

Abstract
BCR‐ABL1 gene fusion associated with additional DNA lesions involves the pathogenesis of chronic myelogenous leukemia (CML) from a chronic phase (CP) to a blast crisis of B lymphoid (CML‐LBC) lineage and BCR‐ABL1 + acute lymphoblastic leukemia (BCR‐ABL1 + ALL). The recombination‐activating gene RAG1 and RAG2 (collectively, RAG) proteins that assemble a diverse set of antigen receptor genes during lymphocyte development are abnormally expressed in CML‐LBC and BCR‐ABL1 + ALL. However, the direct involvement of dysregulated RAG in disease progression remains unclear. Here, we generate human wild‐type (WT) RAG and catalytically inactive RAG‐expressing BCR‐ABL1 + and BCR‐ABL1 − cell lines, respectively, and demonstrate that BCR‐ABL1 specifically collaborates with RAG recombinase to promote cell survival in vitro and in xenograft mice models. WT RAG‐expressing BCR‐ABL1 + cell lines and primary CD34+ bone marrow cells from CML‐LBC samples maintain more double‐strand breaks (DSB) compared to catalytically inactive RAG‐expressing BCR‐ABL1 + cell lines and RAG‐deficient CML‐CP samples, which are measured by γ‐H2AX. WT RAG‐expressing BCR‐ABL1 + cells are biased to repair RAG‐mediated DSB by the alternative non–homologous end joining pathway (a‐NHEJ), which could contribute genomic instability through increasing the expression of a‐NHEJ‐related MRE11 and RAD50 proteins. As a result, RAG‐expressing BCR‐ABL1 + cells decrease sensitivity to tyrosine kinase inhibitors (TKI) by activating BCR‐ABL1 signaling but independent of the levels of BCR‐ABL1 expression and mutations in the BCR‐ABL1 tyrosine kinase domain. These findings identify a surprising and novel role of RAG in the functional specialization of disease progression in BCR‐ABL1 + leukemia through its endonuclease activity.
Keywords: alternative non–homologous end joining pathway, BCR‐ABL1 signaling, recombination‐activating genes RAG1 and RAG2, tyrosine kinase inhibitors, γ‐H2AX
BCR‐ABL1 associates with RAG to promote BCR‐ABL1+ cell survival in vitro and in vivo. The endonuclease activity of RAG drives BCR‐ABL1+ cells to choose the a‐NHEJ pathway in response to DNA damage. RAG stimulates BCR‐ABL1 signaling to reduce TKI therapeutic efficacy, albeit independently of BCR‐ABL1 expression and mutations in the BCR‐ABL1 kinase domain.

1. INTRODUCTION
Lymphocytes express a diverse repertoire of antigen receptors, which can recognize a large variety of foreign pathogens. Functional antigen receptor genes are assembled during lymphocyte development by V(D)J recombination, which is normally initiated by the binding of the RAG complex to target recombination signal sequences (RSS) at the antigen receptor loci to generate DNA double‐strand breaks (DSB). 1 The RSS consist of a heptamer (consensus 5′‐CACAGTG) and a nonamer (consensus 5′‐ACAAAAACC) separated by a degenerate spacer of either 12 or 23 base pairs (bp). The heptamer serves as the cleavage target and the nonamer provides an important binding site for RAG1. Efficient DNA cleavage and recombination require synapsis of one 12‐RSS and one 23‐RSS. Thereafter, the broken DNA ends are shuttled into the classical nonhomologous end‐joining (c‐NHEJ) pathway for DNA repair, resulting in the juxtaposition of the two coding gene segments and precise joining of the RSS. 2 , 3 , 4 The introduction of DSB activates several components of c‐NHEJ, including DNA‐PKcs, XRCC4, and DNA‐Ligase 4, which orchestrate the DNA damage response. 5 Numerous studies support the fact that RAG targeting and choice of c‐NHEJ not only contribute to the high fidelity of V(D)J recombination and genome integrity but also relate to the development of lymphoid malignancies due to aberrant V(D)J recombination and genomic instability. 6 , 7 , 8 , 9
RAG1 and RAG2 genes, which are well‐conserved in humans and mice, are physically linked in the genome and are coordinately expressed in developing lymphocytes. 10 , 11 Human RAG1 is composed of 1,043 amino acids, whereas its mouse orthologue lacks three amino acids at the amino terminus. It contains three active site residues (D603, D711, and E965) that coordinate divalent metal ions and play an indispensable role in DNA cleavage activity. Human RAG2 comprises 527 amino acids and has no catalytic activity but is an accessory factor that strongly promotes DNA cleavage by RAG1. 11 , 12 , 13 , 14 A C‐terminal plant homeodomain (PHD) tethers RAG2 to tri‐methylated histone H3 lysine 4 (H3K4me3). Consequently, RAG2 localization to chromatin mirrors the deposition of H3K4me3, exhibiting no particular preference for the immunoglobulin and T cell receptor loci over the thousands of other highly expressed genes. 15 , 16 However, RAG1 targeting outside of antigen receptor loci is much more complicated. RAG1 binding is driven by the presence of RSS‐like motifs (cryptic RSS, or cRSS) and chromatin accessibility marked by H3K4me3 and H3K27Ac. 9 , 17 Thus, RAG proteins potentially pose a widespread threat to the lymphocyte genome. 18 , 19
Chronic myeloid leukemia (CML) and BCR‐ABL1 + acute lymphoblastic leukemia (BCR‐ABL1 + ALL) are characterized by the Philadelphia chromosome encoding BCR‐ABL1. 20 , 21 If not efficiently treated, CML follows a triphasic clinical course with an initial indolent chronic phase (CML‐CP) followed by an intermediate accelerated phase and eventually a blast crisis of myeloid (CML‐MBC) or B lymphoid (CML‐LBC) lineage. 22 CML‐CP can be effectively treated with tyrosine kinase inhibitors (TKI), but resistance to TKI shortens responses in CML blast crisis and BCR‐ABL1 + ALL. 23 An increasing body of evidence demonstrates that the genomic instability often occurs in CML‐LBC and BCR‐ABL1 + ALL, which is indicative of γ‐H2AX accumulation. 24 In general, BCR‐ABL1 is acquired early in leukemogenesis and its aberrant kinase signaling activates downstream targets that lead to transcriptional and epigenetic dysregulation, providing a plausible mechanism by which RAG could be delivered to thousands of genomic sites outside of its biological targets, resulting in secondary genomic lesions, which are believed to accelerate disease progression. 25 , 26 Although RAG targeting has been linked to physiological and pathophysiological consequences, it has not been associated with any beneficial role in cellular outcomes in BCR‐ABL1 + leukemia.
Here, we establish human WT RAG and catalytically inactive RAG‐expressing BCR‐ABL1 + and BCR‐ABL1 − cell lines and xenograft models. We demonstrate that RAG specifically promotes BCR‐ABL1 + cell survival due to RAG‐induced DNA lesions being repaired by the alternative non–homologous end joining pathway (a‐NHEJ). As a result, RAG‐expressing BCR‐ABL1 + cells decrease sensitivities to TKI by activating BCR‐ABL1 signaling but independent of BCR‐ABL1 expression or mutations in BCR‐ABL1 kinase domain. These findings suggest the novel role of RAG endonuclease activity in disease progression of BCR‐ABL1 + leukemia.
