

Abstract
Cyclooxgenases are key enzymes of lipid signaling. They carry out the first step in the production of prostaglandins, important mediators of inflammation, pain, cardiovascular disease, and cancer, and they are the molecular targets for nonsteroidal anti-inflammatory drugs, which are among the oldest and most chemically diverse set of drugs known. Homodimeric proteins that behave as allosterically modulated, functional heterodimers, the cyclooxygenases exhibit complex kinetic behavior, requiring peroxide-dependent activation and undergoing suicide inactivation. Due to their important physiological and pathophysiological roles and keen interest on the part of the pharmaceutical industry, the cyclooxygenases have been the focus of a vast array of structural studies, leading to the publication of over 80 crystal structures of the enzymes in complex with substrates or inhibitors supported by a wealth of functional data generated by site-directed mutation experiments. In this review, we explore the chemical biology of the cyclooxygenases through the lens of this wealth of structural and functional information. We identify key structural features of the cyclooxygenases, break down their active site into regional binding pockets to facilitate comparisons between structures, and explore similarities and differences in the binding modes of the wide variety of ligands (both substrates and inhibitors) that have been characterized in complex with the enzymes. Throughout, we correlate structure with function whenever possible. Finally, we summarize what can and cannot be learned from the currently available structural data and discuss the critical intriguing questions that remain despite the wealth of information that has been amassed in this field.
1. Introduction
Prostaglandins (PGs) and thromoboxane A2 (TXA2) comprise a class of lipid signaling molecules derived from the 20-carbon, tetra-unsaturated fatty acid, arachidonic acid (AA). Biosynthesis of PGs and TXA2 is initiated by the bifunctional cyclooxygenases (COXs). These enzymes catalyze the bis-dioxygenation of AA to form the hydroperoxy-endoperoxide PGG2 at the cyclooxygenase active site followed by the reduction of the hydroperoxide of PGG2 to an alcohol at the peroxidase active site. The final product, PGH2, then serves as a substrate for additional synthases that transform it into an array of lipid mediators, each of which signals through interaction with one or more specific G protein-coupled receptors (Figure 1).1−5
Figure 1.

Prostaglandin (PG) biosynthetic pathway. Bis-dioxygenation and cyclization of arachidonic acid at the cyclooxygenase active site of COX-1 or COX-2 yields PGG2. Reduction of the 15-hydroperoxyl group of PGG2 at the peroxidase active site of COX-1 or COX-2 yields PGH2. PGH2 serves as a substrate for five different synthases, producing four signaling PG products (PGE2, PGI2, PGF2α, and PGD2) or thromboxane A2 (TXA2). PGH2 is chemically unstable under physiological conditions, and in the absence of the synthase enzymes, it is hydrolyzed to a mixture of PGE2 and PGD2.
Polyunsaturated fatty acids such as AA are highly susceptible to nonenzymatic free radical oxygenation due to their readily abstracted hydrogen atoms, which are attached to the allylic carbons located between two double bonds. Thus, nonenzymatic oxygenation of AA can be initiated by abstraction of a hydrogen atom at carbons-7, 10, or 13 followed by mono-, di-, or even trioxygenation to yield a wide range of products.6 The COX enzymes essentially carry out this reaction with regio and stereospecificity, so that one primary product, PGG2, results. The proposed cyclooxygenase reaction mechanism is shown in Figure 2.2 Notable features of this mechanism are its stereochemical complexity and the geometric constraints that must be applied to the substrate to ensure formation of the bicyclic endoperoxide nucleus of PGG2. Although the COX enzymes synthesize PGG2 with a high degree of fidelity, secondary products are also formed in small quantities. These include 11(R)-hydroxy-(5Z, 8Z, 12E, 14Z)-eicosatetraenoic acid (11(R)-HETE), 15(S)-hydroxy-(5Z, 8Z, 11Z, 13E)-eicosatetraenoic acid (15(S)-HETE), and 15(R)-hydroxy-(5Z, 8Z, 11Z, 13E)-eicosatetraenoic acid (15(R)-HETE).4 The occurrence of these minor products supports the mechanism outlined in Figure 2, as they result from the reaction of oxygen with intermediates predicted by the mechanism. Although not routinely formed under physiological conditions, the demonstration that the reaction can also generate an analog of PGG2 bearing its hydroxyl group at carbon-13 rather than carbon-15 further supports the mechanism (Figure 2).7 Notably, the stereochemistry at the 15 position of PGG2 produced by the wild-type COX enzymes is (S). As discussed below, some structural alterations of the enzymes lead to a reversal of configuration at this site or changes in the quantities of minor products formed. It should also be noted that the COX enzymes can oxygenate other polyunsaturated fatty acids and their derivatives with varying degrees of efficiency, as will be discussed in greater detail in the following sections.
Figure 2.
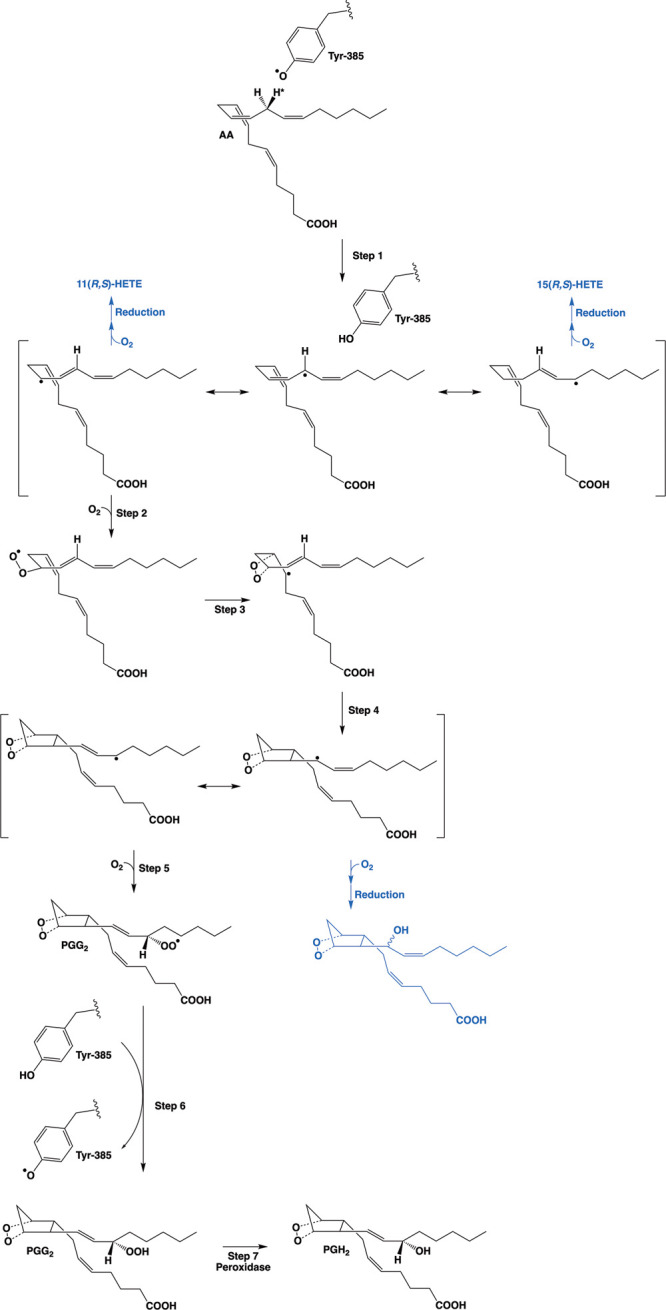
Proposed mechanism of the cyclooxygenase reaction. Activation of the COX enzymes occurs through oxidation of the heme prosthetic group during reduction of a peroxide substrate at the peroxidase active site. Transfer of an electron from Tyr-385 in the cyclooxygenase active site to the heme then generates the catalytic tyrosyl radical. Step 1: the tyrosyl radical abstracts the 13-(pro)-S-hydrogen atom (indicated by an asterisk) from AA. Resonance places the unpaired electron at carbon-13, carbon-11, or carbon-15. Addition of oxygen at the carbon-11 or carbon-15 radical, followed by enzymatic or nonenzymatic reduction, results in the 11(R,S)-HETE or 15(R,S)-HETE minor products (indicated in blue), respectively. Step 2: antarafacial oxygen addition occurs at carbon-11. Step 3: the peroxyl group then attacks carbon-9, forming the endoperoxide ring and placing the unpaired electron at carbon-8. Step 4: bond formation between carbon-8 and carbon-12 generates the five-membered ring of PGG2 and places the unpaired electron on carbon-13. Resonance enables migration of the unpaired electron to carbon-15. Step 5: attack of oxygen at carbon-15 follows, generating a peroxyl radical at that position. Step 6: transfer of a hydrogen atom from Tyr-385 reduces the peroxyl radical to a hydroperoxide, yielding PGG2, and regenerates the tyrosyl radical for a new round of catalysis. Step 7: reduction of PGG2 at the peroxidase active site yields PGH2. Alternatively, attack of oxygen at carbon-13 rather than carbon-15 in step 5 followed by enzymatic or nonenzymatic reduction leads to a PGH2 analog with the hydroxyl group at carbon-13 (shown in blue). Note that the overall mechanism requires a peroxidase turnover to produce the catalytic tyrosyl radical, but once this has occurred, the cyclooxygenase reaction is self-perpetuating.
Across many vertebrate species, there are two COX isoforms, COX-1 and COX-2, which are encoded by the genes ptgs1 and ptgs2, respectively. The two proteins share approximately 60% sequence identity and a nearly superimposable three-dimensional structure. They oxygenate AA with very similar kinetics in vitro, leading to the hypothesis that the major functional difference between them derives from the distinct mechanisms by which expression of their genes is regulated. Specifically, ptgs1 is expressed as a housekeeping gene in most cell types, whereas expression of ptgs2 is inducible by a range of stimulants including inflammatory agents, growth factors, and tumor promoters. The seemingly obvious conclusion from these patterns of expression is that COX-1 provides PGs required for normal cellular homeostasis, whereas COX-2 enables signaling that regulates growth and differentiation or modulates the inflammatory response to infection or injury. These conclusions have generally been shown to be correct although they oversimplify the roles of the two isoforms. Nevertheless, the fact that the COX enzymes are the primary sites of action of the widely used isoform nonselective or COX-2-selective nonsteroidal anti-inflammatory drugs (NSAIDs) suggests an important role (particularly for COX-2) in inflammation.1−5,8−10
Although regulation of gene expression is clearly an important factor that distinguishes the functions of the two COX enzymes, subtle differences in protein structure also lead to distinct patterns of enzyme activity. Specifically, the active site of COX-2 is approximately 25% larger than that of COX-1, and this enables COX-2 to efficiently oxygenate substrates that are oxygenated by COX-1 poorly or not at all. These include bulky arachidonic acid derivatives, such as N-arachidonoyl glycine, N-arachidonoyl alanine, N-arachidonoyl-γ-aminobutyric acid, 2-arachidonyl glyceryl ether (noladin ether), 2-arachidonoyl-lysophosphatidylethanolamine, and 2-arachidonoyl-lysophosphatidylcholine.11−14 However, of the COX-2-selective substrates identified to date, the most thoroughly studied are 2-arachidonoylglycerol (2-AG) and arachidonoyl ethanolamide (AEA), which serve as endogenous ligands for the cannabinoid receptors, CB1 and CB2.15,16 The COX isoforms also differ in their kinetic behavior in ways that are not readily apparent during routine in vitro assays but may be pertinent physiologically. For example, both enzymes initially require activation through reaction with a hydroperoxide (which can be PGG2 or other intracellular hydroperoxides), and both undergo suicide inactivation during the course of catalysis. Notably, COX-2 is more readily activated than COX-1,17,18 providing a mechanism for differential isoform regulation based on intracellular peroxide tone.19 The mechanistic basis for this difference between the two enzymes is not well understood.
The COX isoforms are homodimeric proteins; however, growing evidence indicates that they act as functional heterodimers. Each enzyme requires a heme (Fe3+-protoporphyrin IX) cofactor that rests between the cyclooxygenase and peroxidase active sites and is necessary for both activities. Binding studies indicate that heme binds with high affinity to only one monomer and that full activity is achieved upon incorporation of only one heme molecule per dimer. This suggests the possibility that the heme-bearing monomer acts as the catalytic subunit whereas the second monomer serves a regulatory role. Indeed, evidence for positive and negative allosteric modulation of COX enzymes by nonsubstrate fatty acids, substrates, and NSAIDs is growing. Data also support the hypothesis that some NSAIDs block COX activity competitively by binding in the catalytic subunit, whereas others act noncompetitively by interacting at the allosteric subunit. Furthermore, studies of heterodimers comprising a site-directed mutant subunit paired with a wild-type subunit or two functionally distinct site-directed mutants have confirmed that full catalytic functionality can be achieved with only one active subunit per heterodimer.20−28
Due to the long-standing interest in the multiple physiological and pathophysiological functions of the COX enzymes and their roles as NSAID targets, they are among some of the most thoroughly investigated proteins from a structural, functional, and pharmacological point of view. In this review, we explore the wealth of information that can be gleaned from the numerous publicly available COX crystal structures. We focus primarily on enzyme-ligand interactions at the cyclooxygenase active site with emphasis on structural correlates to enzyme function and questions that remain unanswered. A summary of the crystal structures covered in the review is provided in Table S1, which also provides the structure of the ligand in the cyclooxygenase active site in each case. Information derived from the crystal structures has provided insights leading to hypotheses that are testable by site-directed mutagenesis and functional assays. Similarly, mutagenesis experiments have enabled observations that have been supported or explained by structural data. To facilitate the correlation between structural and functional studies, a summary of COX-1 and COX-2 mutant proteins that have been created and the functional impacts of the mutations is provided in Table S2, although this table is not necessarily fully comprehensive. All figures depicting crystal structure data were created using USCF Chimera software. Stereoscopic figures are supplied in wall-eyed format for print. For those who prefer it, figures in cross-eyed format are provided in the Supporting Information. Supplemental figures are provided in both formats.
2. Overview of COX Crystallization and Structure Elucidation
The 3.1 Å resolution crystal structure of a complex of ovine COX-1 and the NSAID (S)-flurbiprofen was published in 1994 (PDB 1CQE).29 COX-1 is a monotopic membrane protein requiring detergent for solubilization. This publication marks one of the first successful attempts to crystallize and diffract a mammalian membrane protein exhibiting these properties. In the initial attempts at COX structure elucidation, resolution was limited by crystal fragility, sensitivity to experimental conditions, and high solvent content. Structure determination was accomplished through the use of heavy atom derivatives obtained through exposure of the protein to ethylmercurithiosalicylate, K2PtCl4, or Au(CN)2. Confidence in the resulting model was enhanced by the identification of a peroxidase active site bearing similarity to the structures of related heme-containing peroxidases and a cyclooxygenase active site marked by the presence of the flurbiprofen inhibitor surrounded by residues known to be important in cyclooxygenase catalysis.29 In the ensuing two years, attempts to improve resolution focused on the use of inhibitors containing heavy atoms (bromine and iodine), but these approaches yielded limited success.30,31
The first published COX structure to achieve a resolution below 3 Å was of murine COX-2, also complexed with (S)-flurbiprofen (PDB 3PGH).32 Additional COX-2-inhibitor structures were soon reported, driven by an intense interest in the discovery of isoform-selective inhibitors, a primary goal of the pharmaceutical industry at the time. Attempts were also made, however, to obtain structures of complexes containing COX-1 or COX-2 and AA or related fatty acids. These were initially thwarted by enzyme-mediated oxygenation of the substrate, a problem not adequately addressed through the use of an inactivating site-directed mutation.33 Ultimately, substrate-enzyme complex structures were obtained through the replacement of heme in the enzyme with Co3+-protoporphyrin IX, a substitution that had no significant effect on enzyme structure but completely eliminated catalytic activity.34
There are now over 80 structures of COX-1 or COX-2 with or without associated ligands deposited in the Protein Data Bank (Table S1). Clearly, many of the technical issues associated with obtaining high quality crystals have been resolved. In most cases, the enzyme was crystallized in the presence of n-octyl-β-d-glucopyranoside with or without polyethylene glycol or poly(acrylic acid). Frequently, resolution between 1.8 and 2.2 Å has been achieved. The data demonstrate a striking consistency with regard to the three-dimensional structure of the proteins regardless of the absence or presence of bound ligand or the nature of the ligand. The data also provide extraordinary insight into the structural basis for the cyclooxygenase reaction mechanism as well as for the determinants of ligand binding. However, the crystal structure data leave a number of questions unanswered. In nearly all cases, heme, substrates, and/or inhibitors are present in both subunits, and these ligands usually (though not always) exhibit the same or similar binding poses in both subunits. Thus, a structural foundation for half-of-sites activity is incomplete. Furthermore, the apparent lack of a major impact of ligand binding on protein structure revealed in these accumulated data leaves the observer with the impression of a rigid, unyielding protein. Such a view would seem to be inconsistent with the flexibility required to enable formation of the endoperoxide ring system of PGG2 and with the dynamics of subunit-to-subunit communication that likely underlie allosteric modulation. However, as outlined below, the crystal structure data reveal subtle, ligand binding-dependent changes in amino acid side chain position that may have important functional consequences, particularly for inhibitor binding and allostery. Furthermore, the combination of crystal structure data with results from site-directed mutagenesis studies demonstrates how key regions of the active site guide each step of the cyclooxygenase reaction in order to ensure formation of PGG2 with the correct stereochemical outcome.
3. Major Structural Features of the COX Enzymes
The genes for COX-1 and COX-2 encode protein chains of 599 to 604 amino acids, depending on the species of origin and isoform. Removal of the signal sequence renders proteins of 576 and 587 amino acids for COX-1 and COX-2, respectively. As noted above, the sequences of the two isoforms from the same species are 60% identical. However, COX-2 lacks 14 amino acids near its N-terminus that are present in COX-1, and it contains an 18 amino acid insertion near the C-terminus that is missing in COX-1. As a result of the N-terminal deletion, the numbers of the vast majority of amino acids in COX-2 are lower by 14 than those of the corresponding residues in COX-1. For example, the critical catalytic tyrosine residue is number 385 in COX-1 and 371 in COX-2. However, to facilitate comparison between the isoforms, numbering for COX-1 is usually applied to both proteins. We will use that convention here. Both isoforms are uniformly glycosylated at Asn-68, Asn-144, and Asn-410. COX-2 is also variably glycosylated at a fourth site, Asn-594. The monomers exhibit an apparent molecular mass of 68–72 kDa by SDS gel electrophoresis, with greater heterogeneity in the case of COX-2 due to the variable glycosylation.2,4,35,36
The homodimeric COX proteins exhibit a C2 axis of symmetry on X-ray crystallography. Each monomer comprises three primary domains, an epidermal growth factor domain (residues 34–72), a membrane-binding domain (residues 73–116), and the catalytic domain (residues 117–586) (Figure 3). The purpose of the epidermal growth factor domain is uncertain, though it may play a role in subunit–subunit interactions or help to stabilize the conformation of the membrane-binding domain. The small amount of β-sheet in the protein is mostly found there. The membrane-binding domain, comprising four approximately orthogonal α-helices designated A through D, is the major site of sequence variation between the two isoforms. The helices form a surface of hydrophobic residues that embeds into one leaflet of the membrane bilayer. The opposite side of each of the helices is composed of hydrophilic residues. Helix D projects upward from the membrane, extending into the catalytic domain, which contains the heme that separates distinct peroxidase and cyclooxygenase active sites.2,4,29,35,36
Figure 3.

Domain structure of the cyclooxygenases. The top view shows the dimeric protein (COX-1) as observed from the side (i.e., parallel to the plane of the membrane). Each of the bottom views follows rotation of the top view by 90°. To the left, the protein is viewed looking down on the enzyme from above. To the right, the protein is viewed looking up from below. In all cases, the epidermal growth factor domain is gold, the membrane-binding domain is green, the catalytic domain is cyan, heme is sienna, and the flurbiprofen ligand is red. The membrane-binding domain inserts into the top leaflet of the underlying membrane bilayer. From PDB 1CQE.
The catalytic domain is notable for the presence of a seven-helix bundle homologous to those seen in other heme peroxidases (Figure 4). This bundle defines the heme binding site and the peroxidase active site, which sits at the bottom of a shallow cleft, exposing the heme to solvent. The large surface area of the peroxidase active site is consistent with the broad substrate specificity of the COX peroxidase and the ability of the enzyme to accept large substrates such as PGG2. As the primary focus of this review is the structure of the COX enzymes with regard to substrate and inhibitor interactions at the cyclooxygenase active site, no further consideration of the peroxidase site will be given here. Other reviews may be consulted for further detail.2,4,35,36
Figure 4.

Structure of the cyclooxygenase active site. Two wall-eyed stereo views of the COX-1 monomer (A and B) and a close-up view of the cyclooxygenase active site (C) are shown as observed from the side (i.e., parallel to the plane of the membrane). In all cases, Co3+-protoporphyrin IX (an inactive heme analog) is dark brown, and AA is red mesh. The side chains of the constriction residues (Arg-120, Tyr-355, and Glu-524) are displayed and labeled, as are the catalytic residue (Tyr-385) and the target of aspirin-mediated inactivation (Ser-530), which are located at the bend of the L-shaped channel. In A and B, His-388, the proximal heme ligand, and His-207, which serves as the distal heme ligand through a coordinating water molecule, are visible. Helices 2 (residues 195–207, light blue), 8 (residues 378–385, medium blue), 6 (residues 324–354, light green), and 17 (residues 519–535, dark green), which surround the active site, along with helices 5 (residues 295–320, dark purple), 11/12 (residues 444–459, medium purple), and 16 (residues 503–510, light purple) form a bundle that is conserved among a number of peroxidases, with helices 2, 5, 6, 8, and 11/12 involved in binding the heme prosthetic group. From PDB 1DIY.
The cyclooxygenase active site comprises a predominantly hydrophobic, inverted L-shaped channel that penetrates deeply into the catalytic domain above the membrane-binding domain (Figure 4). The four helices of the membrane-binding domain surround a spacious alcove that has been designated the lobby. The top of the lobby is defined by Arg-120, Tyr-355, and Glu-524, which form a constriction that must open to provide access into the active site. It is notable that Arg-120 and Glu-524 are the only charged residues in the cyclooxygenase active site and that Arg-120 and Tyr-355 frequently interact with carboxylic acid or other polar functional groups of substrates or inhibitors. Also of interest is the finding that Arg-120 is located in a portion of helix D (residues 118–122) in the membrane-binding domain. This region is relatively disordered in the absence of ligand (as indicated by poorly defined electron density maps and high thermal parameters) and assumes its helical conformation upon AA binding.32 Molecular dynamics simulations suggest a role for helix D in opening the constriction to allow oxygen to gain access to the active site, particularly in the COX-1 isoform.37 Simulations also suggest that the main active site channel is the most likely route by which oxygen can reach the carbon-11 radical of AA in the correct stereochemistry for addition (Figure 2).38 Above the constriction, the channel proceeds into the center of the protein until it reaches Tyr-385, the critical catalytic residue that initiates the cyclooxygenase reaction (Figure 2). Across from Tyr-385 is Ser-530, the target of acetylation by aspirin, a modification that blocks PGG2 formation. These residues mark the bend in the L-shaped channel that leads to a hydrophobic recess. From this recess, a narrow tunnel exits into the region between the two monomers. The tunnel provides an escape route for water that must be displaced upon ligand binding, but it is too small to allow egress of inhibitors or reaction products. The cyclooxygenase channel is constructed of residues contributed by helices 6 (residues 324–354) and 17 (residues 519–535) near the constriction and helices 2 (residues 195–207) and 8 (residues 378–385) at the hydrophobic recess (Figure 4). Most fatty acids bind in the active site with their carboxyl group interacting with Arg-120 and/or Tyr-355 at the constriction and the remainder of the molecule extended upward, looping around Ser-530 and filling the hydrophobic recess. All interactions between fatty acids and the active site are hydrophobic in nature with the exception of those involving the carboxylate moiety.2,4,29,35,36
The cyclooxygenase active sites of the two isoforms are very similar. A notable exception is the mutation of Ile-523 in COX-1 to valine in COX-2. The presence of the smaller valine residue opens up access to a side pocket region in COX-2 that is primarily responsible for the larger size of its active site channel mentioned above. In addition to a broader substrate specificity, COX-2’s larger active site enables it to retain limited activity in the face of aspirin acetylation and diverse mutations that totally inactivate COX-1. Furthermore, access to the side pocket has been exploited in the development of some classes of COX-2-selective inhibitors.2,4,35,39,40 Details concerning the interactions of substrates and inhibitors in the cyclooxygenase active site as revealed by X-ray crystallography will be the focus of the remainder of the review.
4. Interactions of COX Proteins with Fatty Acids
From an examination of the structures of the reactant (AA) and product (PGG2) of the cyclooxygenase reaction, we can surmise what must be achieved through the interaction of enzyme and substrate at the active site. The substrate must be constrained in a conformation that enables dioxygenation, endoperoxide ring formation, and cyclization of the cyclopentane ring all with the correct stereochemistry. As will be discussed below, crystal structure data provide deep insight into how these goals are achieved.
The L-shaped active site channel can be viewed as comprising three pockets: a proximal binding pocket, a central binding pocket, and a distal binding pocket, that interact with the carboxy-terminus, the central portion, and the omega-terminus of AA, respectively. The primary function of the proximal and distal pockets appears to be stabilization of AA conformation, as most mutations of key amino acids in these regions lead to either loss of catalytic efficiency or a shift to minor monohydroxylated products with or without altered stereochemistry. Crystal structures of some of these mutant enzymes in complex with substrate demonstrate how the alterations lead to suboptimal placement of the fatty acid in the active site. The central binding pocket, which contains the catalytic tyrosine residue, is the site where the chemistry occurs. Key features of this pocket are precise positioning of Tyr-385 for hydrogen atom abstraction, adequate space to allow the formation of the endoperoxide ring structure of PGG2, and strategically placed subpockets that provide access to oxygen molecules for attack at the correct carbon atoms with the appropriate stereochemistry. As noted above, the cyclooxygenase active site accommodates a range of alternative fatty acid substrates of various chain lengths and degrees of saturation. Numerous crystal structures demonstrate how these molecules bind with remarkable retention of the overall structure of the site.
Comparisons of the crystal structures of COX-1 and COX-2 bound to various substrates reveal fascinating differences. As noted above, the larger COX-2 active site allows greater flexibility of substrate binding and accommodation of a wider range of bulkier substrates and mutations. For COX-1, formation of a salt bridge between the fatty acid carboxylate and Arg-120 at the constriction appears to be important for catalysis, whereas COX-2 will tolerate formation of a range of polar interactions between the substrate and constriction residues. Perhaps more intriguing, however, are the findings of different conformations of a number of substrates, including AA, in the two active sites of the COX-2, but not the COX-1 dimer. In most cases, one of the conformations is “inverted” with the carboxylic acid group forming polar interactions with Tyr-385 and Ser-530 in the central binding pocket and the omega-terminus projecting down toward the constriction. Both adoption of alternative substrate conformations and accommodation of bulky substrates by COX-2 are nearly always accompanied by a rotation of Leu-531 away from the constriction residues to provide additional room. This movement has not been observed in the structures of COX-1 with fatty acids. In the discussion that follows, we explore in detail the information provided by the vast structural data available on the interaction of the COX enzymes with substrate and nonsubstrate fatty acids and their derivatives.
4.1. Binding Interactions of AA in the Cyclooxygenase Active Site
The earliest crystal structures of COX enzyme-ligand complexes, which appeared in the mid 1990s, contained an NSAID as the binding partner. It was not until 2000 that the first structure of a COX-2-AA complex was published.33 To prevent oxygenation of AA, the apoenzyme lacking the required heme cofactor was used. In addition, an H207A site-directed mutant of COX-2 was employed for this study. The extremely low peroxidase activity of this enzyme41 was expected to prevent activation of the cyclooxygenase reaction and AA metabolism (Figure 2), should traces of heme be present in the preparation. Although this goal was realized, the binding pose of AA observed in the 2.4 Å resolution structure (PDB 1CVU) is strikingly different from one that would be expected to support the cyclooxygenase reaction. Specifically, the carboxylate of AA is hydrogen-bonded to Tyr-385 and Ser-530 at the bend in the active site channel, while the hydrocarbon chain extends back toward the constriction so that the omega end of the fatty acid interacts with Arg-120. This orientation fails to bring AA’s 13-pro-(S)-hydrogen into proximity with Tyr-385, as is required for initiation of the cyclooxygenase reaction by abstraction of that atom (Figure 2). Despite AA’s unexpected orientation in the active site, the overall structure of the protein is in close agreement with those observed in prior COX-2-NSAID complex crystal structures. The only key differences are a reduced volume of the membrane channel and the rotation of selected amino acid side chains to accommodate the omega end of AA. In particular, Leu-531 adopts a conformation not observed in prior COX structures, and shifts in the orientation of adjacent residues (115–125) facilitate this move. Although these results did not help to explain how COX catalyzes AA oxygenation, they did presage later findings that are relevant to allosteric modulation and inhibitor interactions with the enzyme as will be discussed below.33
Not long after the appearance of the COX-2-AA complex, a structure of COX-1 complexed with AA was published (PDB 1DIY) (Figure 4).34 For this study, an inactive enzyme was constructed by reconstituting apo-COX-1 with Co3+-protoporphyrin IX in place of heme, a procedure that had been used to obtain crystals of enzyme-AA complexes from which low resolution diffraction data had previously been acquired.42 The reported 3.0 Å resolution COX-1-AA complex is notable for its high degree of structural similarity to published COX-NSAID complexes (described in Section 5). AA is bound to the protein in a productive conformation, with its carboxylate forming a salt bridge with Arg-120 and a hydrogen bond to Tyr-355 at the constriction. From there, AA’s hydrophobic chain projects up into the active site channel in an extended L-shaped conformation that loops around Ser-530 and fills the hydrophobic recess. A total of 45 hydrophobic interactions are identified between the protein and substrate. This conformation places the 13-pro-(S)-hydrogen of AA in close proximity to Tyr-385, aligned ideally for abstraction.34
4.1.1. Binding Pockets
As noted above, we have divided the active site channel into three binding “pockets”, each of which comprises residues that contact or surround different portions of the fatty acid chain of AA [Figure 5 (stereoscopic view) and Figure S1 (monoscopic view)]. Although somewhat arbitrary in nature, these pockets also serve as primary interaction sites for distinct structural components of most NSAIDs. The following description is based on the COX-1-AA complex.34 Details of differences between COX-1 and COX-2 will then be outlined.
Figure 5.

