

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most chemoresistant cancers. An understanding of the molecular mechanism by which PDAC cells have a high chemoresistant potential is important for improvement of the poor prognosis of patients with PDAC. Here we show for the first time that disruption of heat shock protein 47 (HSP47) enhances the efficacy of the therapeutic agent gemcitabine for PDAC cells and that the efficacy is suppressed by reconstituting HSP47 expression. HSP47 interacts with calreticulin (CALR) and the unfolded protein response transducer IRE1α in PDAC cells. Ablation of HSP47 promotes both the interaction of CALR with sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2 and interaction of IRE1α with inositol 1,4,5‐triphosphate receptor, which generates a condition in which an increase in intracellular Ca2+ level is prone to be induced by oxidative stimuli. Disruption of HSP47 enhances NADPH oxidase‐induced generation of intracellular reactive oxygen species (ROS) and subsequent increase in intracellular Ca2+ level in PDAC cells after treatment with gemcitabine, resulting in the death of PDAC cells by activation of the Ca2+/caspases axis. Ablation of HSP47 promotes gemcitabine‐induced suppression of tumor growth in PDAC cell‐bearing mice. Overall, these results indicated that HSP47 confers chemoresistance on PDAC cells and suggested that disruption of HSP47 may improve the efficacy of chemotherapy for patients with PDAC.
Keywords: calreticulin, chemoresistance, HSP47, IRE1α, PDAC
Our study shows for the first time that interaction of HSP47 with calreticulin and IRE1α confers chemoresistance on PDAC cells. Disruption of HSP47 in PDAC cells enhances the efficacy of chemotherapy for PDAC‐bearing mice. Our study proposes the possibility of HSP47 being a candidate for a therapeutic target against PDAC.
1. INTRODUCTION
Pancreatic cancer is one of the leading causes of cancer‐related death worldwide. The median survival period of patients with pancreatic ductal adenocarcinoma (PDAC) is still only 5 to 8 mo and the 5‐y survival rate is only c. 5%. 1 , 2 Currently available therapies are not sufficient for treatment of PDAC, and the development of novel therapeutic modalities for PDAC is urgently needed. 3 , 4 PDAC is also one of the most chemoresistant cancers, and the efficacy of therapeutic agents is hindered by multiple intrinsic and acquired mechanisms of resistance before and during chemotherapy. 5 , 6 , 7 Gemcitabine has been widely used as a first‐line drug for PDAC. 8 , 9 , 10 , 11 Gemcitabine is a deoxycytidine nucleotide analog and induces inhibition of DNA synthesis and cell cycle arrest at the G1/S phase by its incorporation into DNA during DNA replication. Gemcitabine has also been reported to trigger oxidative stress by the generation of reactive oxygen species (ROS) by the activity of NAD(P)H oxidase (NOX), which is a major source of the generation of ROS, resulting in induction of apoptosis. 12 , 13 Although gemcitabine remains the therapeutic agent of choice that is used either alone or in combination with nab‐paclitaxel in patients with PDAC, treatment with gemcitabine has not resulted in a marked improvement in survival of patients with PDAC due to the presence of innate or acquired chemoresistant mechanisms in PDAC. 8 , 9 , 10 , 11 It has been widely accepted that reduction of anti‐cancer drug uptake, alteration of the drug target, induction of drug‐detoxifying mechanisms, repair of drug‐induced damage and insensitivity to drug‐induced cell death are responsible for the chemoresistance of PDAC cells. 7 , 14 , 15 However, current therapies targeting the chemoresistance‐associated molecular pathways in PDAC cells have not provided satisfactory results, due in part to rapid upregulation of alternative compensatory pathways. Therefore, an understanding of the precise molecular mechanism by which PDAC cells have tolerance against chemotherapies is an urgent issue for improvement of the poor survival of patients with PDAC.
Heat shock protein 47 (HSP47), a molecular chaperone of collagen, is localized in the endoplasmic reticulum (ER) and it is widely accepted to play a crucial role in correct folding of collagen. 16 , 17 Disruption of HSP47 triggers the accumulation of unfolded pro‐collagen and subsequent activation of ER stress, resulting in delayed cell growth and apoptosis. 18 , 19 , 20 HSP47 is ubiquitously expressed in several types of cancer cells in which the expression level of collagen is extremely low, and increased expression of HSP47 is associated with a high malignant grade of glioma and low overall survival of patients with breast cancer and colon cancer. 21 , 22 , 23 , 24 , 25 , 26 , 27 It has also been reported that the overall survival rate of patients with PDAC in which the expression level of HSP47 is low is higher than that of patients with PDAC in which the expression level of HSP47 is high. 21 , 28 Furthermore, induced expression of HSP47 promotes the ability of cancer cells for tumor growth and metastasis to other organs. 25 , 27 , 29 , 30 Recently, we showed that HSP47 interacts with the unfolded protein response (UPR) transducer IRE1α and maintains cancer cell growth by inhibiting IRE1α. 29 , 31 Furthermore, interaction of HSP47 with IRE1α stimulates the activity of myosin IIA, augmenting the metastatic potential of breast cancer cells. 30 Although the involvement of HSP47 in the malignancy and chemoresistance of cancer cells has been suggested, the precise molecular mechanism by which HSP47 contributes to the tolerance of cancer cells against treatment with therapeutic agents remains unclear. Here we show that disruption of HSP47 improves the efficacy of the therapeutic agent gemcitabine against PDAC cells, while overexpression of HSP47 confers chemoresistance to PDAC cells by regulation of calcium homeostasis by inhibiting interaction of HSP47 with the ER‐resident Ca2+‐binding protein calreticulin (CALR) and with IRE1α, both of which are associated with chemoresistance of PDAC cells. The results of the present study also indicated the possibility of HSP47 being a candidate for a therapeutic target against PDAC.
2. MATERIALS AND METHODS
2.1. Human cell lines
Human PDAC cell lines (PANC1, MIA‐PaCa2, BxPC3, AsPC1, Capan‐2, SW1990, CFPAC1 and Suit2) were purchased from the ATCC. MIA‐PaCa2‐luc cells were purchased from JCRB Cell Bank (National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, Japan). MIA‐PaCa2, MIA‐PaCa2‐luc, BxPC3, AsPC1, Capan‐2, CFPAC1 and Suit2 cells were cultured in DMEM (Sigma‐Aldrich) supplemented with 10% FBS (Thermo Fisher Scientific). SW1990 and PANC1 cells were cultured in RPMI1640 medium (Sigma‐Aldrich) supplemented with 10% FBS.
2.2. Quantitative PCR
Total RNAs were extracted from cancer cells using an RNeasy Mini Kit (Qiagen). Total RNA (1 μg) was used for reverse transcription by High‐Capacity RNA‐to‐cDNA Master Mix (Applied Biosystems). Quantitative PCR was carried out using Power SYBR Green PCR Master Mix (Applied Biosystems) and the following primers against HSP47 (sense: 5′‐TGACCTGCAGAAACACCTGG‐3′, antisense: 5′‐AGGAAGATGAAGGGGTGGTC‐3′), p22phox (sense: 5′‐ TGGACGTTTCACACAGTGGT‐3′, antisense: 5′‐ACCGACAACAGGAAGTGGAG‐3′) and GAPDH (sense: 5′‐GAGTCAACGGATTTGGTCGT‐3′, antisense: 5′‐TTGATTTTGGAGGGATCTCG‐3′).
2.3. Western blot analysis
The cells were then washed with PBS and lysed with a lysis buffer (50 mmol/L Tris, 250 mmol/L NaCl, 25 mmol/L EDTA and 1% NP‐40) supplemented with a protease inhibitor (Roche). Equal amounts of proteins were separated by SDS‐PAGE and transferred to polyvinylidene difluoride membranes (Millipore). After blocking with 5% skim milk in PBS containing 0.05% Tween‐20, the membranes were probed with the following primary antibodies: Myc (Medical & Biological Laboratories), HSP47 (ADI‐SPA‐470, Enzo Life Sciences; ab77609, Abcam), calreticulin (#12238, Cell Signaling Technology), IRE1α (#3294, Cell Signaling Technology), SERCA2 (MAB2636, Millipore), IP3R (ab108517, Abcam), Nox4 (ab133303, Abcam), caspase‐12 (#2202, Cell Signaling Technology), caspase‐9 (#9502, Cell Signaling Technology), caspase‐3 (#9665, Cell Signaling Technology) and GAPDH (ab8245, Abcam). After treatment with primary antibodies, the membranes were incubated with the following secondary antibodies: horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit IgG (#7074, Cell Signaling Technology) and HRP‐conjugated goat anti‐mouse IgG (#7076, Cell Signaling Technology). Proteins were detected by a chemiluminescence method using ECL (GE Healthcare).
2.4. Transfection of siRNA
Human PDAC cells were transfected with control siRNA (siControl, Thermo Fisher Scientific), HSP47‐A siRNA (siHSP47‐A, sense: 5′‐CUACGACGACGAGAAGGAATT‐3′; antisense: 5′‐UUCCUUCUCGUCGUCGUAGTA‐3′, Thermo Fisher Scientific) and HSP47‐B siRNA (siHSP47‐B, sense: 5′‐AGCCCUCUUCUGACACUAATT‐3′; antisense: 5′‐UUAGUGUCAGAAGAGGGCUGG‐3′, Thermo Fisher Scientific) using Lipofectamine RNAiMAX (Thermo Fisher Scientific) and were cultured in Opti‐MEM reduced medium (Thermo Fisher Scientific) for 5 h at 37°C in air. At 5 h after transfection with siRNA, the cells were cultured in RPMI1640 medium (for PANC1 cells) or DMEM (for MIA‐PaCa2 cells and Suit2 cells) supplemented with 10% FBS.
