs derived from umbilical cord blood, bone marrow, fetal liver or peripheral blood HSCs into irradiated neonatal or adult immunodeficient mice. BLT: engraftment of human fetal thymus and liver fragments under the renal capsule of the kidney in irradiated adult immunodeficient mice and IV injection of human CD34+ HSCs from the autologous fetal liver. B, Engraftment of human immune system to mouse models is monitored by flow cytometry to determining the percentage of differentiated human cells in the peripheral blood of the mice. Then Cell line‐derived xenografts or patient‐derived xenografts can be implanted into immunodeficient mice (First tumor engraft). C, Upon characterization and expansion of the first tumor‐xenograft mice, the immunotherapy of interest may be conducted. Findings are then translated and applied to the adapted therapy of the patient. BLT, bone marrow–liver–thymus; B.M, bone marrow; Hu‐PBL, peripheral blood lymphocytes; PBMC, peripheral blood mononuclear cell
TABLE 2.
Summary and comparison of different humanized mouse models
| Hu‐PBL | hu‐HSC | BLT | References | |
|---|---|---|---|---|
| Method, cell source and mice used | i.p. injection of human PBMC. SCID, NOD‐SCID, NSG, NOG | Intrahepatic injection of CD34+ HSC into newborn mice within 72 h of birth Intravenous injection of CD34+ HSC into adults. Rag1−/− gc−/−, Rag2−/− gc−/−, NSG, NOG | Co‐implantation of thymic fragments and human fetal liver under kidney capsule with iv injection of autologous CD34+ HSC. Rag1−/− gc−/−, Rag2−/− gc−/−, NOD‐SCID, NSG | 11, 14, 39, 43 |
| Preconditioning | − | Sub lethal irradiation | Sub lethal irradiation | 43 |
| Human B cell | + | + | + | 124, 127, 128 |
| Human T cell | + | + | + | 124, 129 |
| Human NK cell | − | −/+ with IL15 or Flt3L | − | 61, 124 |
| Human macrophages | − | + | + | 56, 59 |
| Human dendritic cells | − | + specially with Flt3L | + | 56, 62 |
| Neutrophils | − | −/+ with IL3, GM‐CSF and M‐CSF | + | 56, 127 |
| Primary immune response | − | + Humoral and cellular. IgM and weak IgG | + Humoral and cellular. IgM and weak IgG | 123, 130 |
| Advantages |
Easy to prepare and Fast to establish T cells are HLA restricted and functional such as memory T cells |
Easy to prepare Multilineage hematopoiesis Primary immune response Mucosal engraftment |
Multilineage hematopoiesis Primary immune response Presence of human thymus Human HLA‐restricted Mucosal engraftment |
11, 13, 14, 124, 129, 131, 132 |
| Disadvantages |
More prone to GVHD No primary immune response Lack B and myeloid cell engraftment No multilineage hematopoiesis Just suitable for experiments below 3 mo |
Low NK and IL‐15/ IL‐15Rα requirement to increase function No human HLA restriction Immune cells differentiate more than 10 wk |
Poor class switching Possibility of GVHD Surgery needed Requires human fetal tissue Immune cells differentiate more than 10 wk |
11, 13, 14, 129, 131, 132 |
B.M, bone marrow; BLT, bone marrow–liver–thymus; Flt3L, Flt3 ligand; G‐CSF, granulocyte‐colony‐stimulating factor; GM‐CSF, granulocyte/ macrophage colony‐stimulating factor; GVHD, graft versus host disease; HLA, human leukocyte antigen; HSC, hematopoietic stem cell; Hu‐PBL, peripheral blood lymphocytes; IgG immunoglobulin G; IgM immunoglobulin M; IL, interleukin; IL‐15Rα, interleukin‐15 receptor α; NOD, non‐obese diabetic; NSG, NOD‐scid combination; PBMC, peripheral blood mononuclear cell; Rag, recombination activating gene; scid, severe combined immunodeficiency.
4.1. Hu‐PBL model
Immunodeficient mice reconstituted with PBMCs and tumors, are called Hu‐PBL models which are the simplest version of humanization. Generation of Hu‐PBL models starts by isolating PBMCs using Ficoll‐Hypaque gradient centrifugation by which mainly neutrophils are removed. Beside the mature human leukocytes in PBMC inoculum, a few HSCs exist which are unable to colonize the murine host because of the lack of a proper microenvironment. 22 Moreover, human B lymphocytes and myeloid cells are observed at low levels which may be due to the lack of the human cytokines required for their survival. 23 , 24 PBMCs could be transplanted into adult mice intravenously, intraperitoneally, or intrasplenically. The major limitation of this model is that it leads to graft‐versus‐host disease (GVHD). 13 , 23 , 25 The onset of GVHD is directly associated with the degree of human T cell, in particular CD4+ T cell, engraftment as well as prior sublethal irradiation. 23 , 26 , 27 , 28 Therefore, therapeutic‐relevant outcomes evaluation is limited to the weeks after PBMC injection and before the onset of GVHD. 13 , 23 Generating these mouse models have allowed for the identification and characterization of ICIs antibodies. 28 , 29 Furthermore, these “immune‐avatar” mice can be utilized to investigate the immune‐mediated effects of antibodies targeting cancer cell antigens and allow for the infiltration of patient‐derived tumors by lymphocytes. 30
4.2. Hu‐CD34+ model
Injection of CD34+ HSCs isolated from bone marrow (BM), cord blood or fetal liver of the patient allows for generation of various types of human immune cells in the murine host and triggers tolerance against mouse tissues. These cells can be injected intravenously, intraperitoneally, intrafemorally and also intracardially and intrahepatically into neonatal or adult immunodeficient mice 11 , 13 , 31 (Figure 1). Several factors determine the success of engraftment such as HSCs source, route of injection, strain, age and sex of the recipient. For instance, in newborn or mice up to 4 weeks of age T cell development occurs faster compared with adult mice. 32 In a study, Brehm et al 15 evaluated engraftment outcome of different mouse strains and routs of injection in adult or neonatal mice and showed that transplantation into newborn NOD‐scid IL2Rγ‐ and NOD‐Rag1‐ IL2Rγ‐mice resulted in higher levels of human immune cell engraftment compared with BALB/c‐ Rag1‐ IL2Rγ‐ mice. Generation of Hu‐CD34+ mice starts with irradiation of mice that are 5‐12 weeks of age in order to help HSCs engraftment. Then, human CD34+ cells are transplanted into irradiated mice. Around 10‐12 weeks of age, engraftment of the human HSCs in the murine host can be confirmed by assessing for differentiated human CD45+ cells (leukocyte common antigen) in the Peripheral blood of the mice using flow cytometry. 33 If the mice have more than 25% human CD45+ cells in their peripheral blood then the engraftment of human immune system is considered successful. Now the HM could be inserted with specific PDXs and an immunotherapeutic agent could be subsequently applied for testing. Alternative methods to irradiations are busulfan 34 and antibody‐mediated deletion of mouse progenitor cells. 35 In the same context some mouse strains like the NOD, B6. SCID Il2rγ−/−KitW41/W41 (NBSGW) mice support the transplantation of HSCs without irradiation. 36 , 37
