

Abstract
Urea is an important raw material in the chemical industry and is widely used as a nitrogen source in chemical fertilizers. The current industrial urea synthesis not only requires harsh reaction conditions, but also consumes most of the NH3 obtained through artificial synthesis. The conversion of N2 and CO2 into urea through electrochemical reactions under ambient conditions represents a novel green urea synthesis method. However, the large-scale promotion of this method is limited by the lack of suitable electrocatalysts. Here, by means of density functional theory computations, we systematically study the catalytic activity of three experimentally available two-dimensional metal borides (MBenes), Mo2B2, Ti2B2, and Cr2B2 toward simultaneous electrocatalytic coupling of N2 and CO2 to produce urea under ambient conditions. According to our results, these three MBenes not only have superior intrinsic basal activity for urea formation, with limiting potentials ranging from −0.49 to −0.65 eV, but also can significantly suppress the competitive reaction of N2 reduction to NH3. In particular, 2D Mo2B2 and Cr2B2 possess superior capacity to suppress surface oxidation and self-corrosion under electrochemical reaction conditions, rendering them relatively promising electrocatalysts for urea production. Our work paves the way for the electrochemical synthesis of urea.
Subject terms: Electrocatalysis, Density functional theory, Electrocatalysis
The conversion of N2 and CO2 into urea through electrochemical reactions under ambient conditions represents a novel green urea synthesis method. Here, the authors demonstrate that two-dimensional transition metal borides can serve as effective catalysts for electrochemical urea synthesis.
Introduction
Urea, also known as carbamide (CO(NH2)2), was the first organic compound produced from inorganic raw materials. Because its nitrogen content is high (46%) and is readily converted to ammonia (NH3) in the soil, urea is now the most commonly used nitrogen fertilizer in the world, and more urea is manufactured by mass than any other organic chemical1. While over 90% of produced urea is used as fertilizer, it also has important applications in other fields. For example, urea is a raw material for the manufacture of urea-formaldehyde and urea-melamine-formaldehyde resins2. Urea-containing creams are used as topical dermatological products to promote skin hydration3, and large amounts of urea are used for the synthesis of barbiturates4. Since NH3 produced by the hydrolysis of urea can react with nitrogen oxides (NOx) to produce nitrogen, an increasingly important application of urea is to reduce NOx impurities in exhaust gases from diesel and lean-burn natural gas engines5. Therefore, maintaining a sustainable and efficient urea industry is of great importance for the development of human society.
At present, the production of urea in the industry is accomplished mainly through the reaction of NH3 and CO2 under high temperature and high pressure. However, this method is not only relatively energy-consuming but also relies on some complex types of equipment and multicycle processes to improve the conversion efficiency6,7. Remarkably, urea production consumes ~80% of the global NH3, which is mainly derived from the artificial nitrogen reduction reaction (NRR)8. Nevertheless, N2 is a stable molecule, and substantial input energy is required for dissociation of the strong N≡N triple bond, making N2 reduction thermodynamically and kinetically very difficult9. The industrial NRR is still dominated by the traditional Haber–Bosch process, which converts N2 and H2 into NH3 with the assistance of iron-based catalysts under harsh conditions10,11. However, the very large energy consumption and the large amount of the greenhouse gas CO2 emitted by the Haber–Bosch process have aggravated the energy and environmental problems. Therefore, people have been striving for green ammonia synthetic techniques that can be carried out under mild conditions12–14.
Compared to the Haber-Bosch process, the production of NH3 through the electrochemical NRR represents a more efficient and green strategy as it can utilize electricity generated from renewable energy sources and protons directly from water15–20. However, separating and purifying NH3 from an aqueous electrolyte is very difficult, which is detrimental to its further application. Furthermore, most current studies mainly focus on N2 electrochemical reduction to NH3, while further processing of the product is rarely considered. Recently, Jouny et al.21 realized C−N coupling and the production of acetamides with a high rate and selectivity by using NH3 as a nitrogen source. More interestingly, Comer et al.22 demonstrated that during photocatalytic fixation on the surface of TiO2, N2 can have a strong interaction with carbon substitution sites that present as surface-bound radicals, implying the feasibility of direct formation of C−N bonds from the coupling of N2 and carbon-based reagents. Inspired by these pioneering works, Chen et al.23 recently successfully coupled N2 and CO2 in H2O to produce urea using an electrocatalyst consisting of Pd-Cu alloy nanoparticles on TiO2 nanosheets, which opens a new avenue for urea production under mild conditions. However, in addition to the high price, precious metal-based alloy catalysts usually suffer from ambiguous active sites and easy corrosion. Therefore, for the development of emerging electrochemical urea synthesis, inexpensive and efficient electrocatalysts that can fix N2 and CO2 together are highly desirable.
In the past decade, the application of two-dimensional (2D) materials in the field of electrocatalysis have received much attention due to their large specific surface area and more exposed active sites24,25. For example, Li et al.26 theoretically demonstrated that 2D transition metal carbides, namely MXenes, are capable of catalyzing the conversion of CO2 into hydrocarbons. However, the surface metal atoms of MXenes are easily passivated by some functional species (e.g. OH, F, O), thereby degrading the catalytic activity27–29. Recently, several 2D transition metal borides (MBenes), which are boron analogs of MXenes, have also been realized experimentally30–33. In contrast to MXenes, MBenes can be stabilized without the presence of surface passivation groups. Therefore, MBenes provide an ideal platform for the exploration of the catalytic behavior of boron-containing surfaces34–36. Some recent theoretical studies have demonstrated that several types of MBenes could present good activity and selectivity for the NRR37,38. Motivated by the regulated layered configuration and excellent electrical conductivity of MBenes, we wondered whether the direct coupling of N2 and CO2 to produce urea could be realized on some specific MBenes.
In this work, by means of density functional theory (DFT) computations, we show that the electrochemical synthesis of urea on the basal planes of three experimentally realized MBenes, including 2D Mo2B2, Ti2B2, and Cr2B2 is thermodynamically and kinetically favorable. Remarkably, the competitive NRR can be significantly overwhelmed on these three MBenes, suggesting good selectivity. Especially, the surface oxidation/degradation problem can be avoided on the surfaces of Mo2B2 and Cr2B2, endowing these two MBenes with intrinsic activity, selectivity, and a large reaction region toward electrochemical urea synthesis.
