

Abstract
Li‐Fraumeni syndrome (LFS) is a hereditary cancer predisposition syndrome, and the majority of patients with LFS have been identified with germline variants in the p53 tumor suppressor (TP53) gene. In the past three decades, considerable case reports of TP53 germline variants have been published in Japan. To the best of our knowledge, there have been no large‐scale studies of Japanese patients with LFS. In this study, we aimed to identify Japanese patients with TP53 germline variants and to reveal the characteristics of LFS in Japan. We collected reported cases by reviewing the medical literature and cases diagnosed at the institutions of the authors. We identified 68 individuals from 48 families with TP53 germline pathogenic or likely pathogenic variants. Of the 48 families, 35 (72.9%) had missense variants, most of which were located within the DNA‐binding loop. A total of 128 tumors were identified in the 68 affected individuals. The 128 tumor sites were as follows: breast, 25; bones, 16; brain, 12; hematological, 11; soft tissues, 10; stomach, 10; lung, 10; colorectum, 10; adrenal gland, 9; liver, 4; and others, 11. Unique phenotype patterns of LFS were shown in Japan in comparison with those in a large national LFS cohort study in France. Above all, a higher frequency of patients with stomach cancer was observed in Japanese TP53 germline variant carriers. These results may provide useful information for the clinical management of LFS in Japan.
Keywords: Japan, Li‐Fraumeni syndrome, phenocopy, stomach cancer, TP53 germline pathogenic variant
Comparing this study in Japan with a large national Li‐Fraumeni syndrome cohort study in France, the unique phenotype patterns of Li‐Fraumeni syndrome was shown in Japan. Especially, the higher frequency of patients with stomach cancer in Japanese TP53 germline variants carriers was shown.
1. INTRODUCTION
Li‐Fraumeni syndrome (LFS) is an autosomal dominant cancer predisposition syndrome with high penetrance, resulting in a high risk of developing various types of cancers from early childhood to adulthood. LFS was first proposed by Li and Fraumeni in 1969, and the causative gene was identified as the p53 tumor suppressor (TP53) gene in 1990. 1 , 2 , 3 The TP53 database of the International Agency for Research on Cancer (IARC) (http://p53.iarc.fr/) includes data on over 800 families worldwide. 4 Previously published literature review studies, cohort studies, and hospital‐based follow‐up studies have shown that tumors arising from LFS are increasingly diverse, 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 and the diagnostic criteria have changed over time 8 , 15 , 16 , 17 , 18 (Table S1). Recently, TP53 germline variants were discovered through multigene cancer panel testing. 19 , 20 , 21 The interpretation of the pathological significance of TP53 germline variants is sometimes difficult. 22 , 23 However, the detection and precise diagnosis of LFS is now considered more important because the usefulness of whole‐body MRI as a cancer‐screening technique for patients with LFS has been established. 24 , 25 , 26
Since the first report in 1992 that identified TP53 germline variants in two cases from a family survey of children with adrenocortical cancer (ACC) in Japan, a considerable number of studies on families with a TP53 germline variant have been published. 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 However, no large‐scale studies have been conducted on this syndrome in Japan, and the specific clinical and pathological features of cancers occurring in the Japanese population with TP53 germline variants remain unknown. Genetic diagnosis of hereditary breast and ovarian cancers has frequently been conducted in Japan, and the search for TP53 germline variants is considered in patients with breast cancer negative for BRCA1 and BRCA2. 16
In 2016, the Japanese Society for Hereditary Tumors (JSHT) established the LFS Special Committee. Its members, belonging to various specialized fields, came together to determine the characteristics of tumors that most commonly occur in Japanese families with LFS. Therefore, this study aimed to identify Japanese patients with TP53 germline variants and identify the characteristics of LFS in Japan, in order to contribute to TP53 genetic counseling, TP53 germline testing, and healthcare management for LFS. We collected data on the affected TP53 germline variant carriers and their pathological characteristics through a literature review. In addition, the characteristics of tumors occurring in the affected TP53 germline variant carriers tested at our member institutions were examined. The pathogenicity of these identified TP53 germline variants was assessed using widely accepted criteria for TP53 variants. 55 Then, our Japanese study was compared with the French cohort study 17 because the French group had the world's largest series with TP53 germline variants and reported the characteristics of the tumors that occurred in many of the individuals with TP53 germline variants in almost the same period as that of our study. 17 Herein, we describe some findings of the LFS characteristics in Japan that were revealed in this study.
2. MATERIALS AND METHODS
2.1. Families and patients
Information on families with a TP53 germline variant was obtained by searching PubMed (http://www.ncbi.nlm.nih.gov/pubmed), Japan Medical Abstracts Society (http://search.jamas.or.jp), and the IARC TP53 database R.18. 4 We used the keywords “Li‐Fraumeni,” “TP53,” and “Japan” for our search and collected data on families carrying a TP53 germline variant. 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 Data on sex, age at tumor onset, tumor sites, histopathological diagnosis, presence or absence of simultaneous or metachronous cancers, and TP53 variant sites and types were obtained from the studies. Furthermore, unpublished data of the affected TP53 germline variant carriers who underwent genetic testing at three institutions, namely the Nagara Medical Center, Hamamatsu University School of Medicine, and Kindai University, were added. Duplicated cases were excluded from the analysis. In addition, cases found using a next‐generation DNA sequencer were excluded because exhaustive research had the possibility of causing confusion in our phenotype‐conscious study. A compiled list of the families and patients in the order of publication year is presented in Table S2. This study was approved by the Ethics Board of the Japanese Society for Hereditary Tumors. The genetic studies described below were also approved by the ethics boards of the three institutions.
2.2. TP53 molecular analysis
For genetic analysis research in our facilities, written informed consent and patient agreement were obtained before the examination. DNA was collected from peripheral blood samples of patients using the QIAamp DNA Mini Kit (Qiagen), and DNA sequencing analysis of the TP53 gene was performed using Big Dye Terminator Bidirectional Sequencing (Applied Biosystems), as previously reported. 31 , 41 The sequenced reads were aligned to the human reference genome (hg38), and primer sequences are available upon request.
2.3. Interpretation of TP53 germline variants
We assessed the pathogenicity of TP53 germline variants based on the American College of Medical Genomics and Genetics/Association for Molecular Pathology (ACMG/AMP) guidelines for TP53 variants. 55 The TP53 germline variants were evaluated to determine TP53 variant types and their known clinical significance using ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); functional data based on the promoter‐specific transcriptional activity measured in yeast assays 56 ; a functional assay for loss of growth suppression and dominant‐negative activities based on Z‐scores 57 ; population data of the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/), GEnome Medical Alliance Japan Whole Genome Aggregation (GEM‐J WGA), ExAC (http://exac.broadinstitute.org/), human genetic variation database (HGVD) (http://www.hgvd.genome.med.kyoto‐u.ac.jp/), and Tohoku Medical Megabank Organization (ToMMo) (https://www.megabank.tohoku.ac.jp/tommo); and in silico functional prediction algorithm of Align‐GVGD (http://agvgd.hci.utah.edu/), BayesDel, 58 SIFT (http://provean.jcvi.org/index.php), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/). The information was evaluated on December 10, 2020, and the results are summarized in Table S3. After annotation, the families and patients with pathogenic or likely pathogenic variants of the TP53 germline were selected for further analysis.
2.4. Genetic and phenotypic analysis
To analyze the characteristics of the tumor phenotypes, we counted all the tumors and sites affected in patients with the TP53 germline pathogenic and likely pathogenic variants. For cases with multiple primary cancers, each cancer type was counted. Breast cancer also included those on the contralateral side. Based on this information, the effects and codon distribution of the TP53 germline variants, the age at tumor onset, and the frequency distribution of the tumor were compared with data from 322 individuals affected with the TP53 germline variants between 1993 and 2013, which was provided by the French LFS working group in 2015. 17
2.5. Statistical analysis
The statistical significance of the data on age at the first tumor onset in patients (female vs. male) and age at the first tumor onset according to the effect of TP53 germline variants (missense variants vs. others than missense variants and dominant‐negative missense variants vs. others than dominant‐negative missense variants) was evaluated using the two‐tailed Student's t test. Statistical significance was set at P < .05. A chi‐square test of independence was performed by comparing the frequency of tumors associated with TP53 germline variants (our study [n = 128] vs. the French LFS working group study [n = 548]). Statistical significance was set at P < .05.