2.5. Establishment of gene knockout (KO) cells
The gRNA sequence (5′‐TGCGGAGAAGTTGAGCCCCA‐3′) designed for exon 3 of the human HSP47 genome was inserted into the hspCas9‐nickase‐H1‐gRNA SmartNickase vector (System Biosciences). The constructed targeting vector and pcDNA3.1(+)‐Hygro (Promega) were co‐transfected into human PDAC cells using Lipofectamine 2000 (Thermo Fisher Scientific). The treated cells were cultured in RPMI1640 medium (for PANC1 cells) or DMEM (for MIA‐PaCa2 cells and MIA‐PaCa2‐luc cells) supplemented with 10% FBS and hygromycin (800 μg/mL, Thermo Fisher Scientific) to select cell clones stably silencing HSP47 expression. Single‐cell clones were evaluated by DNA sequencing analysis to detect insertions or deletions in the target alleles and to confirm the expression of HSP47 at the protein level by western blotting.
The gRNA sequence (5′‐GTATACAGGCTGCCATCATT‐3′) designed for exon 4 of the human IRE1α genome and the gRNA sequence (5′‐CGGCAAGTTCTACGGTGACG‐3′) designed for exon 2 of the human CALR genome were each inserted into the hspCas9‐nickase‐H1‐gRNA SmartNickase vector. The constructed targeting vector and pcDNA3.1(+)‐Neo (Promega) were co‐transfected into PANC1 cells and HSP47 KO PANC1 cells using Lipofectamine 2000. The treated cells were cultured in RPMI1640 medium supplemented with 10% FBS and G418 (1000 μg/mL, Thermo Fisher Scientific) to select cell clones stably silencing IRE1α or CALR expression. Single‐cell clones were evaluated by DNA sequencing analysis to detect insertions or deletions in the target alleles and to confirm expression of IRE1α or CALR at the protein level by western blotting.
2.6. Establishment of cancer cells overexpressing HSP47‐Myc, HSP47‐GFP, and SERCA2‐Myc
PANC1 cells, HSP47 KO PANC1 cells and HSP47 KO MIA‐PaCa2‐luc cells were transfected with pCMV‐HSP47‐Myc plasmid (Origene), pCMV‐HSP47‐GFP plasmid (Origene) and pCMV‐SERCA2a‐Myc plasmid (Origene). At 48 h after transfection with the plasmid, the treated cells were cultured in DMEM supplemented with 10% FBS and 2 μg/ml puromycin (Thermo Fisher Scientific) for 2 wk to select cell clones stably expressing HSP47‐Myc, HSP47‐GFP, and SERCA2a‐Myc. To confirm HSP47‐Myc expression in the selected cell clones, expression of HSP47‐Myc mRNA and protein was determined by quantitative PCR and western blotting.
2.7. Cell viability assay
PDAC cells were treated with the therapeutic agent gemcitabine (Gemzar, Eli Lilly Japan) at concentrations of 1, 5, 10, 20, 50, 100, 500 and 1000 μmol/L for PANC1 cells and at concentrations of 0.1, 0.5, 1 5, 10, 50, 100, 500 and 1000 nmol/L for MIA‐PaCa2 cells and Suit2 cells. Viability of the treated cells was determined by a dye‐exclusion assay.
2.8. Immunoprecipitation
PANC1 cells stably expressing HSP47‐Myc and SERCA2a‐Myc were lysed with a lysis buffer (50 mmol/L Tris‐HCl pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% NP‐40 and complete mini protease inhibitors (Roche)) on ice. The soluble protein fraction was collected by centrifugation at 12 000 × g for 15 min, and protein quantification was performed using a BCA assay (Thermo Fisher Scientific). Protein aliquots each containing approximately 1 mg of cellular proteins were incubated with anti‐myc‐tag mAb‐magnetic beads (Medical & Biological Laboratories) for 16 h at 4°C, and then the beads were washed with the lysis buffer on a magnetic stand. Proteins were eluted from the beads with 50 mmol/L glycine‐HCl buffer (pH 3.5) containing SDS‐PAGE sample buffer. Eluents were immediately transferred to a fresh tube containing 5 μL of 1 M Tris‐HCl buffer (pH 7.5) in advance to neutralize sample pH.
2.9. Proteomic analysis
To identify co‐immunoprecipitated proteins, immunoprecipitated proteins after concentration by centrifugal filtration (3000 molecular weight cut‐off, PALL Life Science) were subjected to tryptic digestion. Peptides were combined and dried with a centrifugal evaporator (Thermo Fisher Scientific) and then finally re‐dissolved in 10 μL of H2O/acetonitrile/FA 97.9/2/0.1. Extracted peptides were subjected to Nano LC‐MS/MS mass spectrometry (LTQ‐Orbitrap XL, Thermo Scientific) with the Pradigm MS2 LC system (Michrom BioResources) equipped with a 0.1 x 150 mm Zaplous alpha Pep C18 column (AMR). The flow rate was set to 400 nL/min. Separation was performed using a gradient mixture of H2O/acetonitrile/FA 98/2 with 0.1% FA (buffer A) and H2O/acetonitrile/FA 10/90 with 0.1% FA (buffer B), and 5%‐35% buffer B gradient was used until 20 min. The obtained data were processed using the software MaxQuant 1.6.0.1 and Proteome Discoverer 1.4.0.288 (Thermo Fisher Scientific) with the UniProt Homo sapiens proteome database. Mass tolerance of precursors was set at 40 ppm, and MS/MS tolerance was set at 0.5 Da. In a protein search, carbamidomethylation of cysteine and oxidation of methionine were used as a fixed modification. Protein identification was performed with tryptic digestion mode with permission of a maximum of 2 missed cleavages.
2.10. Measurement of intracellular Ca2+ level
At 72 h after treatment with gemcitabine (10 μmol/L for PANC1 cells and 1 nmol/L for MIA‐PaCa2 cells), PDAC cells were incubated in RPMI1640 medium (for PANC1 cells) or DMEM (for MIA‐PaCa2 cells) supplemented with 10% FBS and Fluo 4‐AM (3 μmol/L, Thermo Fisher Scientific) for 30 min. The level of intracellular Ca2+ in the treated cells was determined using flow cytometry (FACS Canto, BD Biosciences) and by calculating the intensity of Fluo‐4.
PDAC cells were incubated in RPMI1640 medium (for PANC1 cells) or DMEM (for MIA‐PaCa2 cells) supplemented with 10% FBS and Fura 2‐AM (25 μmol/L, Thermo Fisher Scientific) for 30 min. After treatment with hydrogen peroxide (H2O2) at a concentration of 2 μg/mL, intracellular Ca2+ levels in PDAC cells were determined by calculating the Fura 2‐AM ratio as the quotient of the detected fluorescence intensities at 510 nm after excitation with 340 nm or 380 nm.
2.11. Detection of NAD(P)H oxidase activity
The treated cells were lysed in an NADH oxidase lysis buffer (BioVision). All samples were stored at −80°C℃ until use. NAD(P)H oxidase (NOX) activity was determined using the NADH Oxidase Activity Assay kit (BioVision).
2.12. Detection of intracellular ROS
PDAC cells were treated with CM‐H2DCFDA (6‐chloromethyl‐2′,7′‐dichlorodihydro fluorescein diacetate, acetyl ester, Molecular Probes) at a concentration of 5 μmol/L for 10 min. Accumulation of intracellular ROS was quantitatively detected by measuring fluorescence intensity (495/527 nm) of CM‐H2DCFDA and was analyzed using NIH ImageJ software.
2.13. Xenograft experiment
Mock:MIA‐PaCa2‐luc cells and HSP47 KO MIA‐PaCa2‐luc cells at a concentration of 1 × 106 cells/50 μL of PBS/animal were orthotopically transplanted into the pancreas of BALB/c nu/nu male mice (7‐9 wk old, CLEA Japan). At 2 wk after transplantation with the cells, gemcitabine at a concentration of 100 mg/kg body weight was intravenously injected once per wk for 4 wk into mice bearing Mock:MIA‐PaCa2‐luc cells and mice bearing HSP47 KO MIA‐PaCa2‐luc cells. Bioluminescence of tumors in mice and in pancreatic tissues was determined by in vivo and ex vivo imaging analyses using an in vivo imaging system (IVIS2000, Xenogen) to examine tumor growth. All animal studies were reviewed and approved by the Experimental Animal Ethics Committee of Hokkaido University. The feeding, maintenance, and use of the animals were performed in accordance with the guidelines of the Experimental Animal Ethics Committee of Hokkaido University.
2.14. Immunohistological analysis
Mice were perfused with 10 N formalin and fixed pancreatic tissues were embedded in paraffin. Paraffin‐embedded pancreatic tissue sections (7 μm in thickness) were deparaffinized with xylene and ethanol. Pancreatic tissue sections were stained by hematoxylin and eosin. For immunohistological staining, after permeabilization with HistoVT‐ONE (Nacalai Tesque) or 10 mmol/L citrate buffer (pH 6.0), tissue sections were blocked with 5% goat serum in PBS. The sections were incubated with an antibody for Ki67 (ab15580, Abcam) and secondary antibodies conjugated with horseradish peroxidase (ab6721, Abcam). After reaction with a 3,3′‐diaminobenzidine) (DAB) substrate (Sigma‐Aldrich), the treated sections were counterstained with hematoxylin. Images were taken with a microscope (BZ‐X800, KEYENCE) and were analyzed using software (BZ‐X800, KEYENCE).
2.15. Statistical analysis
All data are shown as means ± SEM. All experiments in this study were performed in duplicate or triplicate. Differences between groups were tested for statistical significance using Student t test or ANOVA. Statistical significance was determined at P < .05.
