

Abstract
Biological samples such as tissues, blood, or tumors are often complex and harbor heterogeneous populations of cells. Separating out specific cell types or subpopulations from such complex mixtures to study their metabolic phenotypes is challenging because experimental procedures for separation may disturb the metabolic state of cells. To address this issue, we developed a method for analysis of cell subpopulations using stable isotope tracing and fluorescence-activated cell sorting (FACS) followed by liquid chromatography – high resolution mass spectrometry (LC-HRMS). To ensure a faithful representation of cellular metabolism after cell sorting, we benchmarked sorted extraction against direct extraction. While peak areas differed markedly with lower signal for amino acids but higher signal for nucleotides, mass isotopomer distributions from sorted cells were generally in good agreement with those obtained from direct extractions, indicating that they reflect the true metabolic state of cells prior to sorting. In proof-of-principle studies, our method revealed metabolic phenotypes specific to T cell subtypes, and also metabolic features of cells in the committed phase of the cell division cycle. Our approach enables studies of a wide range of adherent and suspension cell subpopulations, which we anticipate will be of broad importance in cell biology and biomedicine.
Keywords: sorted cells, metabolite extraction, FACS, mass spectrometry, metabolomics, cell cycle, T cells, CD8, CD4
Graphical Abstract
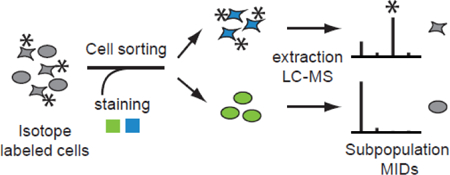
Introduction
The complement of biochemical reactions available to human cells is well charted, but still little is known about the metabolic behavior of specific cell types in their natural environment. While human tissues are complex mixtures of multiple cell types, most of our knowledge derives from bulk measurements of cultured cells. To better understand the variety of metabolic behaviors cells can exhibit, it is of great importance to develop methods for measuring metabolism of subpopulations of cells, separated from such complex mixtures. For example, a great variety of immune cell types are present in human blood samples 1, and solid tumors contain not only cancer cells, but also fibroblasts and infiltrating immune cells 2. Cell cultures can also contain co-existing subpopulations, such as subtypes of different physiological origin present in breast cancer cell lines 3, and even among otherwise identical cells, individual cell-to-cell differences such as cell cycle phases may determine metabolic state.
By far the most versatile and widely used tool for separating cell populations is fluorescence-activated cell sorting (FACS), where individual cells are passed through a capillary and separated into tubes based on fluorescent antibodies detecting endogenous cellular proteins, or fluorescent proteins expressed by engineered cell lines 4. While FACS is commonly used to isolate certain populations of cells which are then re-cultured and analyzed at a later time 5, this approach does not provide information on the metabolic state of the original, complex mixture of cells and would fail to capture transient states like the cell cycle. A few studies have recently attempted metabolomics of subpopulations immediately after FACS separation, including a study on Arabidopsis protoplasts from different root cell types 6, and a report identifying metabolite signatures of CD133+ colon cancer initiating cells 7. This may provide more direct information on the original metabolic state, but is challenging as the FACS procedure may cause significant perturbations of the metabolic state. FACS often necessitates keeping cells in nutrient-poor buffers for the duration of sorting, which can last up to 1 hour until quenching/extraction, depending on cell type. This change of extracellular environment may result in leakage of intracellular metabolites into the buffer, or other metabolic imbalances due to loss of nutrients. Moreover, the temperature or pressure changes inflicted by FACS might cause agitation or stress to the cells. For comparison, in common methods for metabolite extraction, cultured cells are only exposed to buffer solution for a few minutes during removal of spent culture medium and washing. While one report 8 indicated that mRNA levels are minimally disturbed by FACS, it is clearly important to investigate the effects of the FACS procedure on metabolomics data from sorted cells.
Isotope tracing can provide information on enzyme or pathway activity or differences in activity between subpopulations 9. We reasoned that, since the isotope distribution of any given metabolite reflects the cell’s metabolic activity over a longer period of time, isotope distributions should be reasonably robust during FACS. Hence, while metabolite abundances in sorted cells might vary due to technical constraints imposed my FACS, metabolic tracing using stable isotopes might be feasible also in sorted cells. We therefore took an approach where complex cell populations are cultured with isotope-labeled substrates for a period of time, followed by FACS-based separation and measurement of intracellular metabolite isotopomer distributions using liquid chromatography – high resolution mass spectrometry (LC-HRMS) (Figure 1a). The resulting data should reflect the metabolic state of a cell subpopulation during its time in culture, prior to FACS analysis. In this paper, we report on the development of this methodology, benchmark the resulting data against direct cell extraction, and discuss pitfalls and limitations of the approach. We also provide two examples of detection of metabolic differences between subpopulations of adherent human cancer cells, as well as non-adherent, primary human T lymphocytes.