Results
Structural properties and stability of MBenes
Figure 1 shows the schematic structure of a 2D M2B2-type MBene. Unlike many ordinary 2D materials (e.g., graphene, MoS2, MXenes) that possess a hexagonal lattice, our three studied 2D MBenes all present a rectangular lattice with the space group Pmma (No. 51), resulting in an in-plane structural anisotropy. The unit cell of a 2D M2B2-type MBene consists of two metal atoms and two B atoms, and each metal atom or B atom is connected to six neighboring atoms, forming a buckled bilayer structure with metal atoms on the uppermost surface. Compared to MXenes, in which the surface metal atoms bind to three carbon or nitrogen atoms, the higher coordination number of surface metal atoms in M2B2-type MBenes endow them with distinct stability and properties. The optimized lattice parameters and representative bond lengths of Mo2B2, Ti2B2, and Cr2B2 MBenes are summarized in Supplementary Table 1. Moreover, the electronic band structure computations demonstrated that these three MBenes are all metallic (Supplementary Fig. 1), which is beneficial for their electrocatalytic activity. The above results achieved good agreement with previous studies39,40.
Fig. 1. Geometrics of MBenes.
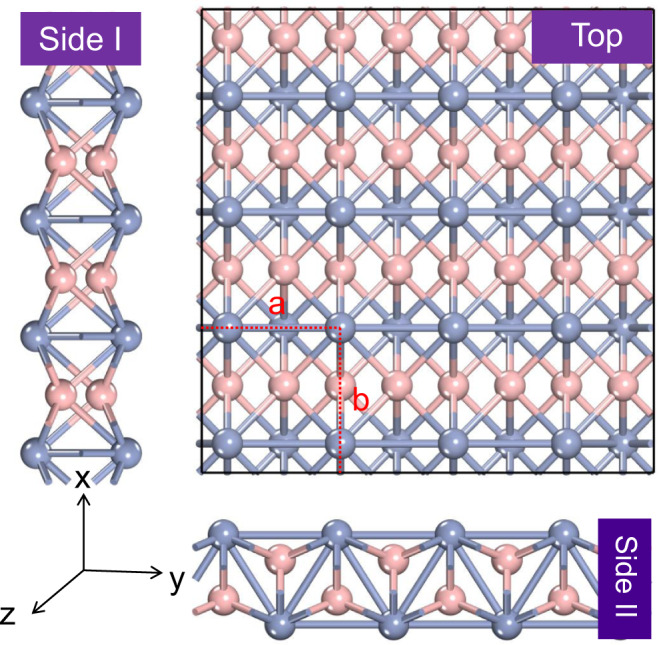
Top and side views of the schematic structure of a M2B2-type MBene. The red dashed lines denote a unit cell. The pink and blue balls represent boron and transition metal atoms, respectively.
Good stability is a prerequisite for the wide utilization of a catalyst. We thus first assessed the stability of 2D Mo2B2, Ti2B2, and Cr2B2 MBenes before revealing their catalytic activity toward urea formation. As shown in Supplementary Fig. 2, no imaginary modes are found in the phonon spectra of the three MBenes, which is indicative of good kinetic stability. The elastic constants of the three monolayers presented in Supplementary Table 2 all meet the criteria for a rectangular 2D structure (C11C22 − C122 > 0, C66 > 0)39, indicating that they are mechanically stable. Moreover, we also performed first-principles molecular dynamics (FPMD) simulations to examine the thermal stability. According to the diagrams presented in Supplementary Fig. 3, no obvious structural deformation can be observed in the structures of the three MBenes after 10 ps FPMD simulations at 300 K, suggesting good thermal stability.
Mechanism of electrochemical urea synthesis
To date, the reaction mechanism for electrochemical N2 and CO2 coupling to produce urea has only been proposed by Chen et al.23. As shown in Fig. 2, the entire reaction can be divided into four stages, namely, the adsorption of N2 and CO2, the reduction of *CO2 to *CO, the coupling of *N2 and *CO into *NCON, and the hydrogenation of *NCON to urea. Specifically, effective adsorption of N2 and CO2 on the surface of the catalyst is the primary condition for electrochemical urea production. Moreover, the adsorbed N2 should not be reduced to NH3, or at least the limiting potential for urea formation () should be lower than that of NH3 formation () to guarantee a high selectivity. Once N2 is adsorbed, the coadsorbed CO2 should be effectively and selectively reduced to *CO. Then, instead of being released or further reduced, the generated *CO should move to the top of adsorbed *N2 to form the tower-like key intermediate *NCON. Finally, the formed *NCON species could be further reduced to urea via four proton-coupled electron transfer (PCET) steps following either the distal or the alternative pathway. This mechanism has been well applied to explain the catalytic activity of the Pd-Cu catalyst. For the investigation of urea formation over the three M2B2-type MBenes, we followed the above-discussed reaction mechanism. The free-energies of reaction intermediates (Supplementary Table 3) and the reaction free-energies (ΔG) of elementary steps for urea production on 2D Mo2B2, Ti2B2, and Cr2B2 were then computed. The optimized atomic configurations of various intermediates along the reaction pathway are displayed in Supplementary Fig. 4, and the corresponding free energy profiles are summarized in Fig. 3.
Fig. 2. Reaction mechanism.

Schematic diagram of the mechanism of urea production through the electrochemical coupling of N2 and CO2. The gray, red, pink, and blue balls represent C, O, N, and H atoms, respectively.
Fig. 3. Catalytic activity of MBenes.