2. MATERIALS AND METHODS
2.1. Patient samples, constructs, and cell lines
Bone marrow (BM) samples were collected at the First Affiliated Hospital of Xi’an Jiaotong University with written informed consent from four CML‐CP and four CML‐LBC patients as described in Table S1. CD34+ BM cells were purified according to the protocol (Miltenyi Biotec, #130‐094‐531), viably frozen, and stored in liquid nitrogen. The study was performed according to the Declaration of Helsinki protocols and was approved by the ethics committee of the First Affiliated Hospital of Xi’an Jiaotong University (Code #19‐975).
The pL‐CRISPR.EFS.PAC was described previously. 27 The human RAG2 single guide RNA (sgRNA) was designed at the CRISPR design website: http://crispr.mit.edu/. sgRNA that does not target the genome was used as a negative control. The sgRNA sequences are listed in Table S2. Lentiviral PWPI vector co–expressing human WT RAG and GFP (WT RAG) or catalytically inactive RAG and GFP (Inactive RAG) were generated and are described in Appendix S1. Lentiviral PWPI vector expressing GFP (Empty), retroviral vector co–expressing human BCR‐ABL1 and hCD4 (MSCV‐BCR‐ABL1‐IRES‐hCD4, MIG‐p210), and retroviral recombination substrate pINV‐12/23 were reported previously. 15 , 28
Three BCR‐ABL1‐positive human cell lines, BV173, K562, and KCL22, and one BCR‐ABL1‐negative human cell line, THP1, were obtained from ATCC. They were maintained in RPMI‐1640 (Hyclone) supplemented with 10% FBS, non–essential amino acids, penicillin‐streptomycin, and β‐mercaptoethanol (50 µM). All cell lines were cultured at 37°C in a 5% CO2 atmosphere and were routinely tested for mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Thermo Scientific, Salem, USA). WT RAG, Inactive RAG, and empty control‐expressing BCR‐ABL1 + K562, KCL22, and BCR‐ABL1 − THP1 cell lines were established and are described in Appendix S1.
2.2. Inversional recombination assay
The retroviral recombination substrate pINV‐12/23 was infected into WT RAG or Inactive RAG‐expressing cells by using the X‐treme GENE HP DNA Transfection Reagent (Roche). pINV‐12/23 recombination was assayed for mCD90 expression using flow cytometry. 29 Alternatively, pINV‐12/23 rearrangement was amplified for coding joints (CJ) by genomic PCR using a and b primers (Figure S1B, Table S3). All PCR products were cloned into the pMD 18‐T vector (#6011, TaKaRa) and sequenced commercially (Sunny).
2.3. Xenotransplant mice models
Non–obese diabetic/severe combined immunodeficiency (NOD/SCID) mice were maintained in specific pathogen‐free facilities at the Xi’an Jiaotong University Laboratory Animal Center. All animal procedures were approved by the Institutional Animal Care and Use Committee of Xi’an Jiaotong University. The mice used in this study were between 8 and 12 weeks old at the beginning of the experiments; 2 × 106 cells were suspended in a mixture of 100 µL 1× PBS and 100 µL high concentration matrix (CORNING Cat#354248). The mixture was then injected at one side of the ilioinguinal region of the mice. Tumor growth was monitored and measured in volume (length × width2 × 0.5) at the indicated time after inoculation. The mice were killed when the largest tumor reached 15 mm in diameter, which served as the clinical end point for euthanasia.
2.4. Immunofluorescence
Cells were affixed to frosted X slides using a cytospin. Cells were then fixed with 4% paraformaldehyde solution for 15 min at room temperature (RT). The cells were washed three times with PBS and then permeabilized with 0.2% Triton X‐100 for 10 min. The slides were blocked with 5% BSA and 10% horse serum in PBST (PBS with 0.2% Triton X‐100) for 1 h at RT and were then incubated with antibodies against γ‐H2AX (1:200; CST #9718) at 4°C overnight. After rinsing with PBST three times, the cells were incubated with an Alexa Fluor 555 secondary antibody (CST, #4413, USA; 1:200) for 45 min at RT. Then, the cells were washed twice and were stained with 1 μg/mL DAPI, followed by imaging using a confocal microscope (Leica SP8 STED) from the core facilities sharing platform at Xi’an Jiaotong University.
2.5. BCR‐ABL1 kinase domain sequence
Total RNA obtained from leukemic cells or xenograft tumors expressing WT and Inactive RAG served as a template for cDNA synthesis, respectively, as recommended by the manufacturer (BioRad iScript cDNA Synthesis Kit). Amplification of the BCR‐ABL1 kinase domain was done by two‐step PCR to exclude amplification of normal ABL1. The first round of PCR used BCR‐specific (exon 13) and ABL1‐specific (exon 9) primers in 10 cycles of amplification. The second round of amplification (30 cycles) focused on the ABL1 kinase domain (exons 4‐6) by high fidelity Pfu DNA polymerase. 30 Later, purified PCR products were cloned into the pMD 18‐T vector and sequenced commercially (Sunny).
2.6. Statistical analysis
Unpaired t tests and ANOVA multiple tests were performed with GraphPad Prism 6.0 (GraphPad Software). Descriptive results were presented as mean ± SEM. Data were considered statistically significant if P‐values were less than 0.05, as indicated.
3. Results
3.1. BCR‐ABL1 collaborates with RAG to increase cell growth in vitro
To explore whether RAG endonuclease was responsible for promoting BCR‐ABL1 + leukemic cell survival, we first created a lentiviral human WT RAG expression vector, in which WT RAG1 and WT RAG2 were co–expressed via the self‐cleavage property of the 2A‐peptide (P2A), and green fluorescence protein gene (GFP) was followed by an internal ribosome entry site (IRES). 31 A lentiviral human catalytically Inactive RAG vector was also generated, in which WT RAG1 was replaced with a D‐to‐A mutation at position 711 (D711A; Figure S1A). The D711A RAG1 mutant is catalytically inactive, which prevents DNA cleavage. 11 The sensitive V(D)J recombination in vitro assay confirmed that ectopic WT RAG induced RSS‐related rearrangement, whereas Inactive RAG lacked DNA cleavage function (Figure S1B,C).