Figure 1.
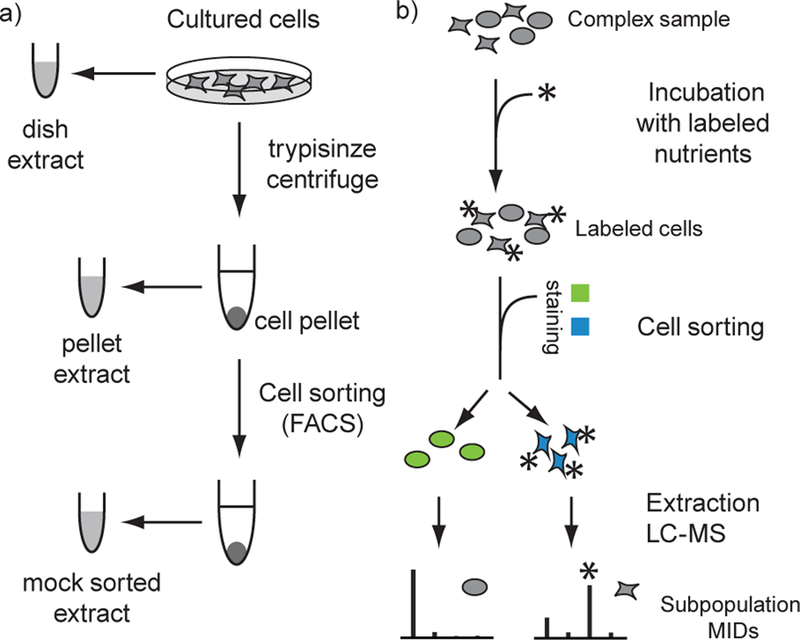
Schematic diagram of the experimental design. a) Comparison of sorted cells populations based on isotope label content. The complex sample is pulse labelled prior to sorting, and differences between subpopulation are reflected in the MIDs. Subpopulations extracts are analyzed separately by LC-HRMS. b) Design of validation experiment. Metabolomics of mock sorted cells were benchmarked with dish and pellet extracts.
Experimental section
Cell culture
HeLa cells were cultured in RPMI-1640 medium (Life Technologies) supplemented with 5% dialyzed fetal bovine serum (dFBS), in 6-well plates for 48h. Cells were plated at a density to achieve ~85% confluence before extraction. FBS (Life Technologies) was dialyzed in SNAKESKIN 10K MWCO dialysis tubing (Nordic Bioloabs, 88245-P). For isotope tracing experiments, cells were cultured in labeled medium containing 40% U-13C-Glucose (Cambridge Isotopes, 40762–22-9 / GLC-018) and 70% U-13C,15N2-Glutamine (Cambridge Isotopes, /CNLM-1275-H-0.1) for 48h, and HBSS (Hank’s Balanced Salt Solution, Sigma, H6648) was replaced by HBSS containing 40% U-13C-Glucose (HBSS*) to avoid washout of 13C isotopes during the sorting procedure.
A mixture of the two main fuels of the cell allows labelling of a larger number of metabolites. We chose an intermediate (40%, 70%) amount of labeling to generate richer MID patterns with intermediate MI fractions, to help evaluate the robustness of MIDs.
Metabolite extraction
Extraction from dish
The wells were first rinsed once with 500µL HBSS and the washing solution was discarded. 60µL HBSS was added and cells were extracted with 540µL 100% methanol pre-cooled on dry ice to obtain a final 90% v/v concentration of methanol. Microplates were then transferred to dry ice and cell material was removed with a cell scraper, transferred to a 1.5 mL tube and stored at −80°C. All experiments were performed in triplicates. Extracts were kept at −80°C until LC-HRMS analysis.
Extraction from pellet
Cells were first rinsed with 500µL HBSS and then detached with 500µL trypsin/EDTA (Life Technologies, 25300062) for 4min at 37°C. Next steps were performed at 4°C to decrease metabolic activities. 1mL HBSS + 5% dialyzed FBS (HBSS-dFBS) was used to deactivate trypsin, and the cells were centrifuged for 3 min at 750g at 4°C. Supernatant was discarded, cells were resuspended in 800µL HBSS-dFBS and the centrifugation step was repeated.