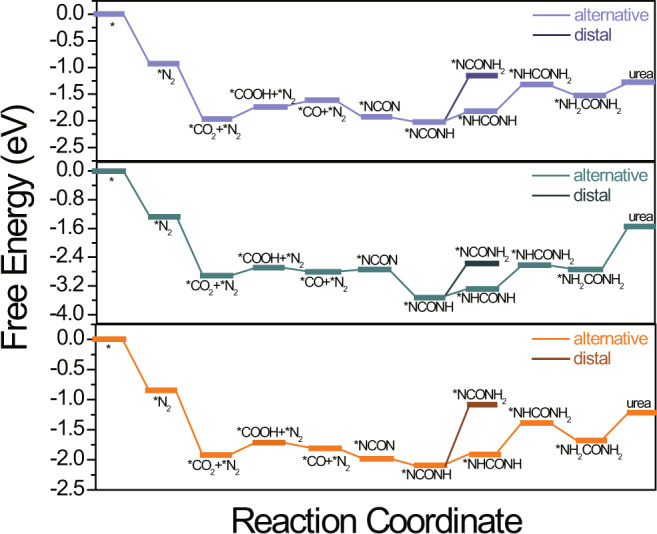
Free energy profiles of electrochemical urea production on Mo2B2, Ti2B2, and Cr2B2.
Electrochemical reactivity of MBenes toward urea production
We first investigated the adsorption of N2 and CO2 on the surfaces of the three MBenes. For the adsorption of N2, both side-on and end-on configurations were considered. According to our computations, the side-on adsorption of N2 on the bridge site of two metal atoms is preferred for all three MBenes (Supplementary Fig. 4). This is because the back-donation of electrons from the d-orbitals of metal atoms to the π* orbitals of N2 could be facilitated by side-on adsorption. The adsorption energies of N2 () on 2D Mo2B2, Ti2B2, and Cr2B2 are −0.93, −1.27, and −0.85 eV, respectively. Due to the strong interaction between N2 and the three MBenes, the bond length of N2 is enlarged from 1.10 Å to 1.21, 1.22, and 1.20 Å on 2D Mo2B2, Ti2B2, and Cr2B2, respectively. Once N2 is adsorbed, the accommodation of a CO2 molecule on its neighboring bridge site is also feasible, with adsorption energies () of −1.04, −1.65, and −1.07 eV for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. Remarkably, the adsorbed CO2 is pronouncedly bent due to the formation of chemical bonds between C and metal atoms. Overall, the above results vividly reveal that both N2 and CO2 can be strongly adsorbed and effectively activated on the surface of our three chosen MBenes.
After disclosing the adsorption behavior of N2 and CO2, we next assessed the feasibility of *CO2 reduction to *CO on the surface of the three MBenes in the presence of coadsorbed N2. Generally, the electroreduction of *CO2 to *CO initiates with the hydrogenation of one O atom through a PCET step, resulting in the formation of a *COOH species. According to our computations, this step is endothermic with a ΔG of 0.24, 0.22, and 0.21 eV for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. Upon the second PCET, the *COOH species can be transformed to a *CO species by releasing a H2O molecule, which is endothermic by 0.10 eV for 2D Mo2B2 but exothermic by −0.11 and −0.08 eV for 2D Ti2B2 and Cr2B2, respectively. Therefore, the key intermediate *CO can be feasibly generated on the surfaces of the three MBenes. The adsorption energies of *CO species on 2D Mo2B2, Ti2B2, and Cr2B2 are −0.69, −1.53, and −0.94 eV, respectively, which are all higher than that of Pd-Cu catalyst (−0.62 eV)23. Due to the low CO yield of Pd-Cu catalyst, we can expect that the *CO desorption can be significantly suppressed for three 2D MBenes, which could facilitate the following coupling process.
As revealed by Chen et al.23 the most important intermediate for urea formation is the tower-like *NCON species, which can be directly produced from the coupling of *CO and *N2. We thus explored the feasibility of forming the *NCON species on the surfaces of the three MBenes using the CI-NEB method. As shown in Supplementary Fig. 5, the coupling of *N2 and *CO into *NCON is exothermic by −0.30 and −0.18 eV on 2D Mo2B2 and Cr2B2, respectively, indicating that the formation of the *NCON species on these two MBenes is energetically favorable. In contrast, due to the relatively strong binding strength of the *CO species, the formation of *NCON on Ti2B2 is slightly endothermic by 0.06 eV. Remarkably, the kinetic barriers for the formation of *NCON on 2D Mo2B2, Ti2B2, and Cr2B2 are 0.58, 0.81, and 0.71 eV respectively, which are comparable to or even lower than that of the Pd-Cu catalyst (0.79 eV)23, indicating that the coupling of *N2 and *CO on these three MBenes is kinetically feasible.
Once the *NCON species is formed, the formation of urea becomes very straightforward, although there exist different sequences of hydrogenation on the two N atoms. Remarkably, the hydrogenation of *NCON to *NCONH via a PCET is exothermic for all three MBenes with a ΔG of −0.09, −0.76, and −0.12 eV for Mo2B2, Ti2B2, and Cr2B2, respectively. Notably, we also considered the hydrogenation of the O atom but found that it is relatively endothermic. When the second H is added to *NCONH, two possible reaction pathways may occur. One is the distal pathway in which the second H is added to the hydrogenated N atom in the last step to form the *NCONH2 species, and the other is the alternative path in which the second H is added to the bare N atom to form *NHCONH species. According to our computations, the formation of the distal product *NCONH2 is endothermic by as high as 0.88, 0.93, and 1.02 eV for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. In contrast, the formation of the alternative product *NHCONH is 0.68, 0.70, and 0.83 eV lower in energy than that of the distal product for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. As a comparison, the distal product is preferred by the Pd-Cu catalyst23. Additionally, we noted that at this stage, the chemical bonding between metal atoms and N atoms is still robust, while the N−N length in *NHCONH is enlarged to 2.35, 2.34, and 2.33 Å for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. Due to the breaking of one M−N bond, the third PCET step to form *NH2CONH is considerably endothermic, with a ΔG of 0.49, 0.65, and 0.52 eV for 2D Mo2B2, Ti2B2, and Cr2B2, respectively. Since the fourth PCET step to form *NH2CONH2 is exothermic for all three M2B2 monolayers, the formation of the *NH2CONH species was identified as the potential-limiting step, and the corresponding were computed to be −0.49, −0.65, and −0.52 V for Mo2B2, Ti2B2, Cr2B2, respectively. Remarkably, the of three MBenes are comparable to or even lower than that of the Pd-Cu catalyst (−0.64 V), which is indicative of superior electrocatalytic reactivity toward urea formation. Moreover, the adsorption energies of *NH2CONH2 on 2D Mo2B2, Ti2B2, and Cr2B2 are −1.28, −1.55, and −1.21 eV, respectively, which are lower than that of the Pd-Cu catalyst (−1.68 eV)23, indicating that the formed urea molecule can be easily released, especially when electrochemical reactions are carried out in flow cells.