Although in the Hu‐CD34+ models all human hematopoietic lineages are represented, but not all are functionally fully developed. 13 For instance, the majority of the human B cells are immature CD5+ B cells, mainly because at the transition phase the process of B cell differentiation is blocked and eventually results in the accumulation of B cell precursors in the spleen. 38 , 39 Similarly, the differentiation of the myelomonocytic lineage is impaired and monocytes are phenotypically immature. 40 In addition, CD4+ T lymphocytes display memory phenotypes, and both T and NK cells have functional impairment. 39 , 41 Mouse thymus supports human T cell development; however, the question of major histocompatibility complex (MHC) restriction is yet to be elucidated. According to Halkias et al 42 human thymocytes show similar behavior in mouse and human thymic environments. Also, they can interact with both HSCs and mouse tissue in HIS mice thymus. Adding to this, Watanabe et al demonstrated that the mouse thymic environment, and not the mouse I‐A MHC molecule, is crucial for the development of human T cells, suggesting that the human CD4+ T repertoire is restricted by human MHC class II molecules and murine MHC. However, these animals have very poor human thymopoiesis. Mentioned limitations may restrict the value of the Hu‐CD34+ model in studies of human immunology and immunotherapy. 43
4.3. BLT model
In the BLT model, implemented human fetal liver and thymus create a human thymic microenvironment that promotes human T cell development and selection. 44 In order to achieve this purpose, pieces of human fetal thymus and liver are transplanted under the kidney capsule. Then, autologous CD 34+ cells that are isolated from the liver are injected which allow HLA restriction. 45 By doing so, human thymopoiesis takes place in engrafted human thymic tissues. More importantly, evidences have shown that BLT models are able to generate potent human immune responses since these animals have the potential to reject allogenic and xenogeneic grafts, trigger HLA‐restricted antigen‐specific human T cell reaction, and produce antigen‐specific human IgM and IgG antibodies with subclass switching upon immunization or xenograft implantation. 46 , 47 , 48 The advantage of this approach would be exclusive positive selection of human T cells in thymus, but on the other hand, high‐affinity T cells against mouse MHCs are not eliminated. As a consequence, the incidence of GVHD in these models is higher than other CD34+HSC engrafted models. However, there are some studies indicative of decreased GVHD in BLT models. As an example, mouse dendritic cells (DCs) can migrate into human thymic grafts in BLT models and take part in educating thymic human T progenitor cell and reduce the incidence of GVHD. In addition, pipetting of human thymic grafts before transplantation and cryopreservation can remove existing human T cell progenitor cells which would further alleviate GVHD. 49 , 50 , 51 Implementation of mesenchymal stem or progenitor cells could further improve the BLT models since they are capable of creating BM environment. 52
Preferably, HM should be generated with the immune system from which PDXs will be produced. Even so, current mouse models are providing a platform for further development of fully personalized and humanized mouse models that can be used for cancer immunology and immunotherapy research. There are several strategies that support the improvement of HM production which will be discussed in the following.
5. IMPROVING HUMANIZED MICE BY OVERCOMING CURRENT LIMITATIONS
Improving current HM models to better represent the human immune system would permit assessment of new biological therapies. Developing humanized have progressed during years; however, some aspects need to be improved like incidence of GVHD, incomplete engraftment of immune cells, lack of human cytokines and growth factors. On the other, HSCs derived from cancer patients are not optimal for repopulating mice. Some areas that require more development for better recapitulating human immune responses are as follow: (a) Innate immune cell development and function, (b) B cell maturation and antibody responses, (c) secondary lymphoid organ development, (d) New robust renewable sources of human cells and tissues for grafts, (e) Development of robust human HLA‐restricted T cell responses, (f) Increase engraftment rate of HSCs, (g) Reduce GVHD, (h) Infrastructure for increasing community access of HM model. Figure 2 summarizes some of the limitations ahead of these improvements as well as some of their related solutions. In the following, we will discuss about some of the improvement strategies for refinement of HM.
FIGURE 2.
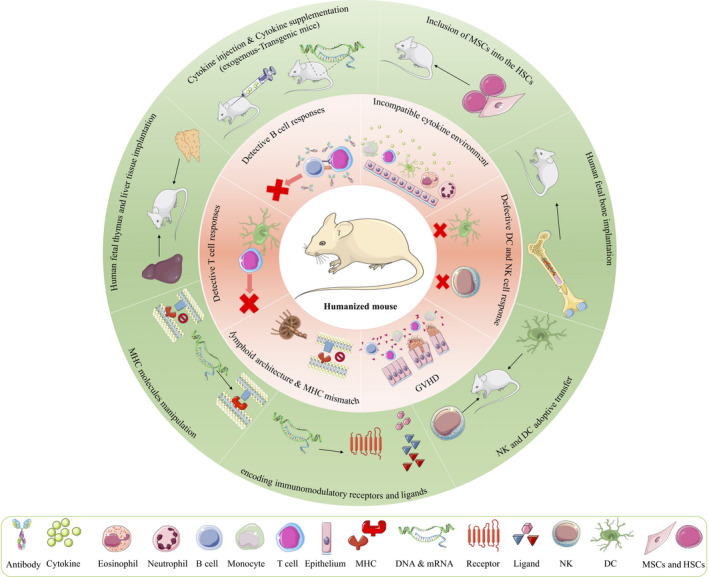
Schema showing the areas that require development and optimization in HM model. The pink ring represents immunological limitations in HM and the green ring provides the possible improvement strategies. CAR, chimeric antigen receptor; DC, dendritic cell; MSCs, mesenchymal stem cells; HSCs, hematopoietic stem cells; MHC, major histocompatibility complex; NK, natural killer; GVHD: graft‐versus‐host disease
5.1. The role of cytokines and growth factors in upgrading HM
Cytokines signaling in the environment of engrafted HSCs and differentiating immune cells directly affects the orderly maturation and thereby trafficking into the tumors on the organism. 53 Several studies focusing on improving the cytokine environment within HM is ongoing. It has been revealed that integrating a population of mesenchymal stem cells (MSCs) into the HSCs destined for engraftment may also modify the eventual reconstitution of the myeloid cell lineage within the HM. According to Chen, Huang, and Womer, co‐culturing of HM BM cells with MSCs, which have shown the evidence of immunoregulatory function and have the potential to produce cytokines and growth factors, improves the viability of newly differentiating DCs. 54 Preliminary studies by Shultz et al 11 by NSG model revealed that administration of IL‐7 increased the production of T cells in HM. Chen et al 55 have shown that administration of IL‐5 and Flt‐3/Flk‐2 cytokines, that are encoded on plasmids expressed in hepatocytes, enhanced the levels of NK cells, while granulocyte‐macrophage colony‐stimulating factor (G‐MCSF) and IL‐4 elevated DCs and macrophage colony‐stimulating factor (M‐CSF) was able to increase the number of macrophages and monocytes detectable within the HM.
