

Abstract
The contribution of the Ubiquitin-Proteasome System (UPS) to mitophagy has been largely attributed to the E3 ubiquitin ligase Parkin. Here we show that in response to the oxidative stress associated with hypoxia or the hypoxia mimic CoCl2, the damaged and fragmented mitochondria are removed by Parkin-independent mitophagy. Mitochondria isolated from hypoxia or CoCl2-treated cells exhibited extensive ubiquitination, predominantly Lysine 48-linked and involves the degradation of key mitochondrial proteins such as the mitofusins MFN1/2, or the import channel component TOM20. Reflecting the critical role of mitochondrial protein degradation, proteasome inhibition blocked CoCl2-induced mitophagy. The five conserved ubiquitin-binding autophagy receptors (p62, NDP52, Optineurin, NBR1, TAX1BP1) were dispensable for the ensuing mitophagy, suggesting that the mitophagy step itself was independent of ubiquitination. Instead, the expression of two ubiquitin-independent mitophagy receptor proteins BNIP3 and NIX was induced by hypoxia or CoCl2-treatment followed by their recruitment to the oxidation-damaged mitochondria. By employing BNIP3/NIX double knockout and DRP1-null cell lines, we confirmed that mitochondrial clearance relies on DRP1-dependent mitochondrial fragmentation and BNIP3/NIX-mediated mitophagy. General antioxidants such as N-Acetyl Cysteine (NAC) or the mitochondria-specific Mitoquinone prevented HIF-1α stabilization, ameliorated hypoxia-related mitochondrial oxidative stress, and suppressed mitophagy. We conclude that the UPS and receptor-mediated autophagy converge to eliminate oxidation-damaged mitochondria.
Keywords: Mitochondria, Proteasome, Ubiquitin, Mitophagy, Hypoxia, HIF-1α, Oxidative stress
Abbreviations: OXPHOS, Oxidative Phosphorylation; ROS, Reactive Oxygen Species; UPS, Ubiquitin Proteasome System; Ub, Ubiquitin; PolyUb, polyubiquitin; MAD, Mitochondria-Associated Degradation; PHD, Prolyl Hydroxylase; NAC, N-Acetyl Cysteine; MitoQ, Mitoquinone; OMM, Outer Mitochondrial Membrane; OCR, oxygen consumption rate
Graphical abstract
Sulkshane P. et al. show that oxidation-induced mitophagy entails two steps: degradation of outer mitochondrial membrane proteins in a ubiquitin-proteasome-dependent manner followed by ubiquitin-independent mitophagy.
Highlights
-
•
Mitochondria-derived ROS contributes to HIF-1α stabilization during hypoxia.
-
•
Oxidation-induced mitophagy entails ubiquitin-dependent and -independent steps.
-
•
PINK1/Parkin & Ub-binding receptors are dispensable for oxidation-induced mitophagy.
-
•
DRP1-dependent fragmentation facilitates oxidation-induced mitophagy.
-
•
BNIP3/NIX partake in hypoxia-induced mitophagy, independent of mitochondrial ubiquitination.
1. Introduction
Mitochondria are central to cellular energy production by oxidative phosphorylation (OXPHOS) and integrate many biosynthetic pathways and intracellular signaling cascades. As a consequence, mitochondrial defects are linked to excessive production of Reactive Oxygen Species (ROS), inability to maintain cellular homeostasis, induction of apoptotic cell death, and therefore underlie several human disorders [1]. Hypoxia, a physiological condition characterized by reduced oxygen tension, has been associated with the generation of free radicals and ROS by electron transport chain (ETC) complexes I and III due to inefficient electron transfer [2,3]. This paradox of increased ROS under low oxygen tension can be explained by insufficient molecular oxygen – the terminal electron acceptor in the ETC – or by hypoxia-induced disassembly of the respiratory chain supercomplexes (respirasomes) [4]. The ROS thus generated can nonspecifically damage various cellular macromolecules and membranes as well as mitochondria themselves, ultimately leading to mitochondrial dysfunction [2]. Hypoxia-associated mitochondrial dysfunction has been implicated in diverse human disorders and pathophysiological conditions including Alzheimer's disease, diabetes, ischemia/reperfusion injury to the cardiac muscles and brain, inflammation, and cancer [4]. Therefore, it is imperative for a cell to remove the mitochondria damaged by hypoxia. Stabilization of the hypoxia-inducible transcription factor-1 alpha (HIF-1α) is pivotal in the adaptive response to hypoxia-induced oxidative stress since the downstream gene expression shift cellular metabolism from oxidative phosphorylation to glycolysis [[5], [6], [7]].
Mammalian cells contain several quality control mechanisms to ensure functional population of mitochondria and eliminate those that are damaged or dysfunctional [8]. Chaperones and mitochondria-resident proteases ensure proper folding of misfolded or aggregated proteins in the mitochondrial matrix in the event of unfolded protein accumulation [9]. Low levels of damage to mitochondrial proteins may be dealt with by internal mitochondrial quality control proteases that selectively degrade damaged mitochondrial proteins [10]. Alternatively, damaged proteins in the mitochondrial outer membrane (OMM) can be selectively removed by the Ubiquitin Proteasomal System (UPS) [11,12]. Recent evidence suggests that the UPS even monitors the import of proteins into mitochondria, aids to remove mislocalized intra-mitochondrial proteins and reinstate flux through clogged import channels [13]. Ultimately, irreversibly damaged mitochondria that are beyond repair may be eliminated as a whole by mitophagy [14,15]. This process probably requires that the damaged mitochondria first undergo fission in a DRP1-dependent manner aided by the degradation of mitofusin proteins. Both mitochondrial fusion and fission processes are mediated by mitochondria-associated GTPases of the dynamin family. Fusion between OMMs is mediated by membrane-anchored mitofusions, MFN1 and MFN2, whereas fission is mediated by a cytosolic family member, DRP1 that is recruited to mitochondria to constrict inner and outer membranes. While there are many reasons for the ensuing mitochondrial dynamics, both fusion and fission contribute to maintenance of oxidative phosphorylation under stressful conditions and elimination of mitochondria damaged by oxidative stress [8]. Consequently, myriad components of the UPS transiently associate with mitochondria under both basal and stressful conditions to maintain mitochondria homeostasis (mitostasis) [[16], [17], [18]].
By sharing ubiquitin as a common signal for tagging the substrates to be degraded [19], both the UPS and autophagy pathways are highly coordinated and complement each other to maintain the cellular homeostasis under stressful conditions [20,21]. Under stressful conditions, specific proteins on the OMM have been shown to selectively undergo ubiquitination followed by proteasome-mediated degradation in a process termed Mitochondria-Associated Degradation (MAD) [12,22,23]. Recent evidence shows that the MAD pathway, but not other mitochondrial quality control systems, is critical for yeast cell survival under conditions of mitochondrial oxidative stress [24]. However, the components of the proteolytic machinery themselves are also susceptible to oxidative damage, which could exacerbate damage to mitochondria [25]. Mitochondrial depolarization leads to PINK1 stabilization and activation at OMM, which in turn recruits Parkin to the damaged mitochondria. Activated Parkin ubiquitinates multiple OMM proteins while PINK1 phosphorylates these polyubiquitin chains leading to a feed-forward signal amplification. These polyubiquitin chains are recognized by a number of ubiquitin-binding autophagy adapter proteins (e.g. p62, NDP52, Optineurin, NBR1, and TAX1BP1), which link the polyubiquitin signal with LC-3, thus enabling engulfment of the damaged mitochondria by autophagosomes [[26], [27], [28]]. Although oxidative stress-induced mitophagy has been largely attributed to the PINK1-Parkin pathway [[29], [30], [31]], recent reports highlight the importance of a Parkin-independent mode of mitophagy [32,33]. For instance, an alternative mechanism, termed receptor-mediated mitophagy, is mediated by certain mitochondrial proteins such as FKBP8, FUNDC1, BNIP3, and NIX which directly interact with LC-3B on the autophagosomal membranes through their LIR motifs, bypassing the need for ubiquitination [26].
In this study, we show that oxidative stress-induced either by hypoxia or by the hypoxia mimic CoCl2 triggers extensive ubiquitination and fragmentation of mitochondria culminating in Parkin-independent receptor-mediated mitophagy. In addition to hypoxia, we also used CoCl2 as a hypoxia mimic as it is known to stabilize HIF-1α in a ROS-dependent manner [34]. Alleviation of the oxidative stress by antioxidants reduced the extent of mitochondrial fragmentation, ubiquitination and also reduced the extent of mitophagy. Although the downstream mitophagy step itself was independent of ubiquitin-binding autophagy receptors, proteasome-dependent upstream processing of ubiquitinated mitochondrial proteins was necessary. We conclude that the UPS and the autophagy pathways cooperate to eliminate oxidation damaged mitochondria.
2. Results
2.1. The hypoxia mimic CoCl2 induces mitochondrial oxidative stress and mitophagy in HeLa cells
We used CoCl2 to induce oxidative stress and compare the fate of damaged mitochondria to that of hypoxia in our experimental system. We first treated HeLa cells with 500 μM CoCl2 and assessed the extent of HIF-1α stabilization and autophagy induction in a time-dependent manner. We observed that CoCl2 stabilized HIF-1α as early as 6 h post-treatment and induced robust autophagy at about 24 h. The autophagy thus induced was associated with apoptotic cell death as evident from Caspase-3 cleavage (Fig. 1A). In order to rule out the potential adverse effects of apoptosis on our evaluation of mitochondrial physiology, in further experiments, we employed a pan-caspase inhibitor ZZ-VAD-FMK, which blocked caspase-3 cleavage and thus prevented cell death induced by CoCl2 (Supplementary figure S1A, S1B, S1C). Since CoCl2 induces a hypoxia-like response, we tested if it limits mitochondrial respiration even under normoxia. We measured the mitochondrial respiratory capacity by using Seahorse Extracellular Flux Analyzer and observed that CoCl2 significantly decreased the overall oxygen consumption rate (OCR), basal respiration, active ATP production, and the maximal respiratory capacity of the mitochondria (Fig. 1B). As evident from the cellular energy generation map, CoCl2-treated cells shifted from aerobic respiration to glycolytic/anaerobic metabolism (Supplementary