(A) Wall-eyed stereo view of AA bound in the cyclooxygenase active site of COX-1 and the side chains that make up the proximal binding pocket (green), the central binding pocket (magenta), and the distal binding pocket (cyan). This view is similar to that used to depict most structures of fatty acids in complex with COX-1 or COX-2 throughout the review. (B) Wall-eyed stereo view of an overlay of the structures of four fatty acids in the active site of COX-1. Fatty acids and amino acid side chains are colored from lightest to darkest in the order of LA, DHLA, EPA, and AA. Notable is the minimal movement of active site residues to accommodate the structural differences among the various fatty acids. Monoscopic views of the individual structures are provided in Figure S1. From PDB 1DIY (A and B) and 1IGZ, 1FE2, and 1IGX (B only).
4.1.1.1. Proximal Binding Pocket (AA Carbons 1–7)
The first seven carbon atoms of AA form interactions with six residues of the enzyme. These are Arg-120, Val-349, Ser-353, Tyr-355, Ile-523, and Ala-527 (Figure 6).34 As noted above, Arg-120 and Tyr-355 participate in the only polar interactions with AA in the productive conformation. The importance of these interactions has been revealed by site-directed mutation studies (Table S2). Specifically, a charge-reversing R120E mutation increases the KM for AA by 100-fold and reduces cyclooxygenase activity by 95%, whereas an R120K mutation increases the KM 20-fold and reduces activity by 85% despite the retention of charge. Interestingly, an R120Q mutation, which replaces the positively charged arginine with neutral glutamine (thus, retaining the capacity for hydrogen bonding), increases the KM for AA by 800-fold while reducing activity by 95%.43,44 These findings confirm the importance of salt bridge formation in the binding of AA to COX-1. Hydrogen bond formation to Tyr-355 is also important, as indicated by the 5-fold increase in KM and 80% reduction in activity observed for the Y355F mutant.43,45,46
Figure 6.

Wall-eyed stereo view of the proximal AA binding pocket as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking downward toward the membrane from above (B). AA is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 1DIY.
Other amino acids in the proximal binding pocket play a significant role in COX catalysis. For example, various mutations of Val-349 in COX-1 alter the product ratio in favor of mono-oxygenated species (11-HETE and/or 15-HETE) and/or change product stereochemistry with or without significantly affecting the enzyme’s catalytic constants. These findings have led to the conclusion that Val-349 plays a role in positioning AA to maximize PGG2 formation.46−48 S353T and I523A mutations in COX-1 also result in increased mono-oxygenated products with only modest effects on overall activity. Similar findings were not reported, however, for S353A, S353G, or I523V mutant COX-1 proteins, which showed activities and product ratios comparable to those of the wild-type enzyme.46
4.1.1.2. Central Binding Pocket (Carbons 7–15)
The central binding pocket surrounds the AA carbons that are directly involved in the cyclooxygenase reaction. The amino acids that interact with AA in this region include Tyr-348, Leu-352, Tyr-385, Trp-387, Gly-526, and Ser-530 (Figure 7).34 As noted above (Figure 2), Tyr-385 is the key catalytic residue that donates a hydrogen atom to the heme during enzyme activation, generating a tyrosyl radical that then abstracts the 13-pro-(S)-hydrogen atom from AA. Predictably, a Y385F mutation completely eliminates cyclooxygenase activity.45 Following hydrogen abstraction at C-13, antarafacial oxygen addition at C-11 occurs prior to endoperoxide ring formation. A pocket in the protein next to C-11 provides access to the oxygen molecule, enabling it to align in the correct orientation. Furthermore, a pocket formed by Phe-381, Leu-384, Trp-387, Phe-518, and Met-522 provides room to accommodate the conformational changes that must occur to enable cyclization (Figure 8A). Thus, the overall structure of the central binding pocket enables the complex chemistry of the cyclooxygenase reaction to occur without significant movement of the substrate.34
Figure 7.

Wall-eyed stereo view of the central AA binding pocket as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking along the axis of the pocket (B). AA is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 1DIY.
Figure 8.

Wall-eyed stereo view of AA bound to the active site of COX-1. (A) A pocket formed by Phe-381, Leu-384, Trp-387, Phe-518, and Met-522 provides space for formation of the endoperoxide ring. (B) Tyr-348, Phe-381, Typ-385, and Ser-530 surround carbon-15 of AA (green) dictating the orientation of oxygen addition at this site. AA is colored by element (with the exception of carbon-15 in B), and its surface is shown as a mesh. Side chains of the residues indicated are displayed, and their surface is shown in solid tan. From PDB 1DIY.
In addition to Tyr-385, both Ser-530 and Tyr-348 possess hydroxyl groups that might be expected to be involved in cyclooxygenase activity; however, S530A and Y348F COX-1 mutations have minimal effects on enzymatic activity, suggesting that the hydroxyl group of neither of these residues is required.46,49−51 Notably, S530C, S530G, S530I, S530L, S530M, S530N, S530T, and S530V COX-1 mutations all result in nearly total elimination of activity, indicating that steric constraints around Ser-530 are important.46,47,51 This is consistent with its strategic location at the bend of the active site channel. Y348L and Y348W COX-1 mutants are also inactive, again likely due to steric constraints.46
Both C-11 and C-12 of AA are located in very close proximity to Trp-387. A W387A COX-1 mutant is totally inactive, as are W387R and W387S, suggesting that both steric bulk and hydrophobicity at this position are important.46 Consistently, W387F and W387L mutants retain some activity but exhibit increased KM’s for AA and a shift to production of mono-oxygenated products. These findings indicate a role for Trp-387 in correct positioning of AA within the active site for cyclization of the peroxyl radical intermediate.46
The apparent role of Val-349 (in the proximal pocket) and Trp-387 in positioning AA for PGG2 formation led Harman et al. to construct a V349A/W387F double mutant COX-1.52 This enzyme produces >84% 11-HETE in contrast to wild-type COX-1, which produces 95% PGG2; however, the combined mutations have little effect on the KM for AA. A crystal structure of V349A/W387F COX-1 in complex with Co3+-protoporphyrin IX and AA (PDB 1U67) obtained at 3.1 Å resolution revealed that the double mutation has minimal impact on overall protein structure or the binding orientation of AA in the cyclooxygenase active site. However, compared to the position of AA in the wild-type enzyme, important differences are observed. Specifically, carbon-3 to carbon-6 of AA move to fill the void created by the V349A substitution, resulting in an ∼3 Å downward shift of the C5–C6 double bond. In addition, a rotation around the C9–C10 bond enables the C11–C12 double bond to move into the space created by the W387F mutation. The results suggest that Val-349 helps to anchor the carboxyl end of AA while Trp-387 exerts a steric effect at C11–C12 that promotes endoperoxide formation.52
4.1.1.3. Distal Binding Pocket (Carbons 15–20)
The omega-terminal tail of AA fills a hydrophobic recess outlined by Phe-205, Phe-209, Val-228, Val-344, Ile-377, Phe-381, Gly-533, and Leu-534 (Figure 9).34 Mutations of Phe-205 and Phe-209 to leucine in COX-1 have only modest effects on activity, but mutation to alanine results in partial inactivation and a shift to greater formation of mono-oxygenated products.46 Thus, it appears that these residues play a role in stabilizing the conformation of AA for PGG2 formation. A similar function may apply to Ile-377, Phe-381, and Leu-534, as mutation of these residues results in a shift toward mono-oxygenation with varying increases in KM and reductions in enzyme activity.46 A G533A mutation in COX-1 completely eliminates cyclooxygenase activity, a finding that can be explained by the close proximity of this residue to C-20 of AA with very little room available for the addition of the alanine methyl side chain.51
Figure 9.

Wall-eyed stereo view of the distal AA binding pocket as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking along the axis of the pocket (B). AA is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 1DIY.
4.1.2. Differences between COX-1 and COX-2
It was not until 2010 that a crystal structure of COX-2 with AA bound in a productive conformation (PDB 3HS5) was published.53 One reason for this is that COX-2 is incompletely glycosylated at Arg-594, leading to the production of a heterogeneous pool of protein in eukaryotic expression systems.54 Use of an N594A mutant COX-2 mitigated this problem, and reconstitution of the apoprotein with Co3+-protoporphyrin IX eliminated enzymatic activity to prevent oxygenation of AA during crystallization. The resulting 2.1 Å resolution structure exhibits the same overall three-dimensional organization as had been previously reported for both COX isoforms. However, despite no obvious differences in the conformations of the two COX-2 monomers, AA is present in a productive conformation in one (designated monomer B) [Figure S2A (monoscopic view) and Figure S3 (stereoscopic view)] and in an inverted conformation in the other (monomer A) [Figure 10 (stereoscopic views) and Figure S4A (monoscopic view)].53
Figure 10.

(A) Wall-eyed stereo view of the structure of AA (nonproductive conformation) bound in the cyclooxygenase active site of COX-2 and the side chains that make up the proximal binding pocket (green), the central binding pocket (magenta), and the distal binding pocket (cyan). Note that AA is observed in two slightly different conformations. (B) Wall-eyed stereo view of an overlay of the structures of the nonproductive (dark gray) and productive (light gray) conformations of AA bound in the cyclooxygenase active site. Residues in the surrounding binding pockets are colored similarly to those in (A) with the lighter and darker colors corresponding to the productive and nonproductive conformations, respectively. Also shown is the marked difference in the position of Leu-531 between the productive conformation (gold) and the nonproductive conformation (sienna). From PDB 3HS5.
In the case of the productive conformation, the structure of the COX-2-AA complex is very similar to that of the COX-1-AA complex but with some exceptions. A notable difference is the absence of any interaction between the carboxylate of AA and Arg-120. Thus, the only hydrophilic interaction is a single hydrogen bond with Tyr-355. In the absence of binding to AA, Arg-120 establishes an interaction with Glu-524, thereby closing and stabilizing the constriction. In addition, to make up for the loss of the salt bridge, AA engages in a larger number of hydrophobic contacts with the enzyme (totaling 53 in all) than the 45 contacts observed in the COX-1-AA structure. Despite these compensatory changes, the reduced polar interaction, along with the 25% larger active site in COX-2, conveys greater flexibility for AA when bound to COX-2 than COX-1.37,53
Perhaps the most striking difference between the COX-1- and COX-2-AA complexes is the inverted conformation of AA in monomer A of the COX-2-AA complex. This conformation is similar to that observed in the structure previously published by Kiefer et al. for apo-COX-2 complexed with AA (see Section 4.1 above).33,53 Thus, the carboxyl group of AA is hydrogen bonded to Tyr-385 and Ser-530. Two water molecules fill the distal binding pocket above Ser-530, while the omega tail of AA projects downward toward Arg-120, abutting Leu-531. As observed by Kiefer et al., Leu-531 adopts a conformation not observed in monomer B in order to provide additional room in the proximal binding pocket for AA in this binding pose (Figure 10B)33,53 It is not clear whether the inverted conformation of AA in the COX-2-AA complex is an artifact of crystallization or a reflection of substrate binding to the enzyme in vivo. However, the finding that one subunit binds AA nonproductively would be consistent with the observed half-of-sites activity discussed above. This is an intriguing hypothesis in light of the fact that this conformation was observed in the apoenzyme, and the allosteric subunit is presumed to lack heme (see Section 1). If the inverted conformation of AA has physiological relevance, then the rotation of Leu-531 to accommodate the fatty acid’s omega tail may be important for COX-2 catalysis. To test this hypothesis, Vecchio et al. obtained a 2.4 Å resolution crystal structure of L531F COX-2 complexed with AA and Co3+-protoporphyrin IX (PDB 3KRK). They found that this mutation eliminates the inverted conformation of AA in monomer A of the enzyme; however binding of AA in that monomer remains suboptimal with regard to the alignment of the 13-(pro)-S-hydrogen for abstraction.53 The mutant enzyme exhibits a 3-fold increase in KM for AA but only a small decrease in maximal activity.53 Other mutations of Leu-531 exhibit only modest effects on AA oxygenation by COX-2.53 In contrast, most mutations of Leu-531 in COX-1 result in a > 90% reduction in maximal activity.46,51 This may be explained by the close approximation of Leu-531 to Arg-120, which helps to stabilize Arg-120’s critical salt-bridge with AA in COX-1.34
An interesting difference between the COX-2- and COX-1-AA complex structures is a dual conformation exhibited by Ser-530 in COX-2.53 This suggests a side-chain flexibility that could facilitate access of AA’s omega tail into the distal binding pocket. This flexibility, along with higher levels of activity exhibited by S530M, S530T, and S530V mutants in COX-2 than COX-1, is consistent with the presence of increased space in that portion of the active site of COX-2 as compared to COX-1. Notably, these mutants all generate 15(R)-HETE as the primary product, indicating a switch to both mono-oxygenation and inversion of stereochemistry from the 15(S)-HETE produced primarily by the wild-type enzyme. The inversion of stereochemistry from (S) to (R) is also observed at the 15-position of PGG2 that is produced by the mutants.47,55 Thus, the more spacious active site of COX-2 accommodates larger side chains at the Ser-530 position without total loss of activity than can be accommodated in COX-1. However, these bulkier side chains displace AA in such a way that they prevent the efficient formation of PGG2.
In an attempt to explain the change in stereochemistry observed with the Ser-530 COX-2 mutant enzymes, which is also observed upon aspirin acetylation of COX-2 (see Section 5.2.10), a 2.2 Å resolution crystal structure of V349I COX-2 in complex with Co3+-protoporphyrin IX and AA (PDB 6OFY) was obtained.56 This enzyme was selected due to its production of mostly 15(R)-HETE and 15(R)-PGH2 as products.47 The findings revealed that AA is bound in an inverted, unproductive conformation in one subunit and a productive conformation in the second. The productive conformation is notable in that AA adopts a C- rather than L-shape, with its omega tail not fully penetrating the distal binding pocket. As a result, AA’s C-terminus extends further into the proximal binding pocket than is seen with the wild-type enzyme, necessitating a rotation of Leu-531 as observed in the nonproductive conformation of AA bound to wild-type COX-2. A single hydrogen bond between the carboxylate of AA and the main chain carboxyl oxygen of Ala-527 is the only polar interaction observed, and the number of hydrophobic contacts is reduced by 50% when compared to those formed with the wild-type active site. Nevertheless, this conformation places the 13-pro-(S)-hydrogen atom close to Tyr-385. Notably, the Cδ1 atom of Ile-349 protrudes into the central binding pocket, where it sterically blocks antarafacial oxygen addition at carbon-15 of AA, as is required to achieve S-stereochemistry at that position.56
Additional mutation studies highlight the differences between the two COX isoforms. Consistent with the absence of salt-bridge formation with Arg-120, an R120Q mutant COX-2 retains full activity. However, an R120E mutant exhibits both an increased KM and a substantial decrease in activity, likely due to loss of the interaction between Arg-120 and Glu-524.57,58 In COX-2, a G533A mutant in the distal binding pocket retains some activity, whereas this mutation completely inactivates COX-1, as discussed above (see Section 4.1.1.3).51,59,60 This may also be a reflection of the larger COX-2 active site that enables the fatty acid to shift position in order to avoid a steric clash with the methyl side-chain of Ala-533 in the mutant. A G533V mutation in COX-2, however, leads to complete loss of AA oxygenation activity, and the basis for this was revealed by a 2.2 Å resolution crystal structure of G533V COX-2 complexed with Co3+-protoporphyrin IX and AA (PDB 3TZI).61 In this structure, AA is observed in essentially the same, C- (rather than L-) shaped conformation in both subunits. Its inability to completely insert its omega tail into the distal binding pocket above Ser-530 forces it downward into the lower portion of the channel, requiring a shift of Leu-531 away from Arg-120 to provide room. The result is that Leu-531 adopts the same conformation as is observed in the nonproductive binding conformation of AA in COX-2, and the carboxylate of AA is shifted > 6.5 Å away from Arg-120. In this conformation, the number of hydrophobic contacts established between AA and the enzyme is reduced to 29 and 25 in monomers A and B, respectively. The displacement of carbon-13 to a position > 6 Å away from Tyr-385 explains the lack of activity of this mutant.61 It should be noted, however, that although reported levels of activity vary, G533A and G533V mutant COX-2 enzymes retain the ability to oxygenate shorter chain fatty acids, particularly α-linolenic acid (αLA) and steridonic acid (SA). Remarkably, a strong retention of activity with eicosapentaenoic acid (EPA) has also been reported for G533A COX-2. Characteristic of all of these fatty acids is the presence of an allylic carbon at the ω-5 position, close to the end of the chain. In contrast, retention of activity is lower with linoleic acid (LA) and AA, each of which possess their terminal allylic carbon at the ω-8 position, requiring deeper insertion of the substrate’s tail into the distal binding pocket.60,61 Similar studies of the activities of G533 COX-1 mutants with alternative fatty acid substrates have not been reported.
A water-mediated hydrogen-bonding network between His-90, Tyr-355, Arg-513, and Glu-524 stabilizes the constriction at the mouth of the COX-2 active site. Arg-513 is one of only three active site residues that is different between COX-1 and COX-2. Residue 513 is histidine in COX-1, and an R513H mutation of COX-2 results in a 2.2-fold increase in the KM for AA while having no effect on kcat.62 A 2.4 Å resolution crystal structure of R513H COX-2 complexed with Co3+-protoporphyrin IX and AA (PDB 3OLT) revealed a loss of hydrogen bonds between Arg-120 and Glu-524 and between Asn-87 and Arg-513. Disruption of these interactions also leads to the loss of six out of seven ordered water molecules that are present in the wild-type crystal structure. The loss of interaction between Asn-87 and Arg-513 decouples helix B of the membrane-binding domain from the base of the catalytic domain. As in the case of the wild-type enzyme, AA binds in a productive conformation in one monomer and an unproductive (inverted) conformation in the second monomer. The productive conformation appears similar to that in the wild-type enzyme, but eight contacts between AA and active site channel residues are lost. These include the hydrogen bond with Tyr-355. The result is greater mobility of AA within the active site that could easily explain the observed increase in KM in the R513H mutant as compared to that of the wild-type enzyme.62
Note that, in contrast to key differences cited above, many COX-2 mutations have similar effects as the same mutations in COX-1 with regard to AA oxygenation (Table S2).
4.2. Interactions of COX-1 with Other Fatty Acids
4.2.1. Linoleic Acid (LA)
LA is a diunsaturated 18-carbon fatty acid that is oxygenated to monohydroxy-octadecadienoic acids by the COX enzymes. LA is a substantially less efficient substrate than AA for both isoforms. A 2.9 Å resolution crystal structure of COX-1 complexed with Co3+-protoporphyrin IX and LA (PDB 1IGZ) was published in 2001.63 The structure reveals a binding pose for LA that is very similar to that of AA (Figures 5B and S1B). LA’s carboxylate forms a salt bridge with Arg-120 and two hydrogen bonds with Tyr-355, and the fatty acid establishes 44 hydrophobic contacts with amino acids lining the active site channel. Despite the absence of a double bond at carbons 1 through 8, the interactions of the carboxyl end of LA are very similar to those of AA. However, from carbon-7 through carbon-18, LA takes a more direct path in the active site channel, placing C-11 rather than C-13 next to the phenolic oxygen of Tyr-385 and enabling the shorter chain of LA to completely fill the distal binding pocket above Ser-530. LA’s greater flexibility, resulting from the absence of a C5–C6 double bond, increases the importance of contacts with residues in the proximal binding pocket. Thus, V349A and V349L mutants have a much greater negative impact on enzyme activity for LA than AA.63 Val-349 forms a part of the wall of the active site channel that stabilizes the fatty acid chain. Mutation to alanine opens a large pocket that enables LA to shift away from its contacts with Ile-523, Gly-526, and Ala-527, likely resulting in a misalignment of the C-11 hydrogen atom with Tyr-385. In contrast, a V349L mutant introduces excess steric bulk into the channel.
4.2.2. Dihomo-γ-linolenic Acid (DHLA)
DHLA is a triunsaturated 20-carbon fatty acid. It is identical in structure to AA except that it lacks the C5–C6 double bond. COX-1 and COX-2 oxygenate DHLA with somewhat less (∼50%) efficiency than AA, producing PGs with one, rather than two double bonds. Thus, a 3.0 Å resolution structure of COX-1 complexed with Co3+-protoporphyrin IX and DHLA (PDB 1FE2) revealed few significant differences from the structure of the COX-1-AA complex (Figures 5B and S1C).64 The carboxylate of DHLA forms a salt bridge with Arg-120 and two hydrogen bonds to Tyr-355 rather than one as seen with AA. In total, DHLA forms 59 hydrophobic contacts with the COX-1 active site, a higher number than was observed for AA. Both fatty acids establish contacts with 19 residues in the active site channel, but the identities of some of the residues are different. Specifically, Ser-353 and Ile-377 form contacts with AA, while Val-228 and Phe-518 form contacts with DHLA. The major differences in conformation between DHLA and AA in the COX-1 active site are observed in the C-terminal portion of the molecule, which is more flexible in DHLA than AA. Thus, the region between carbon-2 and carbon-10 is more compactly coiled in DHLA, a conformation that positions it closer to helix 17 and more distant from helix 6. Mutation studies suggest that Val-349 and Ser-530 play a larger role in positioning DHLA correctly for catalysis than they do for AA, an observation that can, at least partially, be explained by small differences in orientation and interaction of these amino acids with the substrates as observed in the respective crystal structures.64
4.2.3. Eicosapentaenoic Acid (EPA)
EPA is identical to AA in structure with the exception of a fifth double bond at C17–C18. It is a poor substrate for COX-1 and, in fact, can serve as a competitive inhibitor of AA oxygenation for that isoform. It is more readily oxygenated by COX-2 but with only about 30% efficiency as compared to oxygenation of AA.65 A 3.1 Å resolution crystal structure of COX-1 reconstituted with Co3+-protoporphyrin IX and EPA (PDB 1IGX) was published in 2001.63 EPA adopts a binding pose similar to that of AA in the active site, with its carboxyl group interacting with Arg-120 and Tyr-355, its chain projecting upward and looping around Ser-530, and its omega tail filling the distal binding pocket (Figures 5B and S1D). In all, EPA forms 59 hydrophobic interactions along the cyclooxygenase channel. Despite these similarities, however, the lack of flexibility due to the C17–C18 double bond results in a strained conformation for the fatty acid, with carbon-2 through carbon-10 forced into closer contact with residues 520–535 of helix 17 than is observed for AA. Although this increases the hydrophobic contacts between EPA and the enzyme, it also produces a misalignment between EPA’s 13-pro-(S)-hydrogen atom and Tyr-385, thus explaining the poor activity of COX-1 with this substrate. As in the case of LA and DHLA, mutations of Val-349 and Ser-530 have a much larger impact on EPA oxygenation than on AA oxygenation.63
4.3. Interactions of COX-2 with Other Fatty Acids
4.3.1. α-Linolenic Acid (αLA)
αLA is a triunsaturated 18-carbon fatty acid that is reported to be oxygenated by COX-2 with approximately 40% of the efficiency of AA oxygenation.61 A 2.1 Å resolution crystal structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and αLA (PDB 4E1G) reveals nearly identical conformations for both the protein and fatty acid in the two monomers (Figures S2B and S3).61 Notably, αLA makes two hydrophilic contacts with Arg-120 and one with Tyr-355 at the constriction in addition to 51 hydrophobic contacts in the active site channel. As in the case of LA, the absence of double bonds in the first 8 carbons of αLA provides greater flexibility to that region of the molecule, and it takes a more direct course through the active site than AA, allowing it to completely fill the distal binding pocket above Ser-530 despite its shorter length. This trajectory places carbon-11 close to Tyr-385, aligning it appropriately for hydrogen abstraction.61
4.3.2. Eicosapentaenoic Acid (EPA)
As noted above (Section 4.2.3), EPA is oxygenated by COX-2, but with only about 30% efficiency as compared to oxygenation of AA.65 A 2.4 Å resolution crystal structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and EPA (PDB 3HS6) revealed, as in the case of AA, a different binding mode for the fatty acid in each monomer.53 In monomer B, EPA is oriented in a productive conformation, similar to that of AA in COX-2 (Figures S2C and S3). Although the loss of flexibility at the C17–C18 double bond restrains motion of the omega end of EPA, the larger COX-2 active site can accommodate the strained conformation with retention of a favorable orientation of the 13-pro-(S)-hydrogen to Tyr-385. Thus, the respective crystal structures help to explain why EPA is a more efficient substrate for COX-2 than for COX-1. The inverted conformation of EPA in monomer A is highly similar to that observed for AA (Figure S4B). Notably, the rotation of Leu-531 observed in monomer A, which bears the inverted conformation of AA, is present in both monomer A and monomer B of the crystal structure of COX-2 with EPA. This has led to the hypothesis that rotation of Leu-531 in COX-2 may contribute to its broad substrate selectivity (as compared to that of COX-1). As noted above (see Section 4.1.2), Leu-531 is believed to play a role in stabilizing Arg-120 for salt bridge formation in COX-1. Consistently, a similar rotation of this residue has not been observed in any COX-1-substrate complex crystal structures to date.53
4.3.3. Docosahexaenoic Acid (DHA)
DHA is a 22-carbon hexa-unsaturated fatty acid that is oxygenated by COX-2 with poor efficiency, yielding primarily mono-oxygenated products. A 2.6 Å resolution crystal structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and DHA (PDB 3HS7) revealed that, despite its greater length and the inflexibility provided by two additional double bonds, DHA adopts an L-shaped conformation in the cyclooxygenase active site that is strikingly similar to the productive conformation of AA (Figures S2D and S3).53 Unlike the crystal structures of AA and EPA in complex with COX-2, DHA exhibits the same binding pose in both monomers of the dimer. Also, unlike AA and EPA, the carboxylate of DHA forms two salt bridges with Arg-120 in addition to a hydrogen bond with Tyr-355. To fit into the channel, the longer chain of DHA is more compacted than that of AA, enabling it to make 23% more contacts. The constrained conformation of DHA places carbon-15 in close proximity to Tyr-385, enabling hydrogen abstraction at that position. In contrast to EPA, however, rotation of Leu-531 appears to play no role in accommodating DHA binding.53
4.3.4. Palmitic Acid (PA)
PA is a 16-carbon saturated fatty acid that is not a substrate for the COX enzymes but has been reported to stimulate COX-mediated oxygenation of AA via an allosteric mechanism.21,28 A 2.1 Å resolution crystal structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and PA (PDB 3QH0) provided some insight into the mechanism of this effect (Figures S2E and S3).21 The overall structure of the protein is similar in the two monomers of the COX-2 dimer; however, PA is present in only one of the monomers as might be expected for an allosteric modulator. Two molecules of ethylene glycol partially fill the active site in the second monomer. The carboxylate of PA forms two salt bridges to Arg-120 in addition to three hydrogen bonds, one to Arg-120 and two to Tyr-355. From the constriction, PA extends upward into the cyclooxygenase channel, but it does not reach into the distal binding pocket above Ser-530, which is occupied instead by two water molecules. In total, an additional 31 hydrophobic contacts are established between PA and the cyclooxygenase active site.
4.3.5. 13(S)-Methyl-arachidonic Acid (13-Me-AA)
13-Me-AA is an AA derivative that does not serve as a substrate for COX-1 or COX-2 but selectively stimulates 2-AG oxygenation by both COX isoforms.22 In wild-type COX-2, 13-Me-AA stimulates 2-AG oxygenation by increasing kcat and reducing substrate inhibition. It also rescues the 2-AG oxygenating activity of a number of mutant enzymes notable for particularly low activity with that substrate.22 A 2.2 Å resolution crystal structure of COX-2 complexed with Co3+-protoporphyrin IX and 13-Me-AA (PDB 4RUT) reveals similar inverted binding modes for the fatty acid in both monomers (Figure S4C). The carboxyl group of 13-Me-AA is hydrogen bonded to Tyr-385 and Ser-530, and the omega tail adopts multiple conformations above the constriction. In the most prevalent conformation, the 13-methyl group of the ligand is inserted into a pocket formed by Val-349, Tyr-355, and Leu-359. As is seen in the inverted conformation of AA in COX-2, Leu-531 rotates away from Arg-120 in order to provide room for the omega tail of the fatty acid.22
4.3.6. 1-Arachidonoylglycerol (1-AG)
1-AG, the 1-glyceryl ester of AA, is the thermodynamically more stable isomer of the endocannabinoid 2-AG. Both 1-AG and 2-AG are metabolized efficiently by COX-2, with 2-AG preferred over 1-AG, but they are poor substrates for COX-1.66 A crystal structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and 1-AG (PDB 3MDL) was obtained at 2.2 Å resolution.62 The investigators formed the complex by adding 2-AG to the enzyme, but isomerization to 1-AG occurred during crystallization. In the structure, 1-AG exhibits the same overall conformation as is observed for the productive conformation of AA. An inverted conformation for 1-AG is not observed; however, there are subtle differences between the binding poses in the two monomers leading to the conclusion that only one is a productive conformation.
The productive conformation of 1-AG in monomer B places the glycerol moiety close to Arg-120 and Tyr-355 (Figure S5A,C,D). A single hydrogen bond between the glyceryl moiety and Tyr-355, and a second one between the glyceryl moiety and the carbonyl of Ala-527 are the only polar interactions. The hydrocarbon chain extends upward, loops around Ser-530, and fills the distal binding pocket, forming 60 hydrophobic contacts. The 13-(pro)-S-hydrogen atom of 1-AG is in close proximity to Tyr-385, in adequate, though not ideal alignment for abstraction. Notably, 1-AG establishes a contact with Val-116 that is not formed by AA, and AA forms contacts with Val-228 and Val-344 that are not formed by 1-AG. The amino acid side chains in the active site adopt the same overall conformation in the 1-AG complex as in the complex of AA bound productively to COX-2 with the exception of Leu-531, which is rotated away from Arg-120 as seen in the productive conformation of COX-2 with EPA and unproductive complexes with EPA and AA. In the nonproductive conformation, the overall orientation of 1-AG is the same as in the productive conformation, but its omega tail fails to extend fully into the distal binding pocket, resulting in a major misalignment of carbon-13 for hydrogen abstraction (Figure S5B,C,D).62
The structure of COX-2 complexed with 1-AG has been used to explain or predict the effects of various mutations on the oxygenation of 2-AG by the enzyme. It should be noted that the complex with 1-AG may or may not be a good model for the 2-AG binding interaction with COX-2. For example, we do not know if the observed suboptimal alignment of the 13-(pro)-S-hydrogen atom of 1-AG with Tyr-385 also applies to 2-AG. Indeed, this misalignment may explain why 1-AG is the less efficient substrate of the two glyceryl esters.66 Nevertheless, as no structure is available for a complex between COX-2 and 2-AG, the 1-AG complex structure must serve as a surrogate. A hydrogen bond to Tyr-355 is the only polar interaction between AA and COX-2 and one of only two polar interactions between 1-AG and COX-2 in their respective productive conformations. A Y355F mutation of COX-2 increases the KM for AA while reducing kcat by 50%. In contrast, this mutation has no effect or increases kcat for 2-AG while actually decreasing KM and increasing the proportion of mono-oxygenated products. It is possible that the Y355F mutation destabilizes the hydrogen-bonding network at the constriction, thereby facilitating access of 2-AG to the active site while also increasing substrate flexibility once binding has occurred.62,67
The major differences between the active sites of COX-1 and COX-2 result from three amino acid substitutions, Ile-434, His-513, and Ile-523 in COX-1, which are Val-434, Arg-513, and Val-523 in COX-2. These substitutions produce a “side pocket” next to the active site of COX-2 that is a key binding site exploited by many COX-2-selective inhibitors. Mutant studies had suggested that the side pocket, particularly in the region of Arg-513, is also an important determinant of isoform-selective 2-AG oxygenation by COX-2.67 However, the crystal structure demonstrated no interaction between 1-AG and Arg-513, or more generally between the glycerol moiety of 1-AG and the side pocket. Consistently, in these more recent studies, an R513H mutant COX-2 exhibited no significant loss in activity for 1-AG or 2-AG as a substrate.62 The potential role of Arg-513 was further explored in a 2.4 Å resolution crystal structure of R513H COX-2 complexed with Co3+-protoporphyrin IX and 1-AG (PDB 3OLU).62 In this structure, 1-AG adopts a productive conformation in the active site in both monomers; however, the glyceryl moiety is bound differently in each. In monomer B, the overall 1-AG conformation is similar to that of 1-AG in the wild-type enzyme, but the glyceryl moiety is reconfigured to make a hydrophilic contact with Arg-120 instead of Ala-527. The protein conformation in monomer A is notable in that Leu-531 occupies the same position that it occupies when AA is bound in the productive orientation, rather than the position it adopts in the wild-type COX-2–1-AG complex. The glyceryl moiety points away from Leu-531 and forms two hydrophilic contacts with Arg-120. The result is a compression of 1-AG between carbon-1 and carbon-13, enabling it to form an additional 15 contacts with the channel.62
The crystal structure of the wild-type enzyme suggests that rotation of Leu-531 is important to provide adequate room for the binding of 1-AG. Early work on the structural determinants of 2-AG oxygenation showed that an L531A mutation reduces the rate of oxygenation of both AA and 2-AG, with greater effects on AA. A similar but less pronounced pattern was observed for an L531V mutation.67 In later studies, however, L531A COX-2 was observed to cause a similar ∼50% reduction in kcat and KM for both substrates. L531F and L531P mutations had only minimal effects on 2-AG oxygenation.62 These results leave the role of Leu-531 rotation unresolved.
4.3.7. Prostaglandin G2/H2 (PGG2/H2)
As noted above, the first published crystal structure of a COX protein complexed with AA utilized an H207A mutation of apo-COX-2 to ensure inactivity (see Section 4.1). This approach resulted in an inverted conformation of AA in the active sites of both monomers. A second attempt by the same investigators used wild-type apo-COX-2.33 The resulting 3.0 Å resolution structure (PDB 1DDX) revealed AA in an inverted conformation in some monomers and PGG2 or PGH2 in the remaining monomers. The presence of product in the crystals was explained on the basis of a small amount of contaminating heme that was believed to be able to move from one monomer to another in the presence of the high AA concentration used for crystallization. Prior reports had indicated that high AA concentrations lower the affinity of COX enzymes for heme.33
Although this was not the intended result, it provided an opportunity to explore the conformation of PGG2/H2 in the cyclooxygenase active site (Figure 11). The binding mode of PGG2/H2 is consistent with expectations, placing the carboxyl group near Arg-120 and Tyr-355 at the constriction and the omega end in the distal binding pocket above Ser-530 and Tyr-385. Notably, the carboxyl group of PGG2/H2 is displaced upward, so that contacts with Arg-120 and Tyr-355 are not formed. The endoperoxide ring establishes hydrophobic contacts with Phe-381, Leu-384, Tyr-385, and Trp-387. The importance of the interaction with Trp-387 is highlighted by the effects of a W387F mutation, which results in a marked shift toward production of monohydroxy products.46
Figure 11.