3. RESULTS
3.1. Japanese families and patients with LFS selected in this study
By reviewing the literature through 2016, we found a total of 128 Japanese families who met the diagnostic criteria for LFS or Li‐Fraumeni like syndrome (LFL) and/or individuals with a TP53 germline variant: 28 in PubMed, 62 in the Japan Medical Abstracts Society, 24 in the IARC TP53 database, and 14 in our facility. After excluding duplicated cases, 88 Japanese families who met the criteria of LFS/LFL and/or carrying a TP53 germline variant were identified. First, we excluded 29 families not verified for any TP53 germline variants and one family with one Japanese‐born Brazilian boy with p.Arg337His, whose parents were from the Brazilian state of Paraná (Figure 1). Among the remaining 58 families, we identified 38 distinct TP53 germline variants (Table 1 and Table S3). We attempted to interpret each of these 38 variants of the 58 families based on the ACMG/AMP guidelines for TP53 variants; 10 families with 12 individuals carrying p.Val31Ile, p.Asp49His, p.Ser106Arg, p.Ala189Val, p.Arg156His, and p.Glu285Gln were excluded (Table 1 and Table S3). Finally, among the 58 families with TP53 germline variant carriers, 48 families with 68 affected individuals were found to have a germline pathogenic or likely pathogenic variant (Table 1 and Figure 1).
FIGURE 1.
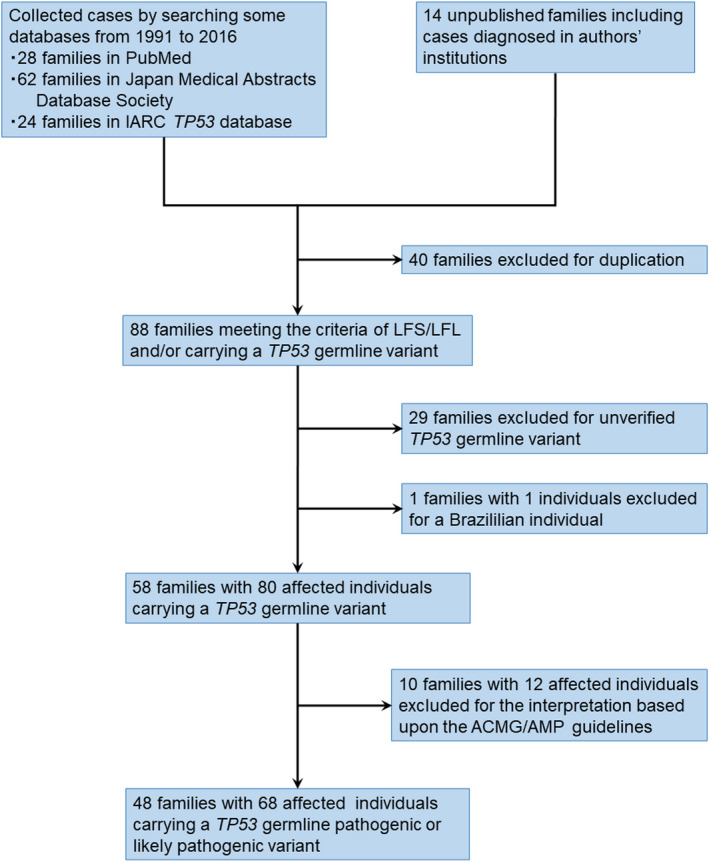
Flow diagram of this study population
TABLE 1.
Summary of each family and individual with TP53 germline pathogenic or likely pathogenic variant in this study
| Family No. | Patient No. | Relationship | Sex | Cancer site | Age (y) | Clinical category | TP53 variant | Clinvar | TA Class | PHANTM | gnomAD | GEM‐J WGA | Align‐GVGD | BayesDel |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Proband | M | AC | 5 | Chompret | c.918_919insG, p.Ala307Hisfs | N/A | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 2 | 2 | Proband | M | CNS | 41 | Chompret | c.857A>C, p.Glu286Ala | LP | NF | DNE_LOF | N/A | N/A | C65 | 0.5335 |
| 3 | Son | M | TM | 19 | ||||||||||
| 3 | 4 | Proband | F | OS | 10 | Chompret | c.844C>T, p.Arg282Trp | P/LP | NF | DNE_LOF | 3.98E‐06 | N/A | C65 | 0.5418 |
| 5 | Father | M | ST | 46 | ||||||||||
| 4 | 6 | Proband | F | OS‐BR‐ST | 13 | Classic LFS, Chompret | c.844C>T, p.Arg282Trp | P/LP | NF | DNE_LOF | 3.98E‐06 | N/A | C65 | 0.5418 |
| 5 | 7 | Proband | F | ST‐LU | 41 | No | c.637C>T, p.Arg213X | P | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 8 | Daughter | F | ST | 20 | ||||||||||
| 6 | 9 | Proband | F | BR‐BR‐LU | 26 | Chompret | c.398T>G, p.Met133Arg | N/A | NF | DNE_LOF | N/A | N/A | C45 | 0.4966 |
| 10 | Sibling | M | HE‐ST | 37 | ||||||||||
| 11 | Sibling | M | CNS | 25 | ||||||||||
| 12 | Sibling | F | BR(bilateral) | 40 | ||||||||||
| 7 | 13 | Proband | M | ST | 44 | No | c.818G>A, p.Arg273His | P | NF | DNE_LOF | 1.59E‐05 | 6.60E‐05 | C25 | 0.5259 |
| 8 | 14 | Proband | M | CNS‐HE | 28 | Chompret | c.725G>A, p.Cys242Tyr | P | NF | DNE_LOF | N/A | N/A | C65 | 0.5405 |
| 9 | 15 | Proband | F | SO | 3 | Classic LFS | c.839G>T, p.Arg280Ile | VUS | NF | DNE_LOF | N/A | N/A | C65 | 0.5969 |
| 16 | Mother | F | CNS | 33 | ||||||||||
| 17 | Sibling | M | CNS‐OS | 2 | ||||||||||
| 10 | 18 | Proband | M | ST | 38 | No | c.818G>A, p.Arg273His | P | NF | DNE_LOF | 1.59E‐05 | 6.60E‐05 | C25 | 0.5259 |
| 19 | Son | M | LI‐HE | 7 | ||||||||||
| 11 | 20 | Proband | F | OS‐BR | 12 | Classic LFS, Chompret | c.524G>A, p.Arg175His | P | NF | DNE_LOF | 3.98E‐06 | 0.0002 | C25 | 0.5462 |
| 12 | 21 | Proband | F | AC‐HE | 3 | Chompret | c.742C>T, p.Arg248Trp | P | NF | DNE_LOF | 3.98E‐06 | N/A | C65 | 0.5336 |
| 13 | 22 | Proband | F | OV‐HE‐SK | 65 | No | c.524G>A, p.Arg175His | P | NF | DNE_LOF | 3.98E‐06 | 0.0002 | C25 | 0.5462 |
| 14 | 23 | Proband | M | OS‐LU | 9 | Chompret | c.16bp dup, p.Asn310X | N/A | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 24 | Father | M | OS | 26 | ||||||||||
| 15 | 25 | Proband | M | LU | 30 | No | c.277‐278delCT, p.Leu93fsX224 | N/A | N/A | N/A | N/A | N/A | N/A | ‐ |
| 16 | 26 | Proband | M | KI‐LU‐ST‐SK‐CO‐BL‐TH | 43 | No | c.733G>A, p.Gly245Ser | C | NF | DNE_LOF | N/A | N/A | C55 | 0.5536 |
| 17 | 27 | Proband | F | AC‐LI‐TH‐BR&BR | 2 | Chompret | c.672+1G>T | P/LP | N/A | N/A | N/A | N/A | N/A | ‐ |
| 28 | Sibling | M | OS | 13 | ||||||||||
| 18 | 29 | Proband | F | SO‐HE‐HE | 10 | No | c.733G>A, p.Gly245Ser | C | NF | DNE_LOF | N/A | N/A | C55 | 0.5536 |
| 30 | Father | M | ST | 44 | ||||||||||
| 19 | 31 | Proband | F | OS‐BR‐CO‐SO‐LU‐BR(bilateral)‐SO | 15 | Chompret | c.659A>G, p.Tyr220Cys | P | NF | DNE_LOF | 7.96E‐06 | N/A | C65 | 0.5625 |