For computational simplicity, the above results on electrochemical steps were all obtained by assuming that the catalyst is charge neutral, and thus, the Fermi-level of the catalyst would change with the variation in adsorbed reaction species. However, in real electrochemical reactions, electron transfer between the catalyst and the electrode to match the Fermi level of the catalyst with the applied electrode potential. In a recent theoretical study, Kim et al.40 revealed that the surface charge can have a substantial effect on the electrochemical activity of graphene-based materials. To this end, we also performed grand-canonical DFT computations to investigate the effect of surface charge on the electrocatalytic reactivity of the three MBenes toward urea formation. Supplementary Fig. 6 presents the free energy of all reaction intermediates of urea formation computed using the constant-potential method (CPM), and those computed from the constant-charge method (CCM) are also listed for comparison. Remarkably, the energy difference between the two methods is within the range of 0.1 eV for all three MBenes, which is similar to the values found in bulk materials but much lower than that of graphene. The less pronounced charge effect on MBenes in comparison to graphene should be attributed to greater thickness.
Discussion
Electrocatalytic selectivity of CO2 reduction on MBenes
The above results demonstrated that the electrochemical production of urea could be feasible on the surfaces of 2D Mo2B2, Ti2B2, and Cr2B2. However, at present we still cannot claim that these three 2D MBenes are qualified catalysts for urea production as some important issues concerning the selectivity are still pending. First, as we discussed above, the *CO species is the key intermediate for the entire reaction. Although we demonstrated that the formation of *CO on the surfaces of the three studied MBenes is feasible via the intermediate *COOH, the transfer of a first proton/electron pair to CO2 could also lead to the formation of *OCHO, which is a key intermediate of formate. Therefore, knowing whether CO2 can be selectively reduced to *CO is very important. To address this concern, we first computed the binding energy of the *OCHO species on three MBenes, and found that the binding energy of *OCHO is generally higher than the binding energy of *COOH. However, this does not mean that the formation of *OCHO is preferred. As revealed by previous studies41,42, many transition metal catalysts bind the *OCHO species more strongly than the *COOH species, but they all have CO, rather than formate, as the major product. Therefore, predicting the selectivity of *CO formation solely based on thermodynamics computations is actually not reliable.
As revealed by Cheng et al.43, the selectivity of CO2 reduction is essentially controlled kinetically, and the formation of *COOH and *OCHO can be achieved via either the Eley–Raideal (ER) mechanism by accepting a H atom from water or via the Langmuir-Hinshelwood (LH) mechanism by accepting a surface-bound H atom (Fig. 4a). To obtain some deep insight into the selectivity, we also constructed explicit liquid/solid interfaces for all three MBenes by adding a water layer on their surfaces (Supplementary Fig. 7), and both the ER and LH mechanisms were considered for the formation of *COOH and *OCHO. As shown in Fig. 4b–d, for the formation of the *COOH species, the ER mechanism is preferred with kinetic barriers of 0.38, 0.31, and 0.32 eV for the 2D Mo2B2, Ti2B2, and Cr2B2, respectively. In contrast, the formation of the *OCHO species entails much large kinetic barrier via either the ER mechanism or the LH mechanism. Therefore, on the surfaces of our three chosen 2D MBenes, CO2 would be dominantly reduced to *CO rather than formate.
Fig. 4. Selectivity of CO2 electroreduction.

a Schematic diagram of the ER and LH mechanisms of CO2 electroreduction to *COOH or *OCHO. Kinetic pathways for the electroreduction of CO2 on the surfaces of (b) Mo2B2, c Ti2B2, and d Cr2B2.
The second concern regarding the *CO species is whether it could be further reduced to *CHO or *COH under the working conditions of urea production. To address this question, we also computed the free energy of *CO reduction to *CHO and *COH for 2D Mo2B2, Ti2B2, and Cr2B2 (Supplementary Table 4). The limiting potential for the formation of *CHO or *COH on the three MBenes is at least −1.22 V, which is much higher than the . Therefore, the formation of *CHO or *COH would be significantly suppressed under the working potential of urea production.
Moreover, we are also aware of that there would be competition between *CO adsorption and adsorption/dissociation of H2O molecule in the realistic aqueous environment. According to our results, the kinetic barrier for the direct dissociation of one H2O molecule on 2D Mo2B2, Ti2B2, and Cr2B2 is 0.63, 0.53, and 0.72 eV, respectively (Supplementary Fig. 8), indicating the formation of surface bounded *H and *OH is also feasible for three MBenes. While the *H species can serve as the proton source to react with the reaction intermediates via the LH mechanism, there could exist the adsorption competition between *CO and *OH species on three MBenes. Therefore, we then plotted the curves of equilibrium surface coverages of these two species as a function of electrode potential by performing microkinetic simulations. As shown in Supplementary Fig. 9, the population of *OH and *CO is potential dependent. Specifically, the surfaces of three MBenes would be predominately covered by OH* species under low electrode potential. When the electrode potential is higher than −0.31, −0.64, and −0.22 V, respectively, the coverage of CO* on 2D Mo2B2, Ti2B2 and Cr2B2 begins to increase, while the coverage of OH* begins to decrease. Since the critical potential of *CO/*OH adsorption for each MBene is lower than the respective , it can be expected that surface active sites of 2D MBenes, especially Mo2B2 and Cr2B2, would be mainly covered by *CO rather than *OH under working potentials.
