

Abstract
The BRAF V600E mutation occurs in approximately 10% of patients with metastatic colorectal cancer (CRC) and constitutes a distinct subtype of the disease with extremely poor prognosis. To address this refractory disease, we investigated the unique metabolic gene profile of BRAF V600E‐mutated tumors via in silico analysis using a large‐scale clinical database. We found that BRAF V600E‐mutated tumors exhibited a specific metabolic gene expression signature, including some genes that are associated with poor prognosis in CRC. We discovered that BRAF V600E‐mutated tumors expressed high levels of glycolytic enzyme enolase 2 (ENO2), which is mainly expressed in neuronal tissues under physiological conditions. In vitro experiments using CRC cells demonstrated that BRAF V600E‐mutated cells exhibited enhanced dependency on ENO2 compared to BRAF wild‐type cancer cells and that knockdown of ENO2 led to the inhibition of proliferation and migration of BRAF V600E‐mutated cancer cells. Moreover, inhibition of ENO2 resulted in enhanced sensitivity to vemurafenib, a selective inhibitor of BRAF V600E. We identified AP‐1 transcription factor subunit (FOSL1) as being involved in the transcription of ENO2 in CRC cells. In addition, both MAPK and PI3K/Akt signaling were suppressed upon inhibition of ENO2, implying an additional oncogenic role of ENO2. These results suggest the crucial role of ENO2 in the progression of BRAF V600E‐mutated CRC and indicate the therapeutic implications of targeting this gene.
Keywords: BRAF V600E‐mutated colorectal cancer, ENO2, FOSL1, glycolysis, metabolic genes
There was no significant difference in the expression of ENO1 in BRAF V600E‐mutated CRC and other types of CRC, but ENO2 levels were significantly higher in BRAF V600E‐mutated CRC.

1. INTRODUCTION
Approximately 10% of patients with metastatic colorectal cancer (CRC) have BRAF V600E mutations. 1 BRAF V600E‐mutated CRC is more common in the right side of the colon and in older patients, and is associated with a poor prognosis. 2 , 3 , 4 In BRAF V600E‐mutated CRC, the constitutive activation of BRAF is independent of extracellular factors and activates a signaling pathway that leads to cell proliferation. 5 , 6 , 7 Inhibition of BRAF alone using a selective antibody leads to paradoxical activation of the MAPK pathway through suppression of a negative feedback signal from downstream molecules. 8 , 9 Although combination therapy using antibodies against BRAF, EGFR, and downstream MEK has been shown to improve patients’ prognosis over several months, 10 , 11 , 12 , 13 activation of the MAPK pathway through BRAF V600E‐mutation is extremely powerful and therapeutic approaches for BRAF V600E‐mutated CRC remain challenging.
Cancer metabolism is a key determinant of malignant potential in most solid cancers. 14 Cancer cell metabolism is based on enhancement of the glycolytic system to ensure that the cell can survive under hypoxic conditions and to provide stable energy production. 14 , 15 Glycolysis dependency is caused by overexpression of various glycolytic enzymes due to the dysregulation of oncogenes and tumor suppressors. 16 , 17 BRAF V600E‐mutated tumors require considerable energy to maintain their high proliferative capacity. However, the mechanism by which BRAF V600E mutations affect the alteration of the metabolic system has not yet been sufficiently investigated.
To understand the contribution of the BRAF V600E mutation to the metabolic alterations of CRC, we analyzed a large‐scale database and identified unique metabolic gene expression signatures in BRAF V600E‐mutated CRC. Among genes specifically overexpressed in BRAF V600E‐mutated tumors, we focused on glycolytic enzyme enolase 2 (ENO2). Enolase is a glycolytic enzyme and includes three isoforms: ENO1, ENO2, and ENO3. Under physiological conditions, the gastrointestinal tract depends on ENO1 for energy production, and ENO2 is predominantly expressed in neuronal tissues. 18 , 19 , 20 Although previous studies have shown that ENO2 is overexpressed in some malignancies, including neuroendocrine tumors, small cell lung cancer (SCLC), and breast cancer, 21 , 22 the involvement of ENO2 in the oncogenic context has not yet been investigated in detail. Here, we sought to investigate the role of ENO2 in the progression of BRAF V600E‐mutated CRC and to explore the therapeutic implications of targeting this gene.
2. MATERIAL AND METHODS
2.1. Clinical tissue samples
Primary CRC specimens were collected from 121 patients who underwent surgery at the Department of Gastroenterological Surgery, Osaka University (Suita, Japan) between 2011 and 2012. All patients were diagnosed with CRC based on the clinicopathological criteria described by the Japanese Society for Cancer of the Colon and Rectum. None of the patients received preoperative chemotherapy or radiotherapy. Specimens were fixed in 10% formalin overnight at room temperature, processed through graded ethanol solutions, and embedded in paraffin. The specimens were used appropriately and with the approval of the Ethics Committee of the Graduate School of Medicine, Osaka University. The present study was approved by the Research Ethics Committee of Osaka University (approval ID: 08226) and written informed consent was obtained from all patients included in this study.
2.2. Cell lines and cell culture
The human CRC cell lines RKO, HT29, HCT116, and DLD1 were purchased from the American Type Culture Collection and maintained in DMEM (Sigma‐Aldrich; Merck KGaA) containing 10% FBS (Gibco; Thermo Fisher Scientific) in a humidified incubator at 37°C and 5% CO2.
2.3. RNA interference
Two types of ENO2‐specific siRNA were purchased from Sigma‐Aldrich to knock down ENO2 mRNA. RKO, HT29, HCT116, and DLD1 cells were seeded (2 × 105 in 6 well dish) and transfected with ENO2 or negative control siRNA using Lipofectamine RNAiMax (Thermo Fisher Scientific) at a concentration of 10 nmol/L, according to the manufacturer’s protocol. These cell lines were maintained in a humidified incubator at 37°C and 5% CO2 for 72 hours after transfection. The sequences of the siRNA against ENO2 were: siRNA#1 CCAUUUGACCAGGAUGAUU[dT][dT]; siRNA#2 GGUCACUGAAGCCAUCCAA[dT][dT]. The sequences of the siRNA against FOSL‐1 were: siRNA#1 GACUGACAAACUGGAAGAU[dT][dT]; siRNA#2 CAAACUGGAAGAUGAGAAA[dT][dT].