3. RESULTS
3.1. Silencing and disruption of HSP47 improves the efficacy of gemcitabine for PDAC cells
We first examined the expression of HSP47 in human PDAC cells (PANC1 cells, MIA‐PaCa2 cells, and Suit2 cells) and confirmed the expression of HSP47 in PDAC cells at the mRNA level and protein level (Figure 1A,B). Then, we investigated whether silencing of HSP47 expression affected the viability of PDAC cells after treatment with the therapeutic agent gemcitabine. The expression of HSP47 was significantly inhibited in human PDAC cells transfected with 2 batches (A and B) of HSP47 siRNA (siHSP47) (Figure 1C,D). Consistent with the results of previous studies, 25 , 27 , 29 , 30 silencing of HSP47 expression suppressed in vitro proliferation of PDAC cells (Figure 1E). Treatment of HSP47‐silenced PDAC cells with gemcitabine markedly reduced their cell viability compared with that of their counterparts (Figure 1F). The IC50 values of gemcitabine against PDAC cells treated with siHSP47‐A or siHSP47‐B were significantly lower than those of their counterparts (Figure 1G).
FIGURE 1.
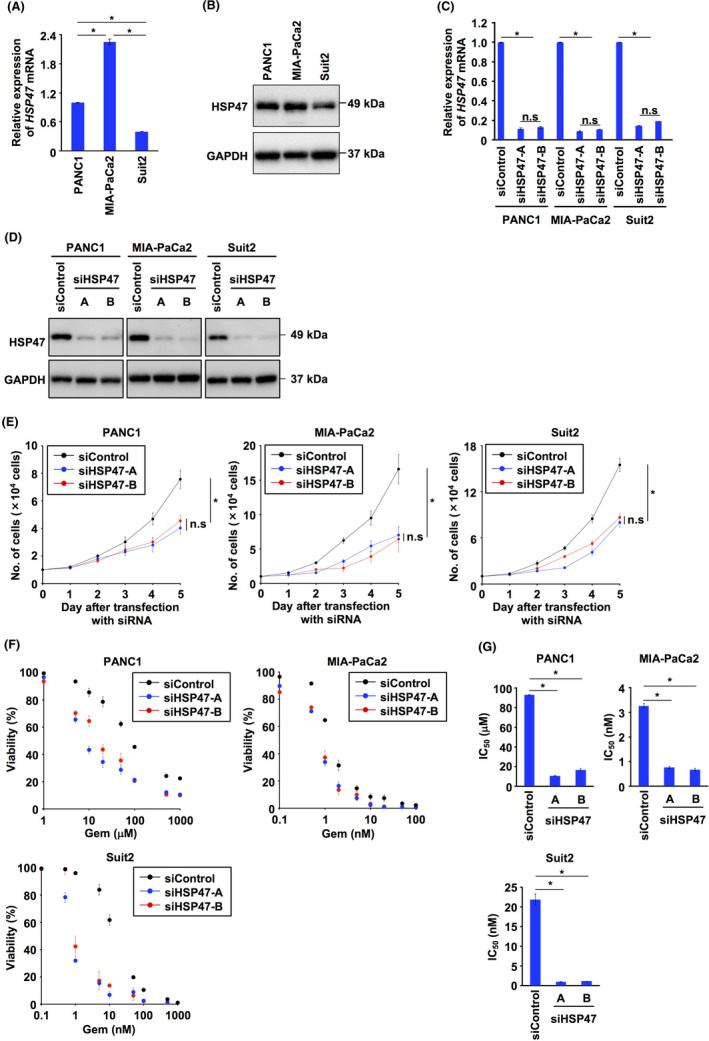
Knockdown of HSP47 dampens the chemoresistance of pancreatic cancer cells. A, Expression of HSP47 mRNA in PDAC cells (PANC1 cells, MIA‐PaCa2 cells, and Suit2 cells). B, Expression of HSP47 protein in PDAC cells. C, Expression of HSP47 mRNA in PDAC cells transfected with control siRNA (siControl, 10 nmol/L) and 2 batches (A and B) of HSP47 siRNA (siHSP47, 10 nmol/L). D, Expression of HSP47 protein in PDAC cells transfected with siControl, siHSP47‐A, and siHSP47‐B. E, In vitro proliferation of PDAC cells transfected with siControl, siHSP47‐A, and siHSP47‐B. F, Viability of HSP47‐silenced PDAC cells after treatment with gemcitabine at each indicated concentration. G, IC50 values of HSP47‐silenced PDAC cells after treatment with gemcitabine at each indicated concentration. *, P < .05. n.s., not significant
We next established HSP47 KO PDAC cells (Figure 2A) and investigated the viability of HSP47 KO PDAC cells after treatment with gemcitabine. Ablation of HSP47 partially inhibited in vitro proliferation of PDAC cells (Figure 2B). The viabilities of HSP47 KO PDAC cells treated with gemcitabine were significantly lower than those of their counterparts (Figure 2C). The IC50 value of gemcitabine against HSP47 KO PDAC cells was also significantly lower than that of their counterparts (Figure 2D). We also investigated the viability of HSP47 KO PDAC cells overexpressing HSP47‐Myc after treatment with gemcitabine (Figure 2E), and we found that overexpression of HSP47‐Myc completely restored the viability of HSP47 KO PDAC cells treated with gemcitabine (Figure 2F). The IC50 value of gemcitabine against HSP47 KO PDAC cells was restored by reconstitution of HSP47‐Myc expression (Figure 2G). Furthermore, the IC50 value of gemcitabine against Mock PDAC cells overexpressing HSP47‐Myc was statistically higher than that against Mock PDAC cells (Figure 2G). These results indicated that HSP47 is involved in the tolerance of PDAC cells against treatment with gemcitabine.
FIGURE 2.
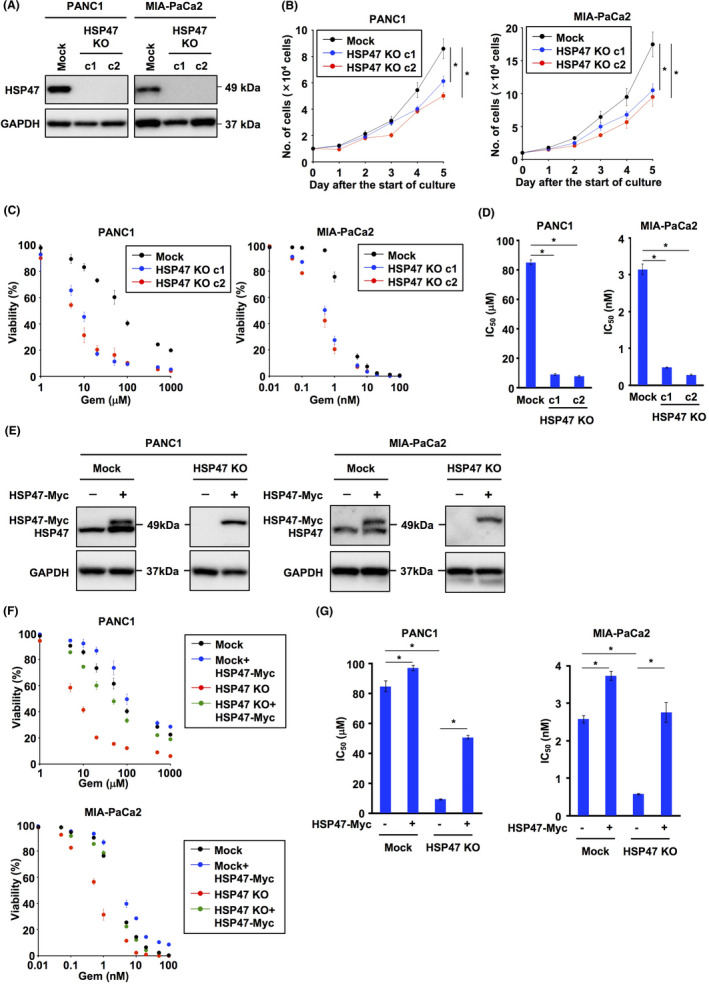
Ablation of HSP47 enhances the sensitivity of pancreatic cancer cells to gemcitabine. A, Expression of HSP47 protein in HSP47 knockout (KO) PDAC cells (PANC1 cells and MIA‐PaCa2 cells). B, In vitro proliferation of HSP47 KO PDAC cells. C, Viability of HSP47 KO PDAC cells after treatment with gemcitabine. D, IC50 values of HSP47 KO PDAC cells after treatment with gemcitabine at each indicated concentration. E, Expression of HSP47 protein in HSP47 KO PDAC cells expressing HSP47‐Myc. F, Viability of HSP47 KO PDAC cells expressing HSP47‐Myc after treatment with gemcitabine. G, IC50 values of HSP47 KO PDAC cells expressing HSP47‐Myc after treatment with gemcitabine at each indicated concentration. *, P < .05
3.2. HSP47 interacts with calreticulin and IRE1α in PDAC cells
To uncover the mechanism by which HSP47 is involved in the chemoresistance of PDAC cells, we investigated HSP47‐interacting proteins in PDAC cells. We purified HSP47‐interacting proteins from the HSP47‐Myc‐overexpressing PDAC cell line PANC1 using anti‐Myc antibody‐conjugated magnetic microbeads and we performed LC‐MS/MS analysis of purified HSP47‐interacting proteins. As shown in Table 1, we identified 6 molecules (IRE1α, β‐subunit prolyl 4‐hydroxylase (P4HB), endoplasmin, CALR, translocon‐associated protein subunit α (SSR1) and mesencephalic astrocyte‐derived neurotrophic factor (MANF)) as HSP47‐interacting proteins localized in the ER. Among the identified HSP47‐interacting proteins, we focused on 2 proteins, the Ca2+‐binding protein CALR and the UPR transducer IRE1α, as both CALR and IRE1α have been reported to be associated with cancer progression. 32 , 33 , 34 Immunoblot analysis showed that CALR and IRE1α proteins were expressed in 8 human PDAC cell lines and in HSP47 KO PDAC cells (HSP47 KO PANC1 cells and HSP47 KO MIA‐PaCa2 cells) (Figure 3A,B). To further confirm the interactions of HSP47 with CALR and IRE1α, we carried out the immunoprecipitation of HSP47‐Myc in PANC1 cells overexpressing HSP47‐Myc and found that HSP47 interacts with CALR and IRE1α in PANC1 cells treated with or not treated with gemcitabine (Figure 3C).