Despite the positive effects of addition of these cytokines on immune cell differentiation and expansion, non‐physiological concentration of them within HM would misdirect cell development and trafficking. Different genetic backgrounds of HM have been genetically engineered to express IL‐3, M‐CSF, G‐MSCF, Thpo or Sirpα. In order to improve the expression of these critical cytokines, Rongvaux et al 56 produced strains of Rag 2(−/−)‐Il2γnull mice called MITRG characterized by targeted knock‐ins of the human genes encoding IL‐3, M‐CSF, GM‐CSF and Thpo. After the engraftment of human CD34+, these MITRGs produced T and B cells, as well as functional NK and myeloid cells, that had the capacity to infiltrate cell‐line derived melanoma tumors and change their growth through a vascular endothelial growth factor (VEGF)‐dependent mechanism. Wunderlich et al 57 developed another transgenic humanized strain. They produced the NSG‐SGM3 (NSGS) mouse, which expresses human SCF, GM‐CSF, and IL‐3, to facilitate the study of acute myeloid leukemia via increased production of mature myeloid cells. Another strain, NOG–IL‐2 Tg mouse, was developed by Katano et al, through inserting a human IL‐2 transgene into a NOG background. The HM generated from this strain produced a diverse set of NK cells with the ability to target both introduced leukemia and lymphoma cells. 58
In the same context, in the NOD background, NSG SGM3 engineered HM were developed by some modifications on NSG mice to express human SCF (c‐kit ligand), GMCSF and IL‐3 genes which were encoded by cDNA constructs that randomly integrate and are driven by a CMV promoter. 59 In addition, NOG‐EXL were developed by engineering NOG mice to ubiquitously express human GMCSF and IL3 genes under control of the SRa promoter. 60 Furthermore, Flavell's group developed SRG‐15 engineered HM which are BALB/cRag2−/− IL2rgc −/− knock‐in for human SIRPα and IL‐15. These HM showed increased development and function of NK cells, CD8+ T cells, and tissue‐resident innate lymphoid cells. 61
Although generation of mouse strains bearing these transgenes can help in creating a functional myeloid lineage, but they are relatively difficult to breed and their development complicate the generation of the large cohorts necessary for immune therapy cancer studies. Different types of DC exist in mice which are not homologous human DCs. Also, in HM the development of DCs and their maturation are not optimal. In order to overcome this, novel HM models based on the BALB/c Rag2 ( −/− ) Il2rg( −/− ) Flt3( −/− ) (BRGF) and NOD. Cg‐Rag1tm1MomIl2rgtm1Wjl/SzJ Flk2/Flt3−/− (NRGF) mice containing a mutated receptor tyrosine kinase Flk2/Flt3 were produced. Development of human DCs in BRGF and NRGF mice are improved upon exogenous administration of human Flt3 ligand (Flt3L) after HCT which results in the marked increase of human NK and T cell population. 62 , 63
5.2. MHC modification and limiting GVHD
Myelopoiesis, a process by which different population of leukocytes are generated, is inevitable and associates with pathology in animal models of GVHD. CD11c+CD14+ is the dominant donor‐derived population of leukocytes in GVHD. It is observed that GVHD‐isolated macrophages are able to stimulate greater activation and proliferation of allogenic T cells, secret higher levels of inflammatory cytokines in steady‐state and mediate direct toxicity. These observations accentuate the function of human macrophages and the potential to prevent and treat GVHD by exploiting their functionality. 64 Another important player in the development of GVHD are T lymphocytes. According to a recent study on a humanized mouse model, donor monocytes are able to activate host skin‐resident T cells and result in GVHD‐like dermatitis. The phenomenon suggests a pathogenic role in development of acute GVHD by host tissue‐resident T cells. 65
Several studies are focusing on the improvement of human T cells and preventing GVHD in HM; achieving this purpose will qualify HM with functional T‐cell receptors (TCRs) that are able to interact with the matched HLA complexes on antigen‐presenting cells. In order to do so, different strategies have been tested based on the genetic manipulation of the MHC molecules. As for Hu‐CD34+ mice, defective T cell function was somewhat associated to the mismatch between human and mouse MHCs. Conducting this refinement relies on the substitution of the mouse MHC I and II by different haplotypes. Danner et al 66 used NOD‐Rag1‐IL2γ−/− mice that expressed human HLA‐DR4 allele and demonstrated that upon engraftment with HLA‐matched HSCs, the immune system in these mice was reconstituted with high numbers of functional B and T cells and also was capable of appropriate response to immune challenge. Patton et al 67 showed that when NSG mice expressing human allele HLA.A2.1 were engrafted by CD34+ cells from a HLA.A2.1 matched donor, they were not as efficiently humanized as NSG controls. They postulated that this could be due to the alloreactivity between mouse and human peptide antigens bound to HLA proteins. 6
In another study by Kim et al 68 a transgenic NRG mouse called “DRAG” was produced that could express HLA‐DR4. This strain of mice which was transplanted with HLA‐DR4+ HSCs was able to develop higher number of CD4+ T cells and also higher concentration IgG and IgM. Similarly, BRGSA2DR2 mice, which are generated from BRGS mice and are able to express human HLA‐A2 and DR2 transgenes, revealed faster development of CD4+ and CD8+ T cells and higher concentration of IgG. 69 Lone's team developed a mouse called “HUMAMICE” that is a combination of both murine MHC deficient and HLA transgene expressing mice. This engineered mouse lacks T and B cell as a result of rag mutation, NK cells due to IL2Rgc and has no residual cytolytic activity because of perforin KO. 70 Transplanting HLA‐matched PBMCs in HUMAMICE reconstituted with human immune cells was not followed by signs of GVHD. Moreover, these mice develop functional human T and B cell as evidence of vaccination with Hepatitis B virus (HBV) showed production of HBV‐specific antibodies. 70 Major limitation ahead of this approach is the difficulty to find HSCs that express a particular combination of HLAs.
Recently by using CRISPR/Cas9 in NOG mice, Ka et al established a novel beta‐2 microglobulin (B2m) KO mouse model. A modified dKO (dKO‐em) mouse model is established by crossing B2m KO mice with I‐Ab KO mice. dKO‐em mice showed high engraftment efficiency as well as no signs of GVHD after the transfer of human PBMC. Moreover, engrafted human PBMCs significantly survived longer in the peripheral blood and spleens of dKO‐em mice, compared with dKO‐tm mice. Thus, dKO‐em mice may count as a promising model for preclinical investigations of novel therapeutics for human diseases. 71
5.3. Other improvement strategies
Generally, HM mice undergo myeloablative conditioning before implementation of human HSCs in order to provide the required space in the host BM niche for the substitution of human HSCs engraftment. 11 Each strain of mice reacts differently to the irradiation. For instance, the scid mice are more sensitive to radiation‐induced DNA damage compared with Rag1null or Rag2null mice. 12 As mentioned earlier, the c‐kit (CD117) mutant mouse was found to be a suitable host for human HSCs engraftment which requires no prior irradiation. Given that c‐kit plays an important role in HSCs maintenance and differentiation, NSGW41 mice that carry the w41 mutation in c‐kit, show reduced HSCs numbers which lead to lower competition and better engraftment of human HSCs. 36 , 72
A growing number of engineered mice are being commercially developed by modifying immunocompetent mice to express one or more fully human genes. Also, by generating “humanized” knock‐ins mice encoding negative or positive immunomodulatory receptors and ligands such as CD47, Programmed death‐ligand 1 (PD‐L1), B and T lymphocyte attenuator (BTLA), CD137, T cell immunoglobulin and mucin domain‐containing protein 3 (TIM3), lymphocyte‐activation gene 3 (LAG‐3), inducible T‐cell costimulatory (ICOS), glucocorticoid‐induced tumor necrosis factor receptor (GITR), OX40, OX40L 43 . These mice are particularly utilized for the investigation of checkpoint combination therapy. Mice expressing “humanized” programmed cell death 1 receptor (PD‐1) or CTLA‐4 molecules have been also beneficial for separating efficacy and autoimmunity induced by anti‐CTLA4 antibodies 73 or characterizing a clinical candidate anti‐PD‐1 antibody. 74
As for autoimmunity complications, Khosravi‐Maharlooei et al investigated the role of thymus in development of multi‐organ autoimmunity in HIS mice. They observed that autoimmunity was developed earlier in HIS mice with a native mouse thymus than thymectomized mice with a thymocyte‐depleted human thymus graft. Structural defect in the native mouse thymus correlated with impairment in the negative selection of transgenic TCR expressing thymocytes with the capacity to recognize self‐antigens. It appears that disease developed in an indirectly and without recognition of antigens on recipient mouse MHC. Even if human thymus grafts have normal structure and negative selection, failure in tolerating human T cells that recognize mouse antigens being presented on HLA molecules may explain the development of autoimmunity. 75 This suggests generating methodologies that bypass human autoimmunity in the next generation of HIS mice.