2.4. Lentivirus vector construction
The full length of human ENO2 cDNA was amplified by PCR and ligated into the CSII‐CMV‐MCS‐IRES2‐Bsd lentivirus vector (provided by Dr Miyoshi, RIKEN‐BRC, Japan 23 and transfected into 293FT cells with packaging and envelope plasmids using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. After 48 hours of incubation, the supernatant was filtered and used for virus transduction to target cells. Stable clones were obtained after antibiotic selection. The overexpression of genes was confirmed by RT‐PCR and western blotting.
2.5. Real‐time quantitative RT‐PCR
Total RNA was extracted from the indicated cells using TRIzol RNA Isolation Reagent (Thermo Fisher Scientific), as previously described. 24 The cDNA was synthesized from 10 ng total RNA using the Rever Tra Ace qPCR RT Master Mix (Toyobo Life Science), according to the manufacturer’s protocol. Quantitative PCR was performed in a Light Cycle 2.0 System (Roche Applied Science) using the Light Cycler FastStart DNA Master SYBR Green I (Roche Applied Science). The amplification conditions were as follows: initial denaturation at 95°C for 10 minutes, followed by 45 cycles of denaturation at 95°C for 10 seconds, annealing at 60°C for 10 seconds, and extension at 72°C for 10 seconds. Data were normalized to the expression of PPIA.
The following primers were used: ENO2(F), 5′‐ACCAGGACTTTGTCAGGGACTA‐3′; ENO2(R), 5′‐TACATTGGCTGTGAACTTGGAC‐3′; FOSL1(F), 5′‐GGCCTCTGACCTACCCTAC‐3′; FOSL1(R), 5′‐CTTCCAGTTTGTCAGTCTCTCCTG‐3′; PPIA(F), 5′‐ATGCTGGACCCAACACAAAT‐3′; PPIA(R), 5′‐TCTTTCACTTTGCCAAACACC‐3′.
2.6. BRAF inhibitor sensitivity assay
RKO, HT29, HCT116, and DLD1 cells were seeded at a density of 3.0 × 103 cells/well in 96‐well plates and pre–cultured for 24 hours. The cells were exposed to various concentrations of vemurafenib (Selleck). The cytotoxic effects of vemurafenib were evaluated using a Cell Counting Kit‐8 (Dojindo), according to the manufacturer’s protocol.
2.7. Western blotting
Total proteins were extracted from cultured cells using radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific). The proteins were separated by 10% SDS‐PAGE and electroblotted onto polyvinylidene fluoride membranes (Merck KGaA) at 300 mA for 60 minutes. After blocking with 3% skim milk at room temperature for 1 hour, the membranes were incubated with primary antibodies at the appropriate concentrations at 4°C overnight: ENO1 (1:1000; #3810, Cell Signaling), ENO2 (1:1000; #9536, Cell Signaling), BRAF (1:1000; #14814, Cell Signaling), FOSL1 (1:1000; #D80B4, Cell Signaling), pERK (1:2000; #4370, Cell Signaling), ERK (1:1000; #4695, Cell Signaling), pAKT (1:2000; #4060, Cell Signaling), and AKT (1:1000; #9272, Cell Signaling). After incubation with secondary antibodies, protein bands were detected using the Amersham Enhanced Chemiluminescence Prime Western Blotting Detection Reagent (GE Healthcare).
2.8. Immunohistochemical staining
The anti–ENO1 rabbit antibody (Anit‐ENO1 #ab227978, Abcam), anti–ENO2 rabbit antibody (Anti–NSE, #ab53025, Abcam), anti–BRAF V600E rabbit antibody (Anti–BRAF mutated V600E #200535, Abcam), and the VECTASTAIN Elite ABC Rabbit Immunoglobulin G Kit (Vector Laboratories) were used for immunohistochemical staining. The slides with antibody were diluted 1:2000 (Anti–ENO1), 1:200 (Anti–BRAFV600E) and 1:100 (Anti–NSE) were incubated overnight at 4°C. We assigned an intensity score of +2 to cytoplasm that was stained as intensely as the positive control, a score of +1 when the cytoplasmic staining was weaker than the positive control, and a score of 0 to unstained cytoplasm.
2.9. Cell proliferation assay
Cells were seeded in 96‐well plates at a density of 3.0 × 103 cells/well. Cell proliferation was assessed 24, 48, and 72 hours after transfection using the Cell Counting Kit‐8, according to the manufacturer’s protocol. We confirmed that the absorbance was compatible with manual cell counting before the experiment.
2.10. Cell migration assay
Cell migration was evaluated using the scratch wound‐healing assay. Cells were seeded in 6‐well plates at a density of 1.0 × 106 cells/well and grown to confluence under standard conditions. A scratch was generated in the cells using a 1000‐μL pipette tip. Cells were cultured under standard conditions in DMEM supplemented only with 1% FBS to prevent proliferation. The images were captured at 0, 48, and 72 hours after scratching using a BZ‐X710 microscope (Keyence Corporation) and analyzed using a BZ‐X analyzer (v. 1.3.0.3; Keyence Corporation). Cell migration was calculated by measuring the average distance between the wound edges at five random sites.
2.11. Data analysis
R programming language v. 4.0.2 and JMPpro 14.0.0 (SAS Institute) were used for statistical analyses. The Cancer Genome Atlas mRNA expression data and clinical information were downloaded from the GDAC Firehose website (http://gdac.broadinstitute.org). “Metabolic genes” refers to genes that are involved in cellular metabolic function, such as glycolysis, oxidative phosphorylation, and small molecule transport, which were downloaded from a previous pivotal report. 25 The differentially expressed genes between BRAF V600E‐mutated CRC and other types of CRC were analyzed using the R package “limma.” Z‐scores were calculated using the survfit function of R package “survival.” Statistically significant differences between groups were determined by Student’s t test and Fisher’s exact probability test. Overall survival (OS) and relapse‐free survival (RFS) rates were calculated using the Kaplan‐Meier method. When P < .05 was obtained, the results indicated a statistically significant difference.
3. RESULTS
3.1. Glycolytic enzyme enolase 2 is specifically upregulated in BRAF V600E‐mutated colorectal cancer cells
To identify specific metabolic gene expression signatures in BRAF V600E‐mutated CRC, we performed unsupervised hierarchical clustering analysis using the Cancer Genome Atlas (TCGA) colorectal adenocarcinoma (COADREAD) dataset (Figure 1A). We searched for metabolic‐related genes that are highly expressed in CRC with poor prognosis and are specifically expressed in BRAF V600E‐mutated CRC. The expression of ENO2 was higher in BRAF V600E‐mutated CRC than in non–BRAF V600E‐mutated CRC or BRAF wild‐type CRC. We focused on the glycolytic enzyme ENO2, which is specifically upregulated in BRAF V600E‐mutated CRC and is significantly associated with poor prognosis (Figure 1B). Gene expression analysis of glycolytic enzymes and tricarboxylic acid cycle enzymes in BRAF V600E‐mutated CRC, non–BRAF V600E‐mutated CRC, and BRAF wild‐type CRC revealed that the expression of ENO2 was significantly higher in BRAF V600E‐mutated CRC than in the other CRC types (Figure 1C).