TABLE 1.
HSP47‐interacting proteins localized in the ER in PANC1 cells
FIGURE 3.
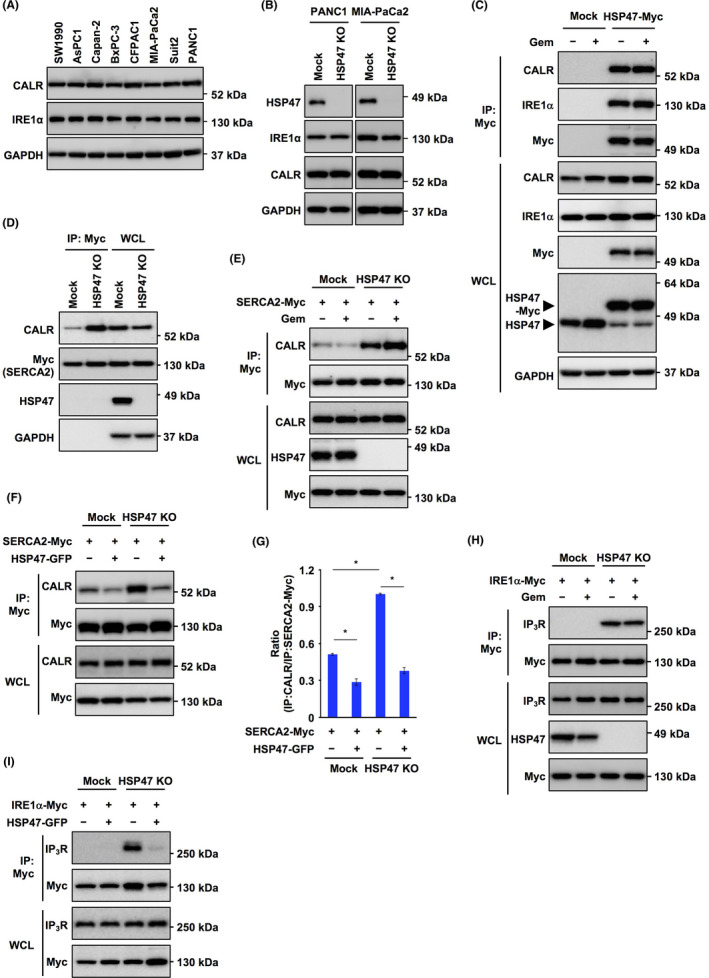
HSP47 interferes with both calreticulin/SERCA2 interaction and IRE1α/IP3R interaction in pancreatic cancer cells. A, Expression of calreticulin (CALR) and IRE1α protein in human PDAC cell lines. GAPDH was used as an internal control. B, Expression of CALR and IRE1α protein in HSP47 KO PDAC cells (HSP47 KO PANC1 cells and HSP47 KO MIA‐PaCa 2 cells). C, Interaction of HSP47 with CALR and IRE1α in PANC1 cells expressing HSP47‐Myc after treatment with gemcitabine (10 μmol/L). D, Interaction of CALR with SERCA2 in HSP47 KO PANC1 cells expressing SERCA2‐Myc. E, Interaction of CALR with SERCA2 in HSP47 KO PANC1 cells expressing SERCA2‐Myc after treatment with gemcitabine (10 μmol/L). F, Interaction of CALR with SERCA2 in HSP47 KO PANC1 cells expressing SERCA2‐Myc and reconstituting HSP47‐GFP expression after treatment with gemcitabine (10 μmol/L). G, The binding ratio of CALR (IP) /SERCA2‐Myc (IP) was determined by using NIH ImageJ software. *, P < .05. H, Interaction of IRE1α with IP3R in HSP47 KO PANC1 cells expressing IRE1α‐Myc after treatment with gemcitabine (10 μmol/L). (I) Interaction of IRE1α with IP3R in HSP47 KO PANC1 cells expressing IRE1α‐Myc and reconstituting HSP47‐GFP expression after treatment with gemcitabine (10 μmol/L)
3.3. HSP47 interferes with both the interaction of CALR with SERCA2 and the interaction of IRE1α with IP3R in PDAC cells
Over 50% of Ca2+ stored in the ER is bound to CALR and CALR serves as a regulator of Ca2+ homeostasis in the ER. 35 CALR interacts with sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase (SERCA) 2, which is prerequisite for inactivation of SERCA2. 36 Therefore, we investigated whether HSP47 affects the interaction of CALR with SERCA2 in PDAC cells. Immunoprecipitation showed that the interaction of CALR with SERCA2 was very weak in PANC1 cells, while the interaction was promoted in HSP47 KO PANC1 cells (Figure 3D). Interaction of CALR with SERCA2 was not enhanced in HSP47 KO PANC1 cells regardless of treatment or no treatment with gemcitabine (Figure 3E). Interaction of CALR with SERCA2 in Mock:PANC1 cells and HSP47 KO PANC1 cells was inhibited by reconstitution of HSP47‐GFP expression (Figure 3F,G).
IRE1α acts as a UPR transducer and plays an important role in cell survival under the conditions of ER stress. 37 , 38 In addition, IRE1α physically interacts with the calcium ion channel inositol 1,4,5‐triphosphate receptor (IP3R) on the ER and its interaction stimulates the release of Ca2+ from the ER, suggesting that IRE1α indirectly maintains intracellular Ca2+ homeostasis. 39 Therefore, we investigated whether HSP47 affected the interaction of IRE1α with IP3R in PDAC cells after treatment with gemcitabine. Immunoprecipitation showed that IRE1α interacted with IP3R in HSP47 KO PANC1 cells but not in Mock:PANC1 cells regardless of treatment or no treatment with gemcitabine (Figure 3H). The interaction of IRE1α with IP3R in HSP47 KO PANC1 cells was completely inhibited by reconstitution of HSP47‐GFP expression (Figure 3I). These results indicated that HSP47 inhibits both the interaction of CALR with SERCA2 and the interaction of IRE1α with IP3R in PDAC cells.
3.4. Disruption of HSP47 enhances the increase in intracellular Ca2+ level in PDAC cells after treatment with gemcitabine
We examined whether disruption of HSP47 affected the level of intracellular Ca2+ in PDAC cells after treatment with gemcitabine. Treatment with gemcitabine induced an increase in intracellular Ca2+ level in Mock:PDAC cells and HSP47 KO PDAC cells (Figure 4A). The degree of increase in intracellular Ca2+ level in HSP47 KO PDAC cells treated with gemcitabine was significantly higher than that in Mock:PDAC cells. The excess increase in intracellular Ca2+ levels in HSP47 KO PDAC cells treated with gemcitabine was clearly inhibited by reconstitution of HSP47 expression. We also investigated the effect of the Ca2+ chelator BAPTA‐AM on the increase in intracellular Ca2+ levels in and the viability of HSP47 KO PDAC cells after treatment with gemcitabine. The increase in intracellular Ca2+ levels in and the death of HSP47 KO PDAC cells after treatment with gemcitabine were inhibited by treatment with BAPTA‐AM (Figure 4B). The IC50 value of gemcitabine for HSP47 KO PDAC cells was significantly lower than that for Mock:PDAC cells, which was markedly restored by treatment with BAPTA‐AM (Figure 4C,D).
FIGURE 4.
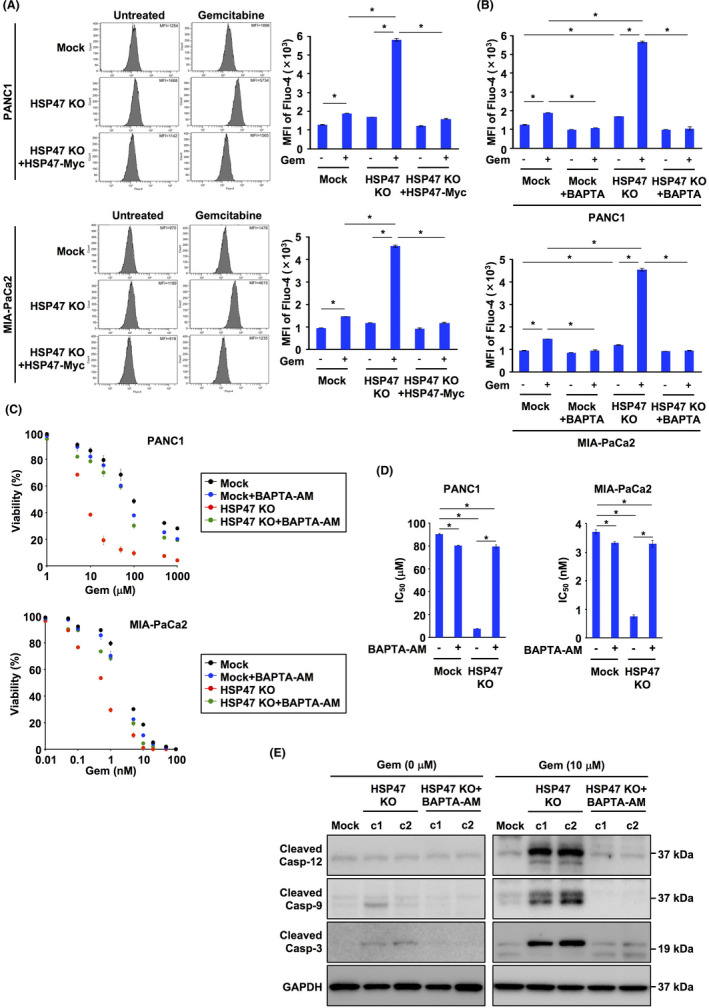
Disruption of HSP47 induces an increase in intracellular Ca2+ levels in pancreatic cancer cells after treatment with gemcitabine. A, Levels of intracellular Ca2+ in PDAC cells (PANC1 cells and MIA‐PaCa2 cells), HSP47 KO PDAC cells (HSP47 KO PANC1 cells and HSP47 KO MIA‐PaCa2 cells) and HSP47 KO PDAC cells expressing HSP47‐Myc after treatment with gemcitabine (10 μmol/L for PANC1 cells and 1 nmol/L for MIA‐PaCa2 cells). Levels of intracellular Ca2+ were determined using flow cytometry (left panel) and calculating the intensity of Fluo‐4 (right panel). B, Levels of intracellular Ca2+ in PDAC cells and HSP47 KO PDAC cells after treatment with BAPTA‐AM (5 μmol/L) and gemcitabine. C, Viability of PDAC cells and HSP47 KO PDAC cells after treatment with BAPTA‐AM and gemcitabine. D, IC50 values of HSP47 KO PDAC cells after treatment with BAPTA‐AM and gemcitabine. E, Activation of the calcium/caspase‐12/caspase‐9/caspase‐3 axis in HSP47 KO PANC1 cells (c1 and c2) after treatment with or without BAPTA‐AM and gemcitabine. *, P < .05
It has been reported that an increase in intracellular Ca2+ concentration triggers activation of the caspase 12/caspase 9/caspase 3 axis, causing apoptosis of cells. 40 Therefore, we investigated the activation of the calcium/caspases axis in HSP47 KO PDAC cells after treatment with gemcitabine. As expected, cleaved caspase 12, cleaved caspase 9 and cleaved caspase 3 were detected in HSP47 KO PANC1 cells treated with gemcitabine (Figure 4E). The activation of the calcium/caspases axis in HSP47 KO PANC1 cells treated with gemcitabine was inhibited by treatment with BAPTA‐AM. These results indicated that gemcitabine promotes the increase in intracellular Ca2+ level, resulting in activation of the calcium/caspase axis and subsequent death of HSP47‐deleted PDAC cells.