6. PRE‐CLINICAL EXPERIENCES OF HUMANIZED MICE
6.1. ICI‐based and monoclonal antibody therapy
Humanized mice engrafted with tumors are being used to better understand how checkpoint blockade interacts with the immune system and also to test the efficacy and effects of immunomodulation agents. Given the relatively simple accessibility and handling of human PBL samples, the Hu‐PBL model is widely utilized in evaluating the interactions between human immune cells such as T cells and NK cells as well as human tumors in vivo. 76 For instance, in a study by Ignacio Melero et al it was observed that this model is useful for investigating the effect of human PD‐1 (Nivolumab) and CD137 (Urelumab) antibodies in in vivo T cell‐mediated anti‐tumor responses. 28 Furthermore, Wang et al 77 reported that PD‐1 targeted immunotherapy can be installed in Hu‐CD34+ humanized NSG mice having CDXs and PDXs partial HLA matched human tumor which indicates the value and efficacy of the Hu‐CD34+ model for cancer immunotherapy investigation. Unusual long serum half‐life of IgGs and Fc domains are due to their rescue and recycling via the neonatal Fc receptor (FcRn). There is a significant difference between rodent and human FcRn reactivity, rendering wild type rodents an inadequate model for studying monoclonal antibody therapy. To overcome this problem with the advance of genetic engineering, mouse models have been established expressing human FcRn, and lacking mouse FcRn protein like NSG FcRn‐/‐ hFcRn Tg model. 78
One of the major limitations associated with using antibodies to target checkpoint inhibitors is the incidence of cytokine release syndrome (CRS). As demonstrated by the TGN1412 (anti‐CD28) clinical trial, screening these antibodies in HM and non‐human primate may not necessarily represent the response of a human immune system. 79 To address this issue, two humanized mouse models were developed for the detection of a CRS. NRG‐AB0 or NSG mice were engrafted with PBMC and injected with muromonabCD3(120), or TGN1412. 80 Utilizing this approach in Hu‐CD34+ and BLT models would allow evaluating innate immune cell reactions to antibodies targeted against these cell populations. 81
As more cancer immunotherapy treatments are tested in clinical trials, an association between immune‐mediated tumor killing responses and CRS are observed by clinicians. Due to these observations, pre‐clinical studies that are capable of recapitulating CRS in HM are gaining more interest. In a recent study, HM were engrafted with a diffuse large B cell lymphoma (WSU‐DLCL2) and treated with either novel CD20‐T‐cell bispecific antibody (TCB) or obinutuzumab (anti‐CD20 monoclonal Ab). 82 CD20‐TCB which contains two CD20 binding domains and one CD3e domain, incited more extensive cytotoxic responses than obinutuzumab. Moreover, administration of CD20‐TCB was associated with enhanced expression of inflammatory cytokines which indicates that CRS responses were not generated by obinutuzumab treatment. Such pre‐clinical experiments on HM highlight the value of them in testing protocols that are designed to maximize both efficacy and safety. The key component of pre‐clinical assessments is the question related to immunotherapy mediated toxicity, especially in the context of CRS, and also the requirement for a related reliable assay. As an example, recently a group at the US Food and Drug Administration (FDA) reported the testing of CRS using monoclonal antibody therapies that are known to elicit strong cytotoxic response in the BLT‐HIS mice. 83 Models that can accurately and reliably predict the induction of CRS by immune therapeutics are scarce. Recently Chunting Ye et al reported the development of a HM model based on the NGS mouse to investigate CRS in vivo. NSG‐MHC‐DKO, PBMC‐engrafted NSG and NSG‐SGM3 mice were employed in order to study cytokine release in response to treatment with monoclonal antibody immunotherapies. Results showed that among the three mouse models, PBMC‐engrafted NGS models are quick, sensitive and reproducible platform for screening novel therapeutics for CRS. 84 More recently, others developed a model for predicting CRS while minimizing GVHD by spleen mononuclear cells (SPMCs). They reported that NSG mice reconstituted with PBMC‐and SPMC better predicted OKT3‐mediated CRS. The SPMC model allows generation of large experimental groups while NSG‐dKO mice are able to mitigate the limitation of early GVHD. 85
6.2. NK cell‐based and cytokine‐based immunotherapy
Another promising cellular immunotherapy for cancer is adaptive NK cell therapy. Recently some progression has been made in stimulating NK and NKT anti‐tumor activity utilizing HM models in various cancers such as glioblastoma, colorectal, ovarian and pancreatic cancer. 86 , 87 , 88 , 89 Moreover, cytokine therapy has caught attentions in the efforts to elicit NK and NKT cell antitumor activities. In a study, human neuroblastoma cell line and human NKT cells expanded ex vivo were injected into HSC‐engrafted NSG mice. 90 It was observed that NKT cells resided within the tumor associated macrophages (TAMs) in the tumor microenvironment. However, the survival and function of NKT was inhibited by CCL20 secreted by TAMs favoring tumor growth. Lie et al showed that transducing NKT with Il‐15 before transferring into mice led to decreased tumor growth as a result of increased NKT survival and suggested a role for IL‐15 cytokine therapy. 90 Immune therapy with IL‐15 was utilized to expand the NK cell populations of Hu‐CD34+ NSG mice implemented with human breast cancer and the result was enhanced proportions of activated CD56+CD27− NK cells. 91
Beside IL‐15, the effect of IL‐12 in stimulating the immune system to attack tumor cells has also been investigated in humanized tumor‐bearing mice. As an example, NHS‐IL12 is an antibody‐IL12 fusion protein targeting the naked histones/DNA complexes that are found in necrotic tissues such as tumors. 92 NHS‐IL12 was utilized in conjunction with antibody‐complexed IL12 (IL2MAB602) or IL‐7 (FcIL7) in the Hu‐SRC‐SCID model of rhabdomyosarcoma. 93 NHS‐IL12/IL2MAB206 enhanced tumor infiltrates of NK cells, T cells and macrophages. 94
6.3. T cell editing and CAR‐based immunotherapy
One of the strategies of targeting immune system to upgrade its anti‐tumor activity is redirecting T cell specificity via transgenic TCR or chimeric antigen receptor (CAR) engineered T cell therapy. CAR‐T cells are MHC independent and thereby can be redirected to any target of interest. 95 Recently, using CAR‐T therapy in HM have been employed to broaden the scope of cancer treatment as well as optimizing the safety and efficacy of CAR manipulation. 96 For instance, CARs targeting mesothelin for mesothelioma, CD44v6 for AML and multiple myeloma and ROR1 for mantle cell lymphoma have been tested in NSG humanized mice. 93 , 97 , 98 Adding co‐stimulatory motifs in the CD3ζ in the intracellular signaling domain increases CAR signaling. A CAR without co‐stimulatory receptor (CCR) is designed for a single antigen while incorporating CCR increased the specificity of CAR for a second antigen. Humanized mice have been extensively used to assess and compare the functions of CCR such as ICOS, CD27, CD28 AND 4‐1BB. 99 , 100 It is documented that CARs incorporated with co‐stimulatory domains are more effective in targeting tumors. In a Hu‐PBL model, combining PSCA‐CAR and PSMA‐CCR prostate antigens eradicated tumor cell lines that expressed both antigens and shaped anti‐tumor immune response to PSCA+PSMA‐ tumors. 101 Jakobsen et al reported that Hu‐PBL model could be employed to assess the efficacy of Bi‐specific TCR‐anti‐CD3 regimen for the treatment of LAGE1‐ and NY‐ESO‐1‐ positive human tumors. 76 Another approach to test the safety of CARs is using mRNA transfection, as opposed to viral transduction, as a mechanism of CAR generation. The advantage of this method is that it does not involve the integration of DNA into the genome, thereby removing the possibility of genomic editing. MRNA transduction has been utilized in developing anti‐CD20 NK cells in Hu‐PBL model targeted to Non‐Hodgkin's Lymphoma 102 or anti‐mesothelin CAR T cells in a Hu‐PBL model targeted to mesothelioma. 93
Another targeted CAR‐based therapy is engineering T cells to express anti‐CD19 CARs. Patients with B cell malignancies showed positive response to CD19‐targeted CAR‐T cell therapy. 103 , 104 , 105 However, many showed severe adverse reaction or relapse after therapy largely due to toxicities of unknown mechanisms. 105 , 106 Development of pre‐clinical models would be beneficial in investigating the underlying mechanisms of relapse and toxicity. PDX models engrafted with human B acute lymphocyte leukemia (ALL) revealed to be useful in evaluating CD19‐targeted human CART cell therapy, however, these models either lack host immunity or involve allo‐ and/or xeno‐immune responses. 107 On the other hand, leukemic HM models have genetically‐identical (autologous) primary B‐ALL and a functional human immune system which make them a better model for CD19‐targeted CAR T cell therapy. 108 Another favorable feature of HM model is that the anti‐CD19 CAR‐expressing human T cells are also autologous to the human components (either normal or malignant human cells) and tolerant to the mouse antigens, thereby do not elicit xeno responses against mouse antigens or allo responses against human.