Wall-eyed stereo view of the structure of PGG2/H2 overlaid with that of AA (productive conformation) bound in the active site of COX-2. Side chains that make up the central binding pocket are shown (light and dark magenta), as are Leu-384 (gold and sienna), proximal binding pocket residues Arg-120 and Tyr-355 (light and dark green), and distal binding pocket residue Phe-381 (dark and light blue). PGG2 (dark gray) and AA (light gray) are colored by heteroatom. In the case of residue side chains, the darker and lighter colors denote positions in the complexes containing PGG2/H2 and AA, respectively. The major shift in the position of Tyr-385 is readily apparent, as is the upward shift in the position of the carboxyl group of PGG2 relative to that of AA. These displacements are enabled by the absence of heme and likely would not occur in the holoenzyme. From PDB 3HS5 (chain B) and 1DDX.
A 60° rotation of Leu-384 (as compared to its orientation in prior COX crystal structures) accommodates the endoperoxide ring. A more notable shift occurs in the position of Tyr-385, which fills a space left vacant by the absence of heme. Thus, Tyr-385 occupies a position that is notably different from its conformation in the majority of previous structures and would not be likely in the case of the holoenzyme (Figure 11). Modeling studies suggest that, if heme were present, the resulting displacement of PGG2/H2 would allow Tyr-385 to occupy the more commonly observed position, and it would enable the formation of contacts between the carboxylate of PGG2/H2 and Arg-120.33
The structure reveals how the placement of key amino acid side chains works to ensure the formation of PGG2. The positioning of Phe-381 forces the addition of O2 at carbon-11 to be antarafacial. The compact environment formed by the central binding pocket around the incipient 5-membered ring (Figure 7), the space provided to allow ring formation (Figure 8A), and the shape of the distal binding pocket (Figure 9) dictate the trans configuration for carbon-8 to carbon-12 bond formation. Similarly, the presence of Tyr-348, Phe-381, Tyr-385, and Ser-530 restricts the stereochemistry of oxygen addition at carbon-15, so that the R-isomer is formed at this position (Figure 8B).33 These observations, combined with the results of site-directed mutagenesis studies described above confirm the importance of the overall structure of the cyclooxygenase active site to ensuring the fidelity of PGG2 formation. Indeed, as noted above, seemingly small changes in protein structure, such as V349A or G533A mutations have a profound effect on enzyme activity and product formation.
5. Interactions of COX Proteins with Inhibitors
As will become evident in the discussion that follows, a remarkably wide variety of small molecules can exploit the cyclooxygenase active site, resulting in inhibition. As could easily be predicted, nearly all inhibitors establish contacts with the enzyme in proximal and central inhibitor binding pockets that overlap substantially with the corresponding fatty acid binding pockets. In contrast, only one inhibitor, ARN-2508, a dual inhibitor of COX-2 and fatty acid amide hydrolase (FAAH), has been shown to utilize the distal fatty acid binding pocket. In addition, the diarylheterocycle class of COX-2-selective inhibitors occupies a pocket to the side of the constriction that is larger in COX-2 than COX-1, a strong determinant of their isoform selectivity. The oxicams create a pocket by forcing the rotation of Leu-531 away from the constriction, as is seen in the binding of bulky substrates to COX-2. It is interesting to note that this rotation occurs in oxicam binding to either isoform, whereas it was not observed in the binding of fatty acids to COX-1.
A common feature of many inhibitors is the presence of a carboxylic acid moiety, leading to the expectation that this functional group would interact with polar residues at the constriction, similar to the carboxylate of AA. This is the case for many but not all inhibitors. Just as AA can bind in an “inverted” conformation in COX-2 through interaction between its carboxylate and central pocket polar residues, some inhibitors do as well. Examples include diclofenac and related aryl acetic acids and the fenamic acids. The oxicams, which are enolic rather than carboxylic acids, employ an alternative mechanism, using hydrogen-bonded water molecules to establish polar contacts at both the constriction and the central pocket.
As is the case for substrate binding, formation of ionic interactions at the constriction is less important for the potency of inhibition of COX-2 than COX-1. This fact has been exploited in the design of COX-2-selective inhibitors through conversion of the carboxylate moiety of some traditional NSAIDs to the corresponding ester or amide. Structural data suggest that these inhibitors may employ the spacious lobby of the enzyme to accommodate the added ester or amide functional groups, which may be quite large. In this regard, it is interesting to note that addition of a carboxylate to the uncharged diarylheterocycle scaffold has been used to reverse compound selectivity from COX-2 to COX-1 in the case of mofezolac. The following discussion reveals how X-ray crystallography coupled with site-directed mutagenesis has provided key insight into the determinants of inhibitor binding, structure–activity relationships, isoform selectivity, and in some cases, kinetic mechanism,
5.1. Overview of COX Inhibitors
The use of cyclooxygenase inhibitors to treat pain and inflammation can be traced back to the civilizations of ancient Egypt and Greece, from which records indicate that plants rich in salicylates were employed as medicines. Particularly notable was the use of willow bark as prescribed for joint pain by Dioscourides, a Greek physician of the Roman army. The active ingredient of willow bark was unknown until the 18th century, however, when salicin, a natural ester of salicyl alcohol, was identified. Later, Kolbe’s publication of the complete synthesis of salicylic acid enabled its marketing as a drug to the general public, although most patients found ingestion of the required large doses of the very bitter compound to be intolerable. This impediment led Felix Hoffman at the Bayer Company to synthesize and promote acetyl-salicylic acid (aspirin) as a more palatable derivative of the drug.68 Following World War II, the availability of animal models of inflammation was exploited to search for more powerful and safer anti-inflammatory medications. These efforts led to the discovery of such drugs as phenylbutazone, indomethacin, and ibuprofen; however, their primary mechanism of action was not understood until papers published in Nature New Biology in 1971 described the ability of aspirin, indomethacin, and sodium salicylate to inhibit the biosynthesis of PGs by guinea pig lung homogenates, human platelets, and perfused canine spleen.69−72 The subsequent purification of COX-1 in 197673 spurred a flurry of activity within the pharmaceutical industry and academia to produce better NSAIDs and to understand their pharmacology and chemical biology. This effort intensified after the discovery of COX-2 in 1991,74,75 as the increased expression of this isoform during inflammation suggested that COX-2-selective inhibitors (also referred to as coxibs) should provide anti-inflammatory activity with fewer side effects than those associated with isoform-nonselective NSAIDs. This hypothesis was based on the assumption that COX-1-derived PGs serve important homeostatic functions, particularly in the gastrointestinal tract. It is now known, however, that NSAID-associated gastrointestinal toxicity results from simultaneous blockade of both isoforms rather than selective blockade of COX-1.76,77 Today there are 27 NSAIDs on the market for clinical use in humans (18 available in the United States, and 9 available in other countries).78 These drugs are provided in multiple forms and are often incorporated along with other drugs in combination therapies. In addition, many more compounds with COX inhibitory activity that are not sold as pharmaceuticals have been described. These inhibitors may be classified on the basis of selectivity, kinetic mechanism, or chemical structure.
5.1.1. Selectivity
The majority of NSAIDs inhibit both COX isoforms, though they may exhibit varying degrees of selectivity for COX-1 or COX-2. As noted above, an aggressive effort to discover highly COX-2-selective inhibitors (coxibs) led to the development and marketing of a number of powerful agents in this class. These inhibitors fulfilled their promise as anti-inflammatory agents with reduced gastrointestinal side effects when compared to those of nonselective NSAIDs. However, concern about cardiovascular or liver toxicity led to the withdrawal of most of these agents from the market. Currently only one, celecoxib, is sold in the United States, although several older “nonselective” NSAIDS (i.e., meloxicam, diclofenac) exhibit COX-2 selectivity comparable to that of celecoxib.79−81 A number of COX-1-selective inhibitors has been reported in the literature, but none of these has been marketed to date in the United States.82−91
Recent evidence indicates that inhibitors may also be categorized on the basis of substrate selectivity. This is defined as the ability to potently and noncompetitively block endocannabinoid oxygenation by COX-2 while competitively inhibiting AA oxygenation with much lower potency. Current data support the hypothesis that these inhibitors block endocannabinoid but not AA oxygenation by binding in the allosteric monomer of COX-2, whereas inhibition of AA oxygenation requires binding in the catalytic subunit.92−95
5.1.2. Kinetic Mechanism
From a kinetic standpoint, the interaction of NSAIDs with the COX enzymes may be classified on the basis of binding affinity and time dependency.39 At one extreme is aspirin, which covalently modifies Ser-530 in the cyclooxygenase active site. Inhibition by aspirin is irreversible and time-dependent. At the other extreme is ibuprofen, a rapidly reversible inhibitor of AA oxygenation of low to moderate potency (IC50 values in the micromolar range). Between the two are the time-dependent, noncovalent inhibitors, such as indomethacin and flurbiprofen. Many of these exhibit potent (IC50 values in the nanomolar range) inhibition that is very slowly reversible. These classifications have become somewhat more complex with the introduction of isoform and substrate selectivity. For example, most potent COX-2-selective inhibitors exhibit time-dependent kinetics against COX-2 while inhibiting COX-1 weakly with rapidly reversible kinetics. Similarly, many rapidly reversible inhibitors of AA oxygenation by COX-2 are more potent, time-dependent inhibitors of endocannabinoid oxygenation.39,93,96
5.1.3. Chemical Structure
Most of the large number of COX inhibitors fall into one of five major chemical classes: phenylpropionic acids, arylacetic acids, fenamic acids, oxicams, and diarylheterocycles. This classification is useful in that compounds from similar classes generally interact with the cyclooxygenase active site in a similar binding pose. However, structural class does not necessarily predict kinetic behavior or selectivity for isoform or substrate.
5.2. Interaction of NSAIDs with the Cyclooxygenase Active Site
5.2.1. Binding Pockets
With such a wide range of chemical structures, it is not surprising that NSAIDs exhibit many different modes of binding to the cyclooxygenase active site. However, nearly all inhibitors interact with two or more binding pockets that will be described below. Depending on the size and orientation of an inhibitor, the number of binding pocket residues with which it establishes contacts will vary.
5.2.1.1. Proximal Binding Pocket
Nearly all inhibitors interact with the proximal binding pocket that lies near the constriction and overlaps with the proximal binding pocket for AA. Amino acids that form this pocket include Arg-120, Val-349, Ser-353, Tyr-355, Ile-523 (COX-1), Val-523 (COX-2), and Ala-527. Many inhibitors contain carboxylic acid groups that, like the carboxylate of AA, establish polar interactions with Arg-120 and/or Tyr-355 at the constriction. Nearly all inhibitors also form hydrophobic interactions with other residues in this pocket.
5.2.1.2. Central Binding Pocket
The central inhibitor binding pocket, which is formed by Leu-352, Leu-384, Tyr-385, Trp-387, Phe-381, Phe-518, Met-522, Gly-526, and Ser-530 is so named because it overlaps with the central binding pocket of AA. The majority of inhibitors extend into this binding pocket, establishing hydrophobic contacts. In some cases, polar interactions are established with Tyr-385 and/or Ser-530.
5.2.1.3. COX-2 Side Pocket
As noted above (see Section 4.3.6), there are three residues in the vicinity of the active site that differ between COX-1 and COX-2. These are Ile-434 (COX-1)/Val-434 (COX-2), Ile-523 (COX-1)/Val-523 (COX-2), and His-513 (COX-1)/Arg-513 (COX-2). The combination of these three substitutions provides access to a pocket to the side of the active site above the constriction in COX-2 that is not readily accessible in COX-1. This pocket, which is lined by His-90, Gln-192, Leu-352, Ser-353, Tyr-355, Arg-513, Ala-516, Phe-518, and Val-523 is exploited as a binding site primarily by the diarylheterocycle class of COX-2-selective inhibitors.
5.2.1.4. Oxicam Pocket
Available crystal structures of oxicams bound to COX-2 or COX-1 reveal a pocket surrounded by Met-113, Val-116, Leu-117, Ile-345, Val-349, Leu-359, Leu-531, Leu-534, and Met-535 into which a portion of the molecule is inserted. Access to this pocket is provided by a rotation of Leu-531 away from the active site constriction.
5.2.2. Phenylpropionic Acid Inhibitors
The first published COX crystal structure was of COX-1 complexed with Fe3+-protoporphyrin IX and the phenylpropionic acid inhibitor (S)-flurbiprofen (PDB 1CQE).29 Over the ensuing 20 years, ten additional structures of COX-1, COX-2, or a mutant COX protein complexed with (S)-flurbiprofen or derivatives/analogs of (S)-flurbiprofen have been published. Thus, this potent, time-dependent, isoform-nonselective inhibitor is the single most thoroughly studied of all of the NSAIDs from a structural point of view. Other phenylpropionic acids for which structural data are available include ibuprofen, naproxen, and their derivatives.
5.2.2.1. (S)-Flurbiprofen and its Derivatives
There are three published crystal structures of COX-1 complexed with Fe3+-protoporphyrin IX and (S)-flurbiprofen (PDB 1CQE at 3.1 Å, PDB 1EQH at 2.7 Å, PDB 3N8Z at 2.9 Å) [Figure S6A (stereoscopic view) and Figure S6B (monoscopic view), based on 1EQH].27,29,97 In all cases, only the (S)-enantiomer of flurbiprofen is observed in the active site, even when racemic flurbiprofen was used to generate the complex. This is consistent with the much higher potency of the (S)-enantiomer as compared to the (R)-enantiomer of the inhibitor. The structures indicate the presence of one or two polar interactions (reported as salt bridges or hydrogen bonds) between the carboxylate of flurbiprofen and the guanidinium group of Arg-120 in addition to a hydrogen bond between the flurbiprofen carboxylate and the phenolic hydroxyl of Tyr-355. Salt bridges are also formed between the carboxylate of Glu-524 and Arg-120. These interactions, along with hydrogen bonds involving His-90 and two ordered water molecules form a polar interaction network at the constriction that locks the inhibitor in place.27,29,97
Above the constriction, the fluorophenyl ring of flurbiprofen is surrounded by the proximal inhibitor binding pocket, making hydrophobic contacts with Val-349, Ile-523, Ala-527, and Ser-530 (Figure 12). The phenyl ring inserts into the central inhibitor binding pocket, making hydrophobic contacts with Leu-352, Leu-384, Tyr-385, Trp-387, Gly-526, and Ser-530 (Figure 13). The fluoro substituent lies between the two pockets, nestled in a cleft lined by Leu-352, Phe-518, and Ile-523.27,29,97
Figure 12.

Wall-eyed stereo view of the interaction of the proximal inhibitor binding pocket with (S)-flurbiprofen as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking upward from the membrane (B). (S)-Flurbiprofen is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 1EQH.
Figure 13.

Wall-eyed stereo view of the interaction of the central inhibitor binding pocket with (S)-flurbiprofen as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking upward from the membrane (B). (S)-Flurbiprofen is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 1EQH.
A 2.5 Å resolution structure of (S)-flurbiprofen complexed with COX-2 (PDB 3PGH) was published in 1996.32 This structure showed that the overall placement of the inhibitor in the cyclooxygenase active site is similar for the two isoforms. However, a contact between the fluoro group of flurbiprofen and Ile-523 observed in COX-1 is not established with the shorter side chain of Val-523 of COX-2.32
Additional studies employed structural variants of enzyme or inhibitor. A higher resolution (2.0 Å) structure of COX-1 complexed with (S)-flurbiprofen and Mn2+-protoporphyrin IX (PDB 2AYL) revealed the presence of two conformations of the fluorophenyl ring that differed by a 180° rotation. The investigators concluded that the two conformations likely were present in prior structures but not observed due to lower resolution.98 A crystal structure of COX-1 in complex with Fe3+-protoporphyrin IX and (S)-α-methyl-4-biphenylacetic acid (PDB 1Q4G), a des-fluoro analog of (S)-flurbiprofen, was also notable for its high resolution (2.0 Å). This structure revealed valuable additional information concerning COX protein structure, particularly in the region of the peroxidase active site, but no new insights into protein-inhibitor interactions were reported.99
A structure of the methyl ester of (S)-flurbiprofen in complex with COX-1 (PDB 1HT5, 2.8 Å) revealed a very similar binding pose to that of the parent compound.97 Although the ester group forms polar contacts with Arg-120 and Tyr-355, the methyl substituent comes into close contact with Leu-531, leading to a small perturbation of the hydrogen-bonding network at the constriction. This is accompanied by a slight rotation between the phenyl ring and α-carbon of the inhibitor. A notable observation from this study is that differences in the structures of COX-1 complexed with flurbiprofen or its methyl ester do not readily explain why flurbiprofen is a slow, tight-binding inhibitor, whereas inhibition by the methyl ester is weak and rapidly reversible.97 This conclusion is true in general; with a few exceptions, it has been difficult to use structural data alone to explain differences in inhibitor potency and kinetics.
Both (R)-flurbiprofen and des-methyl-flurbiprofen are classified as substrate-selective inhibitors of COX-2 in that they are poor inhibitors of AA oxygenation but potent inhibitors of 2-AG oxygenation.92,94 In search of a structural foundation for this behavior, a 2.8 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (R)-flurbiprofen (PDB 3RR3) was acquired (Figure 14A).92 Previously published structures of (S)-flurbiprofen complexed with COX isoforms had suggested that (R)-flurbiprofen would bind poorly in the cyclooxygenase active site as a result of a steric clash between the α-methyl group and Tyr-355.27,29,32,97 However, the structure of the COX-2-(R)-flurbiprofen complex demonstrated that this is not the case. In fact, the α-methyl group of (R)-flurbiprofen is directed toward Tyr-355 to a greater degree than that of (S)-flurbiprofen, but a clash is avoided by a shift in the position of the inhibitor’s carboxylate group, α-carbon, and fluorophenyl ring. A consequence of this shift is that (R)-flurbiprofen makes fewer total contacts (42) than does (S)-flurbiprofen (48) with the COX-2 active site, although polar contacts with Arg-120 and Tyr-355 are retained.92 Similar efforts to understand the substrate selectivity of des-methyl-flurbiprofen focused on the analysis of a 2.8 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and this inhibitor (PDB 4FM5).94 The results showed that the des-methyl analog binds in the active site in a very similar overall conformation as does (S)-flurbiprofen, and it makes the expected polar contacts with Arg-120 and Tyr-355. However, the loss of the methyl group substantially reduces the number of hydrophobic contacts, so the total number of interactions is only 35.94 For both (R)-flurbiprofen and des-methyl-flurbiprofen, the reduced number of contacts with the active site explains the lower potency against AA, but it does not readily explain the ability of these molecules to inhibit 2-AG oxygenation with considerably greater potency than they inhibit AA oxygenation.
Figure 14.