FIGURE 1.
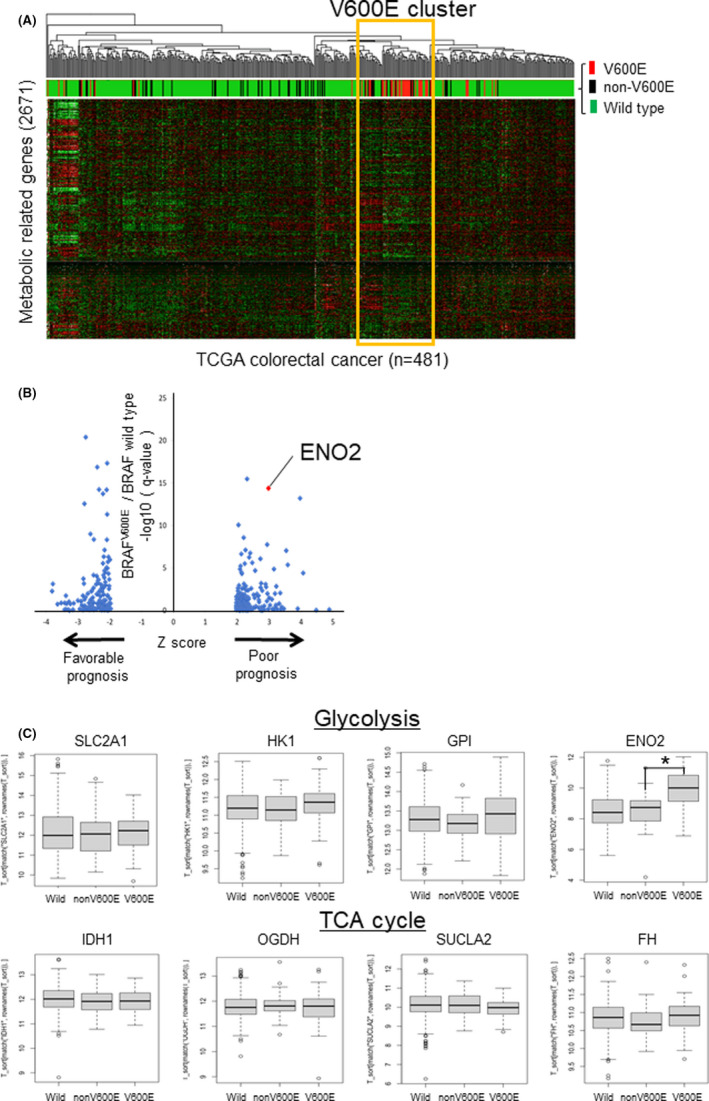
Identification of specific metabolic gene expression signatures of BRAF V600E‐mutated CRC. A, Unsupervised hierarchical clustering using the TCGA COADREAD dataset based on the expression of metabolism‐related genes (n = 2752). The colors of the upper bar represent the BRAF mutation status; red, black, and green represent BRAF V600E‐mutated, non–V600E mutated, and wild‐type tumors, respectively. B, Scatterplot showing the differentially expressed genes between BRAF V600E‐mutated tumors and wild‐type tumors. The horizontal axis indicates the Z‐score, which represents the statistical significance of each gene’s association with patient prognosis. The vertical axis indicates the q‐value (−log 10) and the statistical significance of the comparison between BRAF V600E‐mutated tumors and wild‐type tumors. Only genes with |Z‐score| > 1.96 are shown. C, Expression of representative metabolic‐related genes according to BRAF mutation status. (Upper) Glycolytic enzyme. (Lower) tricarboxylic acid cycle (TCA) enzymes (*P < .0001)
To clarify the clinicopathological significance of ENO2, we performed immunohistochemical staining for ENO2. We confirmed the expression of ENO1 and ENO2 in 9 cases of BRAF V600E‐mutated CRC, 10 cases of RAS‐mutated CRC, and 10 cases of BRAF/RAS wild‐type CRC that were already diagnosed by genetic testing. The results indicate that ENO2 was frequently overexpressed in BRAF V600E‐mutated CRC than in other CRC types. A comparison of the expression of ENO1 and ENO2 in BRAF V600E‐mutated CRC and the other CRC showed that there was no significant difference in the expression of ENO1 between the two groups, but that ENO2 levels were significantly higher in BRAF V600E‐mutated CRC (Figure 2A,B). Similar results were obtained in an analysis of the Cancer Cell Line Encyclopedia (CCLE) database. We found no significant difference in the expression of ENO1 based on BRAF mutation status. While the expression of ENO2 was significantly higher in BRAF V600E‐mutated CRC (Figure S1A). Further bioinformatical analysis using the Project Achilles dataset showed that BRAF V600E‐mutated CRC has more dependency on ENO2 for cell proliferation ability (Figure S1B).
FIGURE 2.