Furthermore, BLT mice can be modified to as a TCR transgenic HM for studies of human T cell adaptive immunotherapy. 109 A melanoma antigen (MART‐1)‐specific TCR transgenic HM model is developed by cotransplanting autologous human CD34+ FLCs transduced with lentiviral vectors containing HLA‐A∗0201 restricted MART‐1 specific TCR genes and HLA‐A∗0201+ human fetal thymic tissues into sub‐lethal irradiation pre‐conditioned NSG mice. 110 Upon employing this model it was revealed that anti‐melanoma effects mediate by adaptive transfer of human MART‐1 TCR+ T cells was remarkably improved by adding rapamycin for MART‐1 TCR+ human T cell expansion in vitro and simultaneous supplementation with human IL‐15 in vivo. 109 Recently, other uses of HM have also shown that exosomes derived from phosphoantigen‐expanded Vδ2‐T cells (Vδ2‐T‐Exos) contained MHC class I and II, CD80, CD86, TRAIL, FasL and NKG2D. Administration of Vδ2‐T‐Exos could effectively control BV‐associated tumors in Rag2‐/‐γc‐/‐ and HM. Given that the expansion of Vδ2‐T cells and ex vivo preparation of autologous Vδ2‐T‐Exos from cancer patients in large scale is challenging, the antitumor activity of allogeneic Vδ2‐T‐Exos was explored in humanized mouse cancer models. 111
Our focus was on pre‐clinical experiments of immunotherapy but HM revolutionized the diagnostic and therapeutic approaches. Moreover, HM are providing a suitable platform for studies of human infectious disease like human immunodeficiency virus, GVHD, regenerative medicine, allergies, and immunity.
7. FUTURE
Humanized mice models are powerful tools for immunotherapy research in the era of cancer immunotherapy. Despite advances in establishing HM, they do not entirely recapitulate a functional human immune system. Thus, efforts for improving HM are ongoing. Scientists from different biomedical disciplines are testing innumerable strategies such as reducing graft rejections, boosting human cell reconstitution, improving human‐specific responses and supporting critical immune cell subsets. Moreover, there are some noticeable obstacles which need to be solved soon. For example, if the HM is engrafted with an immune system from one person and the tumor from another person, then the formed immune response might be as the result of tissue incompatibility, rather than being reflective of the tested treatment. One of the solutions for the issue of MHC incompatibility is using induced pluripotent stem cell (iPSC) technology. This method allows for the use of patient‐specific iPSC which reduces the chance of tissue incompatibility and also provide a renewable source of autologous cells. Still, more comprehensive and functional immune systems need to be generated in HM. More specifically, there is an ongoing need to identify new approaches providing the platform for autologous experiments of engrafted immune cell and diseased tissue from the same individual. Thereby, allowing for more accurate understanding of disease progression and treatment efficacy.
Considering the complexity of T cell development, even if it is possible to recreate the human thymic environment, establishing the same TCR repertoire of that particular patient seems very unlikely. Eventually, the utmost purpose is the formation of specific anti‐tumor immune responses by these human‐derived T cells. Analyzing immunosuppressive cells such as T regulatory cells and M2 macrophages could also help in drawing the whole picture of the interplay between the patient's immune system and the tumor that has managed to grow.
More recently, it has been reported that microbiota, particularly in the gut, affects the efficacy of cancer immunotherapy. Given that germ‐free mice lack microbes, researchers have established human microbiota‐associated mice developed from these mice by fecal microbiota transplantation. 112 , 113 Importance of the microbiota calls for further research on the effect of microbiota on biological responses. Another challenge in the development of HM is the incomplete cross‐compatibility between the murine stroma and transplanted human hematopoietic cells. Recently, complementary strategies have been developed to supplement in vivo xenotransplantation models such as in vivo utilization of three‐dimensional human BM organoids and ex vivo deployment of bioreactor models. 114 Cancer‐associated fibroblasts are normaly observed within the stroma of various cancers, including lung, breast, colon, and pancreatic carcinomas. 115 Recently reported that, in preclinical mouse models, fibroblast‐activating protein a targeting OMTX705 represents a novel a model for cancer immunotherapy study. 116 In conclusion, as humanized PDX are evolving and refining to better represent the human biological system, they are considered as an appropriate platform in personalized medicine and cancer immunotherapy.
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ACKNOWLEDGMENT
This work was supported by Zhejiang Provincial Science and Technology Projects (grant no. LGD19H160001 to KTJ, LGF18H160029 to JLD), and National Natural Science Foundation of China (No. 81672430 to XZM).
Jin K‐T, Du W‐L, Lan H‐R, et al. Development of humanized mouse with patient‐derived xenografts for cancer immunotherapy studies: A comprehensive review. Cancer Sci. 2021;112:2592–2606. 10.1111/cas.14934
Ke‐Tao Jin, Wen‐Lin Du, and Huan‐Rong Lan contributed equally to this work.
Contributor Information
Jin‐Lin Du, Email: dujinlin@zju.edu.cn.
Xiao‐Zhou Mou, Email: mouxz@zju.edu.cn.