Wall-eyed stereo views of overlays of the structures of (R)- and (S)-flurbiprofen (A) and (R)- and (S)-naproxen (B), bound in the cyclooxygenase active site of COX-2 and the side chains that make up the proximal binding pocket (light/dark green) and the central binding pocket (pink/magenta). In each case, structures related to the (R)-enantiomer (tan) are shown in the lighter color and those related to the (S)-enantiomer (sienna) are shown in the darker color. From PDB 3PGH and 3RR3 (A) and PDB 3NT1 and 3Q7D (B).
A number of site-directed mutation studies have explored the importance of various enzyme–inhibitor interactions to flurbiprofen’s potency. R120E and R120Q mutations in both COX-1 and COX-2 markedly decreased or eliminated inhibition by flurbiprofen.43,44,57,58 These findings confirm the importance of the interaction between the carboxylate of the inhibitor and the guanidinium group of Arg-120 and suggest that salt-bridge formation is required for full inhibitor potency. A W387F mutation in COX-2 had only a minor effect on flurbiprofen potency, suggesting that hydrophobic interactions with this amino acid were either retained with the smaller phenylalanine residue or of minor importance.100 Similarly an S530A mutation in COX-1 and S530M mutation in COX-2 had only minimal effects on potency.49,55 A V523I mutation in COX-2, which converts this residue to that found in COX-1, had no significant effect on flurbiprofen-mediated inhibition, consistent with the inhibitor’s strong potency for both isoforms.101
The endocannabinoid AEA triggers anti-inflammatory signaling and is cytoprotective in the gastrointestinal tract. This led to the hypothesis that the combination of an inhibitor of FAAH (the primary AEA degradative enzyme) with an NSAID would produce increased anti-inflammatory activity with reduced gastrointestinal toxicity. Support for this hypothesis was obtained through the synthesis and evaluation of ARN-2508, a carbamate-linked conjugate of the FAAH inhibitor URB597 and flurbiprofen. This compound exhibited high anti-inflammatory activity while protecting the gastrointestinal epithelium in mice.102 The kinetic behavior of ARN-2508 was similar to that of flurbiprofen with regard to enantio, isoform, and substrate selectivity; however, the time required to achieve inhibition was greater for ARN-2508 than for flurbiprofen. A 2.3 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (S)-ARN-2508 (PDB 5W58) revealed that the flurbiprofen moiety of the molecule adopts a binding pose very similar to that of (S)-flurbiprofen (Figure S7).103 However, the carbamate group forms hydrogen bonds with Tyr-385 and Ser-530 in the central inhibitor binding pocket, and the alkyl chain derived from URB597 extends into the region corresponding to the AA distal binding pocket. To date, ARN-2508 is the only COX inhibitor shown to interact with this region of the active site. A Y355F mutation of COX-2 only mildly reduced the enzyme’s sensitivity to ARN-2508, suggesting that polar interactions at this site were not critical to potency. Mutation of Ser-530 to alanine was notable for markedly increasing the rate of ARN-2508-mediated enzyme inactivation, while having little effect on potency. This finding suggests that Ser-530 may serve as a kinetic impediment to binding. A G533L mutation, which blocks access to the distal AA binding pocket, resulted in substantially reduced sensitivity to inhibition.103
5.2.2.2. (S)-Iodosuprofen
Early attempts to obtain crystal structures of COX-inhibitor complexes were often thwarted by the difficulties arising from working with fragile crystals, poor reproducibility, and poor resolution. Consequently, an attempt was made to obtain the structure of COX-1 complexed with Fe3+-protoporphyrin IX and iodosuprofen. The addition of an iodine atom to the thiophene ring of the NSAID suprofen resulted in a mild loss of inhibitory potency but offered the presence of a heavy atom to be used in localizing the inhibitor under conditions of poor resolution. The resulting 3.5 Å resolution crystal structure (PDB 1PGE) provided considerable data on the binding interaction of the inhibitor with the COX-1 active site (Figure S6C).30 As in the case of flurbiprofen, only the (S)-enantiomer is seen, despite the presence of both enantiomers during crystallization. The carboxylate of (S)-iodosuprofen is close to the constriction, forming polar interactions with Arg-120 and Tyr-355. A hydrogen bond between the ketone group that joins the two aromatic rings and the hydroxyl group of Ser-530 is the only other polar interaction. The phenyl ring is located in the proximal inhibitor binding pocket, and hydrophobic interactions with Val-349, Leu-531, Ile-523, and Ala-527 can be observed. The iodo-thiophene ring extends upward into the central binding pocket, forming contacts with Leu-352, Leu-384, Tyr-385, Trp-387, and Gly-526. The α-methyl group interacts with a hydrophobic cleft lined by leucine and valine residues, similar to what is observed in the COX-1-(S)-flurbiprofen complex.30
5.2.2.3. (S)-Ibuprofen
(S)-Ibuprofen is a weak, rapidly reversible inhibitor of AA oxygenation by both COX-1 and COX-2, as well as a much more potent inhibitor of 2-AG oxygenation by COX-2.93 A 2.6 Å resolution crystal structure of COX-1 complexed with Fe3+-protoporphyrin IX and (S)-ibuprofen (PDB 1EQG) published in 2001 revealed polar interactions between the carboxylate of the inhibitor and both Tyr-355 and Arg-120 at the constriction (Figure S6D).97 The network of hydrogen bonds observed in the structure of (S)-flurbiprofen bound to COX-1(see Section 5.2.2.1) is recapitulated in the COX-1-(S)-ibuprofen complex. As seen with the fluorophenyl ring of (S)-flurbiprofen, the aromatic ring of ibuprofen lies in the proximal inhibitor binding pocket. The isobutyl substituent extends upward toward the central inhibitor binding pocket, but due to its relatively small size (compared to that of the phenyl ring of (S)-flurbiprofen), it makes few contacts there. An interesting difference between the structures of (S)-flurbiprofen and (S)-ibuprofen complexed to COX-1 is the rotameric state of the Ser-530 side chain. A possible explanation for this is that Ser-530 can establish a hydroxyl-aromatic hydrogen bond with the phenyl ring of (S)-flurbiprofen that is not possible with (S)-ibuprofen.97
A 1.8 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (S)-ibuprofen (PDB 4PH9) was acquired with the goal of learning the structural basis for substrate-selective inhibition of 2-AG oxygenation.104 The binding pose of the inhibitor, which is present in both monomers, is essentially the same as that observed in the COX-1-(S)-ibuprofen complex. Contacts with Val-349, Val-523, Gly-526, Ala-527, and Ser-530 in the proximal pocket and with Trp-387, Met-522, and Gly-526 in the central pocket were reported. The α-methyl group forms contacts with Val-349 and Leu-359. As might be predicted, R120A and Y355F mutations resulted in a marked reduction in potency for (S)-ibuprofen. However, incubation of the Y355F mutant enzyme with (S)-ibuprofen led to thermal stabilization, suggesting that the protein retains some capacity for inhibitor binding. This is not the case for the R120A mutant. The structure did not convey any new insights into the basis for substrate-selective inhibition.104
Studies intended to explore the structural basis for the rapid reversibility of some inhibitors led to the discovery that V89W/H90W and V89W/S119W double mutant COX-2 enzymes demonstrated a marked increase in sensitivity to inhibition by ibuprofen. These mutations also converted ibuprofen from a rapidly reversible to slow tight-binding inhibitor.105 The V89W mutation was primarily responsible for this effect, with some additional contribution from the H90W mutation. To better understand these observations, a 2.8 Å resolution crystal structure of H90W COX-2 complexed with Fe3+-protoporphyrin IX and (S)-ibuprofen (PDB 4RS0) was acquired. It is notable that the heme was not resolved in this structure despite its presence during crystallization, so it is not included in the data submitted to the PDB. The data revealed no significant change in the binding pose of the inhibitor in the active site or in overall protein conformation except in the region of Trp-90. The orientation of this residue directs it toward the protein surface where the nitrogen atom of the indole ring interacts with a solvent molecule. Hydrophobic contacts are established between the side chain of Trp-90 and those of Arg-513, Pro-514, and Thr-94. The mutation does not significantly alter the structure of the lobby or the active site, but it does cause minor changes to the side pocket that alter thermodynamic stability in that region. The absence of any major effect on the binding pose of (S)-ibuprofen or its established inhibitor-protein interactions suggests that the mutation’s effect on inhibitor kinetics is likely to result from changes in binding dynamics.105
5.2.2.4. (S)-Naproxen and Its Derivatives
(S)-Naproxen is a time-dependent inhibitor of moderate potency for both COX-1 and COX-2. Despite its widespread use as an over-the-counter pain medication, the structural determinants of (S)-naproxen-dependent COX inhibition were not explored until 2010 with the publication of a 1.7 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (S)-naproxen (PDB 3NT1) (Figure S6E).100 The structure revealed that, as in the case of other inhibitors of its class, (S)-naproxen binds with its carboxylate group forming polar interactions with Arg-120 and Tyr-355 at the constriction and its α-methyl group directed toward Val-349 and Leu-359. The naphthyl group extends upward into both the proximal and central inhibitor binding pockets. Interactions with Leu-352, Gly-526, and Ala-527 were specifically noted. An interesting finding was the conformation of Leu-352, which is different from that observed in most other COX-inhibitor complex structures. The methoxy group of (S)-naproxen points toward the apex of the active site where the channel bends sharply prior to ending in the AA distal binding pocket. There, it interacts with Tyr-385 and Trp-387.100
The crystal structure data are consistent with the results of site-directed mutagenesis studies. Loss of potency against R120A and Y355F COX-2 mutants demonstrates the need for polar interactions with these residues. However, the finding that the R120Q mutant was inhibited with greater potency than the wild-type enzyme suggests that the interaction need not be ionic in nature. A V349A mutation had no effect on (S)-naproxen potency, but V349I and V349L mutants were inhibited with greater potency, possibly due to their ability to establish stronger hydrophobic contacts with the α-methyl group. Consistently, 2-des-methyl-naproxen exhibited lower potency than (S)-naproxen for wild-type COX-2, as well as the V349I and V349L mutant enzymes. Inconsistently, however, the α-ethyl analog of (S)-naproxen was inactive. The α-dimethyl analog was also inactive.100
Replacing the methoxy group of naproxen with ethoxy or hydroxy groups resulted in a loss in potency, suggesting the importance of interactions with Tyr-385 and/or Trp-387 at the top of the active site channel. Consistently, a W387F mutant COX-2 was poorly inhibited by (S)-naproxen. Interestingly, this is the only inhibitor currently known for which the W387F mutation affects potency. Activity was restored for the mutant by substitution of naproxen’s methoxy group with an ethyl or a methylthio group. These analogs were also active against wild-type COX-2 but exhibited poor activity against COX-1.100
To further explore the impact of the methylthio substitution on naproxen, a 2.3 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and 6-methylthio-(S)-naproxen (PDB 3NTB) was acquired. The binding pose of the methylthio analog is highly similar to that of naproxen, although it does not penetrate as deeply into the active site as does the parent compound. The sulfur and oxygen atoms of the methylthio group of methylthio-naproxen and methoxy group of naproxen, respectively, are very closely aligned in their crystal structures. The largest deviations between the two structures are at the carboxylate groups of the inhibitors. Also notable is the observation that the conformation of Leu-352 in the COX-2-methylthio-naproxen complex is different from that in the COX-2-naproxen complex, more closely resembling that observed in the majority of COX-2-inhibitor complexes. As a result, methylthio-naproxen does not form contacts with Leu-352 that are observed for naproxen. It does, however, form a contact with Val-523 that is not observed in the COX-2-naproxen structure.100
(R)-Naproxen is a poor inhibitor of AA oxygenation but a potent inhibitor of 2-AG oxygenation.92 In an attempt to understand the structural basis for this substrate selectivity, a 2.4 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (R)-naproxen (PDB 3Q7D) was obtained (Figure 14B).92 The data revealed an almost identical binding pose for (R)- and (S)-naproxen in the COX-2 active site, including similar interactions with Arg-120, Val-349, Leu-352, Tyr-355, Tyr-385, Trp-387, Gly-526, and Ala-527. As might be predicted, differences are noted in the region of the α-methyl group which, in the case of the (R)-enantiomer, establishes a contact with Ser-353 that is not observed for the (S)-enantiomer whereas the (S)- but not the (R)-enantiomer interacts with Leu-359. Another important difference between the two structures is the position of Arg-120 and Tyr-355, as a shift of those residues must occur to accommodate the α-methyl group of the (R)-enantiomer. The shift increases the hydrogen bonding distance between the carboxylate of the inhibitor and Tyr-355, a change that might lead to a reduction in binding energy for (R)-naproxen. This could help to explain its poor inhibitory potency for AA oxygenation, but it does not clarify how this same inhibitor can potently block oxygenation of 2-AG.92
5.2.3. Arylacetic Acid Inhibitors
COX-inhibitor complex crystal structures are available for a number of arylacetic acid inhibitors, including indomethacin, diclofenac, lumiracoxib, and alclofenac. In addition, structures have been obtained for complexes of COX-1 or COX-2 with a number of amides of indomethacin, most of which are selective COX-2 inhibitors. A particularly interesting observation within this class of inhibitors is the difference in orientation of the carboxyl group between indomethacin and inhibitors of similar structure to that of diclofenac, as will be described below. Note that a COX-1-selective inhibitor (mofezolac) is an arylacetic acid; however, it also can be classified as a diarylheterocycle and will be discussed with that class.
5.2.3.1. Indomethacin
Indomethacin is a slow, very tight-binding inhibitor of both COX-1 and COX-2, with a slightly higher potency for COX-1. An early attempt to obtain a crystal structure of indomethacin with COX-1 employed an analog in which an iodobenzoyl group replaced the chlorobenzoyl substituent. This alteration, which resulted in a small increase in potency of the inhibitor, was designed to improve the likelihood of identifying its location in the case of poor resolution data. In fact, the approach proved to be valuable, as only the iodine atom of the inhibitor could be clearly identified in the 3.5 Å resolution structure that was obtained of COX-1 complexed to Fe3+-protoporphyrin IX and iodoindomethacin.30 The overall rigidity of the compound and limited space in the COX-1 active site enabled the researchers to propose two models for the binding pose of the inhibitor, one (PDB 1PGG) in which the carbon-to-nitrogen bond between indomethacin’s indole ring and chlorobenzoyl group is in the (Z) configuration and the second (PDB 1PGF) in which the configuration of this bond is (E). Both models suggest a possible hydrogen bond between the carbonyl linking the phenyl and indole rings and Ser-530. Both also suggest a polar interaction between the carboxyl group and Arg-120, and the (E) model predicts a hydrogen bond between the carboxylate and Tyr-355. Hydrophobic contacts between indomethacin’s indole ring and Val-349, Leu-359, Ala-527, and Leu-531 of COX-1 are also predicted. Although the structural data alone did not allow a choice between the two models, the relative potencies of isosteres of (E)- versus (Z)-indomethacin suggest that the (E) configuration is the more likely.30
A 2.9 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and indomethacin (PDB 4COX) enabled more complete visualization of the inhibitor in the cyclooxygenase active site (Figure S8A).32 Consistent with the modeled structure of the COX-1-iodoindomethacin complex, this structure places the carboxylate moiety of indomethacin near the constriction, establishing polar contacts with Arg-120 and Tyr-355. The indole ring binds in the proximal inhibitor binding pocket and makes hydrophobic contacts with Val-349, Ser-353, Tyr-355, Val-523, and Ala-527 (Figure 15). The chlorobenzoyl ring, which is in the E-configuration with respect to the indole, projects into the central inhibitor binding pocket, where it interacts with Phe-381, Leu-384, Tyr-385, and Trp-387 (Figure 16). The carbonyl group bridging the two ring systems forms a hydrogen bond with Ser-530, and the methoxy group protrudes into a cavity adjacent to Ser-353, Tyr-355, and Val-523. The 2′-methyl group of the indole ring also interacts with a hydrophobic pocket, in this case lined by Val-349, Ala-527, Ser-530, and Leu-531.32
Figure 15.

Wall-eyed stereo view of the interaction of the proximal inhibitor binding pocket with indomethacin as observed from the side (i.e., parallel to the plane of the membrane) (A) or from above (B). Indomethacin is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 4COX.
Figure 16.

Wall-eyed stereo view of the interaction of the central inhibitor binding pocket with indomethacin as observed from the side (i.e., parallel to the plane of the membrane) (A) or looking upward from the membrane (B). Indomethacin is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 4COX.
The structural data are generally consistent with the results of site-directed mutation studies. R120E and R120A mutations in COX-1 and/or COX-2 resulted in a loss of potency for indomethacin.50,55,57 R120Q and Y355F mutations in COX-2 have been reported to be totally resistant to indomethacin or to show the same sensitivity as the wild-type enzyme to the inhibitor.106,107 These results support a requirement to establish a polar interaction with one or both of these residues, although there is no current explanation for the discrepancies. An S530A mutation in COX-2 had little effect on indomethacin potency, suggesting that the hydrogen bond between this residue and the carbonyl of the inhibitor is not a major determinant of binding energy.50 A V349A mutation in COX-2 increased inhibitor potency, while a V349I mutation had little effect and a V349L mutation reduced potency. These findings have been explained on the basis of the effects these mutations have on the 2′-methyl binding pocket. Mutation to a smaller side chain deepens the pocket and increases binding affinity, whereas mutation to a bulkier chain has the opposite effect.108 Also supporting this interpretation, removal of the 2′-methyl group converts indomethacin from a highly potent tight-binding COX inhibitor to 2′-des-methyl-indomethacin, a weak, rapidly reversible inhibitor.108
5.2.3.2. Alclofenac
Alclofenac is a time-dependent, moderately potent inhibitor of both COX isoforms. A 2.7 Å resolution crystal structure of COX-1 complexed to Fe3+-protoporphyrin IX and alclofenac (PDB 1HT8) demonstrated that, similar to most carboxylic acid-containing NSAIDs, this inhibitor binds in the enzyme active site with its carboxyl group oriented toward the constriction where it forms polar interactions with Arg-120 and Tyr-355 (Figure S8B). Alclofenac’s aromatic ring lies in the proximal inhibitor binding pocket, establishing hydrophobic interactions with Val-349, Ser-353, Tyr-355, Ile-523, and Ala-527. The aliphatic substituent of alclofenac extends upward into the central binding pocket and establishes contacts with Leu-352, Leu-384, Tyr-385, Trp-387, and Ser-530.97
5.2.3.3. Diclofenac
Diclofenac is a time-dependent, potent inhibitor of both COX isoforms that exhibits significant COX-2 selectivity. Early studies using site-directed mutant enzymes revealed that, in contrast to most other carboxylate-containing inhibitors, R120A, R120E, and Y355F mutations of COX-1 and/or COX-2 had minimal effect on diclofenac potency, whereas S530A eliminated sensitivity to the inhibitor.44,50,57 These results suggested that polar interactions between diclofenac’s carboxylate and Arg-120 or Tyr-355 were unlikely to contribute significantly to inhibitor binding and had been replaced by an interaction with Ser-530’s hydroxyl group. A 2.9 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and diclofenac (PDB 1PXX) supported this hypothesis [Figure S9A (stereoscopic view) and Figure S9B,C (monoscopic views)].50 Rather than being oriented toward the constriction, the carboxylate of diclofenac is located high in the active site channel, forming polar interactions with Tyr-385 and Ser-530. The aromatic ring that bears the acetate moiety lies in the central inhibitor binding pocket, establishing hydrophobic contacts with Leu-352, Leu-384, Tyr-385, Trp-387, Met-522, and Gly-526. The chlorine-bearing aromatic ring lies in the proximal binding pocket, interacting with Val-349, Val-523, Ala-527, Ser-530, and Leu-531, and one of the chlorine substituents occupies the same pocket into which the 2′-methyl group of indomethacin inserts (see Section 5.2.3.1).108 No contacts are established with Arg-120 or Tyr-355 at the constriction.50 Note that the binding of diclofenac’s carboxyl group to Tyr-385 and Ser-530 is reminiscent of the binding of AA’s carboxylate to these residues in the nonproductive conformation observed in the crystal structure of AA complexed with COX-2 (see Section 4.1.2).
5.2.3.4. Lumiracoxib
Although reported to be the most potent COX-2-selective inhibitor in vivo, lumiracoxib is a relatively weak, time-dependent, COX-2-selective inhibitor in vitro.109 It also demonstrates substrate selectivity in that it blocks endocannabinoid oxygenation with much higher potency than it blocks AA oxygenation.95 Attempts to obtain crystals of apo-COX-2 or the holo-enzyme (COX-2 complexed with Fe3+-protoporphyrin IX) in complex with lumiracoxib were successful only in the case of the apoenzyme. The 2.4 Å resolution crystal structure of the apo-COX-2-lumiracoxib complex (PDB 4OTY) reveals that, like its structural analog diclofenac, lumiracoxib binds in the cyclooxygenase active site with its carboxylate directed toward and forming hydrogen bonds with Tyr-385 and Ser-530 (Figure S9D,E).95 Consistently, an S530A mutation in COX-2 eliminated susceptibility to lumiracoxib-mediated inhibition.109 The phenyl ring that bears the inhibitor’s acetic acid moiety lies in the central inhibitor binding pocket, where its methyl substituent inserts into an alcove formed by Phe-381, Leu-384, Trp-387, Phe-518, and Met-522. Formation of this pocket requires a rotation of Leu-384 away from the active site, toward Leu-503. The movement is possible in COX-2 but precluded in COX-1 due to the presence of the larger phenylalanine residue at the 503 position (Figure S10). This observation helps to explain the poor potency of lumiracoxib for COX-1 and for an L503F mutant COX-2 enzyme. As in the case of diclofenac, the halogen-containing ring of lumiracoxib binds in the proximal binding pocket, interacting with Val-349, Val-523, Ala-527, Ser-530, and Leu-531, and its chlorine atom inserts into the same pocket into which the 2′-methyl group of indomethacin inserts (see Section 5.2.3.1).95,108 Formation of a weak hydrogen bond between the chlorine atom of lumiracoxib and the hydroxyl group of Ser-530 might explain why lumiracoxib analogs bearing a methyl group or hydrogen atom in this position exhibit reduced potency for COX-2. As in the case of diclofenac, lumiracoxib forms no contacts with Arg-120 or Tyr-355.95
As described above for (S)-ibuprofen (see Section 5.2.2.3), V89W/H90W and V89W/S119W double mutant COX-2 enzymes demonstrated a marked increase in sensitivity to inhibition by lumiracoxib.105 The V89W mutation was primarily responsible for this effect, with some additional contribution from the H90W mutation. A 2.8 Å resolution crystal structure of apo-V89W COX-2 complexed with lumiracoxib (PDB 4RRX) and a 2.6 Å resolution crystal structure of apo-H90W COX-2 complexed with lumiracoxib (PDB 4RRZ) provided some insight into the structural basis for the effects of these mutations. In both cases, the presence of the mutation did not lead to a significant change in the overall conformation of the protein or the binding pose of the inhibitor in the cyclooxygenase active site. For the V89W mutation, the Trp-89 side chain fills a gap between helices B and D of the membrane-binding domain, converting a half-closed “donut hole” into a completely closed one and blocking a potential exit channel for the inhibitor. Thus, this mutation appears to substantially restrict egress from the active site. As in the case of the complex of holo-H90W COX-2 with (S)-ibuprofen, the bulky Trp-90 side chain does not cause a significant alteration in the structure of the active site or lobby, suggesting that its effects on lumiracoxib potency are due to changes in the dynamics of inhibitor binding.105
5.2.3.5. Indomethacin Amides and Esters
As discussed above (see Sections 4.1.1.1 and 4.1.2), formation of an ionic interaction between the carboxylate of AA and Arg-120 appears to be highly important for the catalytic activity of COX-1 but not COX-2. This led to the hypothesis that converting nonselective carboxylate-containing NSAIDs into their ester or amide derivatives could be a means to easily derive COX-2-selective inhibitors. This approach proved successful for a wide range of esters and amides of indomethacin and meclofenamic acid.106,110,111 An exception was noted, however, for α-substituted ethanolamides, in which case the (R)-enantiomer exhibited the expected COX-2 selectivity, but the (S)-enantiomer was unselective. This was observed over a range of α-substituents, and a structural basis for the unexpected finding was sought. Hence, a 2.7 Å resolution crystal structure of COX-1 complexed to Fe3+-protoporphyrin IX and indomethacin-(S)-α-ethyl-ethanolamide (PDB 2OYU), and a 2.8 Å resolution crystal structure of COX-1 complexed to Fe3+-protoporphyrin IX and indomethacin-(R)-α-ethyl-ethanolamide (PDB 2OYE) were obtained.112
In the case of the (R)-enantiomer complex, the binding pose of the indomethacin portion of the molecule is very similar to that of indomethacin in the previously reported crystal structure (Figure S11A). The chlorobenzoyl group binds near Tyr-385 and the ethanolamide near Arg-120 and Tyr 355. Hydrogen bonds are established between Arg-120, Glu-524, and the hydroxyl group of the inhibitor’s ethanolamide moiety. To establish this hydrogen bonding network, the side chain of Arg-120 adopts a conformation that is rarely seen in COX-1-inhibitor complex crystal structures. The geometric configuration of the carbon-to-nitrogen bond between indomethacin and the α-ethyl-ethanolamide moiety could not be assigned with certainty, but the finding that the (Z)-isomer could make an additional hydrogen bond with Glu-524 suggests that it is preferred.112
The (S)-enantiomer complex is notable for the strikingly different binding pose of the indomethacin moiety in the cyclooxygenase active site (Figure S11B). The chlorobenzoyl group is directed toward the mouth of the channel, placing the methoxy group near Tyr-385. The ethanolamide binds in a pocket comprising His-90, Gln-192, Leu-517, Phe-518, and Ile-523. The position of this pocket is similar to that of the side pocket of COX-2 that is exploited by the diarylheterocycle class of COX-2-selective inhibitors (see Sections 5.2.1.3 and 5.2.6). This orientation enables formation of hydrogen bonds between the hydroxyl of the inhibitor’s ethanolamide group and both His-90 and Gln-192. In addition, the carbonyl that joins the chlorobenzoyl group to the indole ring forms a hydrogen bond with Arg-120. Hydrophobic interactions are established with Ile-517, Phe-518, and Ile-523. As in the case of the (R)-enantiomer, the (Z) configuration of the carbon-to-nitrogen bond between indomethacin and the α-ethyl-ethanolamide moiety enables the establishment of an additional hydrogen bond, in this case between the amide nitrogen of the inhibitor and the backbone carbonyl of Leu-352. Also of note, the configuration of the carbon-to-nitrogen bond between the indole and chlorobenzoyl groups is (Z) in this structure as opposed to (E) for the (R)-enantiomer and unmodified indomethacin. Modeling studies indicate that the (R)-enantiomer is unable to adopt this binding conformation due to potential steric clashes between the α-ethyl moiety and residues in the side pocket. Thus, the results suggest that the ability of the (S)-enantiomer to assume this particular binding pose within the COX-1 active site enables it to effectively inhibit the enzyme, whereas the (R)-enantiomer retains COX-2 selectivity.112
Structure–activity relationship studies of indomethacin amides indicated that a wide range of chemical entities could be attached to the NSAID nucleus to create a potent COX-2-selective inhibitor.106,110,111 This led to the hypothesis that linking a chemotherapeutic or imaging agent to indomethacin could serve as a means to target the agent to cancer cells that express unusually high levels of COX-2. The hypothesis was tested with the creation of an indomethacin-podophyllotoxin conjugate that exhibited increased COX-2-dependent antitumor activity in vivo but not in cell cultures. A 2.1 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and the indomethacin-podophyllotoxin conjugate (PDB 4OTJ) revealed that the indomethacin portion of the molecule adopts a binding pose very similar to that of indomethacin in COX-2 (Figure S8C).107 The amide nitrogen of the conjugate forms a hydrogen bond to Tyr-355 at the constriction, and the 10-atom linker that connects indomethacin to podophyllotoxin passes through the constriction into the lobby region. Podophyllotoxin is assumed to be located in the lobby but was not visualized in the diffraction data. Computational modeling places this portion of the molecule underneath the D helix of the membrane-binding domain and predicts interactions with Lys-83, Val-89, His-90, Tyr-115, and Ser-119.107
The effects of site-directed mutations on the potency of the indomethacin-podophyllotoxin conjugate were similar to their effects on the potency of indomethacin, with differences that were mostly quantitative rather than qualitative. Thus, V349L, V523I, and S530A mutations resulted in greater losses of activity for the conjugate than for indomethacin. As in the case of indomethacin, Y355F resulted in a total loss of activity of the conjugate, but R120Q increased the conjugate’s potency while having no effect on the potency of indomethacin. These findings are consistent with the similarity of binding between the conjugate and its parent molecule.107
The crystal structure of the complex of COX-2 with the indomethacin-podophyllotoxin conjugate supported a long-held hypothesis that moieties attached to the carboxylate of indomethacin via amide or ester linkages will bind in the lobby of the cyclooxygenase active site with the linker passing through the constriction. Stronger support for this hypothesis came from studies of two indomethacin-dansyl conjugates that were synthesized as part of an effort to discover COX-2-targeted optical imaging agents. The two molecules differed only in the length of the linker joining indomethacin to the dansyl moiety, with conjugates 1 and 2 employing 1,2-diaminoethane and 1,4-diaminobutane, respectively. Crystal structures of both conjugate 1 (PDB 6BL4, Figure 17A) and conjugate 2 (PDB 6BL3, Figure 17B) complexed with COX-2 and Fe3+-protoporphyrin IX were obtained at 2.2 Å resolution.113 In both cases, the indomethacin portion of the molecules adopts a binding pose similar to that of the parent compound in COX-2. A notable exception is the failure of the inhibitor to establish a contact with Arg-120 and displacement of that residue to provide room for the linker to pass through the constriction. In the case of the complex with conjugate 1, two highly ordered water molecules establish contacts with residues in the constriction to compensate for the loss of contact with Arg-120. One water molecule is observed in the complex containing conjugate 2. The dansyl moieties of the two conjugates adopt very similar poses in the lobby region, establishing hydrophobic contacts with Val-89 and Leu-93, and a hydrogen bond links an oxygen atom of the dansyl sulfonyl group to the side chain of Ser-119.113
Figure 17.