| 20 | 32 | Proband | F | PA | 12 | No | c.733G>A, p.Gly245Ser | C | NF | DNE_LOF | N/A | N/A | C55 | 0.5536 |
| 21 | 33 | Proband | M | CNS‐CO | 20 | No | c.584T>C, p.Ile195Thr | VUS | NF | DNE_LOF | N/A | N/A | C55 | 0.5174 |
| 22 | 34 | Proband | F | AC | 20 | Chompret | c.581T>C, p.Leu194Pro | VUS | NF | DNE_LOF | N/A | N/A | C65 | 0.6021 |
| 23 | 35 | Proband | F | CNS | 12 | No | c.637C>T, p.Arg213X | P | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 24 | 36 | Proband | F | AC | 31 | Chompret | c.530C>G, p.Pro177Arg | C | NF | DNE_LOF | N/A | N/A | C65 | 0.4485 |
| 25 | 37 | Proband | M | SO‐SO | 1 | Chompret | c.818G>A, p.Arg273His | P | NF | DNE_LOF | 1.59E‐05 | 6.60E‐05 | C25 | 0.5259 |
| 26 | 38 | Proband | M | OS | 17 | Classic LFS | c.818G>A, p.Arg273His | P | NF | DNE_LOF | 1.59E‐05 | 6.60E‐05 | C25 | 0.5259 |
| 39 | Sibling | F | HE | 10 | ||||||||||
| 27 | 40 | Proband | F | LU‐SO‐SO | 28 | Classic LFS, Chompret | c.637C>T, p.Arg213X | P | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 41 | Son | M | CO | 11 | ||||||||||
| 42 | Sibling | F | CO | 28 | ||||||||||
| 43 | Sibling | F | BR‐BR | 29 | ||||||||||
| 28 | 44 | Proband | F | OS&LI | 8 | No | c.722C>T, p.Ser241Phe | LP | NF | DNE_LOF | 3.98E‐06 | N/A | C65 | 0.545 |
| 29 | 45 | Proband | F | OS | 15 | Chompret | c.672+1G>T | P/LP | N/A | N/A | N/A | N/A | N/A | ‐ |
| 30 | 46 | Proband | M | SO | 2 | Classic LFS | c.997delC, p.Arg333Valfs*12 | N/A | N/A | N/A | N/A | N/A | N/A | ‐ |
| 47 | Mother | F | OS | 11 | ||||||||||
| 31 | 48 | Proband | M | HE‐CO‐LI‐CO‐OS | 12 | No | c.742C>T, p.Arg248Trp | P | NF | DNE_LOF | 3.98E‐06 | N/A | C65 | 0.5336 |
| 32 | 49 | Proband | M | AC | 8 M | Chompret | c.473G>A, p.Arg158His | P/LP | NF | DNE_LOF | 3.98E‐06 | N/A | C25 | 0.5337 |
| 33 | 50 | Proband | M | OS‐UK | 14 | Chompret | c.711G>A, p.Met237Ile | C | NF | DNE_LOF | 3.98E‐06 | N/A | C0 | 0.4419 |
| 51 | Son | M | CNS | 9 | ||||||||||
| 34 | 52 | Proband | M | AC | 3 | Chompret | c.375+1G>A | LP | N/A | N/A | N/A | N/A | N/A | ‐ |
| 35 | 53 | Proband | M | SO | 2 | Chompret | c.736A>G, p.Met246Val | P | NF | DNE_LOF | N/A | N/A | C15 | 0.371 |
| 54 | Sibling | M | AC | 9 | ||||||||||
| 36 | 55 | Proband | F | BR‐BR‐LU | 40 | Chompret | c.743G>A, p.Arg248Gln | P | NF | DNE_LOF | 1.19E‐05 | 0.00013 | C35 | 0.4738 |
| 37 | 56 | Proband | F | OS‐BR‐CO | 16 | Chompret | c.743G>A, p.Arg248Gln | P | NF | DNE_LOF | 1.19E‐05 | 0.00013 | C35 | 0.4738 |
| 38 | 57 | Proband | F | CO | 17 | No | c.1009C>T, p.Arg337Cys | P/LP | NF | notDNE_LOF | N/A | N/A | C45 | 0.3165 |
| 58 | Mother | F | OS‐BR‐CNS | 39 | ||||||||||
| 39 | 59 | Proband | F | SO‐BR | 21 | Chompret | c.1009C>T, p.Arg337Cys | P/LP | NF | notDNE_LOF | N/A | N/A | C45 | 0.3165 |
| 40 | 60 | Proband | F | AC | 2 | Chompret | c.514G>T, p.Val172Phe | LP | NF | unclass | N/A | N/A | C45 | 0.5464 |
| 41 | 61 | Proband | M | ST | 42 | No | c.541C>T, p.Arg181Cys | C | PF | notDNE_LOF | N/A | N/A | C65 | 0.4693 |
| 42 | 62 | Proband | M | CNS | 5 | Chompret | c.817C>T, p.Arg273Cys | P/LP | PF | DNE_LOF | 1.20E‐05 | N/A | C65 | 0.5537 |
| 43 | 63 | Proband | M | CNS | 1 | Chompret | c.928_929ins14bp (GCATCCTCTCCCAG), p.Asn310Serfs*30 | N/A | N/A | N/A | N/A | N/A | N/A | ‐ |
| 44 | 64 | Proband | F | BR‐BR | 32 | Chompret | c.743G>A, p.Arg248Gln | P | NF | DNE_LOF | 1.19E‐05 | 0.00013 | C35 | 0.4738 |
| 45 | 65 | Proband | M | CNS‐TH‐LU | 21 | Chompret | c.473G>A, p.Arg158His | P/LP | NF | DNE_LOF | 3.98E‐06 | N/A | C25 | 0.5337 |
| 46 | 66 | Proband | F | BR‐BR‐LU‐CO | 31 | Chompret | c.586C>T, p.Arg196X | P | N/A | notDNE_LOF | 3.98E‐06 | N/A | N/A | ‐ |
| 47 | 67 | Proband | M | HE | 1 | No | c.637C>T, p.Arg213X | P | N/A | notDNE_LOF | N/A | N/A | N/A | ‐ |
| 48 | 68 | Proband | F | BR‐BR | 32 | No | c.749C>T, p.Pro250Leu | C | NF | DNE_LOF | N/A | N/A | C35 | 0.6029 |
Age indicates age at first tumor onset. M in age indicates month.
Abbreviations: AC, adrenal cortex; BL, bladder; BR, breast; C, conflicting interpretations of pathogenicity; CNS, brain; CO, colorectum; F, female; GEM‐J WGA, GEnome Medical Alliance Japan Whole Genome Aggregation; gnomAD, Genome Aggregation Database; HE, hematological; KI, kidney; LFS, Li‐Fraumeni syndrome; LI, liver; LP, likely pathogenic; LU, lung; M, male; N/A, not available; NF, nonfunctional; OS, bones; OV, ovary; P, pathogenic; PA, pancreas; PF, partially functional; PHANTM, Phenotypic Annotation of TP53 Mutations; SK, skin; SO, soft tissues; ST, stomach; TA, transactivation; TH, thyroid; TM, thymus; UK, unknown origin; VUS, variant of uncertain significance.