REFERENCES
- 1. Wege AK. Humanized mouse models for the preclinical assessment of cancer immunotherapy. BioDrugs. 2018;32:245‐266. [DOI] [PubMed] [Google Scholar]
- 2. Yang YF, Zou JP, Mu J, et al. Enhanced induction of antitumor T‐cell responses by cytotoxic T lymphocyte‐associated molecule‐4 blockade: the effect is manifested only at the restricted tumor‐bearing stages. Cancer Res. 1997;57:4036‐4041. [PubMed] [Google Scholar]
- 3. Azizi G, Jamee M, Yazdani R, et al. CTLA‐4 expression in CD4+ T cells from patients with LRBA deficiency and common variable immunodeficiency with no known monogenic disease. J Investig Allergol Clin Immunol. 2018;28:422. [DOI] [PubMed] [Google Scholar]
- 4. Hoos A, Ibrahim R, Korman A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37:533‐546. [DOI] [PubMed] [Google Scholar]
- 5. Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann Oncol. 2016;27:1190‐1198. [DOI] [PubMed] [Google Scholar]
- 6. Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized mouse xenograft models: narrowing the tumor‐microenvironment gap. Cancer Res. 2016;76:6153‐6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shultz LD, Schweitzer PA, Christianson SW, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz‐scid mice. J Immunol. 1995;154:180‐191. [PubMed] [Google Scholar]
- 8. Takenaka K, Prasolava TK, Wang JCY, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313‐1323. [DOI] [PubMed] [Google Scholar]
- 9. Cao X, Kozak CA, Liu YJ, Noguchi M, O'Connell E, Leonard WJ. Characterization of cDNAs encoding the murine interleukin 2 receptor (IL‐2R) gamma chain: chromosomal mapping and tissue specificity of IL‐2R gamma chain expression. Proc Natl Acad Sci USA. 1993;90:8464‐8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugamura K, Asao H, Kondo M, et al. The interleukin‐2 receptor γ chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Ann Rev Immunol. 1996;14:179‐205. [DOI] [PubMed] [Google Scholar]
- 11. Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz‐scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477‐6489. [DOI] [PubMed] [Google Scholar]
- 12. Pearson T, Shultz LD, Miller D, et al. Non‐obese diabetic–recombination activating gene‐1 (NOD–Rag 1 null) interleukin (IL)‐2 receptor common gamma chain (IL 2 rγnull) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol. 2008;154:270‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175‐3182. [DOI] [PubMed] [Google Scholar]
- 14. Traggiai E, Chicha L, Mazzucchelli L, et al. Development of a human adaptive immune system in cord blood cell‐transplanted mice. Science. 2004;304:104. [DOI] [PubMed] [Google Scholar]
- 15. Brehm MA, Cuthbert A, Yang C, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rγnull mutation. Clin Immunol. 2010;135:84‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian H, Lyu Y, Yang YG, Hu Z. Humanized rodent models for cancer research. Front Oncol. 2020;10:1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao D, Chen Y. Organoid development in cancer genome discovery. Curr Opin Genet Dev. 2015;30:42‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izumchenko E, Paz K, Ciznadija D, et al. Patient‐derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595‐2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao H, Korn JM, Ferretti S, et al. High‐throughput screening using patient‐derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318‐1325. [DOI] [PubMed] [Google Scholar]
- 20. Shultz LD, Brehm MA, Garcia‐Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645‐658. [DOI] [PubMed] [Google Scholar]
- 22. Zhou ZF, Peng F, Li JY, Ye YB. Intratumoral IL‐12 gene therapy inhibits tumor growth in a HCC‐Hu‐PBL‐NOD/SCID murine model. Onco Targets Ther. 2019;12:7773‐7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. King MA, Covassin L, Brehm MA, et al. Human peripheral blood leucocyte non‐obese diabetic‐severe combined immunodeficiency interleukin‐2 receptor gamma chain gene mouse model of xenogeneic graft‐versus‐host‐like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157:104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shultz LD, Pearson T, King M, et al. Humanized NOD/LtSz‐scid IL2 receptor common gamma chain knockout mice in diabetes research. Ann N Y Acad Sci. 2007;1103:77‐89. [DOI] [PubMed] [Google Scholar]
- 25. Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature. 1988;335:256‐259. [DOI] [PubMed] [Google Scholar]
- 26. Moser J, van Ark J, van Dijk MC, et al. Distinct differences on neointima formation in immunodeficient and humanized mice after carotid or femoral arterial injury. Sci Rep. 2016;6:35387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naserian S, Leclerc M, Thiolat A, et al. Simple, reproducible, and efficient clinical grading system for murine models of acute graft‐versus‐host disease. Front Immunol. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanmamed MF, Rodriguez I, Schalper KA, et al. Nivolumab and urelumab enhance antitumor activity of human T lymphocytes engrafted in Rag2‐/‐IL2Rγnull immunodeficient mice. Cancer Res. 2015;75:3466‐3478. [DOI] [PubMed] [Google Scholar]
- 29. Fisher TS, Kamperschroer C, Oliphant T, et al. Targeting of 4–1BB by monoclonal antibody PF‐05082566 enhances T‐cell function and promotes anti‐tumor activity. Cancer Immunol Immunother. 2012;61:1721‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang DK, Moniz RJ, Xu Z, et al. Human anti‐CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol Cancer. 2015;14:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishikawa F, Yasukawa M, Lyons B, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chainnull mice. Blood. 2005;106:1565‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brehm MA, Bortell R, Diiorio P, et al. Human immune system development and rejection of human islet allografts in spontaneously diabetic NOD‐Rag1null IL2rgammanull Ins2Akita mice. Diabetes. 2010;59:2265‐2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pearson T, Greiner DL, Shultz LD. Creation of “humanized” mice to study human immunity. Curr Protoc Immunol. 2008;81(1):11‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayakawa J, Hsieh MM, Uchida N, Phang O, Tisdale JF. Busulfan produces efficient human cell engraftment in NOD/LtSz‐Scid IL2Rgamma(null) mice. Stem Cells. 2009;27:175‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody‐based clearance of hematopoietic stem cell niches. Science. 2007;318:1296‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McIntosh BE, Brown ME, Duffin BM, et al. Nonirradiated NOD, B6.SCID Il2rγ‐/‐ Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports. 2015;4:171‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato‐lymphoid system mice. Haematologica. 2016;101:5‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi MI, Medina KL, Garrett K, et al. Relatively normal human lymphopoiesis but rapid turnover of newly formed B cells in transplanted nonobese diabetic/SCID mice. J Immunol. 2001;167:3033‐3042. [DOI] [PubMed] [Google Scholar]
- 39. Watanabe Y, Takahashi T, Okajima A, et al. The analysis of the functions of human B and T cells in humanized NOD/shi‐scid/gammac(null) (NOG) mice (hu‐HSC NOG mice). Int Immunol. 2009;21:843‐858. [DOI] [PubMed] [Google Scholar]
- 40. Gille C, Orlikowsky TW, Spring B, et al. Monocytes derived from humanized neonatal NOD/SCID/IL2Rγ(null) mice are phenotypically immature and exhibit functional impairments. Hum Immunol. 2012;73:346‐354. [DOI] [PubMed] [Google Scholar]
- 41. André MC, Erbacher A, Gille C, et al. Long‐term human CD34+ stem cell‐engrafted nonobese diabetic/SCID/IL‐2R gamma(null) mice show impaired CD8+ T cell maintenance and a functional arrest of immature NK cells. J Immunol. 2010;185:2710‐2720. [DOI] [PubMed] [Google Scholar]
- 42. Halkias J, Yen B, Taylor KT, et al. Conserved and divergent aspects of human T‐cell development and migration in humanized mice. Immunol Cell Biol. 2015;93:716‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De La Rochere P, Guil‐Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized mice for the study of immuno‐oncology. Trends Immunol. 2018;39:748‐763. [DOI] [PubMed] [Google Scholar]
- 44. Smith DJ, Lin LJ, Moon H, et al. Propagating humanized BLT mice for the study of human immunology and immunotherapy. Stem Cells Dev. 2016;25:1863‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Melkus MW, Estes JD, Padgett‐Thomas A, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST‐1. Nat Med. 2006;12:1316‐1322. [DOI] [PubMed] [Google Scholar]