Wall-eyed stereo views of the structure of indomethacin-dansyl conjugate 1 (A) and indomethacin-dansyl conjugate 2 (B) bound in the cyclooxygenase active site of COX-2. The side chains that make up the proximal binding pocket (green), the central binding pocket (magenta), and membrane-binding domain residues that interact with the dansyl moiety (cyan) are shown. From PDB 6BL4 (A) and 6BL3 (B).
Site-directed mutagenesis studies generally supported conclusions drawn from the crystal structure data. Mutation of Arg-120 to alanine resulted in a marked increase in potency for both dansyl conjugates, in contrast to a dramatic loss of potency for indomethacin. This is consistent with the observation that the dansyl conjugates do not form a polar contact with Arg-120 as is observed with indomethacin, and they suggest that the requirement for displacement of the Arg-120 side chain is a substantial impediment to conjugate binding. Mutation of Ser-530 to alanine had a minimal effect on the potency of dansyl conjugate 2, suggesting that, as in the case of indomethacin, a hydrogen bond observed between the indomethacin moiety of the conjugate and the Ser-530 hydroxyl group does not make a major contribution to binding energy. In contrast, the S530A mutation reduced the potency of dansyl conjugate 1, suggesting that the hydrogen bond plays a significant role in the binding of that molecule. For both conjugates, mutation of Val-89 to tryptophan resulted in reduced potency, likely by introducing steric bulk in a portion of the lobby where the inhibitors contact the enzyme. This mutation did not affect the potency of indomethacin. Somewhat unexpectedly, an S119A mutation had little effect on the potency of the dansyl conjugates despite the fact that both establish a hydrogen bond with the hydroxyl group of this residue. Finally, a V523I mutation that reduces access to the COX-2 side pocket had no significant effect on potency of the dansyl conjugates, consistent with the absence of interaction between the inhibitors and this pocket as observed in the crystal structure data.113
The structural data described above confirmed that binding of at least some indomethacin esters and amides to COX-2 requires occupation of the constriction by the linker. Thus, the finding that mutation of Leu-472 in COX-2 to the corresponding methionine in COX-1 markedly reduces COX-2-selective inhibition of many of these inhibitors is particularly intriguing. X-ray crystal structure data reveal no significant differences in backbone configuration or side chain packing between the two isoforms in this region. However, molecular dynamics simulations suggested that the presence of Leu-472 at this site enables low-frequency dynamical motions in the constriction that promote opening of the site, thereby facilitating occupation of the constriction by the linker.114
5.2.3.6. Indomethacin Carbaborane Analogs
Replacement of the chlorobenzoyl moiety of indomethacin with a carbaborane substituent produces a COX-2-selective inhibitor. In the case of indomethacin methyl ester, this substitution increases both potency and COX-2 selectivity.115,116 To evaluate the impact of the carbaborane moiety on enzyme binding, a 2.3 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and the nido-dicarbaborate derivative of indomethacin methyl ester (PDB 4Z0L) was obtained (Figure S8D).116 The data reveal that the indole ring of the compound is located in the proximal inhibitor binding pocket, as is seen with indomethacin, but its orientation is flipped with respect to the parent compound. Instead of interacting with constriction residues, the methyl ester of the indole ring points upward toward the central inhibitor binding pocket and establishes a hydrogen bond with Ser-530. Unlike the chlorobenzoyl moiety of indomethacin that utilizes the central binding pocket, the carbaborane substituent projects into the oxicam binding pocket (see Sections 5.2.1.4 and 5.2.4) to which it gains access by a movement of Leu-531. There it interacts with Met-113, Val-116, Leu-117, Leu-351, and Leu-359. It also forms hydrophobic interactions with the carbon atoms of the side chain of Arg-120. The carbonyl group that joins the indole ring to the carbaborane moiety interacts with the side chain of Arg-120.116
5.2.3.7. Zomepirac Analogs
Zomepirac is a nonselective COX inhibitor that was marketed briefly but then withdrawn due to unpredictable severe anaphylaxis observed in some patients. Later studies demonstrated that analogs of zomepirac in which the carboxylate was replaced with various substituents were COX-2-selective inhibitors. The mechanism for this may be the same as that underlying the COX-2 selectivity of indomethacin esters and amides described above (see Section 5.2.3.5), although this was not proposed at the time that crystal structures of two of these analogs were published.
One of the analogs was RS104897, a zomepirac derivative bearing a p-iodophenyl ring attached via an acyl sulfonamide linker. A 3.3 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and RS104897 (PDB: not deposited) revealed that the zomepirac portion of the molecule occupies the active site of the enzyme with the chlorophenyl ring in the central inhibitor binding pocket, the carbonyl joining the chlorophenyl and pyrrole rings forming a hydrogen bond with Ser-530, and the pyrrole ring occupying the proximal inhibitor binding pocket. The nitrogen and oxygen atoms of the acyl sulfonamide moiety form a hydrogen-bonding network with Arg-120, Tyr-355, and Glu-524. The p-iodophenyl ring extends past the constriction into the lobby. The inhibitor makes no contacts with any residues that are unique to COX-2.117
The second analog was RS57067, a zomepirac derivative bearing a pyridazinone substituent attached via a methylene linker. A 2.9 Å resolution crystal structure of COX-2 complexed to Fe3+-protoporphyrin IX and RS57067 (PDB: not deposited) was also obtained. In this complex, the zomepirac portion of the molecule occupies essentially the same position as described above, but rather than projecting into the lobby, the pyridazinone ring forces an opening of the constriction. Compared to most other COX-inhibitor crystal structures, Arg-120 is displaced by 2.3 Å, causing a portion of helix D to unwind. As a result, Arg-120 does not interact with the carboxylate of Glu-524 but rather with the carbonyl oxygens of the backbones of Glu-524 and Phe-470. A hydrogen-bonding network still forms, but in this case, the participating residues are Tyr-355, Arg-513, and Glu-524. In COX-1, histidine occupies position 513 and may not be able to participate in this network. This could contribute to the COX-2 selectivity of the inhibitor.117
5.2.4. Oxicams
The term “oxicam” refers to NSAIDS of the enolic acid class of 4-hydroxy-1,2-benzothiazine 3-carboxamides. Structurally distinct from all other NSAIDs, they are widely prescribed anti-inflammatory agents that inhibit both COX isoforms but exhibit varying degrees of selectivity for COX-2. Studies using site-directed mutants of COX-2 revealed that R120A, Y355F, and S530A mutations all resulted in partial or total loss of sensitivity to one or more oxicams.50 Computational modeling suggested an interaction between the sulfonyl dioxide moiety and both Tyr-385 and Ser-530, explaining some, but not all of the mutation study results.118,119 To gain further insight into the interaction of these compounds with the COX enzymes, crystal structures were obtained for two oxicams in complex with COX-2 and one in complex with COX-1.
5.2.4.1. Isoxicam
Isoxicam is a nonselective COX inhibitor. A 2.0 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and isoxicam (PDB 4M10) reveals the inhibitor in an overall planar conformation, positioned between helices 6 and 17 of the protein [Figure S12A (stereoscopic view) and Figure S12B,C (monoscopic views)].120 Isoxicam’s thiazine ring lies in the proximal inhibitor binding pocket, with the 3-carboxamide substituent extending up into the central inhibitor binding pocket. The fused benzene ring occupies the oxicam pocket (Figure 18), to which it gains access by a movement of Leu-531 away from the location it occupies in most COX-2- inhibitor complex crystal structures (Figure 19). In contrast to predictions from computational modeling, isoxicam’s sulfonyl dioxide moiety lies above the constriction. It makes no obvious contacts with the enzyme; however, a clash between one of its oxygen atoms and Val-116 causes helix D to move over, helping to make room for the inhibitor’s benzyl ring. This clash also causes a twist in the conformation of the thiazine ring. Polar contacts between enzyme and inhibitor are established through interactions with two water molecules. The first of these forms a tetrahedrally coordinated hydrogen-bonding network that links the oxygen of the carboxamide and the nitrogen in the thiazine ring to Arg-120 and Tyr-355 in the constriction (Figure 20A,B). The second, trigonally coordinated network links the nitrogen atom of the inhibitor’s isoxazole substituent to Tyr-385 and Ser-530 (Figure 20A,C).120
Figure 18.

(A) Wall-eyed stereo view of the interaction of the oxicam binding pocket with isoxicam as observed from the side (i.e., parallel to the plane of the membrane), and comparison of the size of the oxicam binding pocket (B) with the comparable region in the structure of COX-2 complexed to naproxen (C). The difference in the position of Leu-531 is clearly visible, as is the marked difference in the size of the pockets between the two complexes. Isoxicam and naproxen are colored by element, and their surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. From PDB 4M10 and 3NT1.
Figure 19.

(A) and (B) Wall-eyed stereo views of the structure of isoxicam overlaid with that of (S)-flurbiprofen bound in the COX-2 active site along with the residues that form the oxicam pocket. Isoxicam is shown in sienna, and the associated amino acid residues are in dark blue. (S)-Flurbiprofen is in tan, and the associated amino acids are in light blue. Note the large difference in the conformation of Leu-531 between the two structures. This rotation is necessary to provide access to the oxicam pocket. Note also the difference in the positions of Met-113, Val-116, and Leu-117 that results from the movement of helix D. From PDB 4M10 and 3PGH.
Figure 20.

Wall-eyed stereo view of isoxicam bound in the cyclooxygenase active site of COX-2, and the hydrogen -bonded water molecules through which the inhibitor establishes polar contacts with the side chains of residues in the proximal binding pocket (A and C) and the central binding pocket (B and C). Side chains are colored in green (proximal binding pocket) or magenta (central binding pocket) with the exception of Arg-120, Tyr-355, Tyr-385, and Ser-530, which are colored by heteroatom on a sienna background. Isoxicam is colored by atom. The coordinated water molecule is shown as a red sphere. From PDB 4M10.
The crystal structure helps to explain the results of prior site-directed mutation data and structure–activity relationship data for the oxicam inhibitor class. Oxicams are unusual among the NSAIDs in that they establish polar interactions both at the constriction, with Arg-120 and Tyr-355, and at the top of the channel, with Tyr-385 and Ser-530. These interactions were revealed by the site-directed mutant data but were not explained by the results of computational modeling, which failed to predict the presence of the two key water molecules that are required to establish these interactions. That latter failure is not surprising in light of the fact that oxicams were the first COX inhibitors found to engage water in a functional manner to establish a critical binding interaction with the enzyme. Formation of the hydrogen-bonding network with Tyr-385 and Ser-530 also helps to explain the requirement for a heteroatom at the 1′-position of the carboxamide substituent. Hydrophobic contacts between the 2-methyl group of isoxicam and Val-349, Tyr-355, and Leu-359 of the enzyme explain the requirement for a methyl group at this position.120
The crystal structure data also suggest that oxicams are unusual among the NSAIDs in that binding induces significant conformational changes in the enzyme active site. Movement of Leu-531 and helix D, relative to their positions in other COX-NSAID crystal structures, must occur to accommodate the fused isoxicam ring structure (Figure 19). The result of this movement is the opening of the remarkably spacious oxicam binding pocket that is not present in the structures of COX enzymes with most other inhibitors (Figure 18B,C). As described above, a similar movement of Leu-531 has been reported in the case of the nido-dicarbaborate derivative of indomethacin methyl ester to COX-2, in the binding of 1-AG and 13-Me-AA to COX-2, in the productive conformation of EPA and the nonproductive conformations of both AA and EPA bound to COX-2, in the binding of AA to G533V COX-2, and in the binding of AA to V349I COX-2. Note that in all of these cases, Leu-531 shifts in order to provide room for a bulky substrate or inhibitor, although the pocket formed upon oxicam binding is considerably larger than those formed in the other examples due to the added movement of helix D.120
5.2.4.2. Meloxicam
Although meloxicam inhibits both COX isoforms, it has a 5-fold greater potency against COX-2 than COX-1. A 2.4 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and meloxicam (PDB 4M11) indicates an overall binding pose very similar to that of isoxicam described above (Figure S12D,E).120 The major difference lies in the orientation of the carboxamide substituent, which could not be clearly determined in the case of meloxicam. The findings suggest the presence of two conformations. In the first, the methyl group on the thiazole ring points in the direction of the mouth of the active site, and the nitrogen atom in the thiazole ring participates in the hydrogen-bonding network with water, Tyr-385, and Ser-530. The sulfur of the thiazine ring establishes hydrophobic contacts with Phe-518 and Val-523. In the second conformation, the methyl group on the thiazole ring binds in a pocket formed by Leu-384, Trp-387, Phe-518, and Met-522. The sulfur atom in the thiazole ring is oriented toward and interacts with the hydrogen-bonding water molecule.120
To explore the basis for the COX-2 selectivity of the inhibitor, a 2.4 Å resolution crystal structure of COX-1 complexed with Fe3+-protoporphyrin IX and meloxicam (PDB 4O1Z) was obtained.120 The structure revealed an inhibitor binding pose that is essentially the same as that observed in the complex with COX-2, including the water-mediated hydrogen bond networks and the dual conformations of the thiazole ring. Remarkably, the shift of Leu-531 to provide access to the oxicam pocket is also visible, denoting the first time that such a movement of Leu-531 was reported in a COX-1-ligand crystal structure. These findings did not immediately explain the basis for the inhibitor’s isoform selectivity. Site-directed mutagenesis studies focused on three amino acids, Ile-434, His-513, and Ile-523 in COX-1 that are different in COX-2 (Val-434, Arg-513, and Val-523). A V434I/R513H/V523I triple mutant COX-2 enzyme displayed sensitivity to meloxicam similar to that of COX-1. This loss of inhibitor potency could be completely recapitulated with a single V434I mutation, while a V523I mutation had no effect. The crystal structure demonstrated no interaction between meloxicam and His-513 in COX-1. These results suggest that the primary explanation for the substrate selectivity of meloxicam is the single amino acid change at position 434. Close inspection of the crystal structures indicates that isoleucine in this position, as is found in COX-1, pushes Phe-513 in toward the active site channel, reducing space needed for inhibitor binding.120
5.2.5. Fenamic Acids
The fenamic acid class of NSAIDs includes molecules that all share an N-phenylanthranilic acid (fenamic acid) scaffold. These are nonselective COX inhibitors that vary with regard to substrate-selective inhibition of endocannabinoid oxygenation by COX-2. To explore the basis for these differences, crystal structures of four of these inhibitors in complex with COX-2 were obtained.121
5.2.5.1. Mefenamic Acid
Mefenamic acid is a substrate-selective inhibitor of 2-AG oxygenation. A 2.3 Å resolution crystal structure of Δ586–612 COX-2 complexed with Co3+-protoporphyrin IX and mefenamic acid (PDB 5IKR) reveals that the inhibitor binds with the carboxylate projecting upward in the active site, forming hydrogen bonds with Tyr-385 and Ser-530 as seen with diclofenac and lumiracoxib [Figure S13A (stereoscopic view) and Figure S13B,C (monoscopic views)]. This conformation places the carboxylate-containing ring in the central inhibitor binding pocket, enabling hydrophobic interactions with Leu-352, Tyr-385, Trp-387, Met-522, Gly-526, Ser-530, and Leu-531. The dimethyl-substituted aromatic ring interacts primarily with Val-349, Tyr-355, Val-523, Ala-527, and Ser-530 in the proximal inhibitor binding pocket. In total, mefenamic acid forms 61 interactions with the COX-2 active site. No polar interactions with Arg-120 or Tyr-355 at the constriction are observed.121 Our laboratory has obtained essentially the same results from the diffraction of crystals comprising COX-2 complexed with Fe3+-protoporphyrin IX and mefenamic acid (unpublished data).
5.2.5.2. Flufenamic Acid
Flufenamic acid is a substrate-selective inhibitor of 2-AG oxygenation. A 2.5 Å resolution crystal structure of Δ586–612 COX-2 complexed with Co3+-protoporphyrin IX and flufenamic (PDB 5IKV) indicates that this inhibitor adopts a binding pose in the COX-2 active site that is very similar to that of mefenamic acid, engaging the same amino acids to form a total of 59 contacts (Figure S13D,E).121
5.2.5.3. Tolfenamic Acid
Tolfenamic acid is a substrate-selective inhibitor of 2-AG oxygenation. A 2.4 Å resolution crystal structure of Δ586–612 COX-2 complexed with Co3+-protoporphyrin IX and tolfenamic acid (PDB 5IKT) shows that this inhibitor binds in the COX-2 active site in a conformation very similar to that of mefenamic acid, engaging the same amino acids, with the addition of Val-116, to form a total of 68 contacts (Figure S14A,B).121
5.2.5.4. Meclofenamic Acid
Meclofenamic acid is a highly potent time-dependent inhibitor of both AA and 2-AG oxygenation that is not substrate selective. A 2.4 Å resolution crystal structure of Δ586–612 COX-2 complexed with Co3+-protoporphyrin IX and meclofenamic (PDB 5IKQ) indicates that this inhibitor adopts a binding pose in the COX-2 active site that is very similar to that of mefenamic acid, engaging the same amino acids, with the addition of Ser-353, to form a total of 58 contacts (Figure S14C,D).121
Site-directed mutagenesis studies demonstrated that an S530A mutation had no effect on the sensitivity of COX-1 or COX-2 to flufenamic acid or meclofenamic, respectively.49,50 This finding was supported by thermal shift binding assays that showed no effect of the S530A mutation on binding affinity of any of the four fenamic acids for which crystal structure data were obtained. In contrast, a Y385F mutation reduced the binding affinity of all of the fenamic acids as determined by thermal shift assay. A Y385F/S530A double mutation reduced binding affinity more than the Y385F mutation alone. These findings confirm the importance of the inhibitor interaction with Tyr-385 and suggest that the interaction with Ser-530 is not critical to binding unless other interactions are disrupted.121
The data do not reveal an obvious difference in binding conformation that would explain the observation that meclofenamic acid is the only one of the class tested that does not exhibit substrate-selective inhibition of 2-AG. Data have been provided that suggest that substrate-selective inhibition by these inhibitors is, at least in part, dependent on their ability to block peroxide-dependent enzyme activation.121
5.2.6. Diarylheterocycles
The diarylheterocycle acid class of NSAIDs includes compounds comprising a five- or six-membered heterocyclic ring bearing two adjacent aromatic substituents. Although the original compounds in this class, such as phenylbutazone, were nonselective COX inhibitors, the ones that have received the most attention have been COX-2-selective.39 In fact, all of the inhibitors designed to be COX-2-selective that reached the commercial market in the United States were members of this class, although only one (celecoxib) remains due to concerns regarding cardiovascular toxicity.80 COX-2 selectivity in this class is associated with the presence of a sulfonamide or methylsulfone substituent on one of the aromatic rings. COX-1-selective diarylheterocycles have also been reported.
5.2.6.1. SC-558
The first structural data on a complex of COX-2 with a member of the diarylheterocycle inhibitor class was reported in 1996 with the publication of 3.0 and 2.8 Å resolution crystal structures of COX-2 complexed with Fe3+-protoporphyrin IX and SC-558 (PDB 1CX2 and 6COX, respectively).32 The data reveal that the bromophenyl ring of the inhibitor occupies the central inhibitor binding pocket, where it interacts with Phe-381, Leu-384, Tyr-385, Trp-387, Phe-518, and Ser-530 [Figure S15A (stereoscopic view) and Figure S15B,C (monoscopic views)]. The pyrazole ring occupies the proximal binding pocket, with the trifluoromethyl substituent surrounded by Met-113, Val-116, Val-349, Tyr-355, Leu-359, and Leu-531. The phenylsulfonamide group occupies the COX-2 side pocket. As noted above, this region branches off from the active site and is more accessible in COX-2 than COX-1 (Figure 21). The increased accessibility is provided by substitution of Ile-523 in COX-1 with the smaller Val-523 in COX-2 in addition to a change in the position of the segment of amino acids from Leu-352 to Tyr-355 relative to its position in most structures of COX-2 in complex with inhibitors of other classes. A gate into the side pocket is formed by the packing of Val-434 against Phe-518. The larger side chain of Ile-434 in COX-1 (as opposed to Val-434 in COX-2) interferes with opening of this gate (Figure 22). Within the side pocket, the inhibitor interacts with His-90, Gln-192, Leu-352, Ser-353, Tyr-355, Ala-516, Val-523, and Phe-518. It also establishes a hydrogen bond with Arg-513, which is likely not possible with the smaller histidine residue located in this position in COX-1. Thus, three amino acid substitutions in COX-2 relative to COX-1, V434I, R513H, and V523I, are believed to be primarily responsible for the isoform selectivity of the diarylheterocycle class of inhibitors.32 A single V523I mutation in COX-2 alone, however, produced an enzyme that exhibited a sensitivity to these inhibitors comparable to that of COX-1.101 Similarly an I523V mutation in COX-1 markedly increased the enzyme’s sensitivity to this class of inhibitors. An H513R mutation alone had little effect, but an H513R/I523V double mutation increased sensitivity of COX-1 to the diarylheterocycle class of COX-2 inhibitors more than the I523V single mutation. For three out of four of the COX-2 selective inhibitors tested in one study, I523V alone increased potency without conferring time-dependency, whereas the double mutant exhibited both potent and time-dependent inhibition.122 A possible explanation of this behavior is that the smaller Val-523 side chain in COX-2 must initially provide access to the side pocket, where a subsequent interaction with Arg-513 is then responsible for time dependency, a model that is supported by molecular dynamics studies.123 The substrate selectivity of the diarylheterocycles has been frequently attributed to an inability of the inhibitors to access the side pocket in COX-1. However, as will be described below (see Section 6.1.2) conformational rearrangements in its active site enable COX-1 to accommodate binding of the diarylheterocycle celecoxib. Therefore, it is more likely that the structural differences noted between the two enzymes play a greater role in stabilizing the enzyme–inhibitor interaction than allowing access into the binding pocket.
Figure 21.

Wall-eyed stereo view of the interaction of the COX-2 side pocket with the phenylsulfonamide group of SC-558 as seen from two different views. SC-558 is colored by element, and its surface is shown as a mesh. Side chains of the residues comprising the pocket are displayed, and their surface is shown in solid tan. In (B), the phenylsulfonamide group of SC-558 is completely surrounded by the pocket, so that only the sulfonamide can be seen. From PDB 6COX.
Figure 22.

(A) Wall-eyed stereo view of the structure of SC-558 bound in the cyclooxygenase active site of COX-2 highlighting residues involved in side pocket formation and interactions. Overlaid are the same residues as observed in the structure of (S)-flurbiprofen bound in the cyclooxygenase active site of COX-2. Highlighted are Phe-518, which packs against Val-434 (pink/magenta) to open the side pocket, residues 352–355 (tan/sienna), which move to enlarge the side pocket upon diarylheterocycle binding, Val-523 (light/dark green), which is Ile-523 in COX-1, and other side pocket residues (light/navy blue). In each case, the darker colors correspond to residues in the SC-558-COX-2 structure whereas the lighter colors correspond to the (S)-flurbiprofen-COX-2 structure. (B) Same as (A) except that the structure of the SC-558-COX-2 complex (dark colors) is overlaid with the corresponding residues from the structure of (S)-flurbiprofen bound in the cyclooxygenase active site of COX-1 (light colors). Note the three key residues at positions 434, 513, and 523 that are different between the two isoforms. In particular, Ile-523 in COX-1 encroaches on the side pocket, and Ile-434 in COX-1 prevents the movement of Phe-518 that provides access into the pocket. From PDB 6COX, 1EQH, and 3PGH.
5.2.6.2. Celecoxib
Despite the fact that celecoxib was introduced to the market in the United States in 1999, it was not until 2010 that the crystal structure of a complex of this inhibitor with COX-2 was published. The 2.4 Å resolution structure of COX-2 complexed with Fe3+-protoporphyrin IX and celecoxib (PDB 3LN1) revealed essentially the same inhibitor binding pose as that observed for SC-558 (Figure S15D,E). This is not surprising, as the only difference between the two molecules is substitution of the bromo substituent in SC-558 with a methyl group in celecoxib. As for SC-558, the majority of protein-inhibitor contacts are hydrophobic in nature, though hydrogen bonds between the sulfonamide group and His-90, Gln-192, and Arg-513 in the side pocket are visible.124
Prior kinetic studies had revealed that the binding of celecoxib to COX-2 occurred in a three-step process, rather than the two steps observed for most slow-binding nonselective NSAIDs.125 More recently, molecular dynamics simulations suggested that the most stable conformation of celecoxib in the active site of COX-2 is one in which Leu-531 has rotated away from the constriction, as observed in the structures of oxicams complexed with COX-1 and COX-2 (see Section 5.2.4).123 In support of this hypothesis, L531F and L531W mutations of COX-2 resulted in a 3.4- and 9-fold reduction in potency of celecoxib, respectively.56
5.2.6.3. Rofecoxib
Rofecoxib differs from both SC-558 and celecoxib in that it possesses a methylsulfone rather than a sulfonamide group, and its heterocyclic ring is a 2-furanone rather than a pyrazole. Nevertheless, a 2.7 Å resolution structure of Δ586–612 COX-2 complexed with Co3+-protoporphyrin IX and rofecoxib (PDB 5KIR) reveals an overall binding pose very similar to those of the other two diarylheterocycles (Figure S16A,B). The only hydrophilic interactions among the 42 contacts established with the active site are hydrogen bonds between the methylsulfone oxygen atoms and the side-chain nitrogen atoms of His-90 and Arg-513 in the side pocket.126
5.2.6.4. P6
P6 was discovered as a result of the observation that removal of the sulfonamide group from the COX-2-selective diarylheterocycle inhibitor valdecoxib reversed its isoform selectivity.83 To better understand the foundation of P6’s COX-1 selectivity, a 2.9 Å resolution structure of COX-1 complexed with Fe3+-protoporphyrin IX and P6 (PDB 5U6X) was obtained. The data revealed that the binding pose of P6 in the cyclooxygenase active site differs substantially from those of most diarylheterocycle inhibitors in complex with COX-2. The chlorofuranyl group of P6 points toward the constriction where it forms polar interactions with Tyr-355 and Arg-120 (Figure S17A,B). The isoxazole also lies primarily in the proximal inhibitor binding pocket, forming hydrophobic interactions with the surrounding residues. The phenyl ring extends upward into the central inhibitor binding pocket. A total of 56 hydrophobic contacts are established with 9 active site residues. A high B-factor for P6 suggests that it “wobbles” or partially rotates within the active site channel. This is consistent with the relatively poor potency of this compound, which is a weak, reversible inhibitor of COX-1.127
5.2.6.5. Mofezolac
Mofezolac is a time-dependent slowly reversible COX-1-selective inhibitor that resulted from an effort to optimize the COX-1 inhibitory potency and selectivity of P6. It is currently on the market in Japan for use as an anti-inflammatory drug with low gastrointestinal toxicity. A 2.8 Å resolution structure of COX-1 complexed with Fe3+-protoporphyrin IX and mofezolac (PDB 5WBE) indicated that the inhibitor binds to the enzyme active site in a manner similar to that of most COX-2-selective diarylheterocycles, with its isoxazole group occupying the proximal inhibitor binding pocket (Figure S17C,D). Unlike the COX-2-selective inhibitors of this class, however, mofezolac carries an acetate substituent on the isoxazole ring, enabling it to make polar contacts (1 salt bridge and 3 hydrogen bonds) with Arg-120 and Tyr-355. (Note that the presence of this substituent also places mofezolac in the aryl acetic acid class of inhibitors.) One of mofezolac’s methoxyphenyl rings extends upward into the central inhibitor binding pocket, whereas the other is inserted into the COX-1 equivalent of the COX-2 inhibitor side pocket. In total, the inhibitor establishes 83 hydrophobic interactions with 17 active site residues. Mofezolac’s COX-1 selectivity was attributed to the ability of the inhibitor to establish a larger number of contacts with COX-1 than it could with COX-2 due to the smaller size of the COX-1 active site.127
5.2.7. NS-398
NS-398 was one of the earliest COX-2-selective inhibitors discovered. The presence of a methanesulfonamide group attached to an aromatic ring is reminiscent of the methylsulfone and sulfonamide groups found on the diarylheterocycle class of inhibitors. Thus, it was long assumed that the methansulfonamide group of NS-398 would interact with the COX-2 side pocket and that interactions with Arg-120 would contribute little to binding. Inconsistent with that assumption, however, were observations that R120Q and R120E mutations in COX-2 resulted in a substantial change in sensitivity to NS-398-mediated inhibition.57,58 Furthermore, although a V523I mutation abolished time-dependent inhibition, reversible inhibition by NS-398 was retained.101 These findings suggested that NS-398 interacts with the COX-2 active site in a mode distinct from that of the diarylheterocycles.
To address this question more directly, a 3.0 Å resolution structure of N594A COX-2 complexed with Co3+-protoporphyrin IX and NS-398 (PDB 3QMO) was obtained.128 The structure showed that, indeed, the methanesulfonamide group of the inhibitor is not located in the COX-2 inhibitor side pocket but rather at the mouth of the active site channel where it forms polar interactions with Arg-120 (Figure S16C,D). The methyl group of this substituent occupies a pocket surrounded by Val-116, Arg-120, Leu-531, and Ala-527. The aromatic ring of the inhibitor occupies the proximal inhibitor binding pocket, while the nitro group is directed toward His-90, Arg-513, and Val-523 in the side pocket. The cyclohexane ring projects upward into the central inhibitor binding pocket where it makes contacts with Trp-387, Gly-526, Ala-527, and Ser-530. Thus, the binding pose of NS-398 resembles that of indomethacin more closely than that of the diarylheterocycle class of COX-2 inhibitors.128
5.2.8. Benzopyrans (Chromenes)
Although they never reached the commercial market, a benzopyran class of COX-2-selective inhibitors was reported by Pfizer in 2010. Relatively extensive structure-activity relationship data were revealed, and the series achieved high potency and isoform selectivity. Clinical advance was prevented by poor pharmacokinetic properties. The crystal structures of three analogs in the series in complex with COX-2 were reported.124,129
5.2.8.1. (S)-5c
The compound designated 5c was advanced to early clinical trials before being abandoned due to an excessively long half-life in humans. During the process of compound optimization, a 2.2 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (S)-5c (PDB 3LN0) was obtained.124 Crystallization was performed with the (S)-enantiomer, as it was the most potent and selective of the two. The structure revealed that (S)-5c binds in the COX-2 active site with its carboxylate directed upward to form hydrogen bonds with Tyr-385 and Ser-530, as seen with diclofenac (Figure S18A). This orientation places the carboxylate-containing ring in the central inhibitor binding pocket, packing the trifluoromethyl group against Tyr-385, Trp-387, and Phe-518. The ring bearing the dichloro substituents occupies the proximal inhibitor binding pocket, with the 8-chloro group directed toward but not entering the side pocket. Notably, mutations of residues in COX-2 that provide side pocket access to the corresponding COX-1 residues reportedly had no effect on the potency of 5c, but data were not provided.124
5.2.8.2. (R)-23d
The compound designated 23d, was synthesized in an effort to improve the pharmacokinetics of the series. It differed from 5c by the addition of an isopropyl ether substituent at carbon 7. The presence of this bulky group resulted in a change in relative potency of the enantiomers, so that the (R)-isomer was now the most effective. A 2.2 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (R)-23d (PDB 3NTG)129 revealed that the substituent also induced a 180° rotation of the compound in the enzyme active site [relative to the position of (S)-5c]. The carboxylate now interacts with Arg-120 and Tyr-355 at the constriction, and the trifluoromethyl group forms hydrophobic interactions with Val-349 and Leu-359 (Figure S18B). This places the carboxylate-containing ring in the proximal inhibitor binding pocket with the isopropyl ether-containing ring projecting into the central inhibitor binding pocket where its isopropyl ether and chloro substituents form hydrophobic contacts with Leu-352, Tyr-385, Phe-518, Gly-526, and Ser-530.129
5.2.8.3. (S)-SC-75416
The most potent compound in the series, (S)-SC-75416 differed from (R)-23d by the absence of an 8-chloro substituent and substitution of a t-butyl group for the isopropyl ether. These structural changes resulted in a reversion back to the (S)-enantiomer as the most potent. Consistently, a 2.8 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and (S)-SC-75416 (PDB 3MQE)129 revealed a binding pose for the inhibitor that more closely resembled that of (S)-5c than (R)-23d (Figure S18C). The carboxylate projects upward to form polar contacts with Tyr-385 and Ser-530, while the ring containing the t-butyl substituent occupies the proximal inhibitor binding pocket. To accommodate the bulky t-butyl group, helices of the membrane-binding domain move 0.7 Å away from the active site, and the side chain of Tyr-355 also moves by approximately 1.6 Å relative to its position in most COX-2-inhibitor complex structures.129
5.2.9. Harmalines
A search for more potent substrate-selective inhibitors for use in vivo led to the discovery of a novel class of compounds based on harmaline, a member of the harmala alkaloid family. Six analogs bearing the tricyclic indole harmaline nucleus were synthesized and tested for both COX-2 isoform and 2-AG substrate selectivity. Of these, compound 3 exhibited the highest potency and selectivity by both criteria. To better understand the basis for the substrate selectivity of harmaline compound 3, a 2.7 Å resolution crystal structure of COX-2 complexed with Fe3+-protoporphyrin IX and the inhibitor (PDB 6V3R) was obtained.130 The structure revealed that the bulk of the tricyclic indole nucleus of compound 3 binds in the proximal inhibitor binding pocket; however, to accommodate the large ring system, Leu-531 moves away from the constriction, providing access to the oxicam pocket (see Sections 5.2.1.4 and 5.2.4). The 6-methoxy substituent of compound 3 inserts into this pocket and establishes interactions with Leu-531 and Val-116 (Figure 23A). It is interesting to note that compound 3 possesses a 4-chlorobenzoyl substituent, and the crystal structure reveals that it adopts a binding pose in the central inhibitor binding pocket very similar to that of the 4-chlorobenzoyl substituent of indomethacin (Figure 23B). Unlike indomethacin, however, compound 3 possesses no charged substituents, and its 46 contacts with the active site are all nonpolar in nature. Despite its high degree of COX-2 selectivity, compound 3 does not interact with the side pocket utilized by the diarylheterocycle class of inhibitors (see Section 5.2.6). As in the case of other substrate-selective inhibitors in complex with COX-2 (see Sections 5.2.2.1, 5.2.2.3, and 5.2.5), the crystal structure data reveal the presence of compound 3 in both subunits, and the binding pose is essentially identical in both. Furthermore, there are no obvious binding interactions or protein conformational changes that appear to be fully unique to this inhibitor. Thus, the foundation for substrate selectivity remains unclear.130
Figure 23.