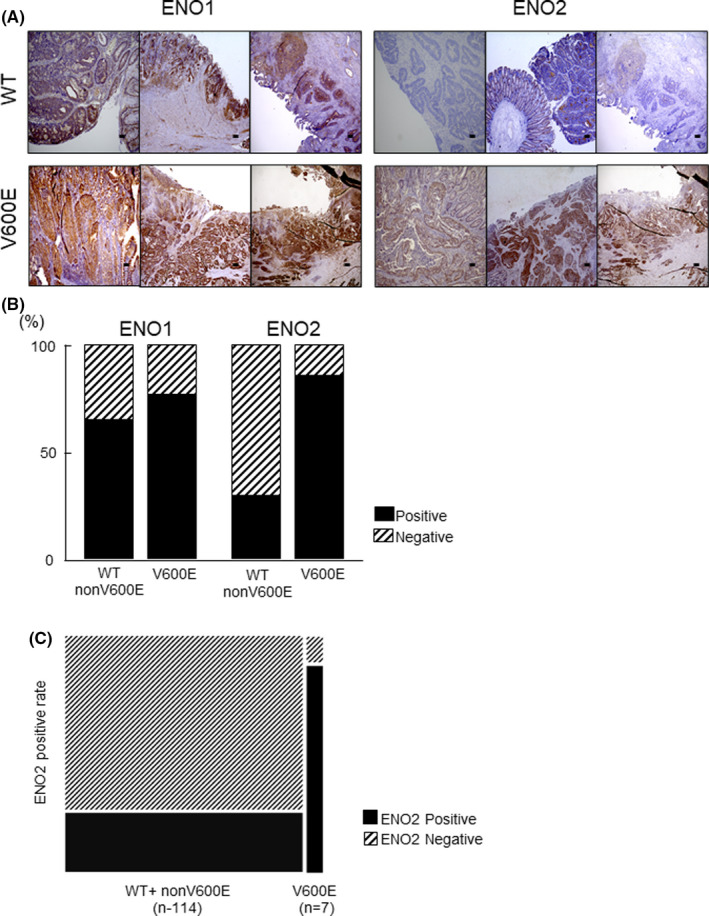
Immunohistochemical analysis of ENO1 and ENO2 in BRAF wild‐type and BRAF V600E‐mutated CRC. A, Representative images of positive and negative immunohistochemical staining with anti–ENO2 antibody in BRAF wild‐type CRC and BRAF V600E‐mutated CRC. Scale bar: 50 µm. B, The positive rate of ENO1 and ENO2 expression in BRAF wild‐type and BRAF V600E‐mutated CRC based on immunohistochemical staining. C, The positive rate of ENO2 expression in BRAF wild‐type+nonV600E and BRAF V600E‐mutated CRC based on immunohistochemical staining in 121 CRC patient
3.2. Glycolytic enzyme enolase 2 expression is associated with poor overall survival and relapse‐free survival in colorectal cancer
To explore the role of ENO2 in the malignant behavior of CRC, we classified CRC patients (n = 121) into three groups based on the ENO2 staining intensity, as described above. We defined a score of 0 as being in the ENO2‐negative group and a score of 1 or 2 as being in the ENO2‐positive group (Figure S2A). BRAF status was defined by immunohistochemical staining using antibody specific for BRAF V600E mutation (Figure S2B). As a result, out of seven cases with BRAF V600E mutation, six cases showed high ENO2 expression (85.7%), whereas only 27.2% (31/114) were ENO2‐positive in cases with BRAF wild and non–V600E mutation (P value < .05) (Figure 2C). A clinicopathological analysis showed that high expression of ENO2 was associated with clinical characteristics, tumor depth, lymph node metastasis, lymphatic invasion, distant metastasis, advanced clinical stage, and BRAF status (Table S1). Kaplan‐Meier overall survival (OS) curves for patients with CRC (n = 121) was performed. The OS rate of patients in the ENO2‐positive group was significantly lower than that of the ENO2‐negative group (*P = .032) (Figure S2C). Kaplan‐Meier relapse‐free survival (RFS) curves were plotted for patients with CRC (n = 112). The RFS rate of patients in the ENO2‐positive group was significantly lower than that of the ENO2‐negative group (**P = .035) (Figure S2D).
3.3. Glycolytic enzyme enolase 2 is involved in proliferation and migration in BRAF V600E‐mutated colorectal cancer cells
To elucidate the mechanism by which ENO2 is involved in the malignant potential of CRC, we performed functional analysis using CRC cell lines with or without BRAF V600E mutations. Knockdown of ENO2 potently inhibited the proliferation and migration ability of CRC cell lines RKO and HT29 possessing BRAF V600E mutations, whereas the effect was modest in the BRAF wild‐type cell lines HCT116 and DLD1 (Figure 3A‐C and Figures S3, S4A,B). We also investigated expression changes of epithelial mesenchymal transition (EMT) markers upon knockdown of ENO2. Although a modest decrease of ZEB1 has observed, other EMT markers exhibit almost no significant changes in expression (Figure S4C).
FIGURE 3.
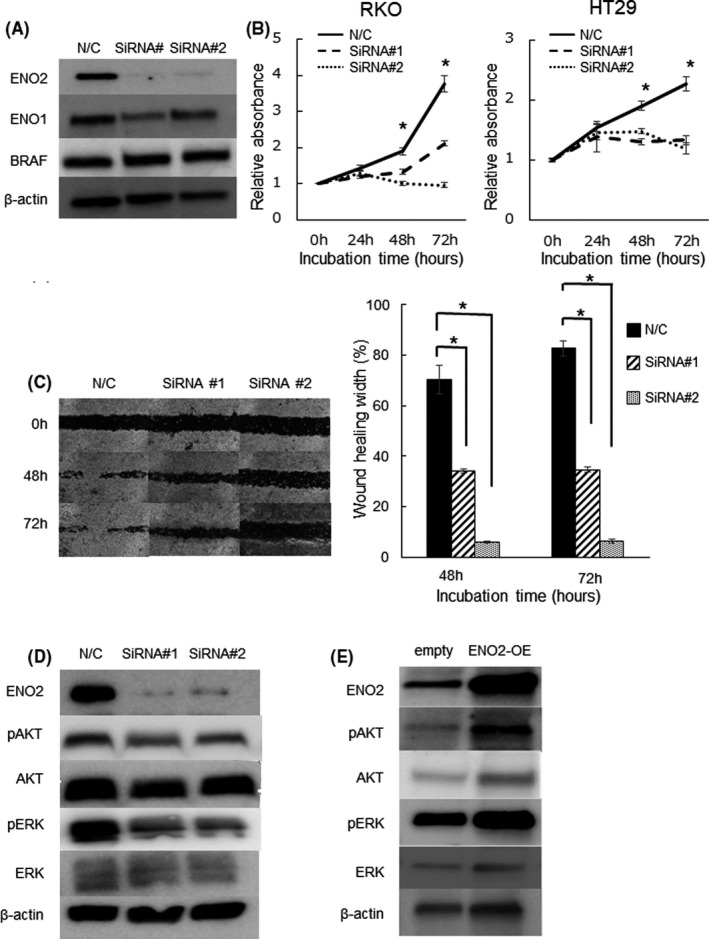
Knockdown of ENO2 attenuates cell proliferation and migration in BRAF V600E‐mutated CRC cells. A, ENO2 protein expression levels in RKO cells transfected with N/C and ENO2 siRNA (siRNA#1 and siRNA#2) in western blot analysis. N/C, negative control. B, Proliferation assay using RKO and HT29 cells transfected with N/C or ENO2 siRNA (*P < .05). C, Representative images of the scratch wound‐healing assay using RKO cells transfected with N/C or ENO2 siRNA. Magnification: 100×. Average distance between wound edges for five different areas at the indicated time points (relative change from the distance at 0 h) (*P < .05). D, Western blot analysis showing the expression of ENO2, phosphorylated AKT, and phosphorylated ERK upon transfected with N/C or ENO2 siRNA. E, Western blot analysis showing the expression of ENO2, phosphorylated AKT, and phosphorylated ERK after overexpression of ENO2 in RKO cells
Cell cycle analysis revealed that knockdown of ENO2 increased the proportion of cells in the G1 phase and decreased the ratio of cells in the S and G2/M phases in RKO cells (Figure S5).