To obtain a pellet extraction, cells were resuspended in 50µL HBSS and extracted by adding 540µL dry ice cold 100% methanol to a final 90% concentration of methanol.
To answer the amino acid leakage question, the obtained pellet is resuspended in HBSS-dFBS + 1mM EDTA and kept in ice for 1h. At the end of the incubation cells were centrifuged at 750g for 3 min. Supernatants were saved and pellets were extracted in 100% methanol. Extracts and saved supernatants were kept at −80°C until LC-HRMS analysis.
Extraction of sorted cells
Extraction of mock sorted cells
Cells cultured in 10cm plates were rinsed with 1.5mL warm HBSS and incubated with 1.5mL trypsin/EDTA solution for 4 min at 37°C. Trypsin was deactivated with 3 mL HBSS-dFBS, and the cell suspension was transferred to 15 mL tubes. Next steps were performed at 4°C. Cells were centrifuged at 750g for 3 min, the supernatant was discarded and the pellet was resuspended in 2mL HBSS-dFBS + 1mM EDTA. Cells were filtered through cell strainers (30µm, BD, 340625) and transferred to a falcon tube tube. Cells were sorted with INFLUX (inFlux v7 Sorter, BD Biosciences) at ~1000 events/sec using a 100µm nozzle and 500,000 cells were collected. Sorted cells were centrifuged at 750g for 3 min at 4°C. The supernatant was discarded and the pellet was resuspended in 50µL ice cold HBSS. Metabolites were extracted by adding 540µL methanol kept in dry ice. Extracts were kept at −80°C until LC-HRMS analysis.
Extraction of cells at different cell cycle phases
The HeLa cells used in this experiment contained a Geminin Fucci probe, which is expressed in early S until late M phase of the cell cycle10, allowing separation of non-fluorescent G1-G0 phase cells from fluorescent S-G2-M phase cells. Cells were cultured as above in 10cm plates for 46h, plated at a density to achieve ~85% confluence after 48h. Before extraction, cells were “pulse labeled” for 2h in RPMI medium containing 40% U-13C-Glucose and 70% U-13C,15N2-Glutamine. Supernatant was then discarded and plates were rinsed with 1.5mL warm HBSS*. Cells were incubated with 1.5mL trypsin/EDTA solution for 4min at 37°C for detachment. Trypsin was deactivated with 3mL HBSS*-dFBS, and cells were transferred to 15mL tubes. The following steps were performed at 4°C. Cells were centrifuged at 750g for 3 min, supernatant was discarded and pellet was resuspended in 2mL HBSS*-dFBS + 1mM EDTA. Cells were filtered and transferred to a falcon tube.
Sorting was performed in INFLUX (inFlux v7 Sorter, BD Biosciences) at ~1000 events/sec, using a 100µm nozzle. The fluorescent signal was detected using a 488nm laser and a 521nm filter, and gating was applied based on the fluorescence signal. A population of 500,000 cells was collected in each tube of G1-G0 and S-G2-M cells, respectively. Sorted cells were centrifuged at 750g for 3min at 4°C. The supernatant was discarded and the pellet was resuspended in 50µL ice cold HBSS* and extracted by adding 540µL dry ice cold methanol. Extracts were kept at −80°C until LC-HRMS analysis.
Extraction of T cells
Peripheral blood mononuclear cells from buffy coats were prepared as described previously11 and pan T cells were isolated by negative magnetic isolation using the pan T cell isolation kit II (Miltenyi Biotec). Cells were activated on anti-CD3-antibody coated plates and with 1 µg/ml anti-CD28 antibody (both from Biolegend) at a concentration of 14.4 × 106 cells/well in 6-well plates, in medium containing 40% U-13C-Glucose and 70% U-13C,15N2-Glutamine and dialyzed FBS. Before sorting, cells were stained with antibodies for CD4 and CD8. A staining mix was prepared with 100μL CD8-eFlour 450 (ebioscience #48–0086-42) and 30μL CD4-APC (ebioscience #17–0048-42 clone OKT4), and completed to 1.5mL with labeled media. ~450uL of the staining mix was used to stain ~30×106 cells, incubated at room temperature for 15min, centrifuged and resuspended in HBSS*-dFBS + 1mM EDTA.