- 46. Rong Z, Wang M, Hu Z, et al. An effective approach to prevent immune rejection of human ESC‐derived allografts. Cell Stem Cell. 2014;14:121‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tonomura N, Shimizu A, Wang S, et al. Pig islet xenograft rejection in a mouse model with an established human immune system. Xenotransplantation. 2008;15:129‐135. [DOI] [PubMed] [Google Scholar]
- 48. Tonomura N, Habiro K, Shimizu A, Sykes M, Yang Y‐G. Antigen‐specific human T‐cell responses and T cell–dependent production of human antibodies in a humanized mouse model. Blood. 2008;111:4293‐4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang Y, Yang Y‐G, Bai O, Xia J, Hu Z. Long‐term survival and differentiation of human thymocytes in human thymus‐grafted immunodeficient mice. Immunotherapy. 2019;11:881‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoshihara S, Li Y, Xia J, Danzl N, Sykes M, Yang Y‐G. Posttransplant hemophagocytic lymphohistiocytosis driven by myeloid cytokines and vicious cycles of T‐cell and macrophage activation in humanized mice. Front Immunol. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kalscheuer H, Danzl N, Onoe T, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med. 2012;4(125):125ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reinisch A, Etchart N, Thomas D, et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood. 2015;125:249‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659‐702. [DOI] [PubMed] [Google Scholar]
- 54. Chen P, Huang Y, Womer KL. Effects of mesenchymal stromal cells on human myeloid dendritic cell differentiation and maturation in a humanized mouse model. J Immunol Methods. 2015;427:100‐104. [DOI] [PubMed] [Google Scholar]
- 55. Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human‐blood lineage cells in humanized mice. Proc Natl Acad Sci USA. 2009;106:21783‐21788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rongvaux A, Willinger T, Martinek J, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32:364‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wunderlich M, Chou FS, Link KA, et al. AML xenograft efficiency is significantly improved in NOD/SCID‐IL2RG mice constitutively expressing human SCF, GM‐CSF and IL‐3. Leukemia. 2010;24:1785‐1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Katano I, Takahashi T, Ito R, et al. Predominant development of mature and functional human NK cells in a novel human IL‐2‐producing transgenic NOG mouse. J Immunol. 1950;2015(194):3513‐3525. [DOI] [PubMed] [Google Scholar]
- 59. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of human CD4+ Foxp3+ regulatory T cells in human stem cell factor–, granulocyte‐macrophage colony‐stimulating factor–, and interleukin‐3–expressing NOD‐SCID IL2Rγnull humanized mice. Blood. 2011;117:3076‐3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ito R, Takahashi T, Katano I, et al. Establishment of a human allergy model using human IL‐3/GM‐CSF‐transgenic NOG mice. J Immunol. 2013;191:2890‐2899. [DOI] [PubMed] [Google Scholar]
- 61. Herndler‐Brandstetter D, Shan L, Yao Y, et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc Natl Acad Sci USA. 2017;114:E9626–E9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li Y, Mention JJ, Court N, et al. A novel Flt3‐deficient HIS mouse model with selective enhancement of human DC development. Euro J Immunol. 2016;46:1291‐1299. [DOI] [PubMed] [Google Scholar]
- 63. Douam F, Ziegler CGK, Hrebikova G, et al. Selective expansion of myeloid and NK cells in humanized mice yields human‐like vaccine responses. Nat Commun. 2018;9:5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jardine L, Cytlak U, Gunawan M, et al. Donor monocyte–derived macrophages promote human acute graft‐versus‐host disease. J Clin Invest. 2020;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Divito SJ, Aasebø AT, Matos TR, et al. Peripheral host T cells survive hematopoietic stem cell transplantation and promote graft‐versus‐host disease. J Clin Invest. 2020;130(9):4624‐4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Danner R, Chaudhari SN, Rosenberger J, et al. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2RgcKO mice is critical for development and function of human T and B cells. PLoS One. 2011;6:e19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Patton J, Vuyyuru R, Siglin A, Root M, Manser T. Evaluation of the efficiency of human immune system reconstitution in NSG mice and NSG mice containing a human HLA.A2 transgene using hematopoietic stem cells purified from different sources. J Immunol Methods. 2015;422:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim J, Peachman KK, Jobe O, et al. Tracking human immunodeficiency virus‐1 infection in the humanized DRAG mouse model. Front Immunol. 2017;8:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Masse‐Ranson G, Dusséaux M, Fiquet O, et al. Accelerated thymopoiesis and improved T‐cell responses in HLA‐A2/‐DR2 transgenic BRGS‐based human immune system mice. Eur J Immunol. 2019;49:954‐965. [DOI] [PubMed] [Google Scholar]
- 70. Zeng Y, Liu B, Rubio M‐T, et al. Creation of an immunodeficient HLA‐transgenic mouse (HUMAMICE) and functional validation of human immunity after transfer of HLA‐matched human cells. PLoS One. 2017;12:e0173754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ka Y, Katano I, Nishinaka E, et al. Improved engraftment of human peripheral blood mononuclear cells in NOG MHC double knockout mice generated using CRISPR/Cas9. Immunol Lett. 2021;229:55‐61. [DOI] [PubMed] [Google Scholar]
- 72. Rahmig S, Kronstein‐Wiedemann R, Fohgrub J, et al. Improved human erythropoiesis and platelet formation in humanized NSGW41 mice. Stem Cell Reports. 2016;7:591‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lute KD, Kenneth F, Lu P, et al. Human CTLA4 knock‐in mice unravel the quantitative link between tumor immunity and autoimmunity induced by anti–CTLA‐4 antibodies. Blood. 2005;106:3127‐3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Burova E, Hermann A, Waite J, et al. Characterization of the Anti‐PD‐1 antibody REGN2810 and its antitumor activity in human PD‐1 knock‐in mice. Mol Cancer Ther. 2017;16:861‐870. [DOI] [PubMed] [Google Scholar]
- 75. Khosravi‐Maharlooei M, Li H, Hoelzl M, et al. Role of the thymus in spontaneous development of a multi‐organ autoimmune disease in human immune system mice. J Autoimmun. 2021;119:102612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McCormack E, Adams KJ, Hassan NJ, et al. Bi‐specific TCR‐anti CD3 redirected T‐cell targeting of NY‐ESO‐1‐ and LAGE‐1‐positive tumors. Cancer Immunol Immunother. 2013;62:773‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang M, Yao L‐C, Cheng M, et al. Humanized mice in studying efficacy and mechanisms of PD‐1‐targeted cancer immunotherapy. FASEB J. 2018;32:1537‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Proetzel G, Roopenian DCJM. Humanized FcRn mouse models for evaluating pharmacokinetics of human IgG antibodies. Methods. 2014;65:148‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Horvath C, Andrews L, Baumann A, et al. Storm forecasting: additional lessons from the CD28 superagonist TGN1412 trial. Nat Rev Immunol. 2012;12:740. author reply 740. [DOI] [PubMed] [Google Scholar]
- 80. Weißmüller S, Kronhart S, Kreuz D, et al. TGN1412 induces lymphopenia and human cytokine release in a humanized mouse model. PLoS One. 2016;11:e0149093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Walsh NC, Kenney LL, Jangalwe S, et al. Humanized mouse models of clinical disease. Annu Rev Pathol. 2017;12:187‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bacac M, Colombetti S, Herter S, et al. CD20‐TCB with obinutuzumab pretreatment as next‐generation treatment of hematologic malignancies. Clin Cancer Res. 2018;24:4785‐4797. [DOI] [PubMed] [Google Scholar]
- 83. Yan H, Bhagwat B, Sanden D, et al. Evaluation of a TGN1412 analogue using in vitro assays and two immune humanized mouse models. Toxicol Appl Pharmacol. 2019;372:57‐69. [DOI] [PubMed] [Google Scholar]