Next, we performed lentiviral transduction to establish WT RAG, Inactive RAG, and empty control‐expressing BCR‐ABL1 + K562, KCL22, and BCR‐ABL1 − THP1 cell lines, which were in absence of endogenous RAG expression. 32 Approximately 90% of GFP+ cells were sorted after infection. The level of WT RAG expression was similar to that of Inactive RAG in each GFP+ cell (Figure 1A). RAG endonuclease activities were comparable in all three cell lines (Figure S1D). Furthermore, the similar levels of BCR‐ABL1 protein were individually induced into expressing WT RAG, Inactive RAG, and empty control THP1 cell lines (Figure S1E). Cell division data showed that WT RAG‐expressing BCR‐ABL1 + cells grew faster than those expressing Inactive RAG and empty vectors in 72 h. By comparison, cell growth in vitro in the empty vector control, WT, and Inactive RAG‐expressing BCR‐ABL1 − cells exhibited no significant differences (Figure 1B). Moreover, the basal levels of apoptosis genes were comparable between the three cell lines when analyzed using quantitative RT‐PCR (qRT‐PCR) (Figure S2A). The transcription levels of anti–apoptotic genes BCL‐2, BCL‐XL, MCL‐1, BCL‐W, A1, and CIAP2 were 2‐fold to 16‐fold higher in WT RAG‐expressing BCR‐ABL1 + cells than in catalytically Inactive RAG‐expressing cells or empty vector control, respectively (Figure 1C‐E). Meanwhile, the expression of pro–apoptotic genes, including BMF, PUMA, BAD, FAS, and BIK, was 1.4‐fold to 6‐fold lower in BCR‐ABL1 + cells with WT RAG than those with Inactive RAG or empty vector, respectively (Figure S2B‐D). In contrast, neither anti–apoptotic nor pro–apoptotic gene expression demonstrated a significant difference in WT RAG, Inactive RAG, and empty control‐expressing BCR‐ABL1 − THP1 cells. (Figures 1F and S2E). Altogether, these results indicate that the specific cooperation between RAG and BCR‐ABL1 enhances BCR‐ABL1 + cell growth in vitro.
FIGURE 1.
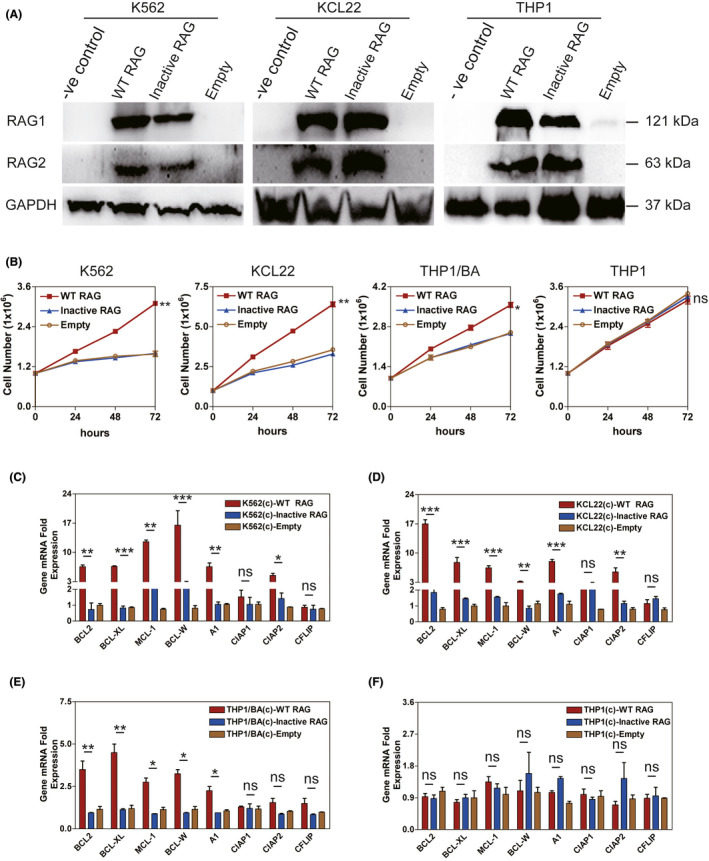
RAG enhances BCR‐ABL1 + cell survival in vitro. A, Western blotting analysis of the wild‐type (WT) RAG and Inactive RAG expression in BCR‐ABL1 + K562 and KCL22 or BCR‐ABL1 − THP1 cell lines infected by lentivirus carrying WT RAG, Inactive RAG, and empty control, respectively. The proteins from K562, KCL22, and THP1 cells without infection (−ve control) were used as negative controls. Blots were stripped and reprobed for GAPDH as a loading control. B, The proliferation rates of the indicated cells were assayed for 72 h. BCR‐ABL1 (BA) proteins were individually introduced into THP1 cell line expressing WT RAG, Inactive RAG, and empty vector control. Five independent experiments were repeated. Data represent the mean ± SEM. C‐F, Transcripts of anti–apoptotic genes in expressing WT RAG, Inactive RAG and empty control K562 cells (C), KCL22 cells (D), THP1/BA cells (E), and THP1 cells (F), respectively. c, cells. Gene mRNA fold expression values have been normalized to the β‐actin, as described in supplemental information, with bars indicating the mean of three independent experiments and error bars representing the SEM. *, **, and *** indicate P < .05, P < .01, and P < .001, respectively. ns, no significant difference [Correction added on 19 May 2021, after first online publication: Figure 1F has been corrected.]
3.2. RAG recombinase accelerates tumor formation induced by BCR‐ABL1 + leukemic cells in a xenograft mouse model
To further investigate tumorigenesis in BCR‐ABL1 + leukemic cells due to RAG‐mediated anti–apoptosis in vivo, equal titers of WT and Inactive RAG‐expressing BCR‐ABL1 + K562 cells were subcutaneously injected into NOD/SCID mice. Tumor growth was monitored by measuring tumor volume weekly after inoculation. Tumors grew faster in WT RAG‐expressing K562 cells than those in Inactive RAG K562 cells (Figure 2A). At the clinical end point for euthanasia, mice injected with WT RAG‐expressing BCR‐ABL1 + cells had increased tumor weights compared to mice inoculated with Inactive RAG‐expressing BCR‐ABL1 + cells (Figures 2B and S3A). Apoptosis in WT RAG‐expressing K562 tumor cells also diminished compared to that in K562 tumor cells with Inactive RAG expression (Figure S3B). The mRNA levels of BCL‐2, BCL‐XL, MCL‐1, BCL‐W, A1, CIAP1, and CIAP2 anti–apoptotic genes were elevated in WT RAG‐expressing K562 tumor cells (Figure 2C). However, the transcription levels of BMF, PUMA, BAD, FAS, and BIM pro–apoptotic genes were reduced in tumors derived from WT RAG‐expressing K562 cells (Figure 2D). In addition, the protein levels of BCL‐2, BCL‐XL, and MCL‐1 had increased more in tumors originating from WT RAG‐expressing K562 cells than in those derived from K562 cells with Inactive RAG. BAD protein levels were reduced in tumors of WT RAG‐expressing K562 cells (Figure 2E). Similar observations were made in tumors from a BCR‐ABL1 + KCL22 cell line transduced with WT or inactive RAG (Figure S3C‐F). There were no differences in the tumor growth, percentage of leukemic cells undergoing apoptosis and apoptosis‐related gene profiles in NOD/SCID mice injected with WT RAG or Inactive RAG‐expressing THP1 cells (Figures 3 and S4). These findings indicate that BCR‐ABL1 by itself creates the environment that allows RAG to be involved in tumorigenesis.
FIGURE 2.