Sorting was performed in INFLUX (inFlux v7 Sorter) at ~1500 events/sec using a 100µm nozzle. Gating was applied based on fluorescence signal and 2.3×106 cells were sorted simultaneously in each tube of CD4 and CD8 cells for each replicate. Sorted cells were centrifuged at 750g, 3 min, at 4°C. The supernatant was discarded and the pellet was resuspended in 50µL ice cold HBSS*. Metabolites were extracted by adding 540µL dry ice cold methanol. Extracts were kept at −80°C until LC-HRMS analysis.
Metabolite measurements
Prior to the LC-HRMS analysis, cell extracts were thawed in ice for 30min, vortexed 15sec and then 100µL were transferred to a spin filter and centrifuged for 10min at 13,000g at 4°C. For the amino acid leakage experiment 100µL of supernatant was dried using a Speed Vacuum system at 30°C for 2h, then were resuspended in 100µL of methanol. 2µL of the isotope labeled standard mix were added to 100µL methanol resuspended supernatant and to 100µL cell extract respectively. For all samples a total of 12.5µL of the filtrate were analyzed by LC-HRMS on a Thermo Ultimate 3000 UHPLC system coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The chromatographic separation of metabolites was performed on a Merck-Sequant ZIC-HILIC column (150 ×4.6mm, 5µm particle size) fitted with a Merck Sequant ZIC-HILIC guard column (20 × 2.1 mm) using a gradient elution of 0.1% formic acid in water (solvent A) and acetonitrile (solvent B). The gradient elution started at 20% of solvent A and increased up to 80% in 17 min. This percentage was maintained for 4min. The flow rate was set at 400µL min-1 and the column temperature and sample tray were held at 23°C and 4°C, respectively.
The Ultimate UHPLC system was coupled to a Q-Exactive instrument (Thermo Fisher Scientific, Bremen, Germany) equipped with a heated electrospray (H-ESI II) ionization source. Nitrogen (purity > 99.995%) was used as sheath gas and auxiliary gas at flow rates of 45 and 10a.u. (arbitrary units), respectively. The ion transfer tube was set at 320°C, the vaporizer temperature at 350°C and the electrospray voltage was set at 4kV in positive mode and −3.5kV in negative mode. A scanning rate of 3 spectrum s−1 with a mass range of m/z 75 – 800 with a mass resolving power of 70,000 Full Width Half Maximum (FWHM) (m/z 200) was used.
Full instrument calibration was performed using a MSCAL5 ProteoMassT LTQ/FT-Hybrid ESI Pos/Neg (Sigma-Aldrich). External mass axis calibration without the use of the specific lock masses was employed. The Xcalibur software version 2.2 (Thermo Fisher Scientific) was used to control the LC/MS system.
Metabolites were annotated by matching accurate mass (mass error < 5ppm) and retention time (+/− 30s) using a reference standard in-house database as previously described 12.
Data analysis
We targeted 85 metabolites within central metabolism that were detectable via analytical standards. Chromatographic peaks were analyzed and quality controlled manually in positive and negative ionization mode. Of these metabolites, 65 peaks with good quality in dish, pellet and mock sorted were selected for further analysis of unlabeled samples.
For clustering analysis and principal component analysis, peak areas were normalized to unit mean for each peak. Hierarchical clustering was done using Euclidean distance and average linkage.
For isotope-labeled samples, this procedure was repeated and a total of 60 peaks were selected. For each metabolite and sample, mass isotopomer (MI) fractions were calculated by dividing the peak area of each isotopomer with total peak areas of all isotopomers. Enrichment of 13C and 15N, respectively, was calculated as
where n is the total number of carbons (or nitrogens, respectively) in the metabolite, and MIx is the MI fraction of x. Enrichment data was clustered using Euclidean distance. All calculations were made with Mathematica v.10 (Wolfram Research).
Results and Discussion
Method development and validation strategy.
To obtain reliable metabolomics data from sorted cells, we strived to minimize the distortion that FACS might cause to cellular metabolism by minimizing the duration of the FACS procedure, keeping cells cold (4°C) during most of the procedure to reduce metabolic activity and using a glucose-containing solution (HBSS, 5.5mM glucose) to support cell viability. In initial experiments, we attempted to sort cells directly into cold extraction solution (methanol) to achieve as rapid metabolic quenching as possible. However, the FACS instrument deposits a significant amount of sheath fluid together with cells into the receiving tube, and this fluid contains high amounts of salt and other contaminants which caused substantial ion suppression during mass spectrometry analysis (data not shown). Therefore, this approach had to be abandoned. We instead turned to a procedure where cells are sorted into tubes containing HBSS, centrifuged, and then extracted (Figure 1b). In our hands, the complete procedure for adherent HeLa cells takes ~1 hour, including trypsinization and collection of a pellet of 500,000 cells. For non-adherent cells trypsinization is not required and the sorting procedure can also be performed faster. In our experiments on lymphocytes, the necessary antibody staining adds 25min, for a total time of 1 hour.