3.4. Glycolytic enzyme enolase 2 activates both PI3K/AKT and MAPK/ERK pathways
PI3K/AKT and MAPK have been reported to be key signaling pathways that promote cell proliferation. 26 , 27 , 28 We found that ENO2 inhibition significantly reduces the phosphorylation levels of AKT and ERK, while overexpression of ENO2 upregulates these phosphorylation levels in colorectal cancer cell lines (Figure 3D,E).
3.5. Glycolytic enzyme enolase 2 is transcriptionally regulated by FOSL1
To clarify the regulatory network underlying the association between the BRAF V600E mutation and ENO2 expression, we administered vemurafenib, a specific inhibitor of mutated BRAF, to RKO cells and examined changes in the expression of ENO2. The expression of ENO2 was decreased by vemurafenib at both the mRNA and protein levels (Figure 4A). Next, we searched for a transcription factor that links BRAF V600E mutation and ENO2 expression. BRAF mutation activates the MAPK/ERK pathway, resulting in an increase in some nuclear transcription factors. We searched for a transcription factor that is specifically upregulated in BRAF V600E‐mutated CRC and is also highly correlated with ENO2 expression. We reanalyzed the TCGA database and found that the FOSL1 gene, a major component of transcription factor complex AP‐1, is a promising candidate. FOSL1 is one of the main targets of BRAF‐MAPK signaling. 29 , 30 , 31 Inhibition of BRAF V600E activity using vemurafenib significantly decreased FOSL1 expression levels in a dose–dependent manner (Figure 4B). FOSL1 is significantly upregulated in BRAF V600E‐mutated colorectal cancer compared with BRAF non–V600E‐mutated and RAS/BRAF wild‐type cancer (Figure 4C). Subgroup analysis revealed that the correlation is specifically higher in BRAF V600E‐mutated colorectal cancer, whereas the correlation is relatively weak in wild‐type and non–V600E‐mutated tumors (Figure 4D and Figure S6A). Furthermore, the correlation with FOSL1 is higher in ENO2 than ENO1 or ENO3 (Figure 4D and Figure S6B,C). To evaluate the molecular association between FOSL1 and ENO2, we subsequently performed in vitro experiments. Knockdown of FOSL1 significantly inhibited the expression of ENO2 depending on the knockdown efficiency of FOSL1 in colorectal cancer cells (Figure 4E). These data indicate a potential close relationship between FOSL1 and ENO2 in BRAF V600E‐mutated colorectal cancer.
FIGURE 4.
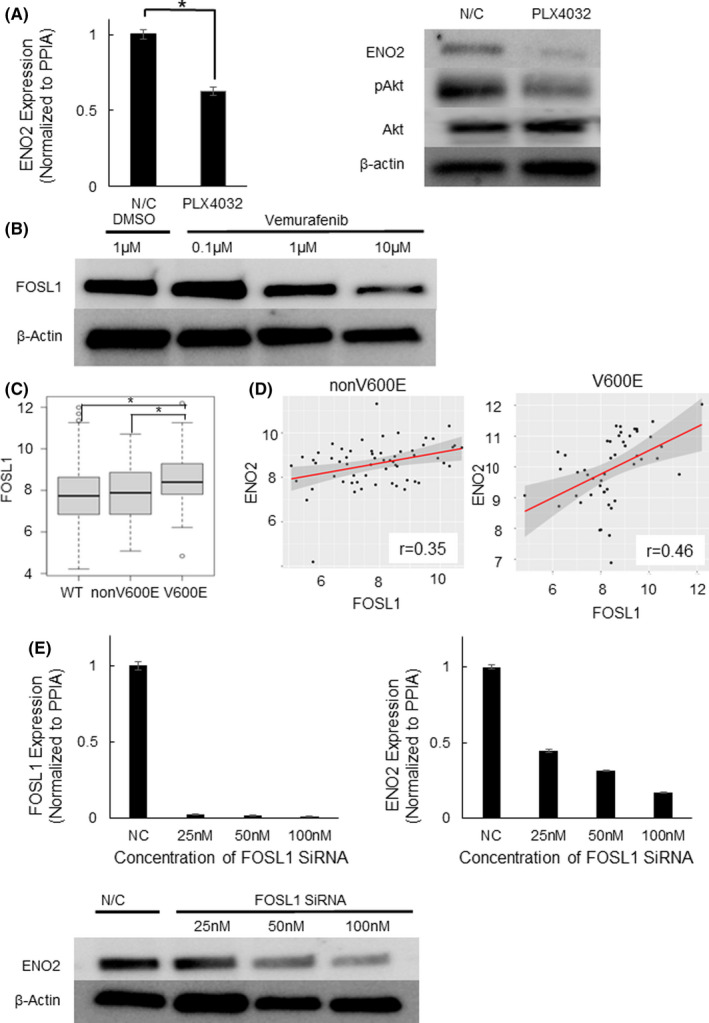
ENO2 expression is transcriptionally regulated by FOSL1. A, Western blot analysis showing ENO2 and phosphorylated AKT expression after administration of N/C or vemurafenib. B, Protein expression levels of FOSL1 treated with DMSO or vemurafenib (0.1, 1, 10 µmol/L). C, Expression levels of FOSL1 according to BRAF mutation status. (TCGA database). D, Correlation between FOSL1 and ENO2 expression in non–V600E mutated cases (left n = 48, r = .35, P = .006) and BRAF V600E‐mutated cases (right n = 60, r = .46, P = .00087) of CRC. E, mRNA and protein expression levels of ENO2 transfected with N/C or FOSL1 siRNA (25, 50 and 100 nmol/L). Each bar represents the mean ± SEM of triplicate measurements. (*P < .05)
3.6. Inhibition of glycolytic enzyme enolase 2 enhances the drug sensitivity to vemurafenib in BRAF V600E0mutated colorectal cancer cells
Vemurafenib was used in the therapeutic treatment of patients with BRAF V600E‐mutated CRC. We confirmed that BRAF V600E‐mutated CRC cells (RKO and HT29) had high sensitivity to vemurafenib, whereas BRAF wild‐type CRC cells (HCT116 and DLD1) did not (Figure 5A). The 50% inhibitory concentration (IC50) values of vemurafenib were as follows: HCT116, not reached; DLD1, not reached; RKO, 11.6 µg; HT29, 3.16 µg. Dose‐response experiments with different concentrations of ENO2 SiRNA were performed (Figure 5B). The absolute cell viable rates are indicated in Figure S7. The data shows that a high concentration of ENO2 siRNA (50 nmol/L) inhibited the cancer cell proliferation more efficiently than a lower concentration (25 nmol/L) of SiRNA. Knockdown of ENO2 improved the sensitivity to vemurafenib in RKO cells, whereas sensitivity was not affected in HCT116 cells with BRAF V600E wild type (Figure 5C). The IC50 values of vemurafenib were as follows: RKO negative control, 11.6 µg; RKO SiRNA25 nmol/L, 7.3 µmol/L; RKO SiRNA50 nmol/L, 5.2 µmol/L; HCT116 negative control, not reached; HCT116 SiRNA#1, not reached; HCT116 SiRNA#2, not reached.