3.2. Potency of the clinical criteria
Of the 48 families, six families (12.5%) met the classic LFS criteria, and 29 (60.4%) met the 2015 version of the Chompret criteria for TP53 testing. Fifteen families (31.3%) had individuals with significant family history in the 2015 version of the Chompret criteria, seven (14.5%) had individuals with multiple primary cancers, 10 (20.8%) had individuals with ACC or choroid plexus cancer (CPC) without a significant family history, and eight (16.7%) had individuals with early diagnosis of breast cancer. Overall, 32 families (66.7%) tested positive for a TP53 germline pathogenic or likely pathogenic variant meeting either classic diagnostic criteria or the 2015 version of the Chompret criteria for TP53 testing (Table 1 and Table S2).
3.3. Patterns of pathogenic or likely pathogenic variants
Among the 48 families with a TP53 germline pathogenic or likely pathogenic variant, 32 distinct TP53 germline variants were identified, corresponding to 23 missense variants, three nonsense variants, two splice variants, and four frameshift variants, including two deletions and two insertions (Figure S1). Among the 48 families, 35 (72.9%) had missense, six (12.5%) nonsense, four (8.3%) frameshift, and three (6.3%) splice variants. Among the 48 families with a TP53 germline pathogenic or likely pathogenic variant, 41 families carried a single‐base substitution of missense and nonsense variants. Among the 41 families, sites of the variants in 39 families (95.1%), except two families with missense variants in codon 337, were located in the DNA‐binding loop of TP53 (Figure S2).
3.4. LFS tumor onset in Japan
Among the 68 individuals affected with TP53 germline variants (34 females and 34 males), a total of 128 tumors developed. The mean age at first tumor onset was 19.9 years (range: 8 months to 65 years), and the median age at first tumor onset was 16.5 years. Of the 68 affected individuals (both sexes), 14 (20.6%) firstly developed cancer by 5 years of age, and 50 (73.5%) and 67 (98.5%) individuals were aged by 30 and 60 years, respectively (Figure S3). Although we found a tendency for low cancer incidence in women during the first decade of life, no significant difference was observed between men and women according to the age at tumor onset (P = .42) (Figure 2).
FIGURE 2.
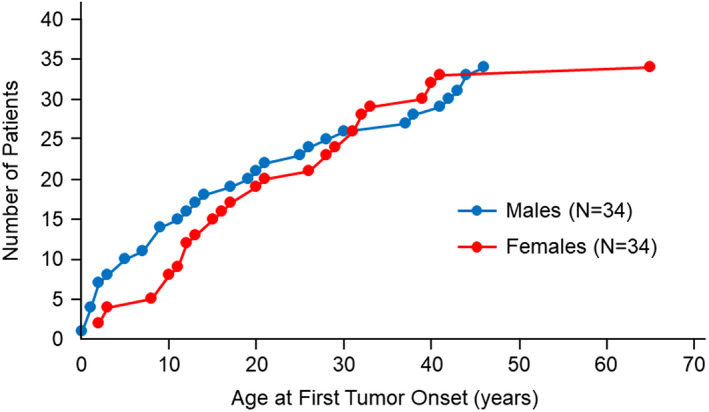
Age at first tumor onset in individuals affected with TP53 germline pathogenic or likely pathogenic variants according to sex (women [n = 34] and men [n = 34]). The X‐axis shows age in years at the first tumor onset. The Y‐axis shows the cumulative number of patients
Multiple primary cancers (n = 2‐7) were observed in 32 of the 68 affected individuals (47.0%). Two individuals (2.9%) had simultaneous cancers at the onset, 30 (44.1%) had second primary cancers, and 14 (20.6%) had third or more primary cancers (Figure S4). The mean and median latency periods to the second tumor occurrence in 30 affected individuals were 6.8 and 5 years, respectively (range: 0‐20 years) (Figure S5).
3.5. LFS tumor distribution in Japan
The distribution of the 128 tumors is summarized in Table 2. The tumor sites were as follows: breast, 25; bones, 16; brain, 12; hematological, 11; soft tissues, 10; stomach, 10; lung, 10; colorectum, 10; adrenal gland, 9; liver, 4; and others, 11 (Table 2).
TABLE 2.
Distribution of tumors in affected individuals with TP53 germline pathogenic or likely pathogenic variant
| Site | Total no. of patients (N = 68) | No. of patients (females/males) (N = 34/34) | Total no. of tumors | No. of patients (0‐18/>18 y) a (N = 36/32) | No. of patients (1st/>2nd) | Mean/median age at tumor onset, y (range) |
|---|---|---|---|---|---|---|
| Breast | 15 | 15/0 | 25 | 0/25 | 8/17 | 31.9/32.0 (19‐47) |
| Bones | 16 | 9/7 | 16 | 13/3 | 14/2 | 16.3/14.5 (8‐39) |
| Brain | 12 | 3/9 | 12 | 5/7 | 11/1 | 20.7/20.5 (1‐51) |
| Hematological | 10 | 5/5 | 11 | 7/4 | 4/7 | 20.3/13.0 (1‐70) |
| Soft tissues | 8 | 5/3 | 10 | 5/5 | 6/4 | 15.7/15.5 (1‐36) |
| Stomach | 10 | 3/7 | 10 | 0/10 | 7/3 | 38.8/43.0 (20‐46) |
| Lung | 10 | 6/4 | 10 | 1/9 | 2/8 | 37.3/38.5 (14‐52) |
| Colorectum | 9 | 5/4 | 10 | 2/8 | 3/7 | 29.6/27.5 (11‐52) |
| Adrenal cortex | 9 | 5/4 | 9 | 7/2 | 9/0 | 8.4/3.0 (0‐31) |
| Liver | 4 | 2/2 | 4 | 3/1 | 2/2 | 13.8/13.75 (7‐26) |
| Thyroid | 3 | 1/2 | 3 | 1/2 | 0/3 | 38.0/35.0 (15‐64) |
| Skin | 2 | 1/1 | 2 | 0/2 | 0/2 | 58.0/58.0 (46‐70) |
| Kidney | 1 | 0/1 | 1 | 0/1 | 1/0 | 43/43 |
| Pancreas | 1 | 1/0 | 1 | 1/0 | 1/0 | 12/12 |
| Ovary | 1 | 1/0 | 1 | 0/1 | 1/0 | 65/65 |
| Thymus | 1 | 0/1 | 1 | 0/1 | 1/0 | 19/19 |
| Bladder | 1 | 0/1 | 1 | 0/1 | 0/1 | 64/64 |
| Unknown origin | 1 | 0/1 | 1 | 0/1 | 0/1 | 30/30 |
| Total | 114 | 62/52 | 128 | 45/83 | 70 b /58 | 26.3/26.0 (0‐70) (19.9 a /16.5 (0‐65)) |
Age at first tumor onset was indicated.
Two of 68 patients had simultaneous cancers at onset.
A total of 25 breast cancers were observed in 15 affected individuals, all female, with mean and median ages of 31.9 and 32 years (range: 19‐47 years) at onset, respectively. Two of the 15 individuals with breast cancer had bilateral primary cancers at onset, and four had contralateral breast cancers during the clinical course. Seventeen out of 25 (68.0%) breast cancers were identified as second or more primary cancers. Four out of 15 (26.7%) individuals with breast cancer had phyllodes breast tumors. Limited histological information, including hormone receptors and/or HER2‐positive breast cancer, was available.