To investigate possible distortion caused by the sorting procedure itself, we first generated metabolite extracts from HeLa cells, passing them through the FACS instrument without selecting any subpopulations (Figure 1b, “mock sorted”). As a baseline for validating the resulting LC-HRMS data, we extracted metabolites from HeLa cells directly from the culture dish after removing medium and washing (Figure 1b, “dish”), which allows very fast (~1 min) quenching of metabolism. In order to investigate the effects of the cell detachment step, we also generated extracts from pellets of detached (trypsinized) and centrifuged cells (Figure 1b, “pellet”). All extracts were analyzed by LC-HRMS using full-scan acquisition mode in both positive and negative ion mode. Metabolites were identified and annotated by matching accurate mass and retention time against analytical standards (see Experimental section).
Peak areas are affected by cell sorting.
We performed a targeted analysis of 87 metabolites within central metabolism, of which 54 metabolites were detectable in cell extracts in positive and/or negative ionization modes, represented by 78 high quality peaks (Table S-1, see Experimental section for details). Of these, 73 peaks (94%) were high quality in the mock sorted extracts, and 65 in all three extract types (Figure 2a). Hence, almost all metabolites that were measureable in dish extracts could also be measured after FACS processing. However, peak areas differed markedly between mock sorted and dish extracts, often by a factor 10 or more, while peak areas from dish and pellet extracts were more similar (Figure 2b).
Figure 2.

Peak area data from the validation experiment. a) Venn diagram of good quality peaks detected in dish, pellet, and sorted extracts, b) Scatter plots of peak areas between dish, pellet and sorted extracts. All replicates are shown and connected by lines, c) Heat map of normalized peak areas, clustered by metabolites and samples. ncl: nucleotides, aa: amino acids. d) Principal component analysis (PCA) of normalized peak areas. PVE: percentage of variance explained. Red: sorted, blue: pellet, black: dish extracted cells. e) CV (Coefficient of Variation) of peak areas of 65 metabolites from plate, pellet and sorted extracts. Metabolites are plotted in increasing order of mean CV of the extracts.
Observed peak area differences may be a result of either matrix effects 13, or actual metabolic changes to the cells, such as loss of metabolites by leakage over the cell membrane 14, which in turn might alter cellular metabolism. We noted a cluster of metabolites, containing mostly amino acids, with highest peak areas in dish extracts, lower in pellet extracts, and lowest in mock sorted extracts (Figure 2c). Amino acids are known to be lost from cells placed in solution lacking amino acids 14, and this pattern is consistent with progressive loss with increasing time in buffer solution. On the other hand, we noted a cluster consisting mainly of nucleotides whose peak areas were highest in mock sorted extracts; this finding is difficult to explain other than by matrix effects. To further investigate possible leakage of amino acids during FACS conditions, we kept cells in HBSS at 4°C for 1h and then performed absolute quantification of amino acids in cells and supernatants against 13C standards. We found substantial amounts of amino acids in supernatants, indicating that about half of cellular amino acids are lost by leakage in these conditions (Table S-2). While substantial, this number is far from the >10 fold peak area differences between dish and mock-sorted extracts (Figure 2), suggesting that most of these differences are due to matrix effects.
We observed good reproducibility of peak areas, as indicated by hierarchical clustering (Figure 2c) and principal component analysis (Figure 2d) which grouped the replicates together. In addition, for dish extracts, 88% of peak areas had CV < 20%, compared to 84% for pellet extracts and 66% for mock sorted extracts (Figure 2e). This increased variability is expected given the additional experimental steps.
Mass isotopomer distributions (MIDs) are robust during cell sorting.
While total peak areas are expected to be altered due to experimental procedures, we investigated isotopomer ratios, which may be more resilient to artifact induced by FACS analysis. MIDs reflect the metabolic state before sorting as well as represent an internal ratio, which is not affected by changes in peak areas, matrix effect or experimental procedures (Figure 1a). To perform isotope tracing, we cultured cells with U-13C-glucose and U-13C,15N-glutamine for 48h, and quantified all combinations of carbon and nitrogen mass isotopomers in the labeled metabolites.