3.5. Ablation of HSP47 promotes NOX‐induced generation of intracellular ROS in PDAC cells after treatment with gemcitabine
Treatment with gemcitabine induces the generation of ROS by the activity of NOX. 12 , 13 Recently, we showed that silencing of HSP47 promoted the generation of intracellular ROS in cancer cells. 29 Therefore, we investigated the level of intracellular ROS in HSP47 KO PDAC cells after treatment with gemcitabine. The level of intracellular ROS in HSP47 KO PDAC cells was higher than that in its counterparts, and the level of ROS was increased by treatment with gemcitabine (Figure 5A). It has been reported that the expression levels of NOX4 in PDAC cells were higher than the expression levels of other NOX family members, NOX1, NOX2, NOX3 and NOX5, 12 , 41 and we also confirmed the expression of NOX4 in PDAC cells and HSP47 KO cells regardless of treatment or no treatment with gemcitabine (Figure 5B). The expression levels of p22phox, a catalytic subunit of the NOX complex, in both Mock:PDAC cells and HSP47 KO PDAC cells treated with gemcitabine were significantly higher than those in the counterparts (Figure 5C). The activity of NOX in Mock:PDAC cells and HSP47 KO PDAC cells was markedly increased by treatment with gemcitabine (Figure 5D). We examined the level of intracellular ROS in HSP47 KO PDAC cells after treatment with gemcitabine and with an inhibitor of NOX activity, diphenyleneiodonium chloride (DPI), and we found that treatment with DPI inhibited gemcitabine‐induced increases of both NOX activity and intracellular level of ROS in HSP47 KO PDAC cells (Figure 5E,F). Therefore, we investigated whether treatment with the ROS scavenger N‐acetyl‐l‐cysteine (NAC) and the inhibitor of NOX activity DPI suppressed the death of HSP47 KO PDAC cells after treatment with gemcitabine. Gemcitabine‐induced death of HSP47 KO PANC1 cells was also ameliorated by treatment with NAC or DPI (Figure 5G). Furthermore, the IC50 value of gemcitabine for HSP47 KO PANC1 cells was significantly lower than that of Mock:PANC1 cells, which were recovered by treatment with NAC or DPI (Figure 5H). These results indicated that disruption of HSP47 promotes NOX‐induced generation of intracellular ROS in PDAC cells after treatment with gemcitabine.
FIGURE 5.
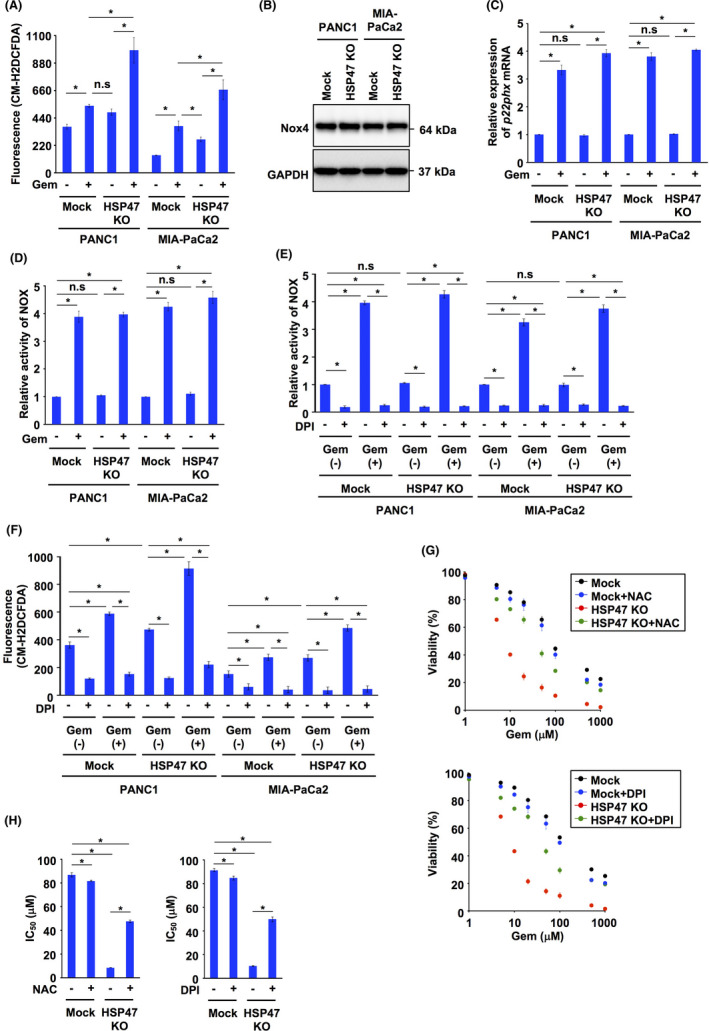
Disruption of HSP47 enhances NOX‐induced generation of ROS in pancreatic cancer cells after treatment with gemcitabine. A, Levels of intracellular ROS in HSP47 KO PDAC cells (HSP47 KO PANC1 cells and HSP47 KO MIA‐PaCa2 cells) treated with gemcitabine (10 μmol/L for PANC1 cells and 1 nmol/L for MIA‐PaCa2 cells). B, Expression of Nox4 protein in HSP47 KO PDAC cells. C, Expression of p22phox mRNA in HSP47 KO PDAC cells treated with gemcitabine. D, Level of NOX activity in HSP47 KO PDAC cells treated with gemcitabine. E, Levels of NOX activity in HSP47 KO PDAC cells treated with the NOX inhibitor DPI (2 μmol/L) and gemcitabine. F, Levels of intracellular ROS in HSP47 KO PDAC cells treated with the NOX inhibitor DPI (2 μmol/L) and gemcitabine. G, Viability of HSP47 KO PANC1 cells after treatment with NAC (10 μmol/L), DPI (2 μmol/L) and gemcitabine. H, IC50 values of HSP47 KO PANC1 cells after treatment with NAC (10 μmol/L), DPI (2 μmol/L) and gemcitabine. *, P < .05. n.s., not significant
3.6. Gemcitabine‐induced generation of ROS triggers an excess increase of intracellular Ca2+ level in HSP47‐depleted PDAC cells
It has been reported that ROS lead to an increase in intracellular Ca2+ concentrations by stimulation of IP3R and inhibition of SERCA2 activity. 42 , 43 , 44 , 45 We showed that disruption of HSP47 induced both the interaction of CALR with SERCA2 and the interaction of IRE1α with IP3R in PDAC cells (Figure 3), and these interactions have been reported to induce inactivation of SERCA2 and stimulation of IP3R, respectively. 36 , 39 Therefore, we hypothesized that depletion of HSP47 generated a condition that is prone to increase intracellular Ca2+ concentration in response to oxidative stimuli such as ROS. Therefore, we examined the effects of NAC and DPI on the increase in intracellular Ca2+ level in HSP47‐depleted PDAC cells treated with gemcitabine. Treatment with gemcitabine induced an increase in intracellular Ca2+ level in HSP47 KO PANC1 cells, which was clearly inhibited by treatments with NAC and DPI (Figure 6A). We also investigated the effect of ROS on the change in the intracellular Ca2+ level of HSP47 KO PDAC cells. Treatment with hydrogen peroxide (H2O2) induced increases in intracellular Ca2+ levels in Mock:PDAC cells and HSP47 KO PDAC cells (Figure 6B). The degree of increase in intracellular Ca2+ level in HSP47 KO PDAC cells after treatment with H2O2 was significantly higher than that in Mock:PDAC cells. Time lapse imaging analysis showed that an increase in intracellular Ca2+ level in HSP47 KO PDAC cells was immediately induced by treatment with H2O2 (Figure 6C). The enhanced increase in intracellular Ca2+ level in HSP47 KO PDAC cells treated with H2O2 was abolished by reconstitution of HSP47‐Myc expression (Figure 6B,C).