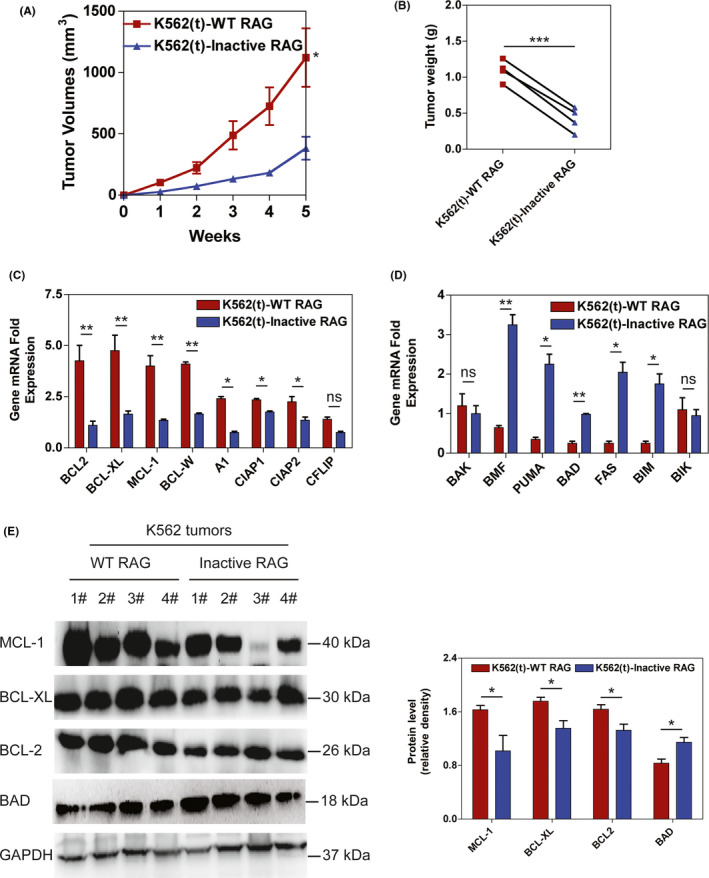
RAG increases tumor formation induced by BCR‐ABL1 + cells in a xenograft mouse model. A, NOD/SCID mice were subcutaneously injected with K562 cells expressing wild‐type (WT) and Inactive RAG, respectively. t, tumors. The tumor volumes were measured at the indicated time points. Bars and error bars represent the mean ± SEM (n = 4). B, Tumors were excised from NOD/SCID mice injected with K562 cell line expressing WT and Inactive RAG. The tumor weights are plotted, where each dot represents a tumor from one mouse. Data represent the mean ± SEM (n = 4). C‐D, Transcripts of anti–apoptotic genes (C) and pro–apoptotic genes (D) in tumor cells from mice injected with K562 cell line transducted by lentiviral WT RAG and inactive RAG, respectively. Bars and error bars represent the mean ± SEM (n = 4). E, Western blotting analysis of MCL‐1, BCL‐XL, BCL2 and BAD proteins in tumors formed by K562 cells expressing WT and Inactive RAG, respectively. The same blot was reprobed with an anti–GAPDH antibody to assess protein loading. The data were quantified by ImageJ software and percentage of MCL‐1, BCL‐XL, BCL2, and BAD level related to GAPDH was presented as mean ± SEM (n = 4). *, **, and *** represent P < .05, P < .01, and P < .001, respectively. ns, no significant difference
FIGURE 3.
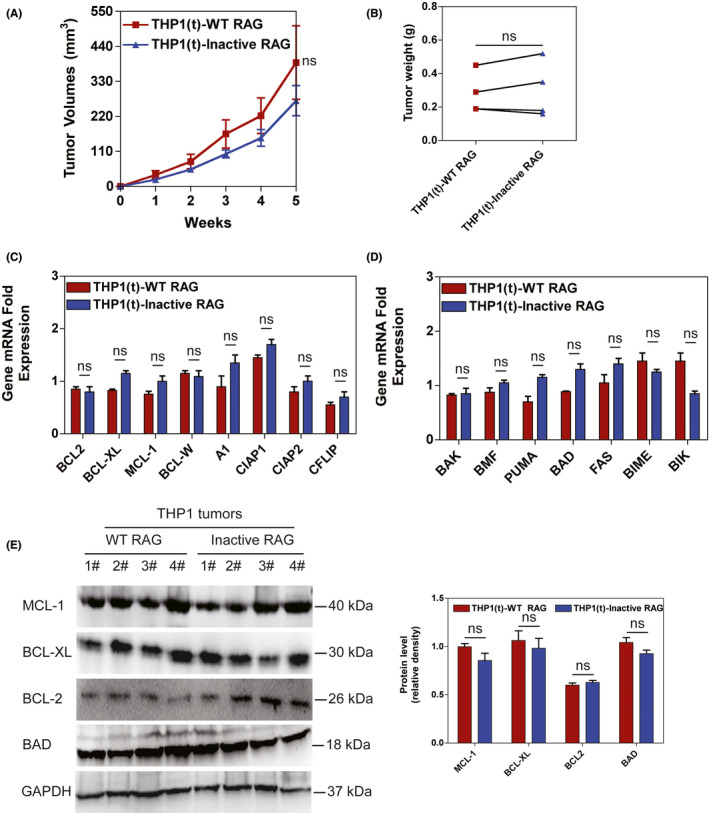
RAG reveals no influence on tumor formation induced by BCR‐ABL1 − cells in a xenograft mouse model. A, NOD/SCID mice were subcutaneously injected with THP1 cells expressing wild‐type (WT) and Inactive RAG, respectively. The tumor volumes were measured at the indicated time points. Bars and error bars represent the mean ± SEM (n = 4). B, Tumors were excised from NOD/SCID mice injected with THP1 cell line expressing WT and Inactive RAG. The tumor weights are plotted, where each dot represents a tumor from one mouse. Data represent the mean ± SEM (n = 4). C‐D, Transcripts of anti–apoptotic genes (C) and pro–apoptotic genes (D) in tumor cells from mice injected with THP1 cell line transducted by lentiviral WT RAG and inactive RAG, respectively. Bars and error bars represent the mean ± SEM (n = 4). E, Western blotting analysis of MCL‐1, BCL‐XL, BCL2, and BAD proteins in tumors formed by THP1 cells expressing WT and inactive RAG. The same blot was probed with an anti–GAPDH antibody to assess protein loading. The data were quantified by ImageJ software and percentage of MCL‐1, BCL‐XL, BCL2, and BAD level related to GAPDH was presented as mean ± SEM (n = 4). ns, no significant difference [Correction added on 19 May 2021, after first online publication: Figure 3A has been corrected.]
3.3. RAG‐induced DSB are repaired by a‐NHEJ in BCR‐ABL1 + cells
To determine whether RAG cleavage activity was linked to DNA lesions, we measured γ‐H2AX accumulation using immunofluorescence. 33 , 34 , 35 We found higher numbers of γ‐H2AX foci per cell in WT RAG‐expressing BCR‐ABL1+ K562 and KCL22 cells compared with Inactive RAG‐expressing cells (Figure 4A,B). However, either WT RAG or Inactive RAG‐expressing BCR‐ABL1 − THP1 cells lacked γ‐H2AX accumulation (Figure 4C). These results suggest that RAG activity causes genomic DNA lesions in BCR‐ABL1 + leukemic cells.
FIGURE 4.