To obtain an overview of the isotopic labeling state of all metabolites, we calculated the 13C and 15N enrichment for each metabolite and visualized this as heat maps (Figures 3a,b) and scatter plots (Figures S-1 a,b). In contrast to peak areas (Figure 2c), we found that for most metabolites, both 15N and 13C enrichment in mock sorted extracts was highly similar to that of dish extracts. Enrichment for 15N appeared particularly robust, likely reflecting slower turnover of amine groups. For example, the MID of glutamate was maintained during cell sorting (Figure 3c), and was consistent with synthesis from U-13C, 15N-glutamine. There were exceptions however: for example, lactate was less labeled in mock sorted extracts than in dish or pellet extracts (Figure 3d). Since glycolysis is a very rapid process, with turnover in minutes 15, the ~1 hour cell sorting procedure might affect the MIDs of glycolytic intermediates. The possible sources of lactate, glucose and glutamine might not lead to less labeled lactate because glucose is labeled and glutamine is not present in HBSS. MIDs of glycolytic intermediates should therefore be interpreted with caution. Since MIDs are calculated as fractions, standard deviation was used to evaluate the reproducibility of MIDs (Figure 3e). In mock sorted extracts, 97% of MIDs exhibited standard deviation below 1%, and 94% below 0.5%, which is considered reliable 16. Taken together, these results indicate that MIDs measured in FACS-sorted cells are highly reproducible, and for the most part reflect the metabolic state of cells prior to sorting.
Figure 3.
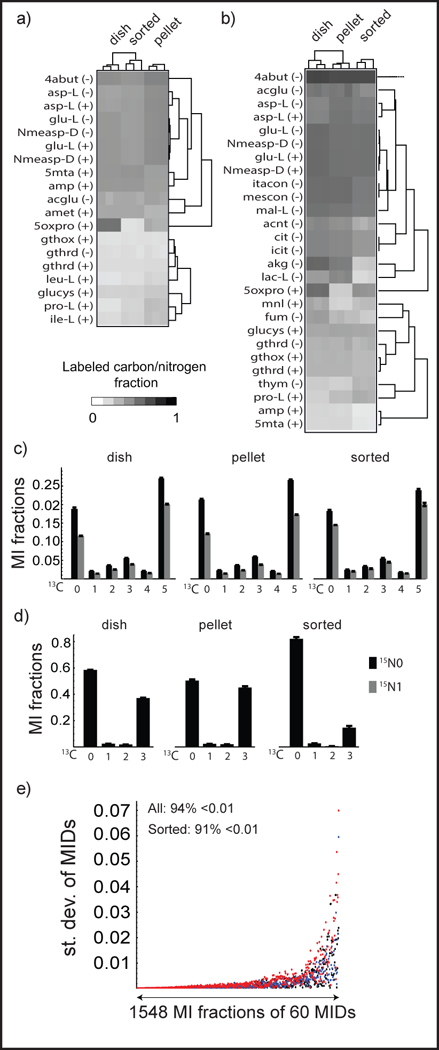
MID data from the validation experiment. (a and b) Heat map of 15N (a) 13C (b) enrichment clustered by metabolites and samples. Unlabeled metabolites, mainly essential nutrients are not shown. c) 13C-15N MIDs of Glutamate. (d) 13C MIDs of Lactate, The error bars are standard deviations of triplicate measurements in (a) and (b). (e) Standard deviation (st.dev) of MI fractions of 60 MIDs. (−) stands for negative mode, (+) stands for positive mode. Descriptions of metabolites can be found in Table S-1.
Detecting metabolic specialization in primary CD8+ and CD4+ T cells.
We next applied our isotope tracing method to investigate differences between T cell subpopulations. We activated T cells in vitro by stimulating the T cell receptor (see methods) and cultured the activated cells for 72 hours in U-13C-glucose and U-13C,15N-glutamine. Cells were then sorted into CD4+ (helper) and CD8+ (cytotoxic) T cell subpopulations by FACS, and LC-HRMS analysis was performed as before. Several metabolites differed in labeling pattern between CD4+ and CD8+ cells. For example, adenosine was more enriched for 13C and 15N in CD8+ cells (Figure 4). The isotope pattern of adenosine in CD8+ cells (Figure 4a) was consistent with de novo purine synthesis: the 13C5 mass isotopomer is likely due to incorporation of 13C5 ribose (derived from glucose), while the 15N isotopomers likely originate from glutamine and glutamine-derived aspartate, which contribute labeled nitrogens to the purine ring. On the other hand, in CD4+ cells adenosine contains mainly a 15N1 isotopomer, which might reflect salvage of (unlabeled) hypoxanthine, which would add one 15N atom from aspartate. These results suggest that in vitro activated CD8+ T cells engage in de novo purine synthesis more than activated CD4+ cells, which appears consistent with generally higher proliferation rates of CD8+ T cells in these cultures (data not shown).