Electrocatalytic selectivity of N2 reduction on MBenes
As reported by previous studies37,38, many 2D MBenes have basal plane activity for N2 electroreduction to NH3. Would the adsorbed N2 molecule also be reduced to NH3 on our three chosen M2B2-type MBenes? To address this question, we investigated the thermodynamics of the electrochemical NRR on 2D Mo2B2, Ti2B2, Cr2B2 to get some deep insight. As shown in Supplementary Fig. 10, the electrochemical NRR on all three M2B2 monolayers is feasible, with values of −0.79, −0.71, and −0.65 V, respectively. Encouragingly, for all three MBenes, is higher than , suggesting that the formation of the NH3 can be greatly suppressed on these three MBenes.
Since kinetic factors play an important role in determining the selectivity, we further computed the kinetic barriers of elementary steps of N2 reduction to urea for three 2D MBenes and compared with those of N2 reduction NH3. Especially, both ER and LH mechanisms were considered for the electrochemical steps. As presented in Supplementary Figs. 11–13, for all three 2D MBenes, the non-electrochemical step of *NCON formation has the biggest kinetic barrier, which is 0.53, 0.77, and 0.70 eV for Mo2B2, Ti2B2, Cr2B2, respectively. Note that the kinetic barriers of *NCON formation predicted from explicit solvent model are quite close to those predicted from implicit solvent model. As a comparison, the first electrochemical step of N2 reduction to NH3, namely N2 reduction to NNH, already has a relatively big kinetic barrier, which is 0.78, 0.80, and 0.76 for 2D Mo2B2, Ti2B2, Cr2B2, respectively (Supplementary Fig. 14). Therefore, the urea synthesis is also kinetically favorable on these three 2D MBenes.
Pourbaix diagrams of MBenes
Finally, another concern that also needs to be addressed is the electrochemical stability of the three MBenes. Although no evidence yet confirm the existence of functional groups on experimentally realized MBenes, we wondered whether the bare surfaces of MBenes could be covered by *O/*OH species in aqueous solution under working conditions. To answer this question, we constructed surface Pourbaix diagrams of the three MBenes to reveal the most stable surface configurations under different equilibrium potentials and pH values (the computational details are given in Supplementary Methods)44,45. As shown in Fig. 5, when the electrode potential is 0 V vs SHE, the basal plane of the 2D Mo2B2 is fully covered by *O species independent of the pH value, whereas the basal planes of the 2D Ti2B2 and Cr2B2 are covered by *O and *OH groups in a strong acid environment. When an electrode potential is applied, the hydrogenation of *O and *OH becomes energetically favorable on the surfaces of all three MBenes. In particular, the minimum potentials required to remove the surface *O/*OH species at pH = 0 () are −0.37, −1.04, and −0.31 V for the 2D Mo2B2, Ti2B2, and Cr2B2 monolayers, respectively. It is worth noting that the of 2D Mo2B2 and Cr2B2 is less negative than the respective , indicating that these two MBenes could possess superior electrochemical stability against surface oxidation under working conditions. In sharp contrast, the of 2D Ti2B2 is far more negative than its and even . As the electrode potential of urea production on the 2D Ti2B2 should not exceed the to guarantee a high selectivity, the surface of the 2D Ti2B2 would inevitably be occupied by *OH/*O species under working conditions, resulting in decreased reactive sites on the basal plane.
Fig. 5. Surface states of MBenes under electrochemical conditions.

Surface Pourbaix diagrams of 2D (a) Mo2B2, (b) Ti2B2, and (c) Cr2B2. The thermodynamically stable states of the surface under SHE and pH values are highlighted by orange (for *O), green (for *O + *OH), and purple (for *OH). The red dashed line represents the limiting potential of urea formation.
Besides the stability of active surfaces, whether the catalyst itself would corrode under electrochemical conditions also needs to be addressed46,47. Taking advantage of the fact that the formation energies are transferable between energy reference systems, we further plotted the whole Pourbaix diagram of the three 2D MBenes as a function of pH and potential at standard conditions to identified their stability window in aqueous solutions by directly combining Gibbs free energies from DFT computations with experimental arbitrary aqueous states. As shown in Fig. 6, a wide passivation region can be identified for Mo2B2 (pH < 7.08) and Cr2B2 (pH < 7.22) at the potential of −0.49 V and −0.52 V, respectively. Using the electrolyte of Pd–Cu catalyst (pH = 6.8)23 as a reference, both 2D Mo2B2 and Cr2B2 can maintain structure integrality under working conditions due to high barriers for solid–liquid phase transformations. In sharp contrast to 2D Mo2B2 and Cr2B2, 2D Ti2B2 has a narrow passivation region (pH < 2.26) when subjected to an external potential of −0.65 V. At high pH region, Ti2B2 would be transformed into Ti(OH)3, indicating that Ti2B2 would be easily corroded under working conditions of urea synthesis, Therefore, 2D Ti2B2 is not a qualified electrocatalyst for urea formation due to its low electrochemical stability.
Fig. 6. Corrosion resistance of MBenes under electrochemical conditions.

Computationally predicted Pourbaix diagrams of (a) Mo2B2, (b) Ti2B2, and (c) Cr2B2 using 10−6 M concentration for aqueous species at 25 °C.
Activity origin of MBenes toward urea production
Due to the good reactivity, selectivity, and stability, 2D Mo2B2 and Cr2B2 have been identified as promising electrocatalysts for urea synthesis. However, at present, the activity origin of these two MBenes is not yet clear. It is known that the activity of an electrocatalyst is essentially governed by its electronic structure. Therefore, in order to get some deeper insights into the specialty of MBenes for urea synthesis, we further computed their electronic density of states (DOS) at the Fermi level and compared with those of some other experimentally realized 2D metallic materials, including four bare MXenes (Ti2C, Mo2C, Ti3C2, Mo3C2) and two transition metal chalcogenides (1T-MoS2, VS2). As shown in Supplementary Fig. 15, the DOS per atom of MXenes are generally much higher than those of 2D Mo2B2 and Cr2B2, implying stronger metallicity and more active surfaces of MXenes. However, this does not mean that MXenes have better electrochemical activity than MBenes, because the active surfaces of MXenes are known to be easily passivated in solvents27–29. Interestingly, the DOS of Ti2B2 are quite close to those of Mo2C and Mo3C2, which could explain the instability of Ti2B2 in aqueous solution. Remarkably, the DOS of Mo2B2 and Cr2B2 are significantly higher than those of 1T-MoS2 and VS2 that are actually inert to urea production according to our test computations. Therefore, the good activity and stability of 2D Mo2B2 and Cr2B2 should be attributed to their moderate metallicity.