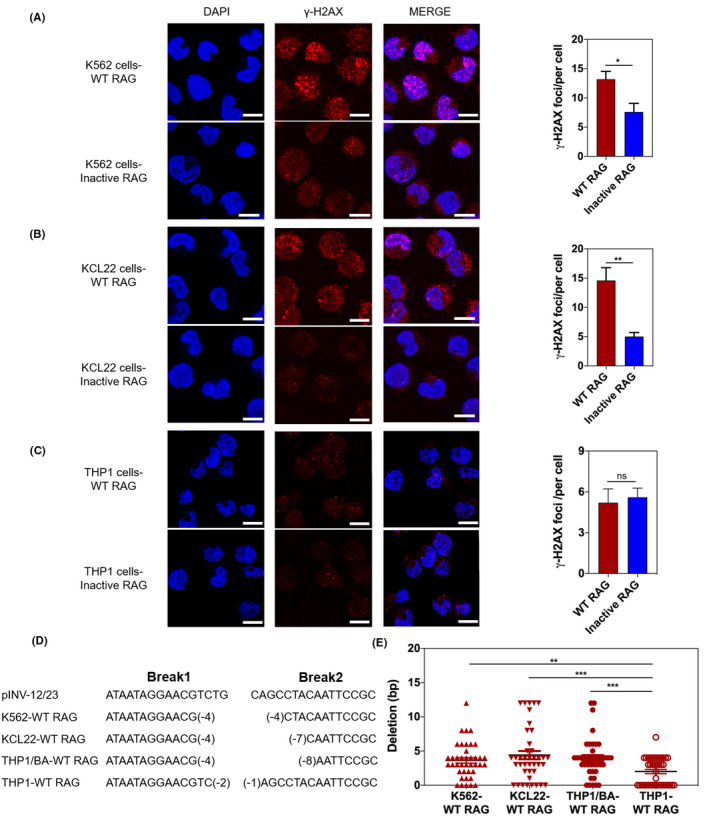
RAG increases γ‐H2AX expression in BCR‐ABL1 + cell lines. A‐C, Immunofluorescence of indicated cells stained with γ‐H2AX antibodies (red) and counterstained with DAPI (blue). Bars, 10 μm. The total number of γH2AX foci/cell was counted for 400 cells. Data are representative of three independent experiments. Bars and error bars represent the mean ± SEM. D, Sequence analysis of nucleotide deletions in coding ends derived from recombination substrate pINV‐12/23 in RAG‐expressing K562, KCL22, THP1/BA, and THP1 cell lines. Italic letters represent sequence derived from pINV‐12/23. Break1 represents coding end sequence flanked 12RSS; Break2 represents coding end sequence close to 23RSS. The representative sequences are presented as indicated. E, Data are summarized from the indicated clones. Bars and error bars represent the mean ± SEM. *, **, and *** indicate P < .05, P < .01, and P < .001, respectively. ns, no significant difference
It was counterintuitive that WT RAG‐expressing BCR‐ABL1 + cells were more prone to anti–apoptosis but displayed γ‐H2AX accumulation. Therefore, we hypothesized that RAG‐expressing BCR‐ABL1 + cells preferred to choose a‐NHEJ to response RAG‐induced DNA lesions, which increased cell survival due to genomic instability. In support of this hypothesis, our findings indicated that RAG activity led to an increase in the expression of several a‐NHEJ molecules and to a reduction in the expression of c‐NHEJ proteins in BCR‐ABL1 + cells (Figure S5). To determine whether RAG‐expressing BCR‐ABL1 + cells preferred to choose a‐NHEJ to response DNA lesions, we introduced a retroviral recombination substrate into expressing WT RAG BCR‐ABL1+ and BCR‐ABL1 − cell lines and examined the substrate rearrangement using genomic PCR with the indicated primers (Figure S1B). We sequenced 160 clones containing PCR fragments derived from the coding joints in the indicated cell lines. Sequence analysis revealed that the a‐NHEJ repair pathway is characterized by coding joint ends with deletions of more than five nucleotides. 36 Our results showed that 26% of coding joint ends had deletions of more than five nucleotides in WT RAG‐expressing BCR‐ABL1 + cells, but only 12% of WT RAG‐expressing BCR‐ABL1 − cells had similar deletion patterns (Figure 4D,E; Table S4). These observations demonstrate that RAG‐mediated DSB are likely to be repaired by a‐NHEJ pathway, albeit with impaired genomic integrity in BCR‐ABL1 + leukemic cells.
3.4. RAG is linked to decreased sensitivities to tyrosine kinase inhibitors by BCR‐ABL1 signaling activation
To investigate whether RAG was involved in TKI resistance, we measured cell viability and IC50 values of BCR‐ABL1 + cells with WT RAG, Inactive RAG, or empty vector control treated with imatinib and dasatinib for 72 h. The data indicated that RAG significantly increased cell viability and the IC50 values of BCR‐ABL1 + cells compared to Inactive RAG and empty vector control (Figures 5A‐D and S6A‐D). qRT‐PCR showed that the levels of BCR‐ABL1 transcripts were not significantly different in either WT or Inactive RAG‐expressing BCR‐ABL1 + cell lines and in xenograft tumors (Figure S6E,F). Sequence analysis of the cDNA corresponding to the ABL1 kinase portion revealed that RAG did not introduce ABL1 kinase domain mutations or deletions (Table S5).
FIGURE 5.
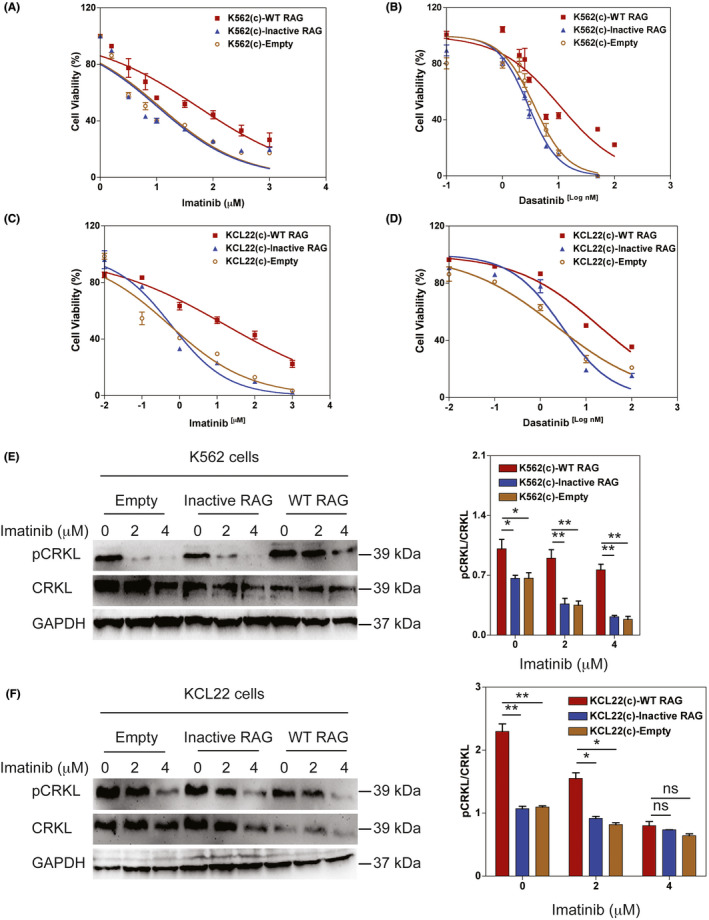
RAG protects against tyrosine kinase inhibitors (TKI)‐induced apoptosis in BCR‐ABL1 + cells. A‐B, Dose‐response curves of wild‐type (WT) RAG, Inactive RAG, and empty vector control‐expressing K562 cells treated with the indicated concentrations of imatinib (A) and dasatinib (B) for 72 h. C‐D, Dose‐response curves of WT RAG, Inactive RAG, and empty vector control‐expressing KCL22 cells treated with the indicated concentrations of imatinib (C) and dasatinib (D) for 72 h. Cell viability was calculated by dividing the viability of the treated cells by that of the untreated control. Data are representative of three independent experiments and are represented as the mean ± SEM. E‐F, Western blotting monitoring the activity of Bcr‐Abl kinase as measured by phosphorylated (p) and total CRKL in WT RAG or Inactive RAG‐expressing K562 (E) or KCL22 cells (F) treated with imatinib at the indicated concentrations for 24 h. The same blot was reprobed with an anti–GAPDH antibody to assess protein loading. Data are representative of three independent experiments. The data were quantified by ImageJ software and the percentage of phosphorylated CRKL level related to total CRKL is presented as mean ± SEM. * and ** represent P < .05 and P < .01, respectively [Correction added on 19 May 2021, after first online publication: Figure 5D has been corrected.]