The mean and median ages of bone tumor onset were 16.3 and 14.5 years (range: 8‐39 years), respectively, and all were histologically classified as osteosarcoma. Fourteen of the 16 (87.5%) individuals with bone tumors developed bone tumor as first primary cancer. In addition, three of them (18.8%) developed tumors at rare sites such as the cranial bone or maxillary sinus. A brain tumor was found in 12 individuals with mean and median ages of 20.7 and 20.5 years (range: 1‐51 years) at onset, respectively, and 11 of 12 (91.7%) individuals with brain tumors developed brain tumor as first primary cancer. Five of 12 (41.7%) individuals with brain tumors had astrocytoma, three (25.0%) individuals had CPC, and none had medulloblastoma. Hematological malignancy was found in 10 individuals with mean and median ages at onset of 20.3 and 13 years (range: 1‐70 years), respectively. Of the 10 individuals with hematological malignancies, five (50.0%) had acute leukemia, three (30.0%) had malignant lymphoma, and three (30.0%) had myelodysplastic syndrome. Six out of 10 (60.0%) individuals with hematological malignancies developed these malignancies as second or more primary cancers. No chromosomal findings were reported in two individuals who developed lymphocytic leukemia. Soft tissue sarcomas were observed in eight individuals at mean and median ages of 15.7 and 15.5 years (range: 1‐36 years) at onset, respectively, and four out of 10 (40.0%) tumors in eight individuals indicated embryonal rhabdomyosarcoma. Intriguingly, a 10‐year‐old girl presented with primary ameloblastic fibrosarcoma, a rare malignant odontogenic tumor. All nine (100%) individuals indicated ACC as their first cancer, with mean and median ages at onset of 8.4 and 3 years (range: 8 months to 31 years), respectively.
Stomach, lung, and colorectal cancers that generally occur at older ages were observed in 10 individuals at a median age of 43 years (range: 20‐46 years), 10 individuals at a median age of 38.5 years (range: 14‐52 years), and nine individuals at a median age of 27.5 years (range: 11‐52 years). The mean age of onset of these stomach, lung, and colorectal cancers was 38.8, 37.3, and 29.6 years, respectively. We found that eight out of 10 (80.0%) lung cancers in 10 individuals were identified as the second or more primary cancers. Liver cancer was also found in four individuals with mean and median ages of 13.8 years (range: 7‐26 years), respectively, and two of four (50%) indicated liver sarcoma.
3.6. Comparing the characteristics of the affected individuals in Japan and in France
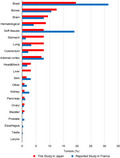
To clarify the characteristics of individuals affected with TP53 germline pathogenic or likely pathogenic variants in Japan, Japanese data were compared with data from the French LFS working group. 17 Both our study and the study of the French LFS working group showed some similarities regarding codon distribution and the effect of the TP53 germline variants, age at the first tumor onset, frequency of patients with multiple primary tumors, and time of occurrence of second tumors. However, a comparison of the frequency distribution of the 128 tumors included in this study with that of the 548 tumors from the French LFS study group showed that the frequency of stomach cancer was significantly higher in Japan (7.8% vs. 1.3%; P = .03). Moreover, a high‐frequency distribution was also observed for lung, colorectal, and liver cancers in Japan (7.8% vs. 3.3%; P = .17, 7.8% vs. 2.2%; P = .08, and 3.1% vs. 0%; P = .08, respectively) (Figure 3). Conversely, the frequency distribution of soft tissue sarcomas was lower in Japan than in France (7.8% vs. 19.0%; P = .03) (Figure 3). In addition, a low‐frequency distribution of breast cancer in Japan was also found (19.5% vs. 31.4%; P = .10) (Figure 3).
FIGURE 3.
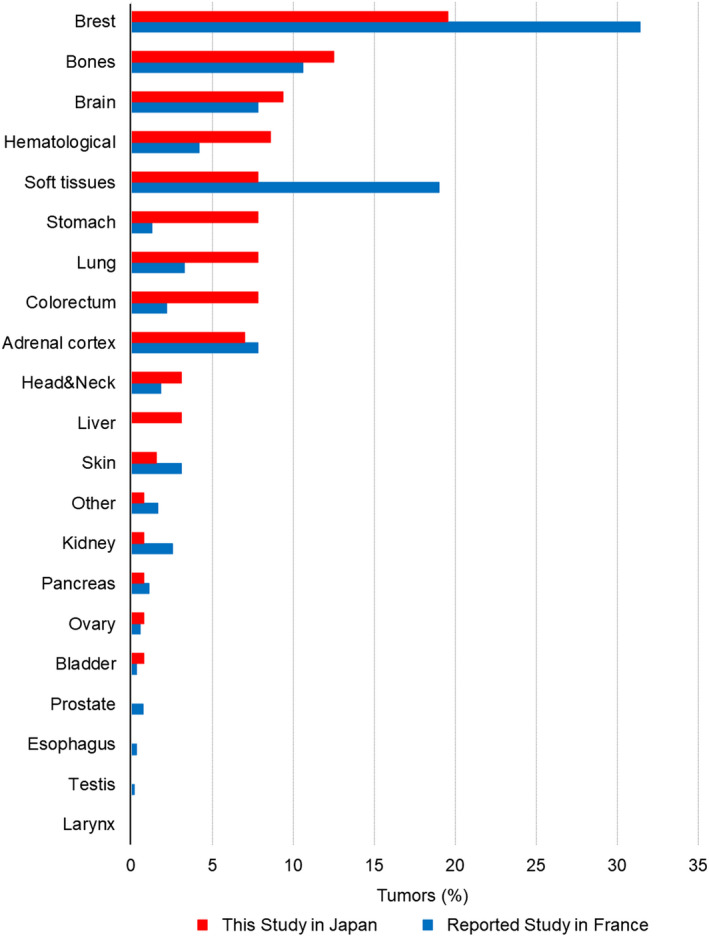
The frequency distribution of tumors associated with TP53 germline variants in our study (n = 128) and the French LFS working group study (n = 548). The X‐axis shows the tumor site. The Y‐axis shows the frequency distribution of the tumor
Moreover, when comparing the mean and median ages at the onset of each tumor between the Japanese and French, the mean and median ages at tumor onset of soft tissue sarcoma, lung cancer, and colorectal cancer in Japan was earlier (16/16 vs. 29/31, 37/39 vs. 42/44, 30/28 vs. 40/40, respectively) than those in France (Table 2). In contrast, the mean and median ages at brain tumor onset in Japan were older (21/21 vs. 15/11) than those in France (Table 2).
3.7. LFS genotype‐phenotype correlations in Japan
Several genotype‐phenotype correlations have been reported in families with TP53 germline variants. One is the correlation between genotype and age at tumor onset. Missense variants have been reported to be associated with earlier tumor onset. 59 , 60 This study of 68 individuals affected with TP53 germline pathogenic or likely pathogenic variants demonstrated that the average age at first tumor onset indicated no significant differences between missense variants (age 21.6 years [n = 48]) and nonsense or other types of variants (age 15.9 years [n = 20]) (P = .15) (Figure S6). In addition, a recent French report indicated that the average age at tumor onset in dominant‐negative missense variants was 5 years earlier than in other types of alterations than the dominant‐negative missense variants. 17 However, based on two systematic studies of dominant‐negative effects, 61 , 62 this study indicated no significant differences between the dominant‐negative missense variants (age 21.5 years [n = 31]) and types of alterations other than the dominant‐negative missense variants (age 18.6 years [n = 37]) (P = .42) (Figure S7).
Moreover, correlations between cancer types and TP53 germline variants at the DNA‐binding site or dominant‐negative missense variants have also been described. 17 , 63 In this study, the TP53 germline pathogenic or likely pathogenic variants at the DNA‐binding site were identified in all 10 (100%) individuals with hematological malignancies, all those (100%) with stomach cancer, and in eight out of nine (88.9%) individuals with colorectal cancer (Table S4). In addition, in Japan, we found that the dominant‐negative missense variants were detected in seven of 10 (70.0%) individuals with hematological malignancies and seven of 10 (70.0%) individuals with stomach cancer (Table S4). In contrast, our study also indicated that seven of nine (77.8%) individuals with ACC and seven of 10 (70.0%) individuals with lung cancer showed other types of alterations than dominant‐negative missense variants (Table S4).