(A) Wall-eyed stereo view of the structure of harmaline compound 3 bound in the cyclooxygenase active site of COX-2. The side chains that make up the proximal binding pocket (green), the central binding pocket (magenta), and the oxicam pocket (cyan) are shown. Note that Val-349 is part of both the proximal pocket and the oxicam pocket. It is colored in cyan. (B) Wall-eyed stereo view of an overlay of the structures of harmaline compound 3 and indomethacin bound in the active site of COX-2, including residues that make up the proximal (light/dark green) and central (pink/magenta) binding pockets. Indomethacin and compound 3 are shown in light and dark gray, respectively, and the lighter residue colors correspond to those in the indomethacin-COX-2 complex. Also shown is Leu-531 (yellow/sienna) which moves away from the constriction to accommodate the tricyclic harmaline nucleus. From PDB 63VR.
5.2.10. Aspirin
Aspirin (acetylsalicylic acid) is the only commercially available NSAID that covalently modifies the COX enzymes. The target of modification is the side chain hydroxyl group of Ser-530, which is acetylated upon aspirin exposure.131,132 Aspirin-dependent acetylation of both COX-1 and COX-2 essentially blocks PG biosynthesis; however, in the case of COX-2, it does not eliminate AA oxygenation, as this isoform remains capable of producing the mono-oxygenated product 15(R)-HETE following aspirin exposure.133 Mutation of Ser-530 to alanine has little effect on enzymatic activity of either COX-1 or COX-2 while rendering the enzymes insensitive to aspirin, indicating that the mechanism of enzyme inhibition is not alteration of a catalytically important functional group.134,135 Instead, as the structural studies discussed below illustrate, the acetyl group physically blocks the active site channel.
5.2.10.1. Bromoacetylated COX-1 in Complex with Salicylic Acid
A very early study of COX-1 utilized 2-bromoacetoxy-benzoic acid, a brominated analog of aspirin, to determine the structural impact of acetylation on the enzyme. The bromo substituent on the acetyl group facilitated its localization in what were, at that time, relatively low resolution data. This approach produced a 3.4 Å resolution crystal structure of bromoacetylated COX-1 complexed with Fe3+-protoporphyrin IX and salicylic acid (PDB 1PTH).31 The data confirmed the presence of the bromoacetyl group on Ser-530 of both dimer subunits and demonstrated that the modification has very little effect on the overall structure of the protein. The bromoacetyl group is present in two rotamers at a ratio of 5:1. The predominant rotamer places the group directly in the active site channel, completely blocking access to the catalytic Tyr-385 residue. The minor rotamer does not completely block the channel, but prevents substrate from aligning correctly for catalysis.31
In this crystal structure, salicylic acid remains bound in the active site. It is located in the proximal inhibitor binding pocket, with its carboxylate forming polar interactions with Arg-120 and Tyr-355.31 This might serve as an initial binding site for aspirin, as suggested by reports that mutations of Arg-120 to alanine, glutamine, or glutamate in COX-1 and/or COX-2 reduce sensitivity to aspirin.43,134 However, this cannot be the position aspirin occupies during its reaction with Ser-530. In fact, mutation studies support the hypothesis that aspirin forms a hydrogen bond network with Tyr-385 and Tyr-348 that facilitates transition state formation.134
5.2.10.2. Acetylated COX-1 in Complex with O-Acetylsalicylhydroxamic Acid (AcSHA)
AcSHA was synthesized as part of an attempt to discover COX inhibitors that covalently modify the enzyme as an alternative to aspirin. The inhibitor was shown to acetylate the enzyme and inhibit PG biosynthesis both in vitro and in vivo. A 3.2 Å resolution crystal structure of a COX-1-Fe3+-protoporphyrin IX complex that had been treated with AsSHA (PDB 1EBV) revealed the presence of an acetyl group on Ser-530 of both subunits and unreacted inhibitor in the enzyme’s active site (Figure S19A,B).135 The position of the acetyl group, which blocks the active site channel below Tyr-385, is similar to that of the bromoacetyl group of bromoacetylated COX-1 that had been previously reported (see above).31 AcSHA binds in the lower portion of the COX-1 active site, predominantly in the proximal inhibitor binding pocket, with the acetylhydroxamate substituent forming polar contacts with Arg-120 and Tyr-355. Nonpolar interactions with Val-349, Leu-352, Phe-518, and Ile-523 are also noted. Consistent with the structural data, S530A and R120Q mutations in COX-1 render the enzyme insensitive to inhibition by AcSHA.135
5.2.10.3. Acetylated COX-2
As noted above, unlike acetylated-COX-1, which retains essentially no activity, acetylated-COX-2 oxygenates AA to form 15(R)-HETE. To understand the structural basis for this difference between the two isoforms, a 2.0 Å resolution crystal structure of a Δ586–612 COX-2-Co3+-protoporphyrin IX complex that had been treated with aspirin was obtained (PDB 5F19).136 The structure revealed the acetyl group on Ser-530 of both subunits, but unlike prior structures of acetylated-COX-1, no salicylic acid is observed in the active site (Figure S19C,D). The acetyl group is positioned below Tyr-385 with which it forms a hydrogen bond, and nonpolar interactions with Val-344, Tyr-348, and Val-349 are evident. In this location, the acetyl group extends far into the active site channel (Figure S19E). Thus, it is not clear how AA can bind properly for 15(R)-HETE formation. This is particularly true in light of previous findings that a G533A mutation in COX-2 abrogates 15(R)-HETE production following acetylation, thereby suggesting that access to the distal AA binding pocket is required.59 A possible explanation for these observations is that Ser-530 can adopt multiple conformations within the COX-2 active site, as is seen upon comparison of crystal structures of the enzyme with various substrates (see Section 4.1.2). Likely, the orientation of the acetylated residue is different when the enzyme is in complex with AA than when the active site channel is empty.136
A 2.4 Å resolution crystal structure of Δ586–612 COX-2 in complex with Co3+-protoporphyrin-IX and salicylic acid (PDB 5F1A) was also obtained.135 In this structure, salicylic acid is bound in the central inhibitor pocket of the active site channel just below Tyr-385. The molecule forms hydrophobic contacts with Leu-384, Try-385, and Trp-387. Although the phenolic hydroxyl group points toward Ser-530, neither it nor the carboxylate establishes any polar interactions.135 This binding pose is different from that observed for salicylate retained in the active site of bromoacetylated-COX-1. As noted above (see Section 5.2.10), in that structure, the salicylate is in the proximal inhibitor binding pocket forming polar interactions with Arg-120 and Tyr-355.31 The conformation of salicylic acid in the central binding pocket is most likely representative of its position during the acetylation reaction. Despite the absence of polar interactions in the crystal structure, prior molecular dynamics simulations suggested that the presence of Arg-513 in COX-2 exerts a destabilizing effect on the transition state during the acetylation reaction. This effect is not observed in COX-1, which has a histidine at this position, a finding used to explain the greater sensitivity of COX-1, as compared to COX-2, to aspirin acetylation.137 Support for this hypothesis was provided through site-directed mutation studies that revealed an increase in sensitivity to aspirin in R513H COX-2 as compared to the wild-type enzyme.56
Whereas the S530A mutation had little effect on activity of either COX-1 or COX-2, most larger mutations in COX-1, including S530I, S530L, S530M, S530N, and S530V, eliminated activity.47,51,64 In COX-2, S530M and S530V mutations led to a reduction, but not elimination, of activity and a shift from PGH2 to 15(R)-HETE as the primary product.47,51 These findings are consistent with the hypothesis that the side chains of the substituted residues act in the same way as the acetyl group in acetylated COX-1 or COX-2. In both enzymes, however, an S530T mutation behaved somewhat differently. In COX-1, it led to 15(R)-HETE production, whereas in COX-2, it resulted in a shift of stereochemistry at the 15-position, so that the major product was 15(R)-PGH2.47,51 To better understand the structural impact of this mutation, a 1.9 Å resolution crystal structure of S530T/N594A COX-2 in complex with Co3+-protoporphyrin-IX (PDB 5FDQ) was obtained.136 The data revealed an overall structure that is nearly identical to that of the wild-type enzyme. The side chain of Thr-530 adopts one of two conformations previously observed for Ser-530 in the crystal structures of COX-2 complexed with AA. This orientation allows ample room for the substrate to access the hydrophobic channel in the upper portion of the active site, explaining the retention of activity by the mutant.136 However, the presence of the extra methyl group in threonine as opposed to serine must influence the substrate’s conformation in such a way as to result in a change in the orientation of oxygen addition at carbon-15. Furthermore, the presence of the larger side chain of Thr-530 is predicted to alter the binding pose of AA in such a way as to require movement of Leu-531 in order to accommodate the C-terminal end of the substrate, as was observed in the crystal structure of a complex of V349I COX-2 with AA56 (see Section 4.1.2). To test this hypothesis, the effects of L531F and L531N mutations on products formed following aspirin acetylation were explored. Unexpectedly, both of these mutations markedly reduced the potency of aspirin, with the L531F mutant enzyme exhibiting total resistance to the inhibitor. These observations were explained on the basis of a postulated interference by the substituted side chains on the initial binding interaction of aspirin in the cyclooxygenase active site.56
6. Structures Addressing the Mechanism of Allostery
As noted previously, the COX enzymes are structural homodimers that behave as functional heterodimers. Current evidence supports the hypothesis that in solution, heme binds with high affinity to only one subunit of the enzyme and that this subunit serves as the catalytic site while the second subunit modulates enzyme activity via allosteric interactions. In general, crystal structure data have provided limited insight into the basis for differential binding of ligands or heme to the two subunits, half-of-sites activity, or subunit-to-subunit communications. In most cases, heme is observed in both monomers of the holoenzyme, and ligands are bound in a similar pose to both. Notable exceptions are the complexes of various substrates with COX-2 in which the ligand is bound productively in one monomer and nonproductively in the other (see Sections 4.1.2 and 4.3.2). These findings clearly indicate that each subunit retains the capability of interacting with heme, substrates, and inhibitors, even if the relative affinities or binding modes of the two subunits are different in solution. The implication is that substrates and inhibitors may modulate enzyme activity through interaction with either or both subunits and that the effects of ligand binding to each subunit may be different.
It is important to note that allostery is, by its very nature, a dynamic process, whereas crystallography can only capture the image of a static structure. Furthermore, if multiple conformations are present in a crystal, the crystallographic results may simply provide an average representation. Thus, crystallography is inherently limited in its ability to explore this phenomenon. Furthermore, the crystallization process will selectively capture those protein conformations that lend themselves best to stable crystal formation. It is likely that symmetrical structures, such as those with ligands similarly bound in both subunits, would be favored. Supporting this hypothesis are experiments described below in which crystallization was carried out at ligand:dimer ratios of 1:1, but ligand occupancy of both subunits in the structure was observed. Consequently, we must interpret results from attempts to explore allostery via crystallography with care. Nevertheless, some attempts have been made to obtain crystal structures in which differential binding of ligands to the two subunits has been achieved. The resulting structures provide some potential clues regarding the basis of allostery.
6.1. COX-1 Structures
6.1.1. (S)-Flurbiprofen Complex with a COX-1 Heterodimer
A site-directed mutagenesis study designed to demonstrate half-of-sites activity involved the construction of a COX-1 heterodimer containing one wild-type subunit and one subunit bearing the R120Q mutation.27 This enzyme demonstrated cyclooxygenase activity at levels similar to those of the wild-type homodimer despite the marked reduction in activity that was associated with the R120Q homodimer. The enzyme also retained nearly the same sensitivity to flurbiprofen inhibition as the wild-type homodimer despite the marked reduction in sensitivity to flurbiprofen associated with the R120Q homodimer. These findings were interpreted to mean that the wild-type monomer is responsible for both enzymatic activity and flurbiprofen binding. A 2.8 Å resolution crystal structure of the heterodimer in complex with Fe3+-protoporphyrin-IX and (S)-flurbiprofen (PDB 3N8W) supports this hypothesis in that the inhibitor is consistently present in the wild-type monomer whereas only approximately 25% of the R120Q monomer active sites are occupied. The conformation and orientation of (S)-flurbiprofen in the enzyme active site is consistent with those of previous structures regardless of monomer location. However, a notable finding in the unbound R120Q monomers is that a loop comprising amino acids 121–129 at the dimer interface adopts two different conformations, one that is the same and one that is different from that observed in ligand-bound monomers in this and previous crystal structures (Figure 24).27 This observation is particularly interesting, as prior reports of experiments carried out using the enzyme in solution have suggested that the 121–129 loop may be involved in allosteric communication between the two monomers.26,28
Figure 24.