To determine whether RAG‐regulated TKI failure was due to increased BCR‐ABL1 activity, we measured the level of phosphorylated CRKL following TKI treatment. WT RAG‐expressing BCR‐ABL1 + cells significantly increased phosphorylation of CRKL compared to cells with Inactive RAG (Figure 5E,F). We also showed that the efficiency of imatinib treatment in WT RAG‐expressing K562 cells was 2‐fold lower than in Inactive RAG‐expressing K562 cells (Figure S7A‐C). Consistent with the data, WT RAG‐expressing K562 cells showed decreased cleaved PARP expression levels following imatinib treatment (Figure S7D). The data suggest that the refractoriness to TKI therapy caused by RAG is independent of BCR‐ABL1 expression and BCR‐ABL1 kinase domain mutations but is associated with the activation of BCR‐ABL signaling.
3.5. Primary human CML‐LBC blasts are characterized by high levels of RAG and γ‐H2AX expression
CML‐LBC and BCR‐ABL1 + ALL cells continually express RAG proteins, which could damage DNA during clonal evolution of the diseases. 37 To determine whether RAG mediated genomic DNA lesions in BCR‐ABL1 + patients, we first confirmed that CD34+ CML‐LBC BM cells expressed RAG protein, but there was no RAG expression in CD34+ CML‐CP BM cells with RT‐PCR (Figure 6A). We continued to show that DNA damage indicator γ‐H2AX expression was elevated in CML‐LBC BM cells compared to that in CML‐CP BM cells (Figure 6B), suggesting that endogenous RAG mediates genomic DNA damage in CML‐LBC. Furthermore, we demonstrated the increased expression of the RAG genes correlated with the Δ3‐6 IKZF1 deletion in CML‐LBC patients (Figure 6C), which concurs with previous findings. 37 , 38 To ascertain the exact role of RAG in CML‐LBC, the RAG2 knockout (KO) BV173 cell line originated from CML in lymphoid blast crisis was generated using CRISPR/Cas9‐based gene‐editing technology. The RAG2 KO1 cells exhibited the greatest knockout efficiency compared to those infected with KO2, KO3 RAG2 sgRNA, or non–targeting sgRNA control (named WT; Figure 6D). The significant inhibition of cell growth was revealed in RAG2 KO1 BV173 cells for 72 h (Figure 6E). The data imply that RAG is one of the risk factors for the transformation of CML‐CP to CML‐LBC.
FIGURE 6.
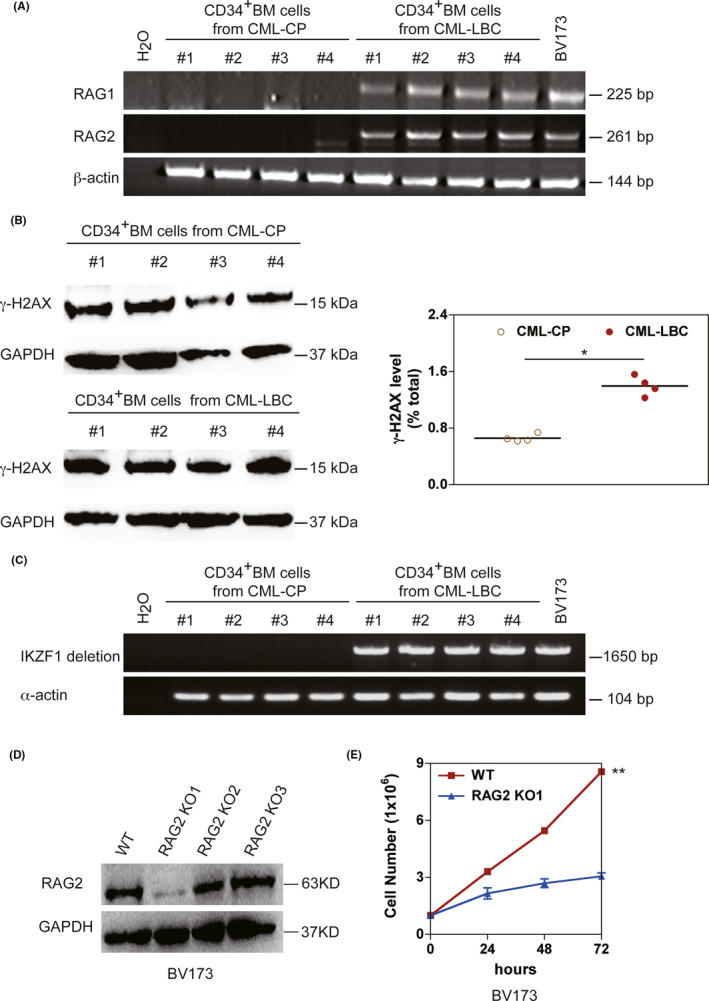
High levels of RAG and γ‐H2AX expressions in CD34+ bone marrow cells derived from primary CML‐CP and CML‐LBC patients. A, The transcripts of RAG1 and RAG2 in CD34+ BM cells derived from CML‐CP and CML‐LBC patients. BV173 cells derived from CML in lymphoid blast crisis served as the positive control for RAG expression. β‐actin was used as a control for cDNA loading. B, Western blotting analysis protein levels of γ‐H2AX in CD34+ BM cells derived from CML‐CP and CML‐LBC patients. The same blot was reprobed with an anti–GAPDH antibody to assess protein loading. The data were quantified by ImageJ software and the percentage of γ‐H2AX level related to GAPDH was presented as mean ± SEM (n = 4). C, Genomic PCR of IKZF1. Δ3‐6 deletion with primers located in intron 2 and intron 6 in CD34+ BM cells derived from CML‐CP and CML‐LBC patients; α‐actin is used as a control for DNA loading. D, RAG2‐knockout BV173 was generated by CRISPR/Cas9‐mediated deletion using three sgRNA (RAG2 KO1‐KO3). The depleting efficiency was confirmed by western blotting. GAPDH was used as protein loading control. E, The proliferation rates of cells derived from BV173 and BV173 RAG2 KO1 were assayed for 72 h. Five independent experiments were repeated. Data represent the mean ± SEM. * and ** indicate P < .05 and P < .01, respectively
4. DISCUSSION
Here, we demonstrate that BCR‐ABL1 cooperates with RAG to enhance BCR‐ABL1+ cell survival in vitro and in vivo. RAG‐expressing BCR‐ABL1 + cells are predominantly biased to selecting the a‐NHEJ pathway in response to RAG endonuclease activity. RAG confers to reduce TKI sensitivity by activating BCR‐ABL1 signaling in BCR‐ABL1 + cells, which is independent of BCR‐ABL1 expression and mutations in the BCR‐ABL1 kinase domain. Thus, our data provide insights into the mechanism of RAG involvement in the pathogenesis of BCR‐ABL1 + leukemia.