4. DISCUSSION
This study analyzed the characteristics of families with a TP53 germline variant in Japan, which was examined by the LFS Special Committee of the Japanese Society for Hereditary Tumors (JSHT) through a literature review until 2016. Target families with LFS also included unpublished families tested at the authors' facilities. Of the Japanese families with LFS, 32 distinct TP53 germline pathogenic or likely pathogenic variants were identified in 48 families with 68 patients. A total of 68 patients developed 128 tumors. Of the 68 patients, 32 (47.0%) had multiple primary cancers. We then compared the genetic and clinical characteristics of these Japanese families with LFS, mainly using data obtained from the French LFS working group study. Some similarities, such as genotype patterns and age at onset, were observed. In addition, our study indicated that the unique phenotypic patterns of LFS, the high frequency of stomach, colorectal, lung, and liver cancers, and the low frequency of breast cancer and soft tissue sarcoma, are observed in Japan.
In this study, we described that the LFS in Japan is mainly characterized by the frequent occurrence of stomach cancer in TP53 germline variant carriers. Recently, Ikenoue et al reported a high incidence of stomach cancer in Lynch syndrome in Japan, with similar characteristics. 64 In addition, Ariffin et al reported the frequent occurrence of stomach cancer in Asian patients, particularly in Japanese patients, with a TP53 germline variant based on the IARC TP53 database R.16 in 2012, and reported that the frequent occurrence of stomach cancer in the general population might phenotypically determine the frequency of this cancer in Asian TP53 germline variant carriers. 65 Ariffin et al suggested that the frequent occurrence of stomach cancer in Asian populations compared with Western populations is due to a combination of dietary (high‐salt diet), environmental (chronic Helicobacter pylori infection), and genetic susceptibility risk factors. 65 Currently, the global frequency of cancer incidence in each population from 2003 to 2007 can be observed using the database managed by the IARC (http://ci5.iarc.fr/CI5‐X/Pages/registry_summary.aspx). 66 The associated results are shown in Figures S8 and S9. We also speculate that the frequent occurrence of stomach cancer in the Japanese general population might partly explain the characteristic patterns in patients with TP53 germline variants in Japan.
Similarly, when we consider the frequency of other cancer incidences, the frequent occurrence of colorectal and liver cancers in the Japanese general population may explain the high‐frequency distribution of colorectal and liver cancers in patients with TP53 germline variants in Japan. In addition, the low‐frequency distribution of breast cancer and soft tissue sarcoma in patients with TP53 germline variants in Japan may also be explained by the low frequency of breast cancer and soft tissue sarcoma in the general Japanese population in comparison with the general French population. In contrast, resolving the reason for the high frequency of lung cancer is difficult because no simple phenocopy was observed in the general population. In our study, eight of 10 patients with lung cancer in TP53 germline variant carriers developed lung cancer as second or more primary cancers. Of the eight patients, three (patients 31, 55, and 66) of five patients with breast cancer had lung cancer after radiation therapy. Therefore, we speculate that the frequent occurrence of lung cancer in Japan may be due to treatment‐related risk factors such as radiation (Table S2).
Children with CPC of the brain and ACC were reported to have a high likelihood of carrying a TP53 germline variant, even in the absence of any family history. 17 , 67 , 68 , 69 , 70 Our analysis also showed that two children with CPC and six patients with ACC without any strong family history of cancer had TP53 germline pathogenic or likely pathogenic variants (Table 1 and Table S2). Furthermore, phyllodes tumors in breast cancer and liver sarcoma in liver cancer were found to be the most frequent. Birch et al and Giacomazzi et al reported the greatest increase relative to the general population rates for phyllodes tumors. 9 , 10 Moreover, in liver sarcoma, one of 16 patients aged 1‐21 years was a member of a family with LFS. 71 Therefore, we believe that at least patients in adolescence with phyllodes tumor and children with liver sarcoma in Japan may be considered for TP53 germline variant testing even in the absence of a distinct family history.
The development of clinical surveillance programs for LFS is ongoing. As a leading study, Villani et al reported a survival rate of 88.8% in patients with TP53 germline variants undergoing comprehensive screening compared with a 59.6% survival rate in those without surveillance. 26 They developed a practical surveillance protocol based on the available data, their own experience, and detection strategies for patients with sporadic LFS component tumors. 25 We speculate that there may be individual unusual LFS characteristics in different countries and regions, as they show different tumor characteristics in the general population of different countries and regions. Our study indicated the frequent occurrence of stomach, colorectal, and liver cancers, the frequent occurrence of lung cancer as second or more primary cancers, and the occurrence of distinctive tumor tissues, such as the phyllodes tumor and liver sarcoma, which may provide useful information for clinical LFS management in Japan. Furthermore, in our study, 6 of 15 (40.0%) patients with breast cancer showed simultaneous or metachronous bilateral primary cancers. This finding may also provide benefits for screening to reduce an elevated risk of contralateral breast cancer or considering the option of bilateral mastectomies, which are consistent with those of patients with breast cancer carrying BRCA 1/2 germline pathogenic variants. 72
The findings of this study have several limitations. First, this study must be interpreted based on the method of collection of study subjects. Although previous reports on the spectrum and frequency of cancers in patients with TP53 germline variants have provided detailed information through registration or follow‐up studies, 8 , 17 our analysis was mainly based on a heterogeneous group of collected cases through searching databases and self‐analyzed cases. Thus, there is ascertainment bias in our study, and an old piece of information was not available in detail. However, we believe that most cases of TP53 germline variants have been reported from inquiring about LFS experiences in Japan before the widespread use of next‐generation DNA sequencing. In addition, the LFS Special Committee–related parties have been consulted for the majority of patients with LFS in Japan and have performed genetic testing. Next, in our study, the data of 128 cancers between 1992 and 2016 were compared with those of the French LFS working group comprising 548 cancers between 1993 and 2013. In this respect, we do not expect this limitation to have biased our results because both studies had some similarities, such as codon distribution and effect of TP53 germline variants, age at the first tumor onset, frequency of patients with multiple primary tumors, and time of occurrence of second tumors. In the future, the variable phenotype, diverse tumor spectrum, diagnostic tumor tissue patterns, and other characteristics should be further investigated by accumulating many cases with TP53 germline variants through patient registration, which will result in better LFS management.
In conclusion, we described TP53 variant patterns and tumor characteristics in patients with TP53 germline variants in Japan. Although we could recognize some unique findings of the LFS characteristics in Japan through this study, these need to be further investigated through clinical cohort studies and basic research on LFS. These findings will provide clues for clinical management measures, such as genetic counseling, genetic testing, and healthcare management.
DISCLOSURE
The authors declare to have no potential conflict of interest to disclose.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGMENTS
Professor Hiroshi Yagata (Saitama Medical University), who passed away before this paper was submitted, made a great contribution to the research on breast cancer occurring in Li‐Fraumeni syndrome and its surveillance. We would like to express our deep appreciation for him. In addition, we greatly appreciate the patients and their families who cooperated with us for this study. We also thank Dr J. Toguchida (Kyoto University), Dr T. Oka (Asahikawa Medical University), Dr M. Tokunaga (Kagoshima Municipal Hospital), Dr T. Kawaguchi (Kumamoto University), Dr M. Tsurusawa (Aichi Medical University), Dr T. Yonemoto (Chiba Cancer Center), Dr M. Yano (Akita University), Dr H. Tsuchiya (Kanazawa University), Dr R. Fukano (Kyusyu Cancer Center), Dr M. Kouroki (Kumamoto University), Dr K. Ishiguro (Tottori University), Dr H. Ogi (Kumamoto University), Dr T. Matsubara (Mie University), Dr A. Matsumine (Mie University), Dr J. Yoshimura (Niigata University), Dr T. Iwai (Kagawa Children Hospital), Dr H. Maeda (Fukui University), Dr N. Kagawa (Osaka University), Dr H. Uchida (Kagoshima University), Dr T. Yoshikawa (Fujita Medical University), Dr S. Sugihara (Shikoku Cancer Center), Dr. S. Nakamura (Showa University), Dr. H. Yamagami (Sapporo‐Kosei General Hospital), Dr. M. Yanagimachi (Yokohama City University), Dr. M. Yamauchi (St. Luke’s International Hospital) for providing the clinical information and Dr S. Kojima (Nagoya University), Dr S. Mizutani (Tokyo Medical Dental University), Dr M. Tsurusawa (Aichi Medical University), Dr K. Osafune (Kyoto University), and Dr H. Kaneko (Nagara Medical Center) for providing advice on this study. We also thank the Japanese Society of Hereditary Tumors (JSHT) for their strong support as the backbone of our Li‐Fraumeni Working Group. We would like to thank Editage (www.editage.com) for English language editing.