To summarize, on the basis of comprehensive DFT computations, we have systematically explored the potential of utilizing the three experimentally realized MBenes, Mo2B2, Ti2B2, and Cr2B2, as electrocatalysts for urea synthesis. The activity, selectivity, and stability of the three MBenes under aqueous conditions were carefully studied. Our computations demonstrated that all three MBenes can adsorb N2 and CO2 on their basal planes, and the adsorbed CO2 can be easily reduced to *CO. Afterwards, the key intermediate *NCON can be formed via the coupling of *N2 and *CO, which can be further reduced to urea via four PCET steps. The limiting potentials of urea formation for our three studied MBenes are in the range of −0.49 to −0.65 eV, which are comparable to that of the Pd-Cu alloy catalyst. In particular, it is found that 2D Mo2B2 and Cr2B2 can prevent the problems of active sites blockage and self-corrosion, while 2D Ti2B2 not only has its surface active sites occupied by *OH and *O groups, but also could be easily corroded under reaction conditions. Therefore, 2D Mo2B2 and Cr2B2 can serve as promising catalysts for urea production, which can be attributed to their moderate metallicity. Our work provides a clear roadmap for the design of electrocatalysts for simultaneously fixing N2 and CO2 to produce urea, which could promote more experimental and theoretical efforts on developing 2D electrocatalysts for this challenging reaction.
Methods
DFT computations
Our DFT computations based on first-principles were performed via the Vienna ab initio simulation package (VASP)48. The ion-electron interactions were described with the projector-augmented plane-wave (PAW) method49. Exchange-correlation potentials were expressed by Perdew–Burke–Ernzerhof (PBE) functional with the generalized gradient approximation (GGA)50. A 460 eV cutoff energy for the plane wave expansion was adopted in all the computations. A Monkhorst-Pack k-points setting of 3 × 3 × 1 and 15 × 15 × 1 was used to sample the 2D Brillouin zone for geometry optimizations and electronic structure computations, respectively. We set the x and y directions parallel and the z direction perpendicular to the layer plane, and adopted a vacuum layer length of 20 Å in the z direction. The systems were relaxed until the energy and force reaching the convergence threshold of 10−5 eV and 0.01 eV/Å.
The phonon spectra were computed using the density functional perturbation theory (DFPT), as implemented in the Phonon code51. The ab initio molecular dynamic (AIMD) simulations were performed using the NVT ensemble. The Nosé-Hoover method simulations last 10 ps with a time step of 1.0 fs52. The solvation effects in aqueous solution were considered with the Poisson-Boltzmann implicit solvation model as implemented in VASP (VASP-sol), where the dielectric constant of water was taken as 8053. The climbing-image nudged elastic band (CI-NEB) method54 as implemented in VASP was used to obtain the kinetic barriers.
The grand-canonical DFT computations, which allow the number of electrons to adjust automatically at a fixed electron chemical potential, were performed using the PBE functional as implemented in JDFTx code55 with a cutoff energy of 20 Hartree. The JDFTx code combines electronic DFT with classical DFT and continuum models of liquids for first-principles computations of electrochemical systems. The charge-asymmetric nonlocal determined local electric (CANDLE)56 solvation model as implemented in JDFTx was utilized for describing the electrolyte. Other numerical parameters, including k-point sampling, convergence criteria, etc. are similar to the VASP computations.
Free energy computations
To compute the free energy change (ΔG) of each elementary step of electrochemical urea synthesis, we adopted the computational hydrogen electrode (CHE) model developed by Nørskov et al.57,58 according to which the ΔG of an electrochemical reaction is computed as:
| 1 |
where ΔE is the DFT computed reaction energy, ΔEZPE and ΔS are the zero-point energy difference and the entropy difference between the adsorbed state and the gas phase, respectively, and T is the temperature (298.15 K, in our work). For adsorbed reaction intermediates, their EZPE and S are obtained via vibrational frequencies computations with harmonic approximation and neglecting contributions from the slab, while for molecules these are taken from the NIST database. Moreover, in accordance with the CHE model, the effects of electrode potential (U) and pH can be treated as an energy shift to free energy change in the electrochemical steps:
| 2 |
| 3 |
where kB is Boltzmann constant. In this work, the value of pH was assumed to be zero in free energy computations.
Supplementary information
Acknowledgements
We are grateful for funding support from the National Key R&D Program of China (2019YFA0308000), the Natural Science Foundation of China (No. 21873050), and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author contributions
Y.L. designed the research, X.R.Z., X.C.Z., and Y.J. demonstrated the initial idea and collected all the data. X.R.Z. and Y.L. wrote the paper and all authors commented on it.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. All of the other data are available from the corresponding author upon reasonable request.
Code availability
The computational codes used in this work are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Joseph Montoya and the other anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-24400-5.