Figure 4.
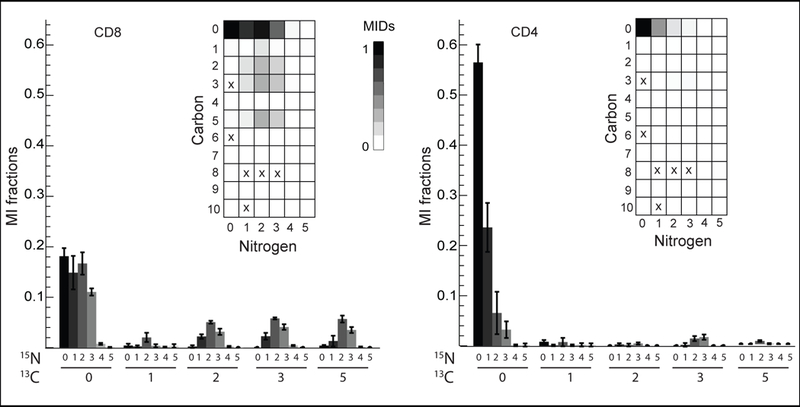
Adenosine is differentially labelled in CD8+ and CD4+ cells. Error bars in bar charts represent standard deviations. Array plots (inset) show MI fractions. ‘x’ stands for missing values, which is noise manually corrected to ‘0’.
Cytidine is differentially labelled in different cell cycle phases.
We also tested our approach to separate HeLa cells by their cell cycle phase, by sorting cells based on a fluorescent protein that is specifically expressed in the S-G2-M phases of the cycle (see methods). Because cell cycle phases last only a few hours, here we “pulse labeled” cells for 2 hours in medium containing U-13C-glucose and U-13C, 15N-glutamine, and separated cells into subpopulations representing the G1-G0 and S-G2-M cell cycle phases, respectively. We anticipated that differences would be more difficult to detect in this case than in the CD4+/CD8+ comparisons, as these subpopulations are likely more similar to each other, and the shorter duration of labeling yields weaker isotopes. Still, a few metabolites showed interesting patterns: for example, cytidine was about 2-fold more enriched for 13C in the S-G2-M subpopulation (Figure 5a), while its labeling pattern was similar in the two subpopulations. As with adenosine, the 13C5 mass isotopomer in cytidine is likely due to ribose. The 15N1 and 15N2 isotopomers are consistent with the known de novo pyrimidine synthesis pathway, where 2 nitrogens are donated by glutamine, while the 13C3 mass isotopomer is consistent with aspartate donating 3 carbons. This data suggests that pyrimidine synthesis is more active in the S-G2-M phase, where DNA synthesis occurs.
Figure 5.
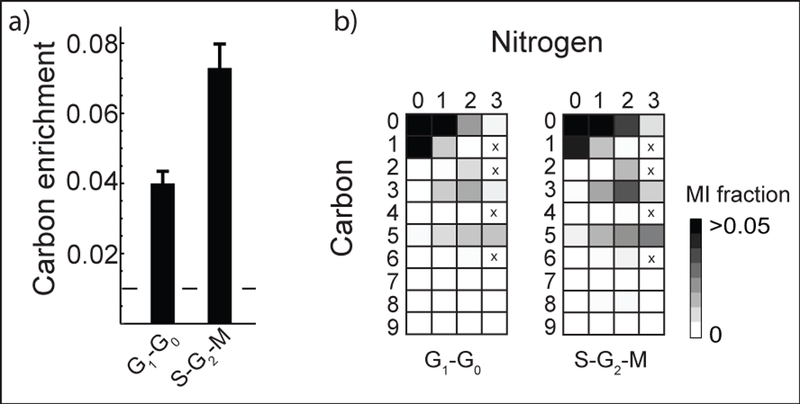
Cytidine is labeled differently in G1–G0 and S-G2-M cells. a) Cytidine 13C enrichment in G1–G0 and S-G2-M phases of the cell cycle. Dashed line stands for carbon enrichment from natural isotope. b) Cytidine MIDs shown as array plots in G1–G0 and S-G2-M phases.