Funato M, Tsunematsu Y, Yamazaki F, et al. Characteristics of Li‐Fraumeni syndrome in Japan: A review study by the special committee of JSHT. Cancer Sci. 2021;112:2821–2834. 10.1111/cas.14919
Funding information
This study was supported in part by a grant from the Japanese Ministry of Education, Science, Sports and Culture (19K07763), the Health and Labor Sciences Research Grants of Research on Measures for Intractable Diseases (H23‐nanchi‐ippan‐069), and Research on Cancer Control (H29‐gantaisaku‐ippan‐002) from the Ministry of Health, Labor, and Welfare.
REFERENCES
- 1. Li FP, Fraumeni JF Jr. Soft‐tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747‐752. [DOI] [PubMed] [Google Scholar]
- 2. Li FP, Fraumeni JF Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty‐four kindreds. Cancer Res. 1988;48:5358‐5362. [PubMed] [Google Scholar]
- 3. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233‐1238. [DOI] [PubMed] [Google Scholar]
- 4. Bouaoun L, Sonkin D, Ardin M, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37:865‐876. [DOI] [PubMed] [Google Scholar]
- 5. Masciari S, Dewanwala A, Stoffel EM, et al. Gastric cancer in individuals with Li‐Fraumeni syndrome. Genet Med. 2011;13:651‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong P, Verselis SJ, Garber JE, et al. Prevalence of early onset colorectal cancer in 397 patients with classic Li‐Fraumeni syndrome. Gastroenterology. 2006;130:73‐79. [DOI] [PubMed] [Google Scholar]
- 7. Ruijs MW, Verhoef S, Rookus MA, et al. TP53 germline mutation testing in 180 families suspected of Li‐Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421‐428. [DOI] [PubMed] [Google Scholar]
- 8. Nichols KE, Malkin D, Garber JE, Fraumeni JF Jr, Li FP. Germ‐line p53 mutations predispose to a wide spectrum of early‐onset cancers. Cancer Epidemiol Biomarkers Prev. 2001;10:83‐87. [PubMed] [Google Scholar]
- 9. Masciari S, Dillon DA, Rath M, et al. Breast cancer phenotype in women with TP53 germline mutations: a Li‐Fraumeni syndrome consortium effort. Breast Cancer Res Treat. 2012;133:1125‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Birch JM, Alston RD, McNally RJ, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621‐4628. [DOI] [PubMed] [Google Scholar]
- 11. Giacomazzi J, Koehler‐Santos P, Palmero EI, et al. A TP53 founder mutation, p.R337H, is associated with phyllodes breast tumors in Brazil. Virchows Arch. 2013;463:17‐22. [DOI] [PubMed] [Google Scholar]
- 12. Holmfeldt L, Wei L, Diaz‐Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hettmer S, Archer NM, Somers GR, et al. Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer. 2014;120:1068‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhukova N, Ramaswamy V, Remke M, et al. Subgroup‐specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927‐2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chompret A, Abel A, Stoppa‐Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250‐1256. [DOI] [PubMed] [Google Scholar]
- 17. Bougeard G, Renaux‐Petel M, Flaman JM, et al. Revisiting Li‐Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33:2345‐2352. [DOI] [PubMed] [Google Scholar]
- 18. Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li‐Fraumeni syndrome cohort. Cancer. 2016;122:3673‐3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genet Med. 2016;18:823‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. LaDuca H, Polley EC, Yussuf A, et al. A clinical guide to hereditary cancer panel testing: evaluation of gene‐specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high‐risk patients. Genet Med. 2020;22:407‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coffee B, Cox HC, Kidd J, et al. Detection of somatic variants in peripheral blood lymphocytes using a next generation sequencing multigene pan cancer panel. Cancer Genet. 2017;211:5‐8. [DOI] [PubMed] [Google Scholar]
- 22. Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, Topper S. Sources of discordance among germ‐line variant classifications in ClinVar. Genet Med. 2017;19:1118‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bittar CM, Vieira IA, Sabato CS, et al. TP53 variants of uncertain significance: increasing challenges in variant interpretation and genetic counseling. Fam Cancer. 2019;18:451‐456. [DOI] [PubMed] [Google Scholar]
- 24. Ballinger ML, Best A, Mai PL, et al. Baseline surveillance in Li‐Fraumeni syndrome using whole‐body magnetic resonance imaging: a meta‐analysis. JAMA Oncol. 2017;3:1634‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Villani A, Tabori U, Schiffman J, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li‐Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12:559‐567. [DOI] [PubMed] [Google Scholar]
- 26. Villani A, Shore A, Wasserman JD, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li‐Fraumeni syndrome: 11 year follow‐up of a prospective observational study. Lancet Oncol. 2016;17:1295‐1305. [DOI] [PubMed] [Google Scholar]
- 27. Sameshima Y, Tsunematsu Y, Watanabe S, et al. Detection of novel germ‐line p53 mutations in diverse‐cancer‐prone families identified by selecting patients with childhood adrenocortical carcinoma. J Natl Cancer Inst. 1992;84:703‐707. [DOI] [PubMed] [Google Scholar]
- 28. Toguchida J, Yamaguchi T, Dayton SH, et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med. 1992;326:1301‐1308. [DOI] [PubMed] [Google Scholar]
- 29. Shiseki M, Nishikawa R, Yamamoto H, et al. Germ‐line p53 mutation is uncommon in patients with triple primary cancers. Cancer Lett. 1993;73:51‐57. [DOI] [PubMed] [Google Scholar]
- 30. Horio Y, Suzuki H, Ueda R, et al. Predominantly tumor‐limited expression of a mutant allele in a Japanese family carrying a germline p53 mutation. Oncogene. 1994;9:1231‐1235. [PubMed] [Google Scholar]
- 31. Saeki Y, Tamura K, Yamamoto Y, Hatada T, Furuyama J, Utsunomiya J. Germline p53 mutation at codon 133 in a cancer‐prone family. J Mol Med. 1997;75:50‐56. [DOI] [PubMed] [Google Scholar]
- 32. Murakawa Y, Yokoyama A, Kato S, et al. Astrocytoma and B‐cell lymphoma development in a man with a p53 germline mutation. Jpn J Clin Oncol. 1998;28:631‐637. [DOI] [PubMed] [Google Scholar]
- 33. Konno K, Iwata N, Agata H, Hirota T, Tsurusawa M, Fujimoto T. A Li‐Fraumeni syndrome family. Jpn J Pediatr Soc. 1999;103:563‐566. [Google Scholar]
- 34. Sugano K, Taniguchi T, Saeki M, Tsunematsu Y, Tomaru U, Shimoda T. Germline p53 mutation in a case of Li‐Fraumeni syndrome presenting gastric cancer. Jpn J Clin Oncol. 1999;29:513‐516. [DOI] [PubMed] [Google Scholar]
- 35. Kimura K, Shinmura K, Hasegawa T, Beppu Y, Yokoyama R, Yokota J. Germline p53 mutation in a patient with multiple primary cancers. Jpn J Clin Oncol. 2001;31:349‐351. [DOI] [PubMed] [Google Scholar]
- 36. Miyaki M, Iijima T, Ohue M, et al. A novel case with germline p53 gene mutation having concurrent multiple primary colon tumours. Gut. 2003;52:304‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yonemoto T, Tatezaki S, Ishii T, Hagiwara Y, Inoue M. Multiple primary cancers in patients with osteosarcoma: influence of anticancer drugs and genetic factors. Am J Clin Oncol. 2004;27:220‐224. [DOI] [PubMed] [Google Scholar]