- 84. Ye C, Yang H, Cheng M, et al. A rapid, sensitive, and reproducible in vivo PBMC humanized murine model for determining therapeutic‐related cytokine release syndrome. FASEB J. 2020;34:12963‐12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Matas‐Céspedes A, Brown L, Mahbubani KT, et al. Use of human splenocytes in an innovative humanised mouse model for prediction of immunotherapy‐induced cytokine release syndrome. Clin Transl Immunology. 2020;9:e1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Geller MA, Knorr DA, Hermanson DA, et al. Intraperitoneal delivery of human natural killer cells for treatment of ovarian cancer in a mouse xenograft model. Cytotherapy. 2013;15:1297‐1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lee SJ, Kang WY, Yoon Y, et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer. 2015;15:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Veluchamy JP, Lopez‐Lastra S, Spanholtz J, et al. In vivo efficacy of umbilical cord blood stem cell‐derived NK cells in the treatment of metastatic colorectal cancer. Front Immunol. 2017;8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ames E, Canter RJ, Grossenbacher SK, et al. Enhanced targeting of stem‐like solid tumor cells with radiation and natural killer cells. OncoImmunology. 2015;4:e1036212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu D, Song L, Wei J, et al. IL‐15 protects NKT cells from inhibition by tumor‐associated macrophages and enhances antimetastatic activity. J Clin Invest. 2012;122:2221‐2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wege AK, Ernst W, Eckl J, et al. Humanized tumor mice–a new model to study and manipulate the immune response in advanced cancer therapy. Int J Cancer. 2011;129:2194‐2206. [DOI] [PubMed] [Google Scholar]
- 92. Strauss J, Heery CR, Kim JW, et al. First‐in‐human phase I trial of a tumor‐targeted cytokine (NHS‐IL12) in subjects with metastatic solid tumors. Clin Cancer Res. 2019;25:99‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhao Y, Moon E, Carpenito C, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053‐9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Schilbach K, Alkhaled M, Welker C, et al. Cancer‐targeted IL‐12 controls human rhabdomyosarcoma by senescence induction and myogenic differentiation. Oncoimmunology. 2015;4:e1014760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Varga J, Lopatin M, Boden G. Hypoglycemia due to antiinsulin receptor antibodies in systemic lupus erythematosus. J Rheumatol. 1990;17:1226‐1229. [PubMed] [Google Scholar]
- 97. Hudecek M, Lupo‐Stanghellini MT, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6‐targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461‐3472. [DOI] [PubMed] [Google Scholar]
- 99. Song DG, Powell DJ. Pro‐survival signaling via CD27 costimulation drives effective CAR T‐cell therapy. Oncoimmunology. 2012;1:547‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Guedan S, Chen X, Madar A, et al. ICOS‐based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124:1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chu Y, Hochberg J, Yahr A, et al. Targeting CD20+ aggressive B‐cell non‐Hodgkin lymphoma by anti‐CD20 CAR mRNA‐modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3:333‐344. [DOI] [PubMed] [Google Scholar]
- 103. Brentjens RJ, Davila ML, Riviere I, et al. CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor‐modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Park JH, Rivière I, Gonen M, et al. Long‐term follow‐up of CD19 car therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378:449‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART‐19 immunotherapy. Cancer Discov. 2015;5:1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Qin H, Cho M, Haso W, et al. Eradication of B‐ALL using chimeric antigen receptor‐expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126:629‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jin CH, Xia J, Rafiq S, et al. Modeling anti‐CD19 CAR T cell therapy in humanized mice with human immunity and autologous leukemia. EBioMedicine. 2019;39:173‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hu Z, Xia J, Fan W, Wargo J, Yang Y‐G. Human melanoma immunotherapy using tumor antigen‐specific T cells generated in humanized mice. Oncotarget. 2016;7:6448‐6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Li Y, Teteloshvili N, Tan S, et al. Humanized mice reveal new insights into the thymic selection of human autoreactive CD8+ T cells. Front Immunol. 2019;10:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang X, Xiang Z, Liu Y, et al. Exosomes derived from Vδ2‐T cells control Epstein‐Barr virus–associated tumors and induce T cell antitumor immunity. Sci Transl Med. 2020;12:eaaz3426. [DOI] [PubMed] [Google Scholar]
- 112. Arrieta M‐C, Walter J, Finlay BB. Human microbiota‐associated mice: a model with challenges. Cell Host Microbe. 2016;19:575‐578. [DOI] [PubMed] [Google Scholar]
- 113. Staley C, Kaiser T, Beura LK, et al. Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome. 2017;5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sommerkamp P, Mercier FE, Wilkinson AC, Bonnet D, Bourgine PE. Engineering human hematopoietic environments through ossicle and bioreactor technologies exploitation. Exp Hematol. 2021;94:20‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392‐401. [DOI] [PubMed] [Google Scholar]
- 116. Fabre M, Ferrer C, Domínguez‐Hormaetxe S, et al. OMTX705, a novel FAP‐targeting ADC demonstrates activity in chemotherapy and pembrolizumab‐resistant solid tumor models. Clin Cancer Res. 2020;26:3420‐3430. [DOI] [PubMed] [Google Scholar]
- 117. Flanagan S. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet Res. 1966;8:295‐309. [DOI] [PubMed] [Google Scholar]
- 118. Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527‐530. [DOI] [PubMed] [Google Scholar]
- 119. Christianson SW, Greiner DL, Hesselton RA, et al. Enhanced human CD4+ T cell engraftment in beta2‐microglobulin‐deficient NOD‐scid mice. J Immunol. 1997;158:3578‐3586. [PubMed] [Google Scholar]
- 120. Bastide C, Bagnis C, Mannoni P, Hassoun J, Bladou F. A Nod Scid mouse model to study human prostate cancer. Prostate Cancer Prostatic Dis. 2002;5:311‐315. [DOI] [PubMed] [Google Scholar]
- 121. Brehm MA, Shultz LD, Luban J, Greiner DL. Overcoming current limitations in humanized mouse research. J Infect Dis. 2013;208:S125–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Her Z, Yong KSM, Paramasivam K, et al. An improved pre‐clinical patient‐derived liquid xenograft mouse model for acute myeloid leukemia. J Hematol Oncol. 2017;10:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Akkina R. New generation humanized mice for virus research: comparative aspects and future prospects. Virology. 2013;435:14‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ali N, Flutter B, Rodriguez RS, et al. Xenogeneic graft‐versus‐host‐disease in NOD‐scid IL‐2Rγ null mice display a T‐effector memory phenotype. PLoS One. 2012;7:e44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Legrand N, Huntington ND, Nagasawa M, et al. Functional CD47/signal regulatory protein alpha (SIRPα) interaction is required for optimal human T‐and natural killer‐(NK) cell homeostasis in vivo. Proc Natl Acad Sci USA. 2011;108:13224‐13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yamauchi T, Takenaka K, Urata S, et al. Polymorphic Sirpa is the genetic determinant for NOD‐based mouse lines to achieve efficient human cell engraftment. Blood. 2013;121:1316‐1325. [DOI] [PubMed] [Google Scholar]
- 127. Jangalwe S, Shultz LD, Mathew A, Brehm MA. Improved B cell development in humanized NOD‐scid IL2Rγnull mice transgenically expressing human stem cell factor, granulocyte‐macrophage colony‐stimulating factor and interleukin‐3. Immun Inflamm Dis. 2016;4:427‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Chen Q, He F, Kwang J, Chan JK, Chen J. GM‐CSF and IL‐4 stimulate antibody responses in humanized mice by promoting T, B, and dendritic cell maturation. J Immunol. 2012;189:5223‐5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Shultz LD, Saito Y, Najima Y, et al. Generation of functional human T‐cell subsets with HLA‐restricted immune responses in HLA class I expressing NOD/SCID/IL2rγnull humanized mice. Proc Natl Acad Sci USA. 2010;107:13022‐13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kuruvilla JG, Troyer RM, Devi S, Akkina R. Dengue virus infection and immune response in humanized RAG2−/− γc−/−(RAG‐hu) mice. Virology. 2007;369:143‐152. [DOI] [PubMed] [Google Scholar]
- 131. Audigé A, Rochat M‐A, Li D, et al. Long‐term leukocyte reconstitution in NSG mice transplanted with human cord blood hematopoietic stem and progenitor cells. BMC Immunol. 2017;18:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lan P, Tonomura N, Shimizu A, Wang S, Yang Y‐G. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108:487‐492. [DOI] [PubMed] [Google Scholar]