Conclusions
We find that peak areas differ markedly between extracts of FACS-sorted cells and direct extractions from cell cultures, although they are generally reproducible between independent samples. Therefore, peak areas from sorted cells should be interpreted with caution. In contrast, MIDs of metabolites generated in stable isotope labeling experiments are generally robust during cell sorting and show excellent reproducibility. Lactate was a notable exception to this rule, suggesting that MIDs of glycolytic intermediates and other metabolites with rapid turnover may be affected by cell sorting, and should be viewed with caution. Our proof-of-principle experiments show that with isotope tracing, it is possible to detect metabolic differences between subpopulations of both adherent and suspension cells, such as lymphocyte subtypes and even between cell cycle phases. We therefore anticipate that this method is broadly applicable to study metabolic phenotypes of cell subpopulations in biology and biomedicine.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Anas Kamleh for valuable help with optimizing mass spectrometry methods, and Annika von Vollenhoven for assistance with cell sorting. This research was supported by the Strategic Programme in Cancer Research (IR, RN); the Swedish Heart-Lung Foundation (CEW, HG); A Marie Curie Intra European Fellowship within the 7th European Community Framework Programme, the Dr. Åke Olsson Foundation and KI research foundations (AS); and Mary Kay Foundation (JW, MJ).
Footnotes
Supporting Information
Table S-1, list of metabolite ID, description, MW, retention times and presence of peaks in each extraction method in positive and negative ionization mode. Table S-2, Percentage of amino acid (AA) leakage in the supernatant. Figure S-1, scatter plots of 13C and 15N enrichment in dish, pellet and mock sorted samples. Figure S-2, CV of MI fractions with values smaller than 0.01 in plate, pellet and sorted extracts. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Gregersen PK Nat. Genet 2012, 44 (5), 478–480. [DOI] [PubMed] [Google Scholar]
- (2).Heppner GH Cancer Res 1984, 44 (6), 2259–2265. [PubMed] [Google Scholar]
- (3).Prat A; Karginova O; Parker JS; Fan C; He X; Bixby L; Harrell JC; Roman E; Adamo B; Troester M; Perou CM Breast Cancer Res. Treat 2013, 142 (2), 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Shapiro HM Pract. Flow Cytom 2003, 257–271. [Google Scholar]
- (5).Hollenbaugh JA; Munger J; Kim B Virology 2011, 415 (2), 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moussaieff A; Rogachev I; Brodsky L; Malitsky S; Toal TW; Belcher H; Yativ M; Brady SM; Benfey PN; Aharoni A Proc. Natl. Acad. Sci. U. S. A 2013, 110 (13), E1232–E1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen K; Liu X; Bu P; Lin C; Rakhilin N; Locasale JW; Data AM; Geo F In 36th Annual International Conference of the IEEE Engineering in Medicince and Biology Society; 2014; pp 4759–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Richardson GM; Lannigan J; Macara IG Cytom. Part A 2015, 87 (2), 166–175. [DOI] [PubMed] [Google Scholar]
- (9).Sauer U Mol. Syst. Biol 2006, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sakaue-Sawano A; Kurokawa H; Morimura T; Hanyu A; Hama H; Osawa H; Kashiwagi S; Fukami K; Miyata T; Miyoshi H; Imamura T; Ogawa M; Masai H; Miyawaki A Cell 2008, 132 (3), 487–498. [DOI] [PubMed] [Google Scholar]
- (11).Gustafsson Sheppard N; Jarl L; Mahadessian D; Strittmatter L; Schmidt A; Madhusudan N; Tegnér J; Lundberg EK; Asplund A; Jain M; Nilsson R Sci. Rep 2015, 5, 15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kamleh MA; Snowden SG; Grapov D; Blackburn GJ; Watson DG; Xu N; Ståhle M; Wheelock CE J. Proteome Res 2015, 14 (1), 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Noack S; Wiechert W Trends Biotechnol 2014, 32 (5), 238–244. [DOI] [PubMed] [Google Scholar]
- (14).Eagle H Science (80-. ) 1959, 168 (3934), 939–949. [Google Scholar]
- (15).Munger J; Bennett BD; Parikh A; Feng X-J; McArdle J; Rabitz H. a; Shenk T; Rabinowitz JD Nat. Biotechnol 2008, 26 (10), 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Antoniewicz MR; Kelleher JK; Stephanopoulos G Anal. Chem 2007, 79 (19), 7554–7559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.