FIGURE 6.
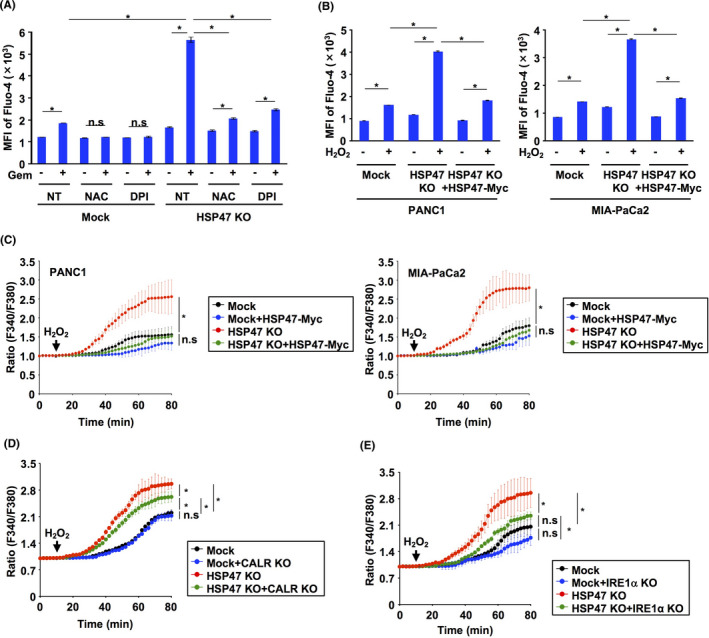
HSP47 alleviates the ROS‐induced increase in intracellular Ca2+ levels in pancreatic cancer cells after treatment with gemcitabine. A, Levels of intracellular Ca2+ in HSP47 KO PDAC cells (PANC1 cells) after treatment with NAC (10 μmol/L), DPI (2 μmol/L) and gemcitabine. Intracellular Ca2+ levels were determined using flow cytometry and calculating the intensity of Fluo‐4. B, Levels of intracellular Ca2+ in Mock:PDAC cells (PANC1 cells and MIA‐PaCa2 cells), HSP47 KO PDAC cells and HSP47 KO PDAC cells expressing HSP47‐Myc at 24 h after treatment with H2O2 (2 μg/mL). C, Changes in intracellular Ca2+ levels in Mock:PDAC cells, HSP47 KO PDAC cells, and HSP47 KO PDAC cells expressing HSP47‐Myc after treatment with H2O2. Changes in intracellular Ca2+ levels were determined by calculating the ratio (F340/F380). D, Changes in intracellular Ca2+ levels in Mock:PANC1 cells, CALR KO PANC1 cells, HSP47 KO PANC1 cells and CALR KO HSP47 KO PANC1 cells after treatment with H2O2 (2 μg/mL). E, Changes in intracellular Ca2+ levels in Mock:PANC1 cells, IRE1α KO PANC1 cells, HSP47 KO PANC1 cells and IRE1α KO HSP47 KO PANC1 cells after treatment with H2O2 (2 μg/mL). * P < .05. n.s: not significant
To further clarify the involvement of both CALR and IRE1α in the increase in intracellular Ca2+ level in HSP47 KO PDAC cells induced by ROS, we examined whether depletion of CALR or IRE1α affected the change of intracellular Ca2+ level in HSP47 KO PDAC cells treated with H2O2. The increase of intracellular Ca2+ level after treatment with H2O2 was not different in Mock:PANC1 cells and CALR KO PANC1 cells (Figure 6D). Conversely, depletion of CALR partially inhibited the increase of intracellular Ca2+ level in HSP47 KO PANC1 cells after treatment with H2O2. The increase of intracellular Ca2+ level after treatment with H2O2 was not also different in Mock:PANC1 cells and IRE1α KO PANC1 cells (Figure 6E). Intriguingly, the increase of intracellular Ca2+ level in HSP47 KO PANC1 cells after treatment with H2O2 was markedly suppressed by depletion of IRE1α (Figure 6E). These results indicated that disruption of HSP47 promotes an increase of intracellular Ca2+ level in PDAC cells in response to oxidative stimuli.
3.7. Disruption of HSP47 enhances the efficacy of chemotherapy with gemcitabine against pancreatic tumors
To determine whether disruption of HSP47 in PDAC cells promoted the chemotherapeutic efficacy of gemcitabine against pancreatic tumor growth, we investigated tumor growth in mice that had been orthotopically implanted with HSP47 KO PDAC cells after treatment with gemcitabine (Figure 7A). Bioluminescence analysis showed that the luminescence level in HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice was significantly lower than that in Mock:MIA‐PaCa2‐luc cell‐transplanted mice (Figure 7B,C). The luminescence levels in Mock:MIA‐PaCa2‐luc cell‐transplanted and HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice were markedly decreased by treatment with gemcitabine. The inhibitory effect of gemcitabine on the luminescence in HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice was significantly greater than that in Mock:MIA‐PaCa2‐luc cell‐transplanted mice (Figure 7D). The luminescence levels of pancreatic tissues from Mock:MIA‐PaCa2‐luc cell‐transplanted and HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice that had been treated with gemcitabine were also lower than those of the counterparts (Figure 7E,F). The inhibitory effect of gemcitabine treatment on the luminescence in pancreatic tissues from HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice was also greater than that in pancreatic tissues from Mock:MIA‐PaCa2‐luc cell‐transplanted mice (Figure 7G). Furthermore, treatment with gemcitabine reduced the tumor volumes and the percentage of Ki67‐positive cells in tumor tissues of Mock:MIA‐PaCa2‐luc cell‐transplanted and HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice (Figure 7H,I). The effects of gemcitabine treatment on tumor volume and percentage of Ki67‐positive cells in tumor tissues of HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice were significantly greater than the effects for Mock:MIA‐PaCa2‐luc cell‐transplanted mice. These results suggested that disruption of HSP47 in PDAC cells augments the chemotherapeutic effect on pancreatic tumors.
FIGURE 7.
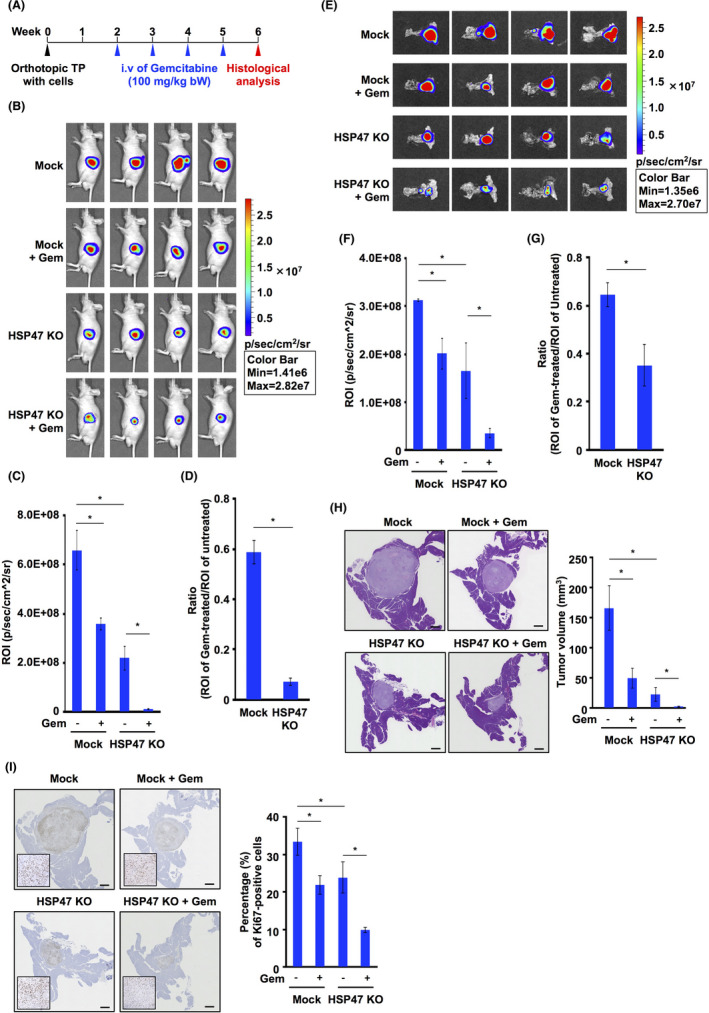
Disruption of HSP47 augments the efficacy of chemotherapy with gemcitabine for pancreatic tumors. A, Scheme of the animal experiment. B, Luminescence of MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice treated with or not treated with gemcitabine. Representative images at 7 d after the final injection of gemcitabine are shown. C, Quantification of luminescence of MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice at 7 d after the final treatment with gemcitabine. D, Effect of gemcitabine treatment on luminescence of MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice at 7 d after the final injection of gemcitabine. E, Luminescence of pancreatic tissues from MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice treated with or not treated with gemcitabine. Representative images of pancreatic tissues at 7 d after the final injection of gemcitabine are shown. F, Quantification of luminescence of pancreatic tissues from MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice at 7 d after the final treatment with gemcitabine. G, Effect of gemcitabine treatment on luminescence of pancreatic tissues from MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice at 7 d after the final injection of gemcitabine. H, Effect of gemcitabine treatment on tumor volumes in MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice. Representative images of pancreatic tissues with HE staining are shown (left panel). Quantification of tumor volume is shown (right panel). (I) Effect of gemcitabine treatment on proliferation index in tumor tissues from MIA‐PaCa2‐luc cell‐transplanted or HSP47 KO MIA‐PaCa2‐luc cell‐transplanted mice. Representative images of Ki67‐positive cells in pancreatic tissues with Ki67 staining are shown (left panel). Quantification of Ki67‐positive cells in tumor tissues is shown (right panel). *, P < .05. Scale bars: 1 mm
4. DISCUSSION
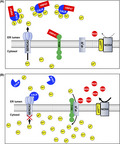
We demonstrated that silencing and ablation of HSP47 improved the efficacy of the therapeutic agent gemcitabine against PDAC cells. The improved efficacy of gemcitabine for HSP47‐depleted PDAC cells was reduced by reconstitution of HSP47 expression. HSP47 interacted with CALR and IRE1α in PDAC cells. Depletion of HSP47 facilitated both the interaction of CALR with SERCA2 and the interaction of IRE1α with IP3R, which generated a condition that is prone to increase the intracellular Ca2+ level in response to oxidative stimuli. Under the conditions of the treatment with gemcitabine, disruption of HSP47 enhanced the NOX‐induced increase in intracellular ROS and the subsequent increase in intracellular Ca2+ levels in PDAC cells, resulting in cell death by activation of the caspase‐12/caspase‐9/caspase‐3 axis (Figure 8). Ablation of HSP47 promoted the inhibitory effect of gemcitabine treatment on increase in tumor volumes in mice orthotopically implanted with PDAC cells. These results indicated that HSP47 functions as a regulator for the tolerance of PDAC cells against chemotherapy.