FIGURE 5.
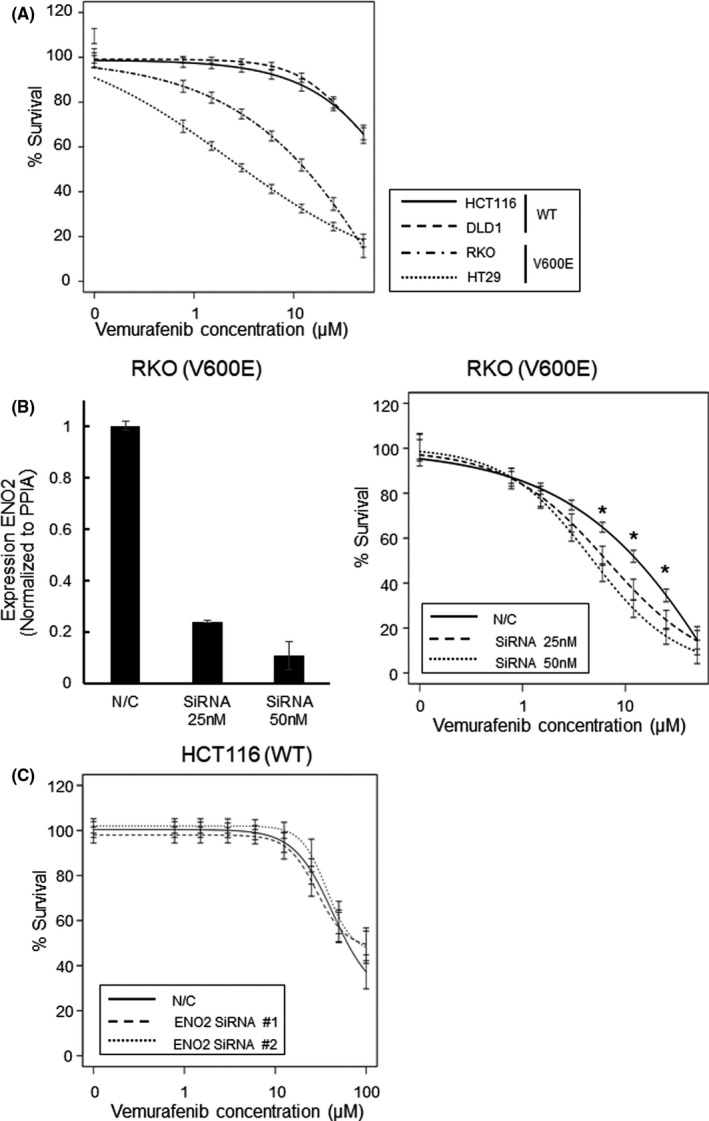
Combinational inhibition of BRAF V600E and ENO2 attenuate proliferation of BRAF V600E‐mutated cells. A, Dose response curve after exposure to vemurafenib in CRC cell lines. B, (Left) mRNA expression levels of ENO2 transfected with N/C or ENO2 SiRNA (25 and 50 nmol/L). (Right) Dose response curve after exposure to vemurafenib in RKO cells transfected with N/C or ENO2 siRNA (25 and 50 nmol/L). (*P < .05). C, Dose response curve after exposure to vemurafenib in HCT116 cells transfected with N/C or ENO2 siRNA. Each bar represents the mean ± SEM of quintuple measurements
4. DISCUSSION
Most solid cancers exhibit the Warberg effect, defined as an enhanced dependency on glycolysis to survive in cancer microenvironments. 2 Although previous studies have shown the overexpression of glycolytic enzymes in BRAF V600E‐mutated tumors, 32 , 33 , 34 the detailed molecular mechanism of how activated BRAF contributes to the activation of the glycolysis pathway remains largely unknown. Among numerous metabolic enzymes, our in silico and immunohistochemical analysis revealed that the glycolytic enzyme ENO2 is specifically overexpressed in BRAF V600E‐mutated CRC. Enolase is an enzyme in the ninth and penultimate step of glycolysis that converts 2‐phosphoglycerate to phosphoenolpyruvate. The enzyme consists of three isoforms: ENO1, ENO2, and ENO3. Under physiological conditions, gastrointestinal tract tissues mainly depend on ENO1 to drive the glycolytic pathway, while ENO2 is predominantly expressed in neuronal tissues and ENO3 is restricted to muscle tissue. ENO1 was reported to be a multifunctional protein and correlated with various biological processes, such as metabolism, hypoxia tolerance, extracellular matrix degradation, metastasis, and allergic responses. It was associated with poor prognosis in patients with lung cancer, pancreatic cancer, and glioma. 35 , 36 , 37 One of the characteristics of alpha‐enolase is that it is expressed on the cell surface of most tumors and acts as a plasminogen‐binding receptor. 38
Previous studies have shown that ENO2 could possibly function as an oncogene even in non–neuronal malignancies, including lung, breast, and prostate cancer. 21 , 22 , 39 , 40 For example, the serum level of neuron‐specific enolase (NSE), which includes ENO2 encoded γ‐enolase, is a reliable tumor marker of small cell lung cancer. 41 , 42 , 43 However, in contrast to ENO1 and ENO2, the functional importance of ENO3 in the pathogenesis of cancer has been unclear. 44 We reported here that ENO2 is also involved in the progression of highly proliferative BRAF V600E‐mutated CRC, and that inhibition of this gene profoundly attenuates the proliferation and invasive ability of CRC cells harboring this mutation. Because EMT marker expressions exhibit almost no significant changes upon inhibition of ENO2, we consider that ENO2 may be involved in migration ability though other mechanisms.