(A) and (B) Wall-eyed stereo views of the structure of (S)-flurbiprofen bound in the cyclooxygenase active site of a wild-type/R12Q COX-1 heterodimer and the side chains that make up the proximal binding pocket (green) and the central binding pocket (magenta). The structures of the two subunits are overlaid, with the wild-type subunit (containing flurbiprofen) shown in the lighter colors. Also shown are residues 121–129, which exist in two conformations in the R120Q subunit (sienna) depending on the presence or absence of bound flurbiprofen. Subunits containing inhibitor exhibit the same conformation as is observed in the wild-type subunit (tan), whereas those lacking inhibitor diverge between Ser-121 and Pro-125. From PDB 3N8W.
6.1.2. Celecoxib Complex with COX-1
Both in vitro and in vivo studies indicate that celecoxib and some other COX-2-selective inhibitors, interfere with the action of aspirin and other NSAIDs on COX-1. Celecoxib is a potent, time-dependent inhibitor of COX-2 and a rapidly reversible inhibitor of COX-1. Consistent with the mechanism proposed for the rapidly reversible inhibition of COX-2-dependent AA oxygenation by ibuprofen and mefenamic acid,93 it was hypothesized that tight binding of celecoxib to the allosteric subunit of COX-1 has no effect on AA oxygenation but interferes with binding of NSAIDs. Actual inhibition of AA oxygenation would then require low affinity binding to the catalytic subunit. To test this hypothesis, a 2.8 Å resolution crystal structure of COX-1 in complex with Fe3+-protoporphyrin-IX and celecoxib (PDB 3KK6) was acquired. The intent of this experiment was to obtain the structure of the enzyme with only the allosteric site occupied with an inhibitor. Hence, the crystallization was carried out with 72 μM each of enzyme dimer and celecoxib. Nevertheless, the data revealed complete occupation of monomer A and 50% occupation of monomer B in the crystallized protein.26
It is the relatively small side chain of Val-523 in COX-2 that allows access of the diarylheterocycle inhibitors to the side pocket. Ile-523 in COX-1 is expected to block that access; however, a 3.1 Å movement of its side chain relative to its position in most other COX-1-inhibitor complexes both provides space for binding celecoxib’s aromatic ring and shifts the position of His-513, Pro-514, and Asn-515 in the side pocket (Figure S20A). As a result, celecoxib is able to bind in the active site of COX-1 in a pose very similar to its orientation in the active site of COX-2. As is seen for both SC-558 and celecoxib complexed to COX-2, the inhibitor makes no contacts with Arg-120. Rather, the trifluoromethyl group abuts Tyr-355. Notably, whereas celecoxib and SC-558 form a hydrogen bond with Arg-513, this interaction is not possible with His-513 of COX-1. Rather, hydrogen bonds are formed between the sulfonamide group of celecoxib and the side chain of Gln-192 and main chain of Leu-352. Also, despite predictions that Ile-434 in COX-1 would not permit adequate movement of Phe-518 to enable binding of a diarylheterocycle inhibitor, such a movement has occurred, and hydrophobic contacts are established between Phe-518 and the inhibitor (Figure S20A).26 Notably, however, the locations of key residues, such as Tyr-385, Phe-518, and His-90, in the COX-1-celecoxib complex are significantly different from those of the corresponding residues in the COX-2-celecoxib complex (Figure S20B). These differences likely contribute to the COX-2 selectivity of the inhibitor.
An interesting observation arising from this structure is the position of the loop of amino acids containing residues 121 through 129 at the dimer interface. This loop is present in two distinct conformations in monomer B, depending on whether or not the active site is occupied with celecoxib (Figure S20C). In the case of occupied monomers, the loop occupies the same position observed in the majority of other COX-1-inhibitor crystal structures in which both monomers are occupied. In the case of unoccupied monomers, a different conformation is noted that brings residues 126 and 127 close to residues 541 and 543 on monomer A. This alternative conformation is similar to that observed in the structure of flurbiprofen complexed to a COX-1 heterodimer (see Section 6.1.1) and may reflect motions of the protein that are relevant to allosteric communications between the subunits.26,28
6.1.3. Nimesulide Complex with COX-1
Nimesulide is an early COX-2-selective inhibitor that is structurally very similar to NS-398. As described above for celecoxib (see Section 6.1.2), nimesulide was found to interfere with the inhibition of COX-1 by some nonselective NSAIDs at concentrations that did not directly inhibit the enzyme. To attempt to further understand this phenomenon, a 2.8 Å resolution crystal structure of COX-1 in complex with Fe3+-protoporphyrin-IX and nimesulide (PDB 3N8X) was obtained.27 In an attempt to limit occupancy of the enzyme to one inhibitor molecule per dimer, crystallization was carried out at dimer:inhibitor ratio of 1:1. Nevertheless, the data indicated that both monomers are occupied by nimesulide. The binding pose of the inhibitor is essentially the same as that observed for NS-398 complexed with COX-2 as described above (see Section 5.2.7). However, a notable steric clash is observed between the inhibitor and Ile-523, which is Val-523 in COX-2.27
6.1.4. Diclofenac Complex with Aspirin-Acetylated COX-1
In vitro studies suggest that only one monomer of COX-1 or COX-2 is acetylated upon incubation with aspirin in solution.137 As outlined above (see Section 5.2.10), in the case of COX-1, acetylation completely inactivates the enzyme, but COX-2 retains some activity, producing markedly reduced amounts of PGH2 and increased levels of 15(R)-HETE compared to those of the unmodified enzyme. Diclofenac completely inhibits COX-2 upon binding of a single molecule per dimer; however, it is a very poor inhibitor of the S530A mutant COX-2, as a hydrogen bond interaction between its carboxylate and Ser-530’s hydroxyl group is a strong contributor to binding affinity. Thus, it is expected that acetylation of Ser-530 by aspirin would interfere with diclofenac-mediated inhibition. This is true in the case of 15(R)-HETE production on which diclofenac has minimal effect. However, diclofenac completely blocks production of PGH2 by aspirin-acetylated COX-2.138 These observations suggest the possibility that 15(R)-HETE is produced by the acetylated subunit, which is not affected by diclofenac, while PGH2 is produced by the unacetylated subunit, where diclofenac can still bind. Note, however, that this interpretation is inconsistent with the widely accepted view that heme is bound to only one subunit of COX (see Section 1).
These observations suggested that a complex of diclofenac with acetylated COX should provide a structure in which differential binding has occurred. This hypothesis was supported by a 2.6 Å resolution crystal structure of aspirin-acetylated COX-1 in complex with Fe3+-protoporphyrin-IX and diclofenac (PDB 3N8Y).27 In this structure, diclofenac is present in 100% of monomer A subunits but only about 20% of monomer B subunits. The remaining monomer B subunits appear to have salicylate bound in the active site. An acetyl group is visible on the side chain of Ser-530 in monomer B but not monomer A. The overall binding pose of diclofenac in monomer A is essentially the same as that previously reported for diclofenac in COX-2 (described above). The pose of salicylate differs from that observed in the previously reported low resolution structure (see Section 5.2.10.1),26,28 in that the carboxylate and hydroxyl group of the salicylate are not ion paired with Arg-120 but rather form polar interactions with the acetylated serine residue. Notably, relatively few differences are visible in the overall protein conformation between the two monomers. Residues 510–526 in monomer A exhibit two alternate conformations, one of which is observed in most crystal structures of liganded-COX-1 and a second that is seen in the unliganded enzyme (see Section 7.1). In monomer B, only the conformation commonly observed in structures of the liganded protein is visible.
6.2. COX-2 Structures
6.2.1. S121P Mutant COX-2
As described above (see Section 6.1.1 and 6.1.2), studies with COX-1 had suggested that a loop of amino acids from 121 to 129 at the dimer interface may play a role in transmitting signals from the allosteric to the catalytic monomer of the COX enzymes. This loop is in close proximity to Arg-120 at the enzyme’s constriction. Arg-120 plays a key role in forming the hydrogen bond network with Glu-524 and Tyr-355 that defines the constriction. In addition, Arg-120 forms polar interactions with many carboxylate-containing allosteric modulators of COX (fatty acids and inhibitors), suggesting that interactions between ligands and this amino acid in the allosteric site may cause structural alterations in the 121–129 loop, thereby transmitting a signal to the catalytic site. Mutation studies to test this hypothesis yielded S121P COX-2, which exhibited increased baseline activity with AA and reduced responsiveness to carboxylate-containing allosteric stimulators and inhibitors.139
To understand the foundation for the effects of this mutation, a 2.4 Å resolution crystal structure of S121P COX-2 in complex with Co3+-protoporphyrin-IX (PDB 5JVY) was acquired.138 The data revealed no significant difference in overall structure between the mutant and the wild-type protein, including in the conformation of the 121–129 loop. The major finding was that the mutation, which is located at the C-terminus of helix D of the membrane-binding domain disrupts the structure of the helix, causing it to partially unwind. As a result, Arg-120 becomes disordered and fails to participate in the hydrogen-bonding network at the constriction, leading to an “open” conformation.139
6.2.2. S121P Mutant COX-2 in Complex with (S)-Flurbiprofen
The binding of (S)-flurbiprofen to both COX-1 and COX-2 involves a polar interaction with Arg-120. Thus, it is conceivable that the disordered side chain of Arg-120 observed in the unliganded structure of S121P COX-2 (see Section 6.2.1) would result in an altered binding pose for the inhibitor. To test this hypothesis, a 2.6 Å resolution crystal structure of S121P COX-2 in complex with Co3+-protoporphyrin-IX and (S)-flurbiprofen (PDB 5JVZ) was obtained.139 The results showed the inhibitor in the COX-2 active site in essentially the same conformation and orientation as is observed in the wild-type enzyme, with its carboxylate forming polar interactions with Arg-120. Thus, it appears that inhibitor binding restores order to Arg-120 despite the presence of the disrupting mutation. This finding is consistent with the results of binding studies that suggest no loss of affinity for the inhibitor as a result of the mutation, even though the efficacy of inhibition observed is reduced.139
6.2.3. S121P Mutant COX-2 in Complex with Celecoxib
The binding of celecoxib with COX-2 occurs in the absence of an interaction with Arg-120. To find out if celecoxib could affect the conformation of Arg-120 in the S121P COX-2 mutant (see Section 6.2.1), a 2.8 Å resolution crystal structure of S121P COX-2 in complex with Co3+-protoporphyrin-IX and celecoxib (PDB 5JW1) was acquired. The data revealed that celecoxib binds in the active site of the mutant in essentially the same pose as it binds in the active site of the wild-type enzyme. However, binding of this inhibitor did not restore order to Arg-120 as was seen in the case of (S)-flurbiprofen.139
7. Miscellaneous Structures of COX Lacking an Active Site Ligand
The overwhelming majority of crystal structure studies have focused on COX in complex with an active site ligand. One reason for this is that obtaining high resolution data for unliganded structures has been challenging. Another reason is that a primary goal of most studies has been to understand the interaction of the enzyme with substrates or inhibitors. However, some crystal structures of the enzyme lacking an active site ligand have been published. A notable finding is that the overall conformation of the protein in these structures is very similar to that of structures in which a ligand is present. This observation disputed the long-held hypothesis that binding of time-dependent inhibitors to the COX active site would likely result in a major protein conformational change.
7.1. COX-1 Structures
In conjunction with studies intended to explore the basis for the allosteric communication between the COX subunits, a 3.0 Å resolution crystal structure of COX-1 in complex with Fe3+-protoporphyrin-IX (PDB 3N8V) was obtained.27 Careful analysis of the data suggested the presence of merohedral twinning in the protein crystals, and appropriate subsequent data analysis led to the conclusion that structural differences between the subunits of the protein dimer were visible. In this case, monomer B was devoid of a ligand, while monomer A contained a single molecule of glycerol. In addition, a shift in the position of residues 510–515 in the region of the side pocket was noted in monomer B. These residues occupied a position closer to the active site of monomer A, a conformation distinct from that observed in the majority of other COX crystal structures. The positions of residues in the region of the dimer interface, however, were comparable to those observed in the majority of crystal structures in which both monomers of COX contain an active site ligand.27
7.2. COX-2 Structures
A 3.0 Å resolution crystal structure of COX-2 in complex with Fe3+-protoporphyrin-IX (PDB 5COX) was reported in 1996 in conjunction with additional structures of the enzyme in complex with inhibitors. Due to poor resolution, the structure was not extensively analyzed. The mouth of the active site channel was noted to be more restricted, and helix D of the membrane-binding domain was more highly disordered in this structure than in structures of the enzyme bearing an active site ligand.32
The first structure of COX-2 lacking both an active site ligand and a heme prosthetic group was published in 2015. The 2.6 Å resolution data (PDB 4RRW) were acquired as part of a study of the effects of lobby mutants on inhibitor binding. Thus, a 2.4 Å resolution crystal structure of H90W COX-2 (PDB 4RRY) was also reported as were structures of this mutant in complex with (S)-ibuprofen and lumiracoxib described above (see Sections 5.2.2.3 and 5.2.3.4). The data revealed no exceptional differences in enzyme folding resulting from the absence of heme in the wild-type enzyme. Similarly, the mutant is essentially folded in the same way as the wild-type protein with the exception of the region immediately surrounding Trp-90 as described above (see Section 5.2.2.3).105
A 2.4 Å resolution crystal structure of S121P COX-2 in complex with Co3+-protoporphyrin-IX (PDB 5JVY) but lacking an active site ligand has been published.139 This structure is described in greater detail in Section 6.2.1.
8. Structure-Function Correlations
The structural analysis of a large number of COX-ligand complexes has been exceedingly helpful in understanding many fundamental features of COX enzymology. For example, structural data reveal how the enzyme enables reaction catalysis and guarantees correct stereochemistry. The data have successfully predicted and/or explained the results of many site-directed mutation studies. Similarly, they have predicted and/or explained the structure–activity relationships of a vast number of inhibitors and have provided key insights into the basis of isoform selectivity of many inhibitors. Nevertheless, a seemingly overriding theme that underlies a thorough examination of COX structural data is the high degree of similarity in protein structure regardless of the presence or absence of ligand or the nature of the ligand. This similarity has frustrated many attempts to fully understand the structural basis of certain aspects of COX function, such as ligand binding dynamics, inhibitor kinetics, substrate selectivity, and allostery. Despite these challenges, by combining X-ray crystallography with kinetic studies and site-directed mutagenesis data, we are gaining insights into these more esoteric questions. In the sections below, we highlight progress that has been made in these areas.
8.1. Ligand-Induced Structural Changes
Despite the high degree of consistency in the structure of the COX enzymes regardless of bound ligand, some ligand-specific induced structural changes have been noted. These are defined here as differences in protein conformation between a subgroup of COX-ligand complexes and the majority. A particularly intriguing one is the movement of Leu-531 observed upon binding AA in the unproductive conformation,53 EPA65 and 1-AG62 in both productive and nonproductive conformations, and 13-Me-AA22 to wild-type COX-2. The displacement is also seen in binding AA to G533V and V349I COX-2.56,61 In all of these cases, the size and/or orientation of the ligand or a modification of the active site itself leads to the need for additional space in the proximal binding pocket, and the relative ease with which Leu-531 can undergo this rotation in COX-2 has been used to explain why its substrate specificity is broader than that of COX-1. Indeed, Leu-531 displacement was thought to be impossible in COX-1 because of the role the amino acid plays in stabilizing Arg-120 in that isoform.34 These conclusions have partially been supported by site-directed mutagenesis studies; however, some discrepant results have been reported.22,46,51,53,62,64,67,140 A more recent report has demonstrated rotation of Leu-531 to provide access to the oxicam pocket for both COX-1 and COX-2, challenging the assumption that the amino acid’s position is fixed in COX-1.120 Furthermore, molecular dynamics and free energy simulations studies suggest that rotation of Leu-531 to an “open conformation” is key to the interaction of slow tight-binding inhibitors with both isoforms and that binding of these inhibitors to the open conformation yields the most energetically stable complex despite the fact that this is not supported by the crystal structure data for most of these inhibitors.123,141,142 Clearly, additional work is required to define the role of Leu-531 in both substrate and inhibitor binding.
Movement of key amino acid residues provides access to two inhibitor binding pockets in one or both COX isoforms. As noted above, Leu-531 displacement, accompanied by a movement of Val-116 and the C-terminus of helix D enable binding of the oxicams in a pocket that has only been shown to be accessed by two other inhibitors, the nido-dicarbaborate derivative of indomethacin methyl ester and the harmalines.116,120,130 In the case of the oxicams, the movement of Val-116 results from a steric clash with the sulfonyl dioxide moiety of the inhibitor. The COX-2 side pocket exploited primarily by the diarylheterocycle class of inhibitors is the second example.32 In this case, a movement of the segment of amino acids from Leu-352 to Thr-355 combined with the smaller side chain of Val-523 in COX-2 (as compared to Il-523 in COX-1) provides access to the pocket. In addition, a gate formed by the packing of Val-434 against Phe-518 must open to allow access into the pocket. It has generally been assumed that the larger Ile-434 in COX-1 interferes with opening of the gate. However, a crystal structure of COX-1 complexed with celecoxib shows that sufficient movement of this residue is possible to provide access of the inhibitor to the side pocket. Similarly, a displacement of Ile-523 along with His-513, Pro-514, and Asn-515 occurs upon binding of celecoxib to COX-1.26
Some additional inhibitors have been noted to induce significant displacement of enzyme residues upon binding. Among them are (S)-SC-75416, which induces a movement of the membrane-binding domain helices away from the active site along with a shift in the position of Tyr-355, and RS57067, the pyridazinone ring of which forces an opening of the constriction that displaces Arg-120 and unwinds part of helix D of the membrane-binding domain.117 In addition, the (R)-enantiomers of naproxen and flurbiprofen induce a shift in the position of Arg-120 and Tyr-355 to accommodate the disfavored stereochemistry of the α-methyl group.92 Finally, as described below (see Section 8.3), the limited number of structures of COX-inhibitor complexes in which one subunit does not contain a ligand but the other does suggests that binding of at least some inhibitors causes movement of residues 121–129 away from the opposing subunit.26,27
8.2. Inhibitor Kinetics
Early investigations tested the hypothesis that high potency and time dependency of COX inhibitors results from major inhibitor-induced changes in protein structure. Structural data have failed to support this hypothesis, however, as clear differences in protein structure are not observed in complexes of COX with highly potent time-dependent inhibitors as opposed to weak reversible ones.8 Nevertheless, a combination of structural, kinetic, and site-directed mutagenesis data has provided a foundation for understanding the time-dependent inhibition of COX by indomethacin. Specifically, it was noted that the 2′-methyl group of the inhibitor binds in a pocket formed by Val-349, Ala-527, Ser-530, and Leu-531. Mutations that obliterate this pocket or removal of the 2′-methyl group of the inhibitor convert indomethacin from a potent tight binding inhibitor to a rapidly reversible, much weaker inhibitor. These findings suggest that the insertion of the 2′-methyl group of indomethacin into the pocket is the time-dependent step that leads to a tightly bound enzyme–inhibitor complex.108
The COX-2-selective diarylheterocycles have generally been classified as weak, reversible inhibitors of COX-1 and potent, time-dependent inhibitors of COX-2.143,144 This may be an oversimplification, as a detailed kinetic study of the binding of SC299, a fluorescent COX-2-selective diarylheterocycle inhibitor, demonstrated time dependency of association to both COX-1 and COX-2.125 This study revealed that binding to both isoforms was rapid, occurring in two steps in the case of COX-1 and three steps in the case of COX-2. Isoform selectivity was attributed to the much slower rate of inhibitor dissociation from COX-2 than COX-1. On the basis of structural data, a model was proposed that correlated the third step of inhibitor binding to COX-2 with insertion of the inhibitor’s benzenesulfonamide group into the side pocket. It had long been assumed that this insertion occurs only with COX-2 and that this is the basis for the time dependency of COX-2-selective diarylheterocycles.32 However, recent structural data indicate that celecoxib can gain access to the side pocket of COX-1.26 These data suggest that insertion into the side pocket alone does not explain the kinetic difference in inhibition of COX-2 versus COX-1 for these inhibitors. An interesting finding in this regard was that some diarylheterocycles exhibited higher potency against an I523V mutant than wild-type COX-1, but time dependency was only observed for an I523V/H513R double mutant enzyme. These observations suggest that hydrogen bond formation with Arg-513, not possible with the smaller His-513 in COX-1, may be a critical determinant for time dependency, a hypothesis supported by molecular dynamics studies.122,123
8.3. Half-of-Sites Activity and Allostery
Evidence for half-of-sites activity of the COX enzymes comes from accumulated data suggesting that heme and many NSAIDs bind to only one of the two subunits and from studies of heterodimers comprising mutant and wild-type monomers or two functionally distinct mutant monomers.20−28 Unfortunately, support for this concept from structural studies has been largely lacking. No COX-1 or COX-2 crystal structures have been reported in which heme is bound to only one subunit, and attempts to limit ligand binding to only one site by using low ligand concentrations or a heterodimer containing a low affinity mutant subunit have been only partially successful.26,27 As noted above, it is possible that the presence of heme and ligand in both subunits favors crystallization. If true, we must conclude that crystal structures do not necessarily reflect the behavior of the enzyme in solution.
Despite the absence of clear evidence of half-of-sites heme or ligand binding in COX crystal structures, the data have provided some intriguing insights into this phenomenon. Among these is the binding of a number of substrates in different orientations, one productive and the other nonproductive, in the subunits of COX-2.53,62 Currently, it is unknown whether this observation is functionally significant or an artifact of crystallization. However, it is fascinating to note that the nonproductive orientation of AA in the COX-2 active site was first obtained in a complex lacking heme, suggesting that the absence of heme (as is proposed for the allosteric monomer of COX) would favor nonproductive binding of AA.33 It is also interesting to note that 13-Me-AA, which is a selective allosteric modulator of 2-AG oxygenation by COX-2 and COX-1 is bound in the active site of COX-2 in an inverted conformation, similar to the nonproductive conformation of AA.22 This observation suggests the possibility that fatty acid binding in this orientation may be correlated with allosteric modulation of COX activity. Of interest in this regard are reports that AA and 2-AG also exert allosteric effects on COX activity, with greater impact on 2-AG than AA oxygenation.25 All three fatty acids, AA, 2-AG, and 13-Me-AA, have been observed to adopt a binding conformation that involves displacement of Leu-531, suggesting that this could be a common theme in COX allostery. Consistently, L531V and L531A mutations eliminated the allosteric effects of 13-Me-AA.22 However, no such binding mode was observed in the crystal structure of PA complexed with COX-2, even though this fatty acid has also been reported to act as an positive allosteric COX modulator,21,28 and the role of Leu-531 in COX activity and regulation has not been fully delineated. Furthermore, nonproductive binding modes of substrates to COX-1 have not been observed, even though this isoform also displays half-of-sites activity and allosteric modulation.
Although attempts to obtain crystal structures of COX complexes containing ligands in only one subunit have not been completely successful, two such structures, one of a COX-1 wild-type/R120Q heterodimer complexed with (S)-flurbiprofen27 and the other of COX-1 complexed with celecoxib,26 have been reported. Both of these structures are notable in that subunits not containing a ligand exhibit a different orientation for residues 121–129 that brings residues Ser-126 and Pro-127 closer to Ser-541 and Ala-543 of the other monomer. The results suggest that movement of these residues may be important in ligand binding and subunit-to-subunit communication, a conclusion consistent with prior functional studies.26,28 Noting that the 121–129 loop is adjacent to Arg-120, which is involved in interactions with many carboxylate-containing COX ligands, led to the hypothesis that ligand interactions with Arg-120 might trigger a conformational change that leads to loop displacement. Consistent with this hypothesis was the finding that an S121P mutation increased baseline activity of COX-2 and reduced responsiveness to carboxylate-containing allosteric modulators. Structural studies of this mutation revealed disruption of helix D of the membrane-binding domain, though order was restored by binding to (S)-flurbiprofen.139 Further work will be required to completely understand the role of the 121–129 loop and Arg-120 in COX allosteric regulation. However, it should be noted that some allosteric modulators of COX-2, for example 13-Me-AA and AA itself, do not interact with Arg-120 according to the relevant crystal structure data.
8.4. Substrate Selectivity
The ability of some weak inhibitors of AA oxygenation to strongly inhibit 2-AG oxygenation by COX-2 has been explained on the basis of allosteric regulation of the enzyme.93 The hypothesis is that tight binding of these inhibitors to the allosteric site blocks 2-AG but not AA oxygenation at the active site, whereas much lower affinity binding of the inhibitor at the catalytic site blocks AA oxygenation. Attempts to explain this behavior on a structural basis have focused on data from complexes of COX-2 with the substrate-selective inhibitors (R)-flurbiprofen, (R)-naproxen, des-methyl-flurbiprofen, and (S)-ibuprofen.92,94,104 In each case, an inhibitor was bound in both subunits with little difference observed in protein conformation or the ligand binding pose between them. Consistently, the structural data provided insight into the lower affinity demonstrated by these inhibitors than their more potent, nonselective counterparts (S)-flurbiprofen and (S)-naproxen, but a basis for substrate selectivity was not readily apparent. If the proposed explanation for this phenomenon is correct, one could argue that it will be necessary to obtain a structure in which inhibitor is present in only one monomer of the enzyme; however, as noted above, this has been a particularly challenging goal.
Another attempt to explain the basis for substrate selectivity focused on the fenamic acids, of which flufenamic acid, tolfenamic acid, and mefenamic acid were found to be substrate-selective inhibitors, whereas meclofenamic acid was not. Crystal structure data revealed similar binding poses for all of these inhibitors, and all were present in both subunits of the enzyme. The structural data did not provide a basis for substrate selectivity of those fenamic acids exhibiting this property or for the higher potency and time-dependency of meclofenamic acid.121
9. Conclusions
The COX enzymes are among the most thoroughly studied membrane proteins from a structural and functional point of view. Thus, it is tempting to assume that all questions regarding these proteins have been answered. Consistently, the pace of new discoveries has slowed, due also to reduced interest in COX inhibitors resulting from the realization that cardiovascular toxicity is a significant issue for many. It may be more correct to argue, however, that it is the easy questions that have been answered, as many intriguing issues remain. For example, if the half-of-sites allosteric model of COX is correct, we must reevaluate the kinetics of enzyme activity and inhibition in light of the fact that both substrates and inhibitors may also be binding to, and competing for, the allosteric as well as the catalytic site. This complicates the study of an enzyme that naturally defies standard kinetics analyses due to its requirement for product activation and catalysis-dependent self-inactivation, despite the fact that standard kinetics analyses have routinely been employed. Similarly, the relative paucity of information concerning the response of the protein to ligand binding gleaned from structural data suggests that these responses may occur at a much subtler level. These considerations suggest that new approaches are needed to gain a full understanding of COX structure–function correlations. Approaches that may yield answers, such as systems-based kinetic modeling,25 or molecular dynamics assessments of enzyme-ligand interactions,114,123,141,142 are being explored.
Acknowledgments
Work in the Marnett laboratory was funded by National Institutes of Health Grant CA89450. Molecular graphics were created using the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Glossary
Abbreviations
- AA
arachidonic acid
- AEA
arachidonoyl ethanolamide
- 1-AG
1-arachidonoylglycerol
- 2-AG
2-arachidonoylglycerol
- COX-1
cyclooxygenase-1 (prostaglandin G/H synthase-1)
- COX-2
cyclooxygenase-2 (prostaglandin G/H synthase-2)
- DHA
docosahexaenoic acid
- DHLA
dihomo-γ-linolenic acid
- EPA
eicosapentaenoic acid
- FAAH
fatty acid amide hydrolase
- HETE
hydroxyeicosatetraenoic acid
- LA
linoleic acid
- αLA
α-linolenic acid
- 13-Me-AA
13(S)-methyl-arachidonic acid
- NSAID
nonsteroidal anti-inflammatory drug
- PA
palmitic acid
- PG
prostaglandin
- PDB
Protein Data Bank
- TXA2
thromboxane A2
Biographies
Carol A. Rouzer received her B.A. in Chemistry from McDaniel College, her Ph.D. in Biomedical Sciences from The Rockefeller University, and her M.D. from the Weill Medical College of Cornell University. After postdoctoral work at the Karolinska Institute, she accepted a position as Research Fellow at the Merck Frosst Center for Therapeutic Research. She moved to McDaniel College, where she was engaged primarily in undergraduate education until she joined the Biochemistry Faculty of Vanderbilt University as Research Professor. Her main research interests have focused on the total metabolism of arachidonic acid in inflammatory and malignant cells. She is the author of over 60 publications.
Lawrence J. Marnett received his B.S. in Chemistry from Rockhurst College and his Ph.D. in Chemistry from Duke University. After postdoctoral training at Karolinska Institute and Wayne State University, he joined the faculty in Chemistry at Wayne in 1975. He moved to Vanderbilt University as Mary Geddes Stahlman Professor of Cancer Research and Professor of Biochemistry and Chemistry. His research interests are in the chemical biology of oxygenation of polyunsaturated fatty acids. Dr. Marnett is a Fellow of the American Chemical Society and the American Association for the Advancement of Science. He was the inaugural winner of both the Founders’ Award of the Division of Chemical Toxicology and the George and Christine Sosnovsky Award for Cancer Research of the Division of Medicinal Chemistry. He is the author of over 550 research publications.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrev.0c00215.
(Table S1) Crystal Structures; (Table S2) site-directed mutations of cyclooxygenase isoforms; (Figures S1 to S20) both wall-eyed and cross-eyed format; and (Figures 1 to 24) cross-eyed format (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Funk C. D. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science 2001, 294, 1871–1875. 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Mechanism of Free Radical Oxygenation of Polyunsaturated Fatty Acids by Cyclooxygenases. Chem. Rev. 2003, 103, 2239–2304. 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- Smith W. Molecular Biology of Prostanoid Biosynthetic Enzymes and Receptors. Adv. Exp. Med. Biol. 1997, 400B, 989–1011. [PubMed] [Google Scholar]
- Smith W. L.; DeWitt D. L.; Garavito R. M. Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu. Rev. Biochem. 2000, 69, 145–182. 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Urade Y.; Jakobsson P. J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Xu L.; Porter N. A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Hecker M.; Ullrich V.; Fischer C.; Meese C. O. Identification of Novel Arachidonic Acid Metabolites Formed by Prostaglandin H Synthase. Eur. J. Biochem. 1987, 169, 113–123. 10.1111/j.1432-1033.1987.tb13587.x. [DOI] [PubMed] [Google Scholar]