FIGURE 8.
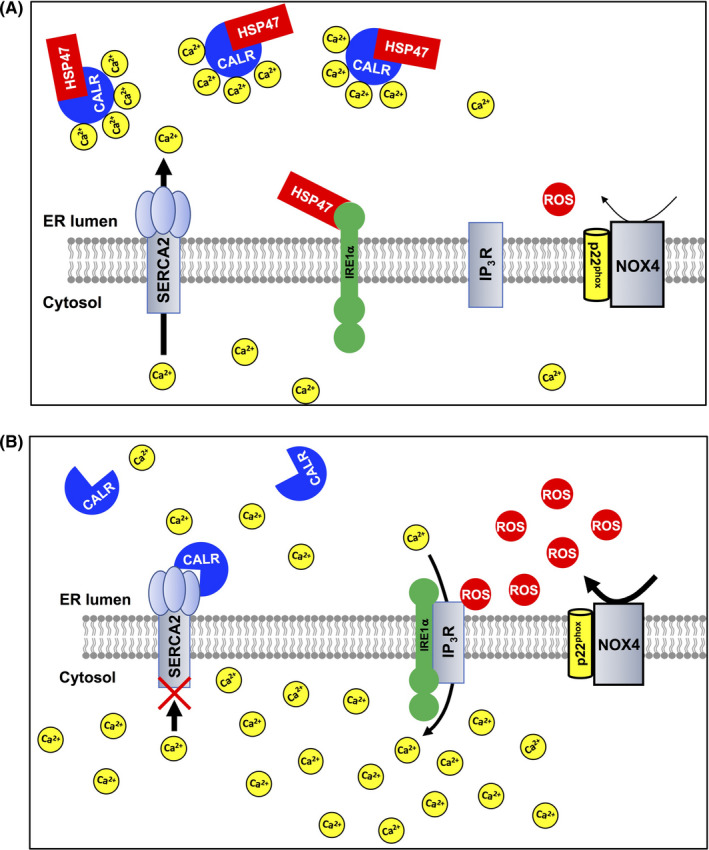
Scheme of HSP47 regulation of the chemoresistance of pancreatic cancer cells. A, In a basal condition, HSP47 interacts with calreticulin (CALR) and IRE1α in PDAC cells. Treatment with gemcitabine induces production of ROS by increased expression of NOX4. However, interaction of HSP47 with both CALR and IRE1α inhibits the SERCA2‐mediated and IP3R‐mediated increase of intracellular Ca2+ level in PDAC cells, resulting in tolerance against chemotherapy. B, In HSP47‐disrupted PDAC cells, CALR interacts with SERCA2, which inhibits activation of SERCA2. IRE1α also interacts with IP3R, which stimulates IP3R activity. These interactions generate a condition that is prone to increase the intracellular Ca2+ level in HSP47‐depleted PDAC cells in response to oxidative stimuli. Treatment of HSP47‐depleted PDAC cells with gemcitabine induces the generation of intracellular ROS and subsequent excess increase of intracellular Ca2+ levels, resulting in death of PDAC cells via activation of the caspase axis
In addition to the function of HSP47 as a collagen‐specific chaperone, 16 , 17 several studies have recently shown that HSP47‐interacting proteins regulated protein maturation and secretion, cell survival, and cell migration and invasion. 29 , 30 , 31 , 32 , 46 , 47 The present study showed that HSP47 interacted with 6 HSP47‐interacting proteins that reside in the ER in PDAC cells. Among the HSP47‐interacting proteins identified in the present study, we focused on CALR and IRE1α, both of which are involved in chemoresistance of cancer cells. Under oxidative stress conditions, interaction of CALR, an ER‐resident Ca2+‐binding protein in the ER, with SERCA2 promoted the inactivation and degradation of SERCA2, causing an excess increase in intracellular Ca2+ concentration and subsequent cell arrest and apoptosis. 35 , 36 Accumulating evidence has also shown that CALR translocates from the ER to the plasma membrane of cancer cells after treatment with chemotherapeutic agents such as anthracyclins and oxaliplatin and acts as an eat‐me signal molecule that facilitates the recognition of stressed cells by phagocytes expressing the CALR receptor CD91, resulting in induction of immunogenic cell death. 48 , 49 , 50 , 51 , 52 Therefore, CALR has been suggested to be a candidate compound as a therapeutic target for tumors. It is well known that IRE1α serves as a serine‐threonine protein kinase and endoribonuclease that represents the most conserved UPR signaling branch, controlling cell fate under ER stress conditions. 37 , 38 In addition to acting as a UPR transducer, IRE1α interacts with IP3R and its interaction promotes the release of Ca2+ from the ER, leading to cell death. 35 The release of Ca2+ from the ER by IP3R is facilitated by ROS under oxidative stress conditions. 42 , 43 It has been shown that targeting the expression or the RNase activity of IRE1α reduced the progression of various forms of cancer due to dysfunction of the UPR and Ca2+ homeostasis. 34 , 39 In the present study, we showed that depletion of HSP47 induced both the interaction of CALR with SERCA2 and the interaction of IRE1α with IP3R and triggered an excess increase in intracellular Ca2+ levels under oxidative stress conditions induced by gemcitabine‐induced production of ROS, and causing death of PDAC cells. We suggest that HSP47 acts as a regulator of the tolerance of PDAC cells against therapeutic agents, as well as acting as a collagen‐specific chaperone.
Although the chemotherapeutic agent gemcitabine or FOLFIRINOX is usually the recommended first‐line drug for patients with PDAC and is given alone or in combination with other agents, a large number of patients with PDAC are resistant to these therapies. 8 , 9 , 10 , 11 Gemcitabine inhibits DNA synthesis and the cell cycle at the G1/S phase by its incorporation into DNA during DNA replication. It has been reported that gemcitabine treatment increases the activity of NOX and induces upregulation of p22phox expression in PDAC cells by activation of ATR, p53, and NF‐κB that are stimulated by gemcitabine‐induced inhibition of DNA synthesis, resulting in generation of ROS. 12 , 13 , 41 , 53 , 54 , 55 , 56 We also confirmed that treatment with gemcitabine induced an increase in the intracellular level of ROS in PDAC cells by an increase in NOX activity and upregulation of p22phox expression. Intriguingly, the present study showed that the gemcitabine‐induced increase in intracellular ROS caused an increase in intracellular Ca2+ levels in PDAC cells. Calcium functions as a second messenger and plays an important role in cell proliferation, cell survival and the chemoresistance of cancer cells by activation of calcium/calmodulin‐dependent protein kinase (CamK) and Akt. 40 , 57 , 58 It has been reported that treatment with a Ca2+ chelator and inhibition of CamK and Akt improved the efficacy of therapeutic agents for tumors. 50 Conversely, an excess increase in intracellular Ca2+ concentration triggered cell death by activation of the calcium/caspases axis. 40 The present study showed that the increase in intracellular Ca2+ level induced by treatment with gemcitabine was markedly augmented by ablation of HSP47 expression, causing death of PDAC cells by activation of the calcium/caspase‐12/caspase‐9/caspase‐3 axis. Our study suggested that HSP47 is involved in maintaining the survival of PDAC cells by relieving the gemcitabine/ROS‐induced increase in intracellular Ca2+ concentration.
In conclusion, we demonstrated that HSP47 interfered with both CALR/SERCA2 interaction and IRE1α/IP3R interaction and reduced the excess increase of intracellular Ca2+ level by stimulation of gemcitabine/NOX‐induced production of ROS, conferring chemoresistance on PDAC cells. Therefore, our study suggested that the targeted disruption of HSP47 in combination with chemotherapy may improve the overall survival of patients with pancreatic tumors.
DISCLOSURE
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by Nitto Denko Corporation (Osaka, Japan). This work was also supported in part by the Grant‐in‐Aid for Scientific Research C from MEXT Japan Society for the Promotion of Science (KAKENHI18K07287 to AY).
Yoneda A, Minomi K, Tamura Y. Heat shock protein 47 confers chemoresistance on pancreatic cancer cells by interacting with calreticulin and IRE1α. Cancer Sci. 2021;112:2803–2820. 10.1111/cas.14976
Funding information
Japan Society for the Promotion of Science, (Grant/Award Number: 'KAKENHI18K07287').