Glycolytic enzyme enolase 2 is a gene that not only functions as a glycolysis enzyme but also functions as a crucial activator of other oncogenic pathways. 45 , 46 Recent evidence has shown that the C‐terminal domain of ENO2, which is not necessary for enzymatic activity, could directly activate the mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MAPK/ERK) signaling pathway, leading to cell differentiation and proliferation. 47 In colorectal cancer, ENO2 has been reported to be correlated with MAPK signaling and ERK signaling based on GSEA analysis. 48 In our own experiments using BRAF V600E‐mutated CRC cells, manipulation of ENO2 expression significantly altered the phosphorylation levels of ERK and AKT, both of which are downstream of receptor tyrosine kinase. These results strongly indicate that ENO2 was involved in the activation of the MAPK/ERK signaling pathway. However, because of the lack of evidence showing the direct molecular binding of these proteins, further investigation is required to reveal the whole mechanism underlying this activation process.
We have revealed that FOSL1 could be a potential mediator connecting activated BRAF and ENO2. FOSL1, which is a member of the activator protein‐1 (AP‐1) transcription factor, is known to be involved in the malignant gene expression network and is itself a downstream target of MAPK pathway. 29 , 30 , 31 We have demonstrated a high correlation between FOSL1 and ENO2 expression in BRAF V600‐mutated CRC. Knockdown experiments revealed that FOSL1 is located in the upstream of ENO2. These results suggested that FOSL1 exerts its malignant potential partially through regulation of ENO2 in BRAF V600E‐mutated CRC cells.
We also explored the possibility of combining a BRAF V600E selective inhibitor with ENO2 suppression for therapeutic application. Enhanced drug sensitivity to the BRAF V600E inhibitor, vemurafenib, was observed upon knockdown of ENO2 in BRAF V600E‐mutated CRC cells but not in BRAF wild‐type cancer cells. Administration of BRAF inhibitor alone has limited efficacy against CRC, mostly because of the reactivation of the MAPK pathway. 49 A previous study indicated that ENO2‐encoded γ‐enolase significantly increased in accordance with the activation of PI3K/AKT and MAPK/ERK pathways in the cancer environment. 47 , 48 , 50 These results suggest that the addition of ENO2 suppression to BRAF inhibition might be an effective therapeutic strategy for BRAF V600E‐mutated CRC.
ENO1 and ENO2 demonstrated different dependences according to tissues and cancer types, which has implications for potential new therapies. ENO2 inhibition is already considered to be a promising therapeutic strategy for glioblastoma, most of which exhibit high dependency on ENO2. 51 Normal gastrointestinal tissues exploit ENO1 as a glycolytic enzyme for energy production. Therefore, selective ENO2 targeting might be a useful therapeutic option for BRAF V600E‐mutated CRC with minimal damage to normal tissues.
Overall, we explored the specific metabolic traits of BRAF V600E‐mutated CRC and revealed the additional activation of ENO2 in the glycolytic system, which is indispensable for the high proliferative ability of this aggressive subtype of CRC. ENO2 could be a novel therapeutic target for BRAF V600E‐mutated CRC.
DISCLOSURE
The authors have the following financial interests. N. Nishida: Institutional endowments were received partially from Yakult Honsha, Chugai Pharmaceutical, and Ono Pharmaceutical. T. Satoh: Institutional endowments were received from Yakult Honsha, Chugai Pharmaceutical, and Ono Pharmaceutical. All other authors declare no conflicts of interest regarding this study.
Supporting information
Fig S1‐S7
Table S1
ACKNOWLEDGMENTS
This work was supported by research grants from the Japanese Society for the Promotion of Science, Grant‐in‐Aid for Scientific Research (C) (18K07970). Partial support was received from Sanofi and Regeneron Pharmaceutical.
Yukimoto R, Nishida N, Hata T, et al. Specific activation of glycolytic enzyme enolase 2 in BRAF V600E‐mutated colorectal cancer. Cancer Sci. 2021;112:2884–2894. 10.1111/cas.14929
REFERENCES
- 1. Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol. 2015;33:1809‐1824. [DOI] [PubMed] [Google Scholar]
- 2. Nagasaka T, Sasamoto H, Notohara K, et al. Colorectal cancer with mutation in BRAF, KRAS, and wild‐type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol. 2004;22:4584‐4594. [DOI] [PubMed] [Google Scholar]
- 3. Jones JC, Renfro LA, Al‐Shamsi HO, et al. Non–V600 BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol. 2017;35:2624‐2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tejpar S, Bertagnolli M, Bosman F, et al. Prognostic and predictive biomarkers in resected colon cancer: current status and future perspectives for integrating genomics into biomarker discovery. Oncologist. 2010;15:390‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signaling. Nat Rev Mol Cell Biol. 2015;16:281‐298. [DOI] [PubMed] [Google Scholar]
- 6. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949‐954. [DOI] [PubMed] [Google Scholar]
- 7. Van Cutsem E, Huijberts S, Grothey A, et al. Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAF V600E–mutant metastatic colorectal cancer: safety lead‐in results from the phase III BEACON Colorectal Cancer Study. J Clin Oncol. 2019;37:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lito P, Pratilas C, Joseph E, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature. 2010;464:427‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600‐mutant colorectal cancer. J Clin Oncol. 2015;33:4023‐4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly K, Brungs D, Szeto E, Epstein RJ. Anticancer activity of combination targeted therapy using cetuximab plus vemurafenib for refractory BRAF (V600E)‐mutant metastatic colorectal carcinoma. Curr Oncol. 2014;21:e151‐e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corcoran RB, Andre T, Atreya CE, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E‐mutant colorectal cancer. Cancer Discov. 2018;8:428‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu M, Yang X, Liu J, et al. Efficacy and safety of BRAF inhibition alone versus combined BRAF and MEK inhibition in melanoma: a meta‐analysis of randomized controlled trials. Oncotarget. 2017;8:32258‐32269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 15. Masoudi‐nejad A, Asgari Y. Metabolic cancer biology: structural‐based analysis of cancer as a metabolic disease, new sights and opportunities for disease treatment. Semin Cancer Biol. 2015;30(21):29. [DOI] [PubMed] [Google Scholar]