BCR‐ABL1 provides the perfect environment for the arrest of maturation and continuous high expression and accumulation of RAG recombinase in CML‐LBC and BCR‐ABL1 + ALL cells. 39 , 40 RAG1 and RAG2 possess their own intrinsic properties and bind various sites throughout the genome. Interestingly, we show that catalytically active RAG is critical for accelerated BCR‐ABL1 + leukemic cell survival and enhances DNA damage in BCR‐ABL1 + cells. K562, KCL22, and THP‐1 cells have been reported to contain the mutant p53 gene. 41 To exclude the possibility that p53 status is involved in the RAG/BCR‐ABL1‐mediated cell growth capacity, we observed that WT p53 BV173 cells expressing endogenous RAG grew faster than those with RAG2 KO, which lacked RAG cleavage activity. 42 , 43 The data demonstrate that RAG activity influences the outcomes in BCR‐ABL1 + B‐ALL independent of the p53 status. Recent studies have shown that DNA DSB signals can initiate transcriptional programs. 44 , 45 In this regard, RAG‐induced DNA breaks regulate lymphocyte migration and homing within specific bone marrow niches through the upregulation of CD62L, CD69, and SWAP70. 46 , 47 Our data are consistent with the notion that RAG‐induced DSB inhibit apoptosis through the upregulation expression of anti–apoptotic genes BCL‐2, BCL‐XL, and MCL‐1 and downregulation of the pro–apoptotic gene BAD in BCR‐ABL1 + cells. Therefore, our data underscore the important roles of RAG in BCR‐ABL1 + leukemia.
RAG‐mediated cleavage events are essential for initiating the induction of DNA repair enzymes during lymphocyte ontogeny. The expression of RAG in developing lymphocytes could lead to increased expression or activity of c‐NHEJ related DNA‐damage repair enzymes, such as DNA‐PKcs, DNA‐Ligase 4, ATM, KU80, and ARTEMIS. 48 , 49 However, expression of oncogenic BCR‐ABL results in increased reactive oxygen species (ROS) which, in turn, causes increased DNA damage. Under these pathophysiological circumstances, we show that the key proteins in the c‐NHEJ pathway, including DNA‐PKcs and DNA‐ligase 4, are downregulated, whereas MRE11 and RAD50 proteins belonging to the a‐NHEJ pathway are upregulated. Furthermore, we confirm that RAG‐induced DNA damage is repaired by the a‐NHEJ pathway in BCR‐ABL1 + cells by using the recombination reporter assays. The data provide the evidence that RAG can lead additional genomic instability and drive disease progression.
The development of TKI has revolutionized the therapy of CML patients, but the failure of TKI often occurs in CML‐LBC and relapsed BCR‐ABL1+ ALL. 50 Our data do not demonstrate that RAG leads to the regulation of BCR‐ABL1 expression and the introduction of kinase mutations. The expression of total CRKL seems to be lower in WT RAG‐expressing BCR‐ABL1 + cells, suggesting that increased CRKL phosphorylation could be secondary to the change in CRKL levels induced by RAG. 45 , 47 , 51 However, previous studies have shown that imatinib triggers apoptosis and leads to G1 arrest and rapid induction of RAG1 and RAG2 expression in v‐Abl pro–B cell lines. 52 , 53 When imatinib resistance is acquired in CML‐LBC and relapsed BCR‐ABL1+ ALL, it ceased to inhibit leukemic cell growth but continues to increase endogenous RAG expression in BCR‐ABL1 + ALL. 40 Therefore, we propose that it might be prudent to treat BCR‐ABL1 + patients with imatinib when the patients already have RAG expression.
Future research will characterize off‐target RAG binding sites in the genome and focus on how these sites mediate oncogenic transformation in BCR‐ABL1 +leukemia.
DISCLOSURE
The authors declare that they have no competing interests.
Supporting information
Fig S1‐S7
Table S1‐S5
Appendix S1
ACKNOWLEDGMENTS
This work was supported by grants (No. 81670157 to YJ; No. 81872026 to YM) from the National Natural Scientific Foundation of China.
Yuan M, Wang Y, Qin M, et al. RAG enhances BCR‐ABL1‐positive leukemic cell growth through its endonuclease activity in vitro and in vivo. Cancer Sci. 2021;112:2679–2691. 10.1111/cas.14939
Meng Yuan and Yang Wang contributed equally to this work as co–first authors.
Contributor Information
Yunfeng Ma, Email: mayunfeng@xjtu.edu.cn.
Yanhong Ji, Email: jiyanhong@xjtu.edu.cn.