- Ricciotti E.; FitzGerald G. A. Prostaglandins and Inflammation. Arterioscler., Thromb., Vasc. Biol. 2011, 31, 986–1000. 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D. L.; Botting R. M.; Hla T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- Smyth E. M.; Grosser T.; Wang M.; Yu Y.; FitzGerald G. A. Prostanoids in Health and Disease. J. Lipid Res. 2009, 50, S423–S428. 10.1194/jlr.R800094-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Zou H.; Yuan C.; Hong Y. H.; Uhlson C. L.; Murphy R. C.; Smith W. L. Interactions of 2-O-Arachidonylglycerol Ether and Ibuprofen with the Allosteric and Catalytic Subunits of Human Cox-2. J. Lipid Res. 2016, 57, 1043–1050. 10.1194/jlr.M067512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Moon S. H.; Jenkins C. M.; Sims H. F.; Gross R. W. Cyclooxygenase-2 Mediated Oxidation of 2-Arachidonoyl-Lysophospholipids Identifies Unknown Lipid Signaling Pathways. Cell Chem. Biol. 2016, 23, 1217–1227. 10.1016/j.chembiol.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Kingsley P. J.; Kozak K. R.; Marnett L. J. Selective Oxygenation of N-Arachidonylglycine by Cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2002, 296, 612–617. 10.1016/S0006-291X(02)00915-4. [DOI] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Turman M. V.; Vila A.; Ball H. L.; Al-Mestarihi A. H.; Di Marzo V.; Marnett L. J. Oxidative Metabolism of Lipoamino Acids and Vanilloids by Lipoxygenases and Cyclooxygenases. Arch. Biochem. Biophys. 2007, 464, 260–268. 10.1016/j.abb.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Non-Redundant Functions of Cyclooxygenases: Oxygenation of Endocannabinoids. J. Biol. Chem. 2008, 283, 8065–8069. 10.1074/jbc.R800005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Endocannabinoid Oxygenation by Cyclooxygenases, Lipoxygenases, and Cytochromes P450: Cross-Talk between the Eicosanoid and Endocannabinoid Signaling Pathways. Chem. Rev. 2011, 111, 5899–5921. 10.1021/cr2002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J. H.; Morrow J. D.; Belosludtsev Y. Y.; Beauchamp D. R.; DuBois R. N.; Falck J. R. The Catalytic Outcomes of the Constitutive and the Mitogen Inducible Isoforms of Prostaglandin H2 Synthase Are Markedly Affected by Glutathione and Glutathione Peroxidase(S). Biochemistry 1995, 34, 3325–3337. 10.1021/bi00010a023. [DOI] [PubMed] [Google Scholar]
- Kulmacz R. J.; Wang L. H. Comparison of Hydroperoxide Initiator Requirements for the Cyclooxygenase Activities of Prostaglandin H Synthase-1 and −2. J. Biol. Chem. 1995, 270, 24019–24023. 10.1074/jbc.270.41.24019. [DOI] [PubMed] [Google Scholar]
- Kulmacz R. J. Cellular Regulation of Prostaglandin H Synthase Catalysis. FEBS Lett. 1998, 430, 154–157. 10.1016/S0014-5793(98)00657-7. [DOI] [PubMed] [Google Scholar]
- Dong L.; Sharma N. P.; Jurban B. J.; Smith W. L. Pre-Existent Asymmetry in the Human Cyclooxygenase-2 Sequence Homodimer. J. Biol. Chem. 2013, 288, 28641–28655. 10.1074/jbc.M113.505503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Vecchio A. J.; Sharma N. P.; Jurban B. J.; Malkowski M. G.; Smith W. L. Human Cyclooxygenase-2 Is a Sequence Homodimer That Functions as a Conformational Heterodimer. J. Biol. Chem. 2011, 286, 19035–19046. 10.1074/jbc.M111.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudalkar S. N.; Nikas S. P.; Kingsley P. J.; Xu S.; Galligan J. J.; Rouzer C. A.; Banerjee S.; Ji L.; Eno M. R.; Makriyannis A.; Marnett L. J. 13-Methylarachidonic Acid Is a Positive Allosteric Modulator of Endocannabinoid Oxygenation by Cyclooxygenase. J. Biol. Chem. 2015, 290, 7897–7909. 10.1074/jbc.M114.634014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmacz R. J.; Lands W. E. Prostaglandin H Synthase. Stoichiometry of Heme Cofactor. J. Biol. Chem. 1984, 259, 6358–6363. [PubMed] [Google Scholar]
- Kulmacz R. J.; Lands W. E. Stoichiometry and Kinetics of the Interaction of Prostaglandin H Synthase with Anti-Inflammatory Agents. J. Biol. Chem. 1985, 260, 12572–12578. [PubMed] [Google Scholar]
- Mitchener M. M.; Hermanson D. J.; Shockley E. M.; Brown H. A.; Lindsley C. W.; Reese J.; Rouzer C. A.; Lopez C. F.; Marnett L. J. Competition and Allostery Govern Substrate Selectivity of Cyclooxygenase-2. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12366–12371. 10.1073/pnas.1507307112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimon G.; Sidhu R. S.; Lauver D. A.; Lee J. Y.; Sharma N. P.; Yuan C.; Frieler R. A.; Trievel R. C.; Lucchesi B. R.; Smith W. L. Coxibs Interfere with the Action of Aspirin by Binding Tightly to One Monomer of Cyclooxygenase-1. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 28–33. 10.1073/pnas.0909765106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu R. S.; Lee J. Y.; Yuan C.; Smith W. L. Comparison of Cyclooxygenase-1 Crystal Structures: Cross-Talk between Monomers Comprising Cyclooxygenase-1 Homodimers. Biochemistry 2010, 49, 7069–7079. 10.1021/bi1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C.; Sidhu R. S.; Kuklev D. V.; Kado Y.; Wada M.; Song I.; Smith W. L. Cyclooxygenase Allosterism, Fatty Acid-Mediated Cross-Talk between Monomers of Cyclooxygenase Homodimers. J. Biol. Chem. 2009, 284, 10046–10055. 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picot D.; Loll P. J.; Garavito R. M. The X-Ray Crystal Structure of the Membrane Protein Prostaglandin H2 Synthase-1. Nature 1994, 367, 243–249. 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- Loll P. J.; Picot D.; Ekabo O.; Garavito R. M. Synthesis and Use of Iodinated Nonsteroidal Antiinflammatory Drug Analogs as Crystallographic Probes of the Prostaglandin H2 Synthase Cyclooxygenase Active Site. Biochemistry 1996, 35, 7330–7340. 10.1021/bi952776w. [DOI] [PubMed] [Google Scholar]
- Loll P. J.; Picot D.; Garavito R. M. The Structural Basis of Aspirin Activity Inferred from the Crystal Structure of Inactivated Prostaglandin H2 Synthase. Nat. Struct. Biol. 1995, 2, 637–643. 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]
- Kurumbail R. G.; Stevens A. M.; Gierse J. K.; McDonald J. J.; Stegeman R. A.; Pak J. Y.; Gildehaus D.; Miyashiro J. M.; Penning T. D.; Seibert K.; Isakson P. C.; Stallings W. C. Structural Basis for Selective Inhibition of Cyclooxygenase-2 by Anti-Inflammatory Agents. Nature 1996, 384, 644–648. 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- Kiefer J. R.; Pawlitz J. L.; Moreland K. T.; Stegeman R. A.; Hood W. F.; Gierse J. K.; Stevens A. M.; Goodwin D. C.; Rowlinson S. W.; Marnett L. J.; Stallings W. C.; Kurumbail R. G. Structural Insights into the Stereochemistry of the Cyclooxygenase Reaction. Nature 2000, 405, 97–101. 10.1038/35011103. [DOI] [PubMed] [Google Scholar]
- Malkowski M. G.; Ginell S. L.; Smith W. L.; Garavito R. M. The Productive Conformation of Arachidonic Acid Bound to Prostaglandin Synthase. Science 2000, 289, 1933–1937. 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- Garavito R. M.; Mulichak A. M. The Structure of Mammalian Cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 183–206. 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Dewitt D. L. Prostaglandin Endoperoxide H Synthases-1 and −2. Adv. Immunol. 1996, 62, 167–215. 10.1016/S0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- Furse K. E.; Pratt D. A.; Porter N. A.; Lybrand T. P. Molecular Dynamics Simulations of Arachidonic Acid Complexes with Cox-1 and Cox-2: Insights into Equilibrium Behavior. Biochemistry 2006, 45, 3189–3205. 10.1021/bi052337p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furse K. E.; Pratt D. A.; Schneider C.; Brash A. R.; Porter N. A.; Lybrand T. P. Molecular Dynamics Simulations of Arachidonic Acid-Derived Pentadienyl Radical Intermediate Complexes with Cox-1 and Cox-2: Insights into Oxygenation Regio- and Stereoselectivity. Biochemistry 2006, 45, 3206–3218. 10.1021/bi052338h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobaum A. L.; Marnett L. J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; Langenbach R. Why There Are Two Cyclooxygenase Isozymes. J. Clin. Invest. 2001, 107, 1491–1495. 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landino L. M.; Crews B. C.; Gierse J. K.; Hauser S. D.; Marnett L. J. Mutational Analysis of the Role of the Distal Histidine and Glutamine Residues of Prostaglandin-Endoperoxide Synthase-2 in Peroxidase Catalysis, Hydroperoxide Reduction, and Cyclooxygenase Activation. J. Biol. Chem. 1997, 272, 21565–21574. 10.1074/jbc.272.34.21565. [DOI] [PubMed] [Google Scholar]
- Malkowski M. G.; Theisen M. J.; Scharmen A.; Garavito R. M. The Formation of Stable Fatty Acid Substrate Complexes in Prostaglandin H(2) Synthase-1. Arch. Biochem. Biophys. 2000, 380, 39–45. 10.1006/abbi.2000.1906. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D. K.; Lecomte M.; Rieke C. J.; Garavito M.; Smith W. L. Involvement of Arginine 120, Glutamate 524, and Tyrosine 355 in the Binding of Arachidonate and 2-Phenylpropionic Acid Inhibitors to the Cyclooxygenase Active Site of Ovine Prostaglandin Endoperoxide H Synthase-1. J. Biol. Chem. 1996, 271, 2179–2184. 10.1074/jbc.271.4.2179. [DOI] [PubMed] [Google Scholar]
- Mancini J. A.; Riendeau D.; Falgueyret J. P.; Vickers P. J.; O’Neill G. P. Arginine 120 of Prostaglandin G/H Synthase-1 Is Required for the Inhibition by Nonsteroidal Anti-Inflammatory Drugs Containing a Carboxylic Acid Moiety. J. Biol. Chem. 1995, 270, 29372–29377. 10.1074/jbc.270.49.29372. [DOI] [PubMed] [Google Scholar]
- Shimokawa T.; Kulmacz R. J.; DeWitt D. L.; Smith W. L. Tyrosine 385 of Prostaglandin Endoperoxide Synthase Is Required for Cyclooxygenase Catalysis. J. Biol. Chem. 1990, 265, 20073–20076. [PubMed] [Google Scholar]
- Thuresson E. D.; Lakkides K. M.; Rieke C. J.; Sun Y.; Wingerd B. A.; Micielli R.; Mulichak A. M.; Malkowski M. G.; Garavito R. M.; Smith W. L. Prostaglandin Endoperoxide H Synthase-1: The Functions of Cyclooxygenase Active Site Residues in the Binding, Positioning, and Oxygenation of Arachidonic Acid. J. Biol. Chem. 2001, 276, 10347–10357. 10.1074/jbc.M009377200. [DOI] [PubMed] [Google Scholar]
- Schneider C.; Boeglin W. E.; Prusakiewicz J. J.; Rowlinson S. W.; Marnett L. J.; Samel N.; Brash A. R. Control of Prostaglandin Stereochemistry at the 15-Carbon by Cyclooxygenases-1 and −2. A Critical Role for Serine 530 and Valine 349. J. Biol. Chem. 2002, 277, 478–485. 10.1074/jbc.M107471200. [DOI] [PubMed] [Google Scholar]
- Thuresson E. D.; Lakkides K. M.; Smith W. L. Different Catalytically Competent Arrangements of Arachidonic Acid within the Cyclooxygenase Active Site of Prostaglandin Endoperoxide H Synthase-1 Lead to the Formation of Different Oxygenated Products. J. Biol. Chem. 2000, 275, 8501–8507. 10.1074/jbc.275.12.8501. [DOI] [PubMed] [Google Scholar]
- DeWitt D. L.; el-Harith E. A.; Kraemer S. A.; Andrews M. J.; Yao E. F.; Armstrong R. L.; Smith W. L. The Aspirin and Heme-Binding Sites of Ovine and Murine Prostaglandin Endoperoxide Synthases. J. Biol. Chem. 1990, 265, 5192–5198. [PubMed] [Google Scholar]
- Rowlinson S. W.; Kiefer J. R.; Prusakiewicz J. J.; Pawlitz J. L.; Kozak K. R.; Kalgutkar A. S.; Stallings W. C.; Kurumbail R. G.; Marnett L. J. A Novel Mechanism of Cyclooxygenase-2 Inhibition Involving Interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. 10.1074/jbc.M305481200. [DOI] [PubMed] [Google Scholar]
- Shimokawa T.; Smith W. L. Prostaglandin Endoperoxide Synthase. The Aspirin Acetylation Region. J. Biol. Chem. 1992, 267, 12387–12392. [PubMed] [Google Scholar]
- Harman C. A.; Rieke C. J.; Garavito R. M.; Smith W. L. Crystal Structure of Arachidonic Acid Bound to a Mutant of Prostaglandin Endoperoxide H Synthase-1 That Forms Predominantly 11-Hydroperoxyeicosatetraenoic Acid. J. Biol. Chem. 2004, 279, 42929–42935. 10.1074/jbc.M403013200. [DOI] [PubMed] [Google Scholar]
- Vecchio A. J.; Simmons D. M.; Malkowski M. G. Structural Basis of Fatty Acid Substrate Binding to Cyclooxygenase-2. J. Biol. Chem. 2010, 285, 22152–22163. 10.1074/jbc.M110.119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival M. D.; Bastien L.; Griffin P. R.; Kargman S.; Ouellet M.; O’Neill G. P. Investigation of Human Cyclooxygenase-2 Glycosylation Heterogeneity and Protein Expression in Insect and Mammalian Cell Expression Systems. Protein Expression Purif. 1997, 9, 388–398. 10.1006/prep.1996.0685. [DOI] [PubMed] [Google Scholar]
- Mancini J. A.; Vickers P. J.; O’Neill G. P.; Boily C.; Falgueyret J. P.; Riendeau D. Altered Sensitivity of Aspirin-Acetylated Prostaglandin G/H Synthase-2 to Inhibition by Nonsteroidal Anti-Inflammatory Drugs. Mol. Pharmacol. 1997, 51, 52–60. 10.1124/mol.51.1.52. [DOI] [PubMed] [Google Scholar]
- Dong L.; Anderson A. J.; Malkowski M. G. Arg-513 and Leu-531 Are Key Residues Governing Time-Dependent Inhibition of Cyclooxygenase-2 by Aspirin and Celebrex. Biochemistry 2019, 58, 3990–4002. 10.1021/acs.biochem.9b00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig G. M.; Francis D. A.; Falgueyret J. P.; Ouellet M.; Percival M. D.; Roy P.; Bayly C.; Mancini J. A.; O’Neill G. P. The Interaction of Arginine 106 of Human Prostaglandin G/H Synthase-2 with Inhibitors Is Not a Universal Component of Inhibition Mediated by Nonsteroidal Anti-Inflammatory Drugs. Mol. Pharmacol. 1997, 52, 829–838. 10.1124/mol.52.5.829. [DOI] [PubMed] [Google Scholar]
- Rieke C. J.; Mulichak A. M.; Garavito R. M.; Smith W. L. The Role of Arginine 120 of Human Prostaglandin Endoperoxide H Synthase-2 in the Interaction with Fatty Acid Substrates and Inhibitors. J. Biol. Chem. 1999, 274, 17109–17114. 10.1074/jbc.274.24.17109. [DOI] [PubMed] [Google Scholar]
- Rowlinson S. W.; Crews B. C.; Goodwin D. C.; Schneider C.; Gierse J. K.; Marnett L. J. Spatial Requirements for 15-(R)-Hydroxy-5z,8z,11z, 13e-Eicosatetraenoic Acid Synthesis within the Cyclooxygenase Active Site of Murine Cox-2. Why Acetylated Cox-1 Does Not Synthesize 15-(R)-Hete. J. Biol. Chem. 2000, 275, 6586–6591. 10.1074/jbc.275.9.6586. [DOI] [PubMed] [Google Scholar]
- Rowlinson S. W.; Crews B. C.; Lanzo C. A.; Marnett L. J. The Binding of Arachidonic Acid in the Cyclooxygenase Active Site of Mouse Prostaglandin Endoperoxide Synthase-2 (Cox-2). A Putative L-Shaped Binding Conformation Utilizing the Top Channel Region. J. Biol. Chem. 1999, 274, 23305–23310. 10.1074/jbc.274.33.23305. [DOI] [PubMed] [Google Scholar]
- Vecchio A. J.; Orlando B. J.; Nandagiri R.; Malkowski M. G. Investigating Substrate Promiscuity in Cyclooxygenase-2: The Role of Arg-120 and Residues Lining the Hydrophobic Groove. J. Biol. Chem. 2012, 287, 24619–24630. 10.1074/jbc.M112.372243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchio A. J.; Malkowski M. G. The Structural Basis of Endocannabinoid Oxygenation by Cyclooxygenase-2. J. Biol. Chem. 2011, 286, 20736–20745. 10.1074/jbc.M111.230367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkowski M. G.; Thuresson E. D.; Lakkides K. M.; Rieke C. J.; Micielli R.; Smith W. L.; Garavito R. M. Structure of Eicosapentaenoic and Linoleic Acids in the Cyclooxygenase Site of Prostaglandin Endoperoxide H Synthase-1. J. Biol. Chem. 2001, 276, 37547–37555. 10.1074/jbc.M105982200. [DOI] [PubMed] [Google Scholar]
- Thuresson E. D.; Malkowski M. G.; Lakkides K. M.; Rieke C. J.; Mulichak A. M.; Ginell S. L.; Garavito R. M.; Smith W. L. Mutational and X-Ray Crystallographic Analysis of the Interaction of Dihomo-Gamma -Linolenic Acid with Prostaglandin Endoperoxide H Synthases. J. Biol. Chem. 2001, 276, 10358–10365. 10.1074/jbc.M009378200. [DOI] [PubMed] [Google Scholar]
- Wada M.; DeLong C. J.; Hong Y. H.; Rieke C. J.; Song I.; Sidhu R. S.; Yuan C.; Warnock M.; Schmaier A. H.; Yokoyama C.; Smyth E. M.; Wilson S. J.; FitzGerald G. A.; Garavito R. M.; Sui D. X.; Regan J. W.; Smith W. L. Enzymes and Receptors of Prostaglandin Pathways with Arachidonic Acid-Derived Versus Eicosapentaenoic Acid-Derived Substrates and Products. J. Biol. Chem. 2007, 282, 22254–22266. 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- Kozak K. R.; Rowlinson S. W.; Marnett L. J. Oxygenation of the Endocannabinoid, 2-Arachidonylglycerol, to Glyceryl Prostaglandins by Cyclooxygenase-2. J. Biol. Chem. 2000, 275, 33744–33749. 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- Kozak K. R.; Prusakiewicz J. J.; Rowlinson S. W.; Schneider C.; Marnett L. J. Amino Acid Determinants in Cyclooxygenase-2 Oxygenation of the Endocannabinoid 2-Arachidonylglycerol. J. Biol. Chem. 2001, 276, 30072–30077. 10.1074/jbc.M104467200. [DOI] [PubMed] [Google Scholar]
- Vane J. R. The Fight against Rheumatism: From Willow Bark to Cox-1 Sparing Drugs. J. Physiol. Pharmacol. 2000, 51, 573–586. [PubMed] [Google Scholar]
- Brune K.; Hinz B. The Discovery and Development of Antiinflammatory Drugs. Arthritis Rheum. 2004, 50, 2391–2399. 10.1002/art.20424. [DOI] [PubMed] [Google Scholar]
- Ferreira S. H.; Moncada S.; Vane J. R. Indomethacin and Aspirin Abolish Prostaglandin Release from the Spleen. Nat. New Biol. 1971, 231, 237–239. 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- Smith J. B.; Willis A. L. Aspirin Selectively Inhibits Prostaglandin Production in Human Platelets. Nat. New Biol. 1971, 231, 235–237. 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- Vane J. R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-Like Drugs. Nat. New Biol. 1971, 231, 232–235. 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- Miyamoto T.; Ogino N.; Yamamoto S.; Hayaishi O. Purification of Prostaglandin Endoperoxide Synthetase from Bovine Vesicular Gland Microsomes. J. Biol. Chem. 1976, 251, 2629–2636. [PubMed] [Google Scholar]
- Kujubu D. A.; Fletcher B. S.; Varnum B. C.; Lim R. W.; Herschman H. R. Tis10, a Phorbol Ester Tumor Promoter-Inducible Mrna from Swiss 3t3 Cells, Encodes a Novel Prostaglandin Synthase/Cyclooxygenase Homologue. J. Biol. Chem. 1991, 266, 12866–12872. [PubMed] [Google Scholar]
- Xie W. L.; Chipman J. G.; Robertson D. L.; Erikson R. L.; Simmons D. L. Expression of a Mitogen-Responsive Gene Encoding Prostaglandin Synthase Is Regulated by Mrna Splicing. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 2692–2696. 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A.; Araki H.; Komoike Y.; Hase S.; Takeuchi K. Inhibition of Both Cox-1 and Cox-2 Is Required for Development of Gastric Damage in Response to Nonsteroidal Antiinflammatory Drugs. J. Physiol. 2001, 95, 21–27. 10.1016/S0928-4257(01)00005-5. [DOI] [PubMed] [Google Scholar]
- Tanaka A.; Hase S.; Miyazawa T.; Takeuchi K. Up-Regulation of Cyclooxygenase-2 by Inhibition of Cyclooxygenase-1: A Key to Nonsteroidal Anti-Inflammatory Drug-Induced Intestinal Damage. J. Pharmacol. Exp. Ther. 2002, 300, 754–761. 10.1124/jpet.300.3.754. [DOI] [PubMed] [Google Scholar]
- Nonsteroidal Antiinflammatory Drugs (Nsaids). https://www.ncbi.nlm.nih.gov/books/NBK548614/ (accessed November 19, 2019).
- Brune K.; Patrignani P. New Insights into the Use of Currently Available Non-Steroidal Anti-Inflammatory Drugs. J. Pain Res. 2015, 8, 105–118. 10.2147/JPR.S75160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C. D.; FitzGerald G. A. Cox-2 Inhibitors and Cardiovascular Risk. J. Cardiovasc. Pharmacol. 2007, 50, 470–479. 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- Marnett L. J.; Kalgutkar A. S. Cyclooxygenase 2 Inhibitors: Discovery, Selectivity and the Future. Trends Pharmacol. Sci. 1999, 20, 465–469. 10.1016/S0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- Calvello R.; Panaro M. A.; Carbone M. L.; Cianciulli A.; Perrone M. G.; Vitale P.; Malerba P.; Scilimati A. Novel Selective Cox-1 Inhibitors Suppress Neuroinflammatory Mediators in Lps-Stimulated N13 Microglial Cells. Pharmacol. Res. 2012, 65, 137–148. 10.1016/j.phrs.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Di Nunno L.; Vitale P.; Scilimati A.; Tacconelli S.; Patrignani P. Novel Synthesis of 3,4-Diarylisoxazole Analogues of Valdecoxib: Reversal Cyclooxygenase-2 Selectivity by Sulfonamide Group Removal. J. Med. Chem. 2004, 47, 4881–4890. 10.1021/jm040782x. [DOI] [PubMed] [Google Scholar]
- Fukai R.; Zheng X.; Motoshima K.; Tai A.; Yazama F.; Kakuta H. Design and Synthesis of Novel Cyclooxygenase-1 Inhibitors as Analgesics: 5-Amino-2-Ethoxy-N-(Substituted-Phenyl)Benzamides. ChemMedChem 2011, 6, 550–560. 10.1002/cmdc.201000462. [DOI] [PubMed] [Google Scholar]
- Imanishi J.; Morita Y.; Yoshimi E.; Kuroda K.; Masunaga T.; Yamagami K.; Kuno M.; Hamachi E.; Aoki S.; Takahashi F.; Nakamura K.; Miyata S.; Ohkubo Y.; Mutoh S. Pharmacological Profile of Fk881(Asp6537), a Novel Potent and Selective Cyclooxygenase-1 Inhibitor. Biochem. Pharmacol. 2011, 82, 746–754. 10.1016/j.bcp.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Kakuta H.; Zheng X.; Oda H.; Harada S.; Sugimoto Y.; Sasaki K.; Tai A. Cyclooxygenase-1-Selective Inhibitors Are Attractive Candidates for Analgesics That Do Not Cause Gastric Damage. Design and in Vitro/in Vivo Evaluation of a Benzamide-Type Cyclooxygenase-1 Selective Inhibitor. J. Med. Chem. 2008, 51, 2400–2411. 10.1021/jm701191z. [DOI] [PubMed] [Google Scholar]
- Kusuhara H.; Fukunari A.; Matsuyuki H.; Okumoto T. Principal Involvement of Cyclooxygenase-1-Derived Prostaglandins in the C-Fos Expression of the Rat Hind Brain Following Visceral Stimulation with Acetic Acid. Mol. Brain Res. 1997, 52, 151–156. 10.1016/S0169-328X(97)00263-5. [DOI] [PubMed] [Google Scholar]
- Liedtke A. J.; Crews B. C.; Daniel C. M.; Blobaum A. L.; Kingsley P. J.; Ghebreselasie K.; Marnett L. J. Cyclooxygenase-1-Selective Inhibitors Based on the (E)-2’-Des-Methyl-Sulindac Sulfide Scaffold. J. Med. Chem. 2012, 55, 2287–2300. 10.1021/jm201528b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T.; Motoyama Y.; Goto T. The Analgesic Effect Profile of Fr122047, a Selective Cyclooxygenase-1 Inhibitor, in Chemical Nociceptive Models. Eur. J. Pharmacol. 2000, 391, 49–54. 10.1016/S0014-2999(00)00051-0. [DOI] [PubMed] [Google Scholar]
- Ono N.; Yamamoto N.; Sunami A.; Yamasaki Y.; Miyake H. [Pharmacological Profile of Mofezolac, a New Non-Steroidal Analgesic Anti-Inflammatory Drug]. Nippon Yakurigaku Zasshi 1990, 95, 63–81. 10.1254/fpj.95.2_63. [DOI] [PubMed] [Google Scholar]
- Smith C. J.; Zhang Y.; Koboldt C. M.; Muhammad J.; Zweifel B. S.; Shaffer A.; Talley J. J.; Masferrer J. L.; Seibert K.; Isakson P. C. Pharmacological Analysis of Cyclooxygenase-1 in Inflammation. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13313–13318. 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan K. C.; Hermanson D. J.; Musee J.; Prusakiewicz J. J.; Scheib J. L.; Carter B. D.; Banerjee S.; Oates J. A.; Marnett L. J. (R)-Profens Are Substrate-Selective Inhibitors of Endocannabinoid Oxygenation by Cox-2. Nat. Chem. Biol. 2011, 7, 803–809. 10.1038/nchembio.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Duggan K. C.; Rouzer C. A.; Marnett L. J. Differential Sensitivity and Mechanism of Inhibition of Cox-2 Oxygenation of Arachidonic Acid and 2-Arachidonoylglycerol by Ibuprofen and Mefenamic Acid. Biochemistry 2009, 48, 7353–7355. 10.1021/bi900999z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windsor M. A.; Hermanson D. J.; Kingsley P. J.; Xu S.; Crews B. C.; Ho W.; Keenan C. M.; Banerjee S.; Sharkey K. A.; Marnett L. J. Substrate-Selective Inhibition of Cyclooxygenase-2: Development and Evaluation of Achiral Profen Probes. ACS Med. Chem. Lett. 2012, 3, 759–763. 10.1021/ml3001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windsor M. A.; Valk P. L.; Xu S.; Banerjee S.; Marnett L. J. Exploring the Molecular Determinants of Substrate-Selective Inhibition of Cyclooxygenase-2 by Lumiracoxib. Bioorg. Med. Chem. Lett. 2013, 23, 5860–5864. 10.1016/j.bmcl.2013.08.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W. L.; DeWitt D. L. Biochemistry of Prostaglandin Endoperoxide H Synthase-1 and Synthase-2 and Their Differential Susceptibility to Nonsteroidal Anti-Inflammatory Drugs. Semin. Nephrol. 1995, 15, 179–194. [PubMed] [Google Scholar]
- Selinsky B. S.; Gupta K.; Sharkey C. T.; Loll P. J. Structural Analysis of Nsaid Binding by Prostaglandin H2 Synthase: Time-Dependent and Time-Independent Inhibitors Elicit Identical Enzyme Conformations. Biochemistry 2001, 40, 5172–5180. 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- Gupta K.; Selinsky B. S.; Loll P. J. 2.0 Angstroms Structure of Prostaglandin H2 Synthase-1 Reconstituted with a Manganese Porphyrin Cofactor. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 151–156. 10.1107/S0907444905036309. [DOI] [PubMed] [Google Scholar]
- Gupta K.; Selinsky B. S.; Kaub C. J.; Katz A. K.; Loll P. J. The 2.0 a Resolution Crystal Structure of Prostaglandin H2 Synthase-1: Structural Insights into an Unusual Peroxidase. J. Mol. Biol. 2004, 335, 503–518. 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- Duggan K. C.; Walters M. J.; Musee J.; Harp J. M.; Kiefer J. R.; Oates J. A.; Marnett L. J. Molecular Basis for Cyclooxygenase Inhibition by the Non-Steroidal Anti-Inflammatory Drug Naproxen. J. Biol. Chem. 2010, 285, 34950–34959. 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierse J. K.; McDonald J. J.; Hauser S. D.; Rangwala S. H.; Koboldt C. M.; Seibert K. A Single Amino Acid Difference between Cyclooxygenase-1 (Cox-1) and −2 (Cox-2) Reverses the Selectivity of Cox-2 Specific Inhibitors. J. Biol. Chem. 1996, 271, 15810–15814. 10.1074/jbc.271.26.15810. [DOI] [PubMed] [Google Scholar]
- Sasso O.; Migliore M.; Habrant D.; Armirotti A.; Albani C.; Summa M.; Moreno-Sanz G.; Scarpelli R.; Piomelli D. Multitarget Fatty Acid Amide Hydrolase/Cyclooxygenase Blockade Suppresses Intestinal Inflammation and Protects against Nonsteroidal Anti-Inflammatory Drug-Dependent Gastrointestinal Damage. FASEB J. 2015, 29, 2616–2627. 10.1096/fj.15-270637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M. C.; Xu S.; Rouzer C. A.; Banerjee S.; Ghebreselasie K.; Migliore M.; Piomelli D.; Marnett L. J. Dual Cyclooxygenase-Fatty Acid Amide Hydrolase Inhibitor Exploits Novel Binding Interactions in the Cyclooxygenase Active Site. J. Biol. Chem. 2018, 293, 3028–3038. 10.1074/jbc.M117.802058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando B. J.; Lucido M. J.; Malkowski M. G. The Structure of Ibuprofen Bound to Cyclooxygenase-2. J. Struct. Biol. 2015, 189, 62–66. 10.1016/j.jsb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobaum A. L.; Xu S.; Rowlinson S. W.; Duggan K. C.; Banerjee S.; Kudalkar S. N.; Birmingham W. R.; Ghebreselasie K.; Marnett L. J. Action at a Distance: Mutations of Peripheral Residues Transform Rapid Reversible Inhibitors to Slow, Tight Binders of Cyclooxygenase-2. J. Biol. Chem. 2015, 290, 12793–12803. 10.1074/jbc.M114.635987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Crews B. C.; Rowlinson S. W.; Marnett A. B.; Kozak K. R.; Remmel R. P.; Marnett L. J. Biochemically Based Design of Cyclooxygenase-2 (Cox-2) Inhibitors: Facile Conversion of Nonsteroidal Antiinflammatory Drugs to Potent and Highly Selective Cox-2 Inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 925–930. 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M. J.; Crews B. C.; Xu S.; Ghebreselasie K.; Daniel C. K.; Kingsley P. J.; Banerjee S.; Marnett L. J. Antitumor Activity of Cytotoxic Cyclooxygenase-2 Inhibitors. ACS Chem. Biol. 2016, 11, 3052–3060. 10.1021/acschembio.6b00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusakiewicz J. J.; Felts A. S.; Mackenzie B. S.; Marnett L. J. Molecular Basis of the Time-Dependent Inhibition of Cyclooxygenases by Indomethacin. Biochemistry 2004, 43, 15439–15445. 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- Blobaum A. L.; Marnett L. J. Molecular Determinants for the Selective Inhibition of Cyclooxygenase-2 by Lumiracoxib. J. Biol. Chem. 2007, 282, 16379–16390. 10.1074/jbc.M609883200. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Marnett A. B.; Crews B. C.; Remmel R. P.; Marnett L. J. Ester and Amide Derivatives of the Nonsteroidal Antiinflammatory Drug, Indomethacin, as Selective Cyclooxygenase-2 Inhibitors. J. Med. Chem. 2000, 43, 2860–2870. 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Rowlinson S. W.; Crews B. C.; Marnett L. J. Amide Derivatives of Meclofenamic Acid as Selective Cyclooxygenase-2 Inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 521–524. 10.1016/S0960-894X(01)00792-2. [DOI] [PubMed] [Google Scholar]
- Harman C. A.; Turman M. V.; Kozak K. R.; Marnett L. J.; Smith W. L.; Garavito R. M. Structural Basis of Enantioselective Inhibition of Cyclooxygenase-1 by S-Alpha-Substituted Indomethacin Ethanolamides. J. Biol. Chem. 2007, 282, 28096–28105. 10.1074/jbc.M701335200. [DOI] [PubMed] [Google Scholar]
- Xu S.; Uddin M. J.; Banerjee S.; Duggan K.; Musee J.; Kiefer J. R.; Ghebreselasie K.; Rouzer C. A.; Marnett L. J. Fluorescent Indomethacin-Dansyl Conjugates Utilize the Membrane-Binding Domain of Cyclooxygenase-2 to Block the Opening to the Active Site. J. Biol. Chem. 2019, 294, 8690–8698. 10.1074/jbc.RA119.007405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkle M. E.; Blobaum A. L.; Moth C. W.; Prusakiewicz J. J.; Xu S.; Ghebreselasie K.; Akingbade D.; Jacobs A. T.; Rouzer C. A.; Lybrand T. P.; Marnett L. J. Conservative Secondary Shell Substitution in Cyclooxygenase-2 Reduces Inhibition by Indomethacin Amides and Esters Via Altered Enzyme Dynamics. Biochemistry 2016, 55, 348–359. 10.1021/acs.biochem.5b01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube M.; Neumann W.; Scholz M.; Lonnecke P.; Crews B.; Marnett L. J.; Pietzsch J.; Kniess T.; Hey-Hawkins E. 2-Carbaborane-3-Phenyl-1h-Indoles--Synthesis Via Mcmurry Reaction and Cyclooxygenase (Cox) Inhibition Activity. ChemMedChem 2013, 8, 329–335. 10.1002/cmdc.201200455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann W.; Xu S.; Sarosi M. B.; Scholz M. S.; Crews B. C.; Ghebreselasie K.; Banerjee S.; Marnett L. J.; Hey-Hawkins E. Nido-Dicarbaborate Induces Potent and Selective Inhibition of Cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. 10.1002/cmdc.201500199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong C.; Miller A.; Barnett J.; Chow J.; Ramesha C.; Browner M. F. Flexibility of the Nsaid Binding Site in the Structure of Human Cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- Kothekar V.; Sahi S.; Srinivasan M.; Mohan A.; Mishra J. Recognition of Cyclooxygenase-2 (Cox-2) Active Site by Nsaids: A Computer Modelling Study. Indian J. Biochem. Biophys. 2001, 38, 56–63. [PubMed] [Google Scholar]
- Park H.; Lee S. Free Energy Perturbation Approach to the Critical Assessment of Selective Cyclooxygenase-2 Inhibitors. J. Comput.-Aided Mol. Des. 2005, 19, 17–31. 10.1007/s10822-005-0098-5. [DOI] [PubMed] [Google Scholar]
- Xu S.; Hermanson D. J.; Banerjee S.; Ghebreselasie K.; Clayton G. M.; Garavito R. M.; Marnett L. J. Oxicams Bind in a Novel Mode to the Cyclooxygenase Active Site Via a Two-Water-Mediated H-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. 10.1074/jbc.M113.517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando B. J.; Malkowski M. G. Substrate-Selective Inhibition of Cyclooxygeanse-2 by Fenamic Acid Derivatives Is Dependent on Peroxide Tone. J. Biol. Chem. 2016, 291, 15069–15081. 10.1074/jbc.M116.725713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E.; Bayly C.; Waterman H. L.; Riendeau D.; Mancini J. A. Conversion of Prostaglandin G/H Synthase-1 into an Enzyme Sensitive to Pghs-2-Selective Inhibitors by a Double His513 --> Arg and Ile523 --> Val Mutation. J. Biol. Chem. 1997, 272, 9280–9286. 10.1074/jbc.272.14.9280. [DOI] [PubMed] [Google Scholar]
- Khan Y. S.; Gutierrez-de-Teran H.; Aqvist J. Molecular Mechanisms in the Selectivity of Nonsteroidal Anti-Inflammatory Drugs. Biochemistry 2018, 57, 1236–1248. 10.1021/acs.biochem.7b01019. [DOI] [PubMed] [Google Scholar]
- Wang J. L.; Carter J.; Kiefer J. R.; Kurumbail R. G.; Pawlitz J. L.; Brown D.; Hartmann S. J.; Graneto M. J.; Seibert K.; Talley J. J. The Novel Benzopyran Class of Selective Cyclooxygenase-2 Inhibitors-Part I: The First Clinical Candidate. Bioorg. Med. Chem. Lett. 2010, 20, 7155–7158. 10.1016/j.bmcl.2010.07.053. [DOI] [PubMed] [Google Scholar]
- Lanzo C. A.; Sutin J.; Rowlinson S.; Talley J.; Marnett L. J. Fluorescence Quenching Analysis of the Association and Dissociation of a Diarylheterocycle to Cyclooxygenase-1 and Cyclooxygenase-2: Dynamic Basis of Cyclooxygenase-2 Selectivity. Biochemistry 2000, 39, 6228–6234. 10.1021/bi992761o. [DOI] [PubMed] [Google Scholar]
- Orlando B. J.; Malkowski M. G. Crystal Structure of Rofecoxib Bound to Human Cyclooxygenase-2. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 772–776. 10.1107/S2053230X16014230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani G.; Panella A.; Perrone M. G.; Vitale P.; Di Mauro G.; Fortuna C. G.; Armen R. S.; Ferorelli S.; Smith W. L.; Scilimati A. Structural Basis for Selective Inhibition of Cyclooxygenase-1 (Cox-1) by Diarylisoxazoles Mofezolac and 3-(5-Chlorofuran-2-Yl)-5-Methyl-4-Phenylisoxazole (P6). Eur. J. Med. Chem. 2017, 138, 661–668. 10.1016/j.ejmech.2017.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchio A. J.; Malkowski M. G. The Structure of Ns-398 Bound to Cyclooxygenase-2. J. Struct. Biol. 2011, 176, 254–258. 10.1016/j.jsb.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. L.; Limburg D.; Graneto M. J.; Springer J.; Hamper J. R.; Liao S.; Pawlitz J. L.; Kurumbail R. G.; Maziasz T.; Talley J. J.; Kiefer J. R.; Carter J. The Novel Benzopyran Class of Selective Cyclooxygenase-2 Inhibitors. Part 2: The Second Clinical Candidate Having a Shorter and Favorable Human Half-Life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. 10.1016/j.bmcl.2010.07.054. [DOI] [PubMed] [Google Scholar]
- Uddin M. J.; Xu S.; Crews B. C.; Aleem A. M.; Ghebreselasie K.; Banerjee S.; Marnett L. J.. Harmaline Analogs as Substrate-Selective Cyclooxygeanse-2 Inhibitors. ACS Med. Chem. Lett. 2020, in press, 10.1021/acsmedchemlett.9b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt D. L.; Smith W. L. Primary Structure of Prostaglandin G/H Synthase from Sheep Vesicular Gland Determined from the Complementary DNA Sequence. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 1412–1416. 10.1073/pnas.85.5.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama C.; Takai T.; Tanabe T. Primary Structure of Sheep Prostaglandin Endoperoxide Synthase Deduced from Cdna Sequence. FEBS Lett. 1988, 231, 347–351. 10.1016/0014-5793(88)80847-0. [DOI] [PubMed] [Google Scholar]
- Lecomte M.; Laneuville O.; Ji C.; DeWitt D. L.; Smith W. L. Acetylation of Human Prostaglandin Endoperoxide Synthase-2 (Cyclooxygenase-2) by Aspirin. J. Biol. Chem. 1994, 269, 13207–13215. [PubMed] [Google Scholar]
- Hochgesang G. P.; Rowlinson S. W.; Marnett L. J. Tyrosine-385 Is Critical for Acetylation of Cyclooxygenase-2 by Aspirin. J. Am. Chem. Soc. 2000, 122, 6514–6515. 10.1021/ja0003932. [DOI] [Google Scholar]
- Loll P. J.; Sharkey C. T.; O’Connor S. J.; Dooley C. M.; O’Brien E.; Devocelle M.; Nolan K. B.; Selinsky B. S.; Fitzgerald D. J. O-Acetylsalicylhydroxamic Acid, a Novel Acetylating Inhibitor of Prostaglandin H2 Synthase: Structural and Functional Characterization of Enzyme-Inhibitor Interactions. Mol. Pharmacol. 2001, 60, 1407–1413. 10.1124/mol.60.6.1407. [DOI] [PubMed] [Google Scholar]
- Lucido M. J.; Orlando B. J.; Vecchio A. J.; Malkowski M. G. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry 2016, 55, 1226–1238. 10.1021/acs.biochem.5b01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J.; Zhou Y.; Xie D.; Zhang Y. Mechanistic Insights into a Classic Wonder Drug--Aspirin. J. Am. Chem. Soc. 2015, 137, 70–73. 10.1021/ja5112964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N. P.; Dong L.; Yuan C.; Noon K. R.; Smith W. L. Asymmetric Acetylation of the Cyclooxygenase-2 Homodimer by Aspirin and Its Effects on the Oxygenation of Arachidonic, Eicosapentaenoic, and Docosahexaenoic Acids. Mol. Pharmacol. 2010, 77, 979–986. 10.1124/mol.109.063115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Yuan C.; Orlando B. J.; Malkowski M. G.; Smith W. L. Fatty Acid Binding to the Allosteric Subunit of Cyclooxygenase-2 Relieves a Tonic Inhibition of the Catalytic Subunit. J. Biol. Chem. 2016, 291, 25641–25655. 10.1074/jbc.M116.757310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak K. R.; Prusakiewicz J. J.; Rowlinson S. W.; Prudhomme D. R.; Marnett L. J. Amino Acid Determinants in Cyclooxygenase-2 Oxygenation of the Endocannabinoid Anandamide. Biochemistry 2003, 42, 9041–9049. 10.1021/bi034471k. [DOI] [PubMed] [Google Scholar]
- Khan Y. S.; Gutierrez-de-Teran H.; Aqvist J. Probing the Time Dependency of Cyclooxygenase-1 Inhibitors by Computer Simulations. Biochemistry 2017, 56, 1911–1920. 10.1021/acs.biochem.6b01006. [DOI] [PubMed] [Google Scholar]
- Shamsudin Khan Y.; Kazemi M.; Gutierrez-de-Teran H.; Aqvist J. Origin of the Enigmatic Stepwise Tight-Binding Inhibition of Cyclooxygenase-1. Biochemistry 2015, 54, 7283–7291. 10.1021/acs.biochem.5b01024. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Williams J. M.; Giannaras J.; Nurnberg S.; Covington M.; Pinto D.; Pick S.; Trzaskos J. M. Mechanism of Selective Inhibition of the Inducible Isoform of Prostaglandin G/H Synthase. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11202–11206. 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet M.; Percival M. D. Effect of Inhibitor Time-Dependency on Selectivity Towards Cyclooxygenase Isoforms. Biochem. J. 1995, 306 (1), 247–251. 10.1042/bj3060247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.