- 16. Nagarajan A, Malvi P, Wajapeyee N. Oncogene‐directed alterations in cancer cell metabolism. Trends Cancer. 2016;2:365‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vermeersch KA, Styczynski MP. Applications of metabolomics in cancer research. J Carcinog. 2013;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marangos PJ, Parma AM, Goodwin FK. Functional properties of neuronal and glial isoenzymes of brain enolase. J Neurochem. 1978;31:727‐732. [DOI] [PubMed] [Google Scholar]
- 19. Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30(142):150. [DOI] [PubMed] [Google Scholar]
- 20. Marangos PJ, Zis AP, Clark RL, Goodwin FK. Neuronal, non–neuronal and hybrid forms of enolase in brain: structural, immunological and functional comparisons. Brain Res. 1978;150:117‐133. [DOI] [PubMed] [Google Scholar]
- 21. Miremadi A, Pinder SE, Lee AHS, et al. Neuroendocrine differentiation and prognosis in breast adenocarcinoma. Histopathology. 2002;40:215‐222. [DOI] [PubMed] [Google Scholar]
- 22. Tapia FJ, Barbosa AJA, Marangos PJ, et al. Neuron‐specific enolase is produced by neuroendocrine tumors. Lancet. 1981;1:808‐811. [DOI] [PubMed] [Google Scholar]
- 23. Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Development of a self‐inactivating lentivirus vector. J Virol. 1998;72(10):8150‐8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sugimura K, Fujiwara Y, Omori T, et al. Clinical importance of a transcription reverse‐transcription concerted (TRC) diagnosis using peritoneal lavage fluids obtained pre– and post‐lymphadenectomy from gastric cancer patients. Surg Today. 2016;46:654‐660. [DOI] [PubMed] [Google Scholar]
- 25. Possemato R, Marks KM, Shaul YD, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi M, Nagata S, Iwasaki T, et al. Dedifferentiation of adenocarcinomas by activation of phosphatidylinositol 3‐kinase. Proc Natl Acad Sci USA. 1999;96:4874‐4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roy HK, Olusola BF, Clemens DL, et al. AKT proto‐oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23:201‐205. [DOI] [PubMed] [Google Scholar]
- 28. Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3'‐kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426‐7429. [PubMed] [Google Scholar]
- 29. Vial E, Marshall CJ. Elevated ERK‐MAP kinase activity protects the FOS family member FRA‐1 against proteasomal degradation in colon carcinoma cells. J Cell Sci. 2003;116:4957‐4963. [DOI] [PubMed] [Google Scholar]
- 30. Young MR, Colburn NH. Fra‐1 a target for cancer prevention or intervention. Gene. 2006;379:1‐11. [DOI] [PubMed] [Google Scholar]
- 31. Gruda MC, Kovary K, Metz R, Bravo R. Regulation of Fra‐1 and Fra‐2 phosphorylation differs during the cell cycle of fibroblasts and phosphorylation in vitro by MAP kinase affects DNA binding activity. Oncogene. 1994;9:2537‐2547. [PubMed] [Google Scholar]
- 32. Yun J, Rago C, Cheong I, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee MH, Lee SE, Kim DW, et al. Mitochondrial localization and regulation of BRAFV600E in thyroid cancer: a clinically used RAF inhibitor is unable to block the mitochondrial activities of BRAFV600E. J Clin Endocrinol Metab. 2011;96:E19‐E30. [DOI] [PubMed] [Google Scholar]
- 34. Parmenter TJ, Kleinschmidt M, Kinross KM, et al. Response of BRAF‐mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014;4:423‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu QF, Liu Y, Fan Y, et al. Alpha‐enolase promotes cell glycolysis, growth, migration, and invasion in non–small cell lung cancer through FAK‐mediated PI3K/AKT pathway. J Hematol Oncol. 2015;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Principe M, Borgoni S, Cascione M, et al. Alpha‐enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J Hematol Oncol. 2017;10:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song Y, Luo Q, Long H, et al. Alpha‐enolase as a potential cancer prognostic marker promotes cell growth, migration, and invasion in glioma. Mol Cancer. 2014;21(13):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saldanha RG, Molloy MP, Bdeir K, et al. Proteomic identification of lynchpin urokinase plasminogen activator receptor protein interactions associated with epithelial cancer malignancy. J Proteome Res. 2007;3:1016‐1028. [DOI] [PubMed] [Google Scholar]
- 39. Soh MA, Garrett SH, Somji S, et al. Arsenic, cadmium, and neuron‐specific enolase (ENO2, γ‐enolase) expression in breast cancer. Cancer Cell Int. 2011;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soh M, Dunlevy JR, Garrett SH, et al. Increased neuron‐specific enolase expression by urothelial cells exposed to or malignantly transformed by exposure to Cd 2+ or As 3+. Toxicol Lett. 2012;212:66‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harding M, McAllister J, Hulks G, et al. Neuron‐specific enolase (Nse) in small cell lung cancer: a tumor marker of prognostic significance? Br J Cancer. 1990;61:605‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jørgensen LG, Osterlind K, Genollá J, et al. Serum neuron‐specific enolase (S‐NSE) and the prognosis in small‐cell lung cancer (SCLC): a combined multivariable analysis on data from nine centres. Br J Cancer. 1996;74:463‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Quoix E, Purohit A, Faller‐Beau M, Moreau L, Oster JP, Pauli G. Comparative prognostic value of lactate dehydrogenase and neuron‐specific enolase in small‐cell lung cancer patients treated with platinum‐based chemotherapy. Lung Cancer. 2000;30:127‐134. [DOI] [PubMed] [Google Scholar]
- 44. Kong KW, Abdul Aziz A, Razali N, et al. Antioxidant‐rich leaf extract of Barringtonia racemosa significantly alters the in vitro expression of genes encoding enzymes that are involved in methylglyoxal degradation III. Peer J. 2016;4:e2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pancholi V. Multifunctional a‐enolase: its role in diseases. Cell Mol Life Sci. 2001;58:902‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan T, Skaftnesmo KO, Leiss L, et al. Neuronal markers are expressed in human gliomas and NSE knockdown sensitizes glioblastoma cells to radiotherapy and temozolomide. BMC Cancer. 2011;11:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hafner A, Obermajer N, Kos J. γ‐Enolase C‐terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem J. 2012;443:439‐450. [DOI] [PubMed] [Google Scholar]
- 48. Pan X, Wu H, Chen G, Li W. Prognostic value of enolase gene family in colon cancer. Med Sci Monit. 2020;26:e922980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Corcoran RB, Ebi H, Turke AB, et al. EGFR‐mediated reactivation of MAPK signaling contributes to insensitivity of BRAF‐mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu CC, Wang H, Wang WD, et al. ENO2 promotes cell proliferation, glycolysis, and glucocorticoid‐resistance in acute lymphoblastic leukemia. Cell Physiol Biochem. 2018;46(4):1525‐1535. [DOI] [PubMed] [Google Scholar]
- 51. Muller FL, Colla S, Aquilanti E, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1