REFERENCES
- 1. Ebert A, Hill L, Busslinger M. Spatial regulation of V‐(D)J recombination at antigen receptor loci. Adv Immunol. 2015;128:93‐121. [DOI] [PubMed] [Google Scholar]
- 2. Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101‐132. [DOI] [PubMed] [Google Scholar]
- 3. Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double‐strand breaks to a specific pathway, suppressing error‐prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171‐184. [DOI] [PubMed] [Google Scholar]
- 4. Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annu Rev Genet. 2011;45:167‐202. [DOI] [PubMed] [Google Scholar]
- 5. Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kirkham CM, Boyes J. Genome instability triggered by the V(D)J recombination by‐product. Mol Cell Oncol. 2019;6:1610323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lieber MR, Yu KF, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair. 2006;5:1234‐1245. [DOI] [PubMed] [Google Scholar]
- 8. Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev. 2003;194:77‐95. [DOI] [PubMed] [Google Scholar]
- 9. Teng G, Maman Y, Resch W, et al. RAG represents a widespread threat to the lymphocyte genome. Cell. 2015;162:751‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol. 2009;21:173‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16:234‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fugmann SD, Schatz DG. Identification of basic residues in RAG2 critical for DNA binding by the RAG1‐RAG2 complex. Mol Cell. 2001;8:899‐910. [DOI] [PubMed] [Google Scholar]
- 13. Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1‐RAG2. Nature. 2015;518:507‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ru H, Chambers MG, Fu TM, Tong AB, Liao MF, Wu H. Molecular mechanism of v(d)j recombination from synaptic RAG1‐RAG2 complex structures (vol 163, pg 1138, 2015). Cell. 2015;163:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matthews AGW, Kuo A, Ramon‐Maiques S, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maman Y, Teng G, Seth R, Kleinstein SH, Schatz DG. RAG1 targeting in the genome is dominated by chromatin interactions mediated by the non–core regions of RAG1 and RAG2. Nucleic Acids Res. 2016;44:9624‐9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen XM, Cui YX, Wang HB, Zhou ZH, Gellert M, Yang W. How mouse RAG recombinase avoids DNA transposition. Nat Struct Mol Biol. 2020;27:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirkham CM, Scott JNF, Wang XL, et al. Cut‐and‐run: a distinct mechanism by which V(D)J recombination causes genome instability. Mol Cell. 2019;74:584–597.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808‐4817. [DOI] [PubMed] [Google Scholar]
- 21. El Fakih R, Jabbour E, Ravandi F, et al. Current paradigms in the management of Philadelphia chromosome positive acute lymphoblastic leukemia in adults. Am J Hematol. 2018;93:286‐295. [DOI] [PubMed] [Google Scholar]
- 22. Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010‐4022. [DOI] [PubMed] [Google Scholar]
- 23. O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513‐526. [DOI] [PubMed] [Google Scholar]
- 24. Heinaniemi M, Vuorenmaa T, Teppo S, et al. Transcription‐coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. Elife. 2016;5:e13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koschmieder S, Vetrie D. Epigenetic dysregulation in chronic myeloid leukaemia: A myriad of mechanisms and therapeutic options. Semin Cancer Biol. 2018;51:180‐197. [DOI] [PubMed] [Google Scholar]
- 26. Waanders E, Scheijen B, van der Meer LT, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B‐cell acute lymphoblastic leukemia support a model of multiclonal evolution. Plos Genet. 2012;8:e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiao J, Lv Z, Zhang P, et al. AID assists DNMT1 to attenuate BCL6 expression through DNA methylation in diffuse large B‐cell lymphoma cell lines. Neoplasia. 2020;22:142‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41:783‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carmona LM, Fugmann SD, Schatz DG. Collaboration of RAG2 with RAG1‐like proteins during the evolution of V(D)J recombination. Genes Dev. 2016;30:909‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klemm L, Duy C, Iacobucci I, et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16:232‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szymczak AL, Workman CJ, Wang Y, et al. Correction of multi‐gene deficiency in vivo using a single ‘self‐cleaving’ 2A peptide‐based retroviral vector (vol 22, pg 589, 2004). Nat Biotechnol. 2004;22:1590. [DOI] [PubMed] [Google Scholar]
- 32. Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR‐ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070‐1079. [PubMed] [Google Scholar]
- 33. Ji Y, Zhang J, Lee AI, Cedar H, Bergman Y. A multistep mechanism for the activation of rearrangement in the immune system. Proc Natl Acad Sci USA. 2003;100:7557‐7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tubbs AT, Dorsett Y, Chan E, et al. KAP‐1 promotes resection of broken DNA ends not protected by gamma‐H2AX and 53BP1 in G(1)‐phase lymphocytes. Mol Cell Biol. 2014;34:2811‐2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. Paving the way for H2AX phosphorylation: chromatin changes in the DNA damage response. Cell Cycle. 2009;8:1494‐1500. [DOI] [PubMed] [Google Scholar]
- 36. Gigi V, Lewis S, Shestova O, et al. RAG2 mutants alter DSB repair pathway choice in vivo and illuminate the nature of ‘alternative NHEJ’. Nucleic Acids Res. 2014;42:6352‐6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mullighan CG, Miller CB, Radtke I, et al. BCR‐ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110‐114. [DOI] [PubMed] [Google Scholar]
- 38. Dong Y, Liu F, Wu C, et al. Illegitimate RAG‐mediated recombination events are involved in IKZF1 Delta3‐6 deletion in BCR‐ABL1 lymphoblastic leukaemia. Clin Exp Immunol. 2016;185:320‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Witkowski MT, Hu YF, Roberts KG, et al. Conserved IKAROS‐regulated genes associated with B‐progenitor acute lymphoblastic leukemia outcome. J Exp Med. 2017;214:773‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomson DW, Shahrin NH, Wang PPS, et al. Aberrant RAG‐mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2020;34:2051‐2063. [DOI] [PubMed] [Google Scholar]
- 41. Shiohara M, Akashi M, Gombart AF, Yang R, Koeffler HP. Tumor necrosis factor alpha: posttranscriptional stabilization of WAF1 mRNA in p53‐deficient human leukemic cells. J Cell Physiol. 1996;166:568‐576. [DOI] [PubMed] [Google Scholar]
- 42. Kuroda J, Kimura S, Segawa H, et al. p53‐independent anti–tumor effects of the nitrogen‐containing bisphosphonate zoledronic acid. Cancer Sci. 2004;95:186‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sliwinski T, Czechowska A, Szemraj J, Morawiec Z, Skorski T, Blasiak J. STI571 reduces NER activity in BCR/ABL‐expressing cells. Mutat Res. 2008;654:162‐167. [DOI] [PubMed] [Google Scholar]
- 44. Bednarski JJ, Nickless A, Bhattacharya D, Amin RH, Schlissel MS, Sleckman BP. RAG‐induced DNA double‐strand breaks signal through Pim2 to promote pre‐B cell survival and limit proliferation. J Exp Med. 2012;209:11‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Helmink BA, Sleckman BP. The response to and repair of RAG‐mediated DNA double‐strand breaks. Annu Rev Immunol. 2012;30:175‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bednarski JJ, Pandey R, Schulte E, et al. RAG‐mediated DNA double‐strand breaks activate a cell type‐specific checkpoint to inhibit pre‐B cell receptor signals. J Exp Med. 2016;213:209‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bredemeyer AL, Helmink BA, Innes CL, et al. DNA double‐strand breaks activate a multi‐functional genetic program in developing lymphocytes. Nature. 2008;456:819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dujka ME, Puebla‐Osorio N, Tavana O, Sang M, Zhu C. ATM and p53 are essential in the cell‐cycle containment of DNA breaks during V(D)J recombination in vivo. Oncogene. 2010;29:957‐965. [DOI] [PubMed] [Google Scholar]
- 49. Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shanmuganathan N, Branford S. The hidden pathogenesis of CML: Is BCR‐ABL1 the first event? Curr Hematol Malig R. 2019;14:501‐506. [DOI] [PubMed] [Google Scholar]
- 51. Adane B, Ye H, Khan N, et al. The hematopoietic oxidase NOX2 regulates self‐renewal of leukemic stem cells. Cell Rep. 2019;27:238–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chow KT, Timblin GA, McWhirter SM, Schlissel MS. MK5 activates Rag transcription via Foxo1 in developing B cells. J Exp Med. 2013;210:1621‐1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Muljo SA, Schlissel MS. A small molecule Abl kinase inhibitor induces differentiation of Abelson virus‐transformed pre‐B cell lines. Nat Immunol. 2003;4:31‐37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1‐S5
Appendix S1