References
- 1.Comer BM, et al. Prospects and challenges for solar fertilizers. Joule. 2019;3:1578–1605. doi: 10.1016/j.joule.2019.05.001. [DOI] [Google Scholar]
- 2.Liu Y, Zhao X, Ye L. A novel elastic urea-melamine-formaldehyde foam: structure and properties. Ind. Eng. Chem. Res. 2016;55:8743–8750. doi: 10.1021/acs.iecr.6b01957. [DOI] [Google Scholar]
- 3.Celleno L. Topical urea in skincare: a review. Dermatol. Ther. 2018;31:e12690. doi: 10.1111/dth.12690. [DOI] [PubMed] [Google Scholar]
- 4.Huang H–M, McDouall JJW, Procter DJ. Radical anions from urea-type carbonyls: radical cyclizations and cyclization cascades. Angew. Chem. Int. Ed. 2018;57:4995–4999. doi: 10.1002/anie.201800667. [DOI] [PubMed] [Google Scholar]
- 5.Seneque M, Can F, Duprez D, Courtois X. NOx selective catalytic reduction (NOx-SCR) by urea: evidence of the reactivity of HNCO, including a specific reaction pathway for NOx reduction involving NO+NO2. ACS Catal. 2016;6:4064–4067. doi: 10.1021/acscatal.6b00785. [DOI] [Google Scholar]
- 6.Barzagli F, Mani F, Peruzzini M. From greenhouse gas to feedstock: formation of ammonium carbamate from CO2 and NH3 in organic solvents and its catalytic conversion into urea under mild conditions. Green. Chem. 2011;13:1267–1274. doi: 10.1039/c0gc00674b. [DOI] [Google Scholar]
- 7.Pérez-Fortes M, Bocin-Dumitriu A, Tzimas E. CO2 utilization pathways: techno-economic assessment and market opportunities. Energy Procedia. 2014;63:7968–7975. doi: 10.1016/j.egypro.2014.11.834. [DOI] [Google Scholar]
- 8.Erisman JW, Sutton MA, Galloway J, Klimont Z, Winiwarter W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008;1:636–639. doi: 10.1038/ngeo325. [DOI] [Google Scholar]
- 9.Service RF. New recipe produces ammonia from air, water, and sunlight. Science. 2014;345:610. doi: 10.1126/science.345.6197.610. [DOI] [PubMed] [Google Scholar]
- 10.Licht S, et al. Ammonia synthesis by N2 and steam electrolysis in molten hydyoxide suspensions of nanoscale Fe2O3. Science. 2014;345:637–640. doi: 10.1126/science.1254234. [DOI] [PubMed] [Google Scholar]
- 11.Qian J, An Q, Fortunelli A, Nielsen RJ, Goddard WA. Reaction mechanism and kinetics for ammonia synthesis on the Fe(111) surface. J. Am. Chem. Soc. 2018;140:6288–6297. doi: 10.1021/jacs.7b13409. [DOI] [PubMed] [Google Scholar]
- 12.Zheng J, et al. Photoelectrochemical synthesis of ammonia on the aerophilic-hydrophilic heterostructure with 37.8% efficiency. Chemistry. 2019;5:617–633. doi: 10.1016/j.chempr.2018.12.003. [DOI] [Google Scholar]
- 13.Li H, Shang J, Ai Z, Zhang L. Efficient visible light nitrogen fixation with BiOBr nanosheets of oxygen vacancies on the exposed (001) facets. J. Am. Chem. Soc. 2015;137:6393–6399. doi: 10.1021/jacs.5b03105. [DOI] [PubMed] [Google Scholar]
- 14.Foster SL, et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018;1:490–500. doi: 10.1038/s41929-018-0092-7. [DOI] [Google Scholar]
- 15.Wan Y, Xu J, Lv R. Heterogeneous electrocatalysts design for nitrogen reduction reaction under ambient conditions. Mater. Today. 2019;27:69–90. doi: 10.1016/j.mattod.2019.03.002. [DOI] [Google Scholar]
- 16.Montoya JH, Tsai C, Vojvodic A, Nørskov JK. The challenge of electrochemical ammonia synthesis: a new perspective on the role of nitrogen scaling relations. ChemSusChem. 2015;8:2180–2186. doi: 10.1002/cssc.201500322. [DOI] [PubMed] [Google Scholar]
- 17.Ling C, et al. New mechanism for N2 reduction: the essential role of surface hydrogenation. J. Am. Chem. Soc. 2019;141:18264–18270. doi: 10.1021/jacs.9b09232. [DOI] [PubMed] [Google Scholar]
- 18.Qing G, et al. Recent advances and challenges of electrocatalytic N2 reduction to ammonia. Chem. Rev. 2020;120:5437–5516. doi: 10.1021/acs.chemrev.9b00659. [DOI] [PubMed] [Google Scholar]
- 19.Suryanto BHR, et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2019;2:290–296. doi: 10.1038/s41929-019-0252-4. [DOI] [Google Scholar]
- 20.Liu H, et al. Homogeneous, heterogeneous, and biological catalysts for electrochemical N2 reduction toward NH3 under ambient conditions. ACS Catal. 2019;9:5245–5267. doi: 10.1021/acscatal.9b00994. [DOI] [Google Scholar]
- 21.Jouny M, et al. Formation of carbon–nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 2019;11:846–851. doi: 10.1038/s41557-019-0312-z. [DOI] [PubMed] [Google Scholar]
- 22.Comer BM, et al. The role of adventitious carbon in photo-catalytic nitrogen fixation by titania. J. Am. Chem. Soc. 2018;140:15157–15160. doi: 10.1021/jacs.8b08464. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 2020;12:717–724. doi: 10.1038/s41557-020-0481-9. [DOI] [PubMed] [Google Scholar]
- 24.Sun, Z., Ma, T., Tao, H., Fan, Q. & Han, B. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chemistry3, 560−587 (2017).