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics 2019. CA Cancer J Clin. 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 2. Yao W, Maitra A, Ying H. Recent insights into the biology of pancreatic cancer. EBioMedicine. 2020;53:102655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peixoto RD, Ho M, Renouf DJ, et al. Eligibility of metastatic pancreatic cancer patients for first‐Line palliative intent nab‐paclitaxel plus gemcitabine versus FOLFIRINOX. Am J Clin Oncol. 2017;40:507‐511. [DOI] [PubMed] [Google Scholar]
- 4. Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379:2395‐2406. [DOI] [PubMed] [Google Scholar]
- 5. Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in pancreatic cancer is driven by stroma‐derived insulin‐like growth factors. Cancer Res. 2016;76:6851‐6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF‐1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeng S, Pöttler M, Lan B, et al. Chemoresistance in pancreatic cancer. Int J Mol Sci. 2019;20:4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi X, Liu S, Kleeff J, et al. Acquired resistance of pancreatic cancer cells towards 5‐Fluorouracil and gemcitabine is associated with altered expression of apoptosis‐regulating genes. Oncology. 2002;62:354‐362. [DOI] [PubMed] [Google Scholar]
- 9. Müerköster SS, Lust J, Arlt A, et al. Acquired chemoresistance in pancreatic carcinoma cells: induced secretion of IL‐1beta and NO lead to inactivation of caspases. Oncogene. 2006;25:3973‐3981. [DOI] [PubMed] [Google Scholar]
- 10. Hung SW, Mody HR, Govindarajan R. Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett. 2012;320:138‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ju HQ, Gocho T, Aguilar M, et al. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther. 2015;14:788‐798. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z, Duan Q, Zhao H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF‐kB/STAT3 signaling cascade. Cancer Lett. 2016;382:52‐63. [DOI] [PubMed] [Google Scholar]
- 14. Grasso C, Jansen G, Giovannetti E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit Rev Oncol Hematol. 2017;114:139‐152. [DOI] [PubMed] [Google Scholar]
- 15. Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers. 2017;9:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito S, Nagata K. Biology of Hsp47 (Serpin H1), a collagen‐specific molecular chaperone. Semin Cell Dev Biol. 2017;62:142‐151. [DOI] [PubMed] [Google Scholar]
- 17. Ito S, Nagata K. Roles of the endoplasmic reticulum‐resident, collagen‐specific molecular chaperone Hsp47 in vertebrate cells and human disease. J Biol Chem. 2019;294:2133‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishida Y, Kubota H, Yamamoto A, et al. Type I collagen in Hsp47‐null cells is aggregated in endoplasmic reticulum and deficient in N‐propeptide processing and fibrillogenesis. Mol Biol Cell. 2006;17:2346‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marutani T, Yamamoto A, Nagai N, et al. Accumulation of type IV collagen in dilated ER leads to apoptosis in Hsp47‐knockout mouse embryos via induction of CHOP. J Cell Sci. 2004;117:5913‐5922. [DOI] [PubMed] [Google Scholar]
- 20. Kawasaki K, Ushioda R, Ito S, et al. Deletion of the collagen‐specific molecular chaperone Hsp47 causes endoplasmic reticulum stress‐mediated apoptosis of hepatic stellate cells. J Biol Chem. 2015;290:3639‐3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maitra A, Iacobuzio‐Donahue C, Rahman A, et al. Immunohistochemical validation of a novel epithelial and a novel stromal marker of pancreatic ductal adenocarcinoma identified by global expression microarrays: Sea urchin fascin homolog and heat shock protein 47. Am J Clin Pathol. 2002;118:52‐59. [DOI] [PubMed] [Google Scholar]
- 22. Hirai K, Kikuchi S, Kurita A, et al. Immunohistochemical distribution of heat shock protein 47 (HSP47) in scirrhous carcinoma of the stomach. Anticancer Res. 2006;26:71‐78. [PubMed] [Google Scholar]
- 23. Yamamoto N, Kinoshita T, Nohata N, et al. Tumor‐suppressive microRNA‐29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. 2013;43:1855‐1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao D, Jiang X, Yao C, et al. Heat shock protein 47 regulated by miR‐29a to enhance glioma tumor growth and invasion. J Neurooncol. 2014;118:39‐47. [DOI] [PubMed] [Google Scholar]
- 25. Zhu J, Xiong G, Fu H, et al. Chaperone Hsp47 drives malignant growth and invasion by modulating an ECM gene network. Cancer Res. 2015;75:1580‐1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mori K, Toiyama Y, Otake K, et al. Proteomics analysis of differential protein expression identifies heat shock protein 47 as a predictive marker for lymph node metastasis in patients with colorectal cancer. Int J Cancer. 2017;140:1425‐1435. [DOI] [PubMed] [Google Scholar]
- 27. Xiong G, Chen J, Zhang G, et al. Hsp47 promotes cancer metastasis by enhancing collagen‐dependent cancer cell‐platelet interaction. Proc Natl Acad Sci USA. 2020;117:3748‐3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y, Zhu L, Wang X. A network‐based approach for identification of subtype‐specific master regulators in pancreatic ductal adenocarcinoma. Genes (Basel). 2020;11:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoneda A, Sakai‐Sawada K, Minomi K, et al. Heat shock protein 47 maintains cancer cell growth by inhibiting the unfolded protein response transducer IRE1α. Mol Cancer Res. 2020;18:847‐858. [DOI] [PubMed] [Google Scholar]
- 30. Yoneda A, Minomi K, Tamura Y. HSP47 promotes metastasis of breast cancer by interacting with myosin IIA via the unfolded protein response transducer IRE1α. Oncogene. 2020;39:4519‐4537. [DOI] [PubMed] [Google Scholar]
- 31. Sepulveda D, Rojas‐Rivera D, Rodríguez DA, et al. Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1α. Mol Cell. 2018;69:238‐252. [DOI] [PubMed] [Google Scholar]
- 32. Sheng W, Chen C, Dong M, et al. Overexpression of calreticulin contributes to the development and progression of pancreatic cancer. J Cell Physiol. 2014;229:887‐897. [DOI] [PubMed] [Google Scholar]
- 33. Genovese G, Carugo A, Tepper J, et al. Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature. 2017;542:362‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Logue SE, McGrath EP, Cleary P, et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat Commun. 2018;9:3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura K, Zuppini A, Arnaudeau S, et al. Functional specialization of calreticulin domains. J Cell Biol. 2001;154:961‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ihara Y, Kageyama K, Kondo T. Overexpression of calreticulin sensitizes SERCA2a to oxidative stress. Biochem Biophys Res Commun. 2005;329:1343‐1349. [DOI] [PubMed] [Google Scholar]
- 37. Gardner BM, Pincus D, Gotthardt K, et al. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5:a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169‐181. [DOI] [PubMed] [Google Scholar]
- 39. Carreras‐Sureda A, Jaña F, Urra H, et al. Non‐canonical function of IRE1a determines mitochondria‐associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol. 2019;21:755‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sergeev IN. Vitamin D and cellular Ca2+ signaling in breast cancer. Anticancer Res. 2012;32:299‐302. [PubMed] [Google Scholar]
- 41. Vaquero EC, Edderkaoui M, Pandol SJ, et al. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643‐34654. [DOI] [PubMed] [Google Scholar]
- 42. Görlach A, Bertram K, Hudecova S, et al. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310‐321. [DOI] [PubMed] [Google Scholar]
- 44. Bidwell PA, Liu GS, Nagarajan N, et al. HAX‐1 regulates SERCA2a oxidation and degradation. J Mol Cell Cardiol. 2018;114:220‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joseph SK, Young MP, Alzayady K, et al. Redox regulation of type‐1 inositol triphosphate receptors in intact mammalian cells. J Biol Chem. 2018;293:17464‐17476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ishikawa Y, Rubin K, Bächinger HP, et al. The endoplasmic reticulum‐resident collagen chaperone Hsp47 interacts with and promotes the secretion of decorin, fibromodulin, and lumican. J Biol Chem. 2018;293:13707‐13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J, Wang S, Zhang Z, et al. Heat shock protein 47 (HSP47) binds to discoidin domain‐containing receptor 2 (DDR2) and regulates its protein stability. J Biol Chem. 2019;294:16846‐16854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Basu S, Binder RJ, Ramalingam T, et al. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303‐313. [DOI] [PubMed] [Google Scholar]
- 49. Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54‐61. [DOI] [PubMed] [Google Scholar]
- 50. Tesniere A, Schlemmer F, Boige V, et al. Immunogenic cell death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482‐491. [DOI] [PubMed] [Google Scholar]
- 51. Krysko DV, Garg AD, Kaczmarek A, et al. Vandenabeele. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860‐875. [DOI] [PubMed] [Google Scholar]
- 52. Raghavan M, Wijeyesakere SJ, Peters LR, et al. Calreticulin in the immune system: ins and outs. Trends Immunol. 2013;34:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okazaki T, Jiao L, Chang P, et al. Single‐nucleotide polymorphisms of DNA damage response genes are associated with overall survival in patients with pancreatic cancer. Clin Cancer Res. 2008;14:2042‐2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage‐induced phosphorylation of p53. Genes Dev. 1999;13:152‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karnitz LM, Flatten KS, Wagner JM, et al. Gemcitabine‐induced activation of checkpoit signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636‐1644. [DOI] [PubMed] [Google Scholar]
- 56. Boudreau HE, Casterline BW, Burke DJ, et al. Wild‐type and mutant p53 differentially regulate NADPH oxidase 4 in TGF‐β‐mediated migration of human lung and breast epithelial cells. Br J Cancer. 2014;110:2569‐2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rokhlin OW, Taghiyev AF, Bayer KU, et al. Calcium/calmodulin‐dependent kinase II plays an important role in prostate cancer cell survival. Cancer Biol. 2007;6:732‐742. [DOI] [PubMed] [Google Scholar]
- 58. Gocher AM, Azabdaftari G, Euscher LM, et al. Akt activation by Ca2+/calmodulin‐dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J Biol Chem. 2017;292:14188‐14204. [DOI] [PMC free article] [PubMed] [Google Scholar]