- 38. Nagasaki K, Horikawa R, Nagaishi J, et al. Virilizing adrenocortical carcinoma invading the right atrium with histological high‐grade malignancy and p53 mutation in a 3‐year‐old child: indication of post operative adjuvant chemotherapy. Clin Pediatr Endocrinol. 2004;13:25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuribayashi K, Matsunaga T, Sakai T, et al. A patient with TP53 germline mutation developed Bowen's disease and myelodysplastic syndrome with myelofibrosis after chemotherapy against ovarian cancer. Intern Med. 2005;44:490‐495. [DOI] [PubMed] [Google Scholar]
- 40. Tamura K, Sakagami T, Tsuda S, et al. A case of multiple primary malignant tumors with germline mutation of the TP53 gene. J Fam Tumor. 2006;6:53‐57. [Google Scholar]
- 41. Yamada H, Shinmura K, Okudela K, et al. Identification and characterization of a novel germ line p53 mutation in familial gastric cancer in the Japanese population. Carcinogenesis. 2007;28:2013‐2018. [DOI] [PubMed] [Google Scholar]
- 42. Izawa N, Matsumoto S, Manabe J, et al. A Japanese patient with Li‐Fraumeni syndrome who had nine primary malignancies associated with a germline mutation of the p53 tumor‐suppressor gene. Int J Clin Oncol. 2008;13:78‐82. [DOI] [PubMed] [Google Scholar]
- 43. Yamada H, Shinmura K, Yamamura Y, et al. Identification and characterization of a novel germline p53 mutation in a patient with glioblastoma and colon cancer. Int J Cancer. 2009;125:973‐976. [DOI] [PubMed] [Google Scholar]
- 44. Momota H, Narita Y, Miyakita Y, Hosono A, Makimoto A, Shibui S. Acute lymphoblastic leukemia after temozolomide treatment for anaplastic astrocytoma in a child with a germline TP53 mutation. Pediatr Blood Cancer. 2010;55:577‐579. [DOI] [PubMed] [Google Scholar]
- 45. Ide H, Terado Y, Tokiwa S, et al. Novel germ line mutation p53–P177R in adult adrenocortical carcinoma producing neuron‐specific enolase as a possible marker. Jpn J Clin Oncol. 2010;40:815‐818. [DOI] [PubMed] [Google Scholar]
- 46. Sugawara W, Arai Y, Kasai F, et al. Association of germline or somatic TP53 missense mutation with oncogene amplification in tumors developed in patients with Li‐Fraumeni or Li‐Fraumeni‐like syndrome. Genes Chromosomes Cancer. 2011;50:535‐545. [DOI] [PubMed] [Google Scholar]
- 47. Kato T, Ishikawa K, Satoh M, Kondo S, Kaji M. Pleomorphic carcinoma of the lung arising in a patient with Li‐Fraumeni syndrome: report of a case. Surg Today. 2011;41:841‐845. [DOI] [PubMed] [Google Scholar]
- 48. Osumi T, Miharu M, Fuchimoto Y, Morioka H, Kosaki K, Shimada H. The germline TP53 mutation c.722 C>T promotes bone and liver tumorigenesis at a young age. Pediatr Blood Cancer. 2012;59:1332‐1333. [DOI] [PubMed] [Google Scholar]
- 49. Piao J, Sakurai N, Iwamoto S, et al. Functional studies of a novel germline p53 splicing mutation identified in a patient with Li‐Fraumeni‐like syndrome. Mol Carcinog. 2013;52:770‐776. [DOI] [PubMed] [Google Scholar]
- 50. Yoshimura J, Natsumeda M, Nishihira Y, et al. Radiation‐induced intracranial osteosarcoma after radiation for acute lymphocytic leukemia associated with Li‐Fraumeni syndrome. No Shinkei Geka. 2013;41:499‐505. [PubMed] [Google Scholar]
- 51. Tanaka S, Ohsumi S, Kiyotou S, et al. A case of early lung cancer detected by surveillance PET/CT for Li‐Fraumeni syndrome. J Fam Tumor. 2017;1:22‐25. [Google Scholar]
- 52. Fujisawa Y, Sakaguchi K, Ono H, et al. Combined steroidogenic characters of fetal adrenal and Leydig cells in childhood adrenocortical carcinoma. J Steroid Biochem Mol Biol. 2016;159:86‐93. [DOI] [PubMed] [Google Scholar]
- 53. Matsuoka T, Shinozaki H, Ozawa H, et al. Juvenile rectal cancer in a family with Li‐Fraumeni syndrome. Jpn J Gastroenterol Surg. 2016;49:1170‐1178. [Google Scholar]
- 54. Yamazaki F, Shima H, Osumi T, Narumi S, Kuroda T, Shimada H. Nodular Lymphocyte‐predominant Hodgkin Lymphoma in a 15‐year‐old boy with Li‐Fraumeni syndrome having a germline TP53 D49H mutation. J Pediatr Hematol Oncol. 2018;40:e195‐e197. [DOI] [PubMed] [Google Scholar]
- 55. Fortuno C, Lee K, Olivier M, et al. ClinGen TP53 variant curation expert panel. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat. 2020; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kato S, Han SY, Liu W, et al. Understanding the function‐structure and function‐mutation relationships of p53 tumor suppressor protein by high‐resolution missense mutation analysis. Proc Natl Acad Sci USA. 2003;100:8424‐8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giacomelli AO, Yang X, Lintner RE, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fortuno C, Cipponi A, Ballinger ML, et al. A quantitative model to predict pathogenicity of missense variants in the TP53 gene. Hum Mutat. 2019;40:788‐800. [DOI] [PubMed] [Google Scholar]
- 59. Bougeard G, Sesboüé R, Baert‐Desurmont S, et al. Molecular basis of the Li‐Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45:535‐538. [DOI] [PubMed] [Google Scholar]
- 60. Palmero EI, Schüler‐Faccini L, Caleffi M, et al. Detection of R337H, a germline TP53 mutation predisposing to multiple cancers, in asymptomatic women participating in a breast cancer screening program in Southern Brazil. Cancer Lett. 2008;261:21‐25. [DOI] [PubMed] [Google Scholar]
- 61. Dearth LR, Qian H, Wang T, et al. Inactive full‐length p53 mutants lacking dominant wild‐type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis. 2007;28:289‐298. [DOI] [PubMed] [Google Scholar]
- 62. Monti P, Perfumo C, Bisio A, et al. Dominant‐negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol Cancer Res. 2011;9:271‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Olivier M, Goldgar DE, Sodha N, et al. Li‐Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643‐6650. [PubMed] [Google Scholar]
- 64. Ikenoue T, Arai M, Ishioka C, et al. Importance of gastric cancer for the diagnosis and surveillance of Japanese Lynch syndrome patients. J Hum Genet. 2019;64:1187‐1194. [DOI] [PubMed] [Google Scholar]
- 65. Ariffin H, Chan AS, Oh L, et al. Frequent occurrence of gastric cancer in Asian kindreds with Li‐Fraumeni syndrome. Clin Genet. 2015;88:450‐455. [DOI] [PubMed] [Google Scholar]
- 66. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev. 2016;25:16‐27. [DOI] [PubMed] [Google Scholar]
- 67. Varley JM, McGown G, Thorncroft M, et al. Are there low‐penetrance TP53 Alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Libé R, Bertherat J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases. Eur J Endocrinol. 2005;153:477‐487. [DOI] [PubMed] [Google Scholar]
- 69. Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GD. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:E119‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gozali AE, Britt B, Shane L, et al. Choroid plexus tumors; management, outcome, and association with the Li‐Fraumeni syndrome: the Children's Hospital Los Angeles (CHLA) experience, 1991–2010. Pediatr Blood Cancer. 2012;58:905‐909. [DOI] [PubMed] [Google Scholar]
- 71. Lack EE, Schloo BL, Azumi N, Travis WD, Grier HE, Kozakewich HP. Undifferentiated (embryonal) sarcoma of the liver. Clinical and pathologic study of 16 cases with emphasis on immunohistochemical features. Am J Surg Pathol. 1991;15:1‐16. [PubMed] [Google Scholar]
- 72. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317:2402‐2416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Table S1
Table S2
Table S3
Table S4