- 25.Zhu X, Li Y. Review of two‐dimensional materials for electrochemical CO2 reduction from a theoretical perspective. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019;9:e1416. doi: 10.1002/wcms.1416. [DOI] [Google Scholar]
- 26.Li N, et al. Understanding of electrochemical mechanisms for CO2 capture and conversion into hydrocarbon fuels in transition-metal carbides (MXenes) ACS Nano. 2017;11:10825–10833. doi: 10.1021/acsnano.7b03738. [DOI] [PubMed] [Google Scholar]
- 27.Gogotsi Y, Anasori B. The rise of MXenes. ACS Nano. 2019;13:8491–8494. doi: 10.1021/acsnano.9b06394. [DOI] [PubMed] [Google Scholar]
- 28.Shahzad F, Iqbal A, Kim H, Koo CM. 2D transition metal carbides (MXenes): applications as an electrically conducting material. Adv. Mater. 2020;32:2002159. doi: 10.1002/adma.202002159. [DOI] [PubMed] [Google Scholar]
- 29.Pang J, et al. Applications of 2D MXenes in energy conversion and storage systems. Chem. Soc. Rev. 2019;48:72–123. doi: 10.1039/C8CS00324F. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Xiang H, Dai FZ, Zhang Z, Zhou Y. First demonstration of possible two-dimensional MBene CrB derived from MAB phase Cr2AlB2. J. Mater. Sci. Technol. 2018;34:2022–2026. doi: 10.1016/j.jmst.2018.02.024. [DOI] [Google Scholar]
- 31.Kota S, et al. Magnetic properties of Cr2AlB2, Cr3AlB4, and CrB powders. J. Alloy. Compd. 2018;767:474–482. doi: 10.1016/j.jallcom.2018.07.031. [DOI] [Google Scholar]
- 32.Wang J, et al. Discovery of hexagonal ternary phase Ti2InB2 and its evolution to layered boride TiB. Nat. Commun. 2019;10:1–8. doi: 10.1038/s41467-018-07882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alameda LT, et al. Multi-step topochemical pathway to metastable Mo2AlB2 and related two-dimensional nanosheet heterostructures. J. Am. Chem. Soc. 2019;141:10852–10861. doi: 10.1021/jacs.9b04726. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T, Zhang B, Peng Q, Zhou J, Sun Z. Mo2B2 MBene-supported single-atom catalysts as bifunctional HER/OER and OER/ORR electrocatalysts. J. Mater. Chem. A. 2021;9:433–441. doi: 10.1039/D0TA08630D. [DOI] [Google Scholar]
- 35.Rezaie AA, et al. Synthesis and Li-ion electrode properties of layered MAB phases Nin +1ZnBn (n = 1, 2) J. Mater. Chem. A. 2020;8:1646–1651. doi: 10.1039/C9TA12937E. [DOI] [Google Scholar]
- 36.Siriwardane EMD, Joshi RP, Kumar N, Çaklr D. Revealing the Formation energy-exfoliation energy-structure correlation of MAB phases using machine learning and DFT. ACS Appl. Mater. Interfaces. 2020;12:29424–29431. doi: 10.1021/acsami.0c03536. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Shang C, Zhou S, Zhao J. MBenes: emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale Horiz. 2020;5:1106–1115. doi: 10.1039/D0NH00242A. [DOI] [PubMed] [Google Scholar]
- 38.Guo X, et al. Establishing a theoretical landscape for identifying basal plane active 2D metal borides (MBenes) toward nitrogen electroreduction. Adv. Funct. Mater. 2021;31:2008056. doi: 10.1002/adfm.202008056. [DOI] [Google Scholar]
- 39.Born, M.; Huang, H. Dynamical theory of crystal lattices, Clarendon, Oxford, 1954.
- 40.Kim D, Shi J, Liu Y. Substantial impact of charge on electrochemical reactions of two-dimensional materials. J. Am. Chem. Soc. 2018;140:9127–9131. doi: 10.1021/jacs.8b03002. [DOI] [PubMed] [Google Scholar]
- 41.Feaster JT, et al. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 2017;7:4822–4827. doi: 10.1021/acscatal.7b00687. [DOI] [Google Scholar]
- 42.Yoo JS, Christensen R, Vegge T, Nørskov JK, Studt F. Theoretical insight into the trends that guide the electrochemical reduction of carbon dioxide to formic acid. ChemSusChem. 2016;9:358–363. doi: 10.1002/cssc.201501197. [DOI] [PubMed] [Google Scholar]
- 43.Cheng T, Xiao H, Goddard WA. Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu(100) surface at 298 K from quantum mechanics free energy calculations with explicit water. J. Am. Chem. Soc. 2016;138:13802–13805. doi: 10.1021/jacs.6b08534. [DOI] [PubMed] [Google Scholar]
- 44.Bajdich M, García-Mota M, Vojvodic A, Nørskov JK, Bell AT. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 2013;135:13521–13530. doi: 10.1021/ja405997s. [DOI] [PubMed] [Google Scholar]
- 45.Sumaria V, Krishnamurthy D, Viswanathan V. Quantifying confidence in DFT predicted surface Pourbaix diagrams and associated reaction pathways for Chlorine Evolution. ACS Catal. 2018;8:9034–9042. doi: 10.1021/acscatal.8b01432. [DOI] [Google Scholar]
- 46.Persson KA, Waldwick B, Lazic P, Ceder G. Prediction of solid-aqueous equilibria: Scheme to combine first-principles calculations of solids with experimental aqueous states. Phys. Rev. B - Condens. Matter Mater. Phys. 2012;85:1–12. doi: 10.1103/PhysRevB.85.235438. [DOI] [Google Scholar]
- 47.Singh AK, et al. Electrochemical stability of metastable materials. Chem. Mater. 2017;29:10159–10167. doi: 10.1021/acs.chemmater.7b03980. [DOI] [Google Scholar]
- 48.Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B. 1993;47:558. doi: 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- 49.Blöchl PE. Projector augmented-wave method. Phys. Rev. B. 1994;50:17953–17979. doi: 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- 50.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 51.Togo A, Oba F, Tanaka I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B. 2008;78:134106. doi: 10.1103/PhysRevB.78.134106. [DOI] [Google Scholar]
- 52.Martyna GJ, Klein ML, Tuckerman M. Nosé-Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 1992;97:2635–2643. doi: 10.1063/1.463940. [DOI] [Google Scholar]
- 53.Mathew K, Sundararaman R, Letchworth-Weaver K, Arias TA, Hennig RG. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014;140:084106. doi: 10.1063/1.4865107. [DOI] [PubMed] [Google Scholar]
- 54.Henkelman G, Uberuaga BP, Jónsson H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000;113:9901–9904. doi: 10.1063/1.1329672. [DOI] [Google Scholar]
- 55.Sundararaman R, et al. JDFTx: Software for joint density-functional theory. SoftwareX. 2017;6:278–284. doi: 10.1016/j.softx.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sundararaman R, Schwarz K. Evaluating continuum solvation models for the electrode-electrolyte interface: challenges and strategies for improvement. J. Chem. Phys. 2017;146:08411. doi: 10.1063/1.4976971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nørskov JK, et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 2004;108:17886–17892. doi: 10.1021/jp047349j. [DOI] [Google Scholar]
- 58.Valdés Á, Qu Z-W, Kroes G-J, Rossmeisl J, Nørskov JK. Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C. 2008;112:9872–9879. doi: 10.1021/jp711929d. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files. All of the other data are available from the corresponding author upon reasonable request.
The computational codes used in this work are available from the corresponding author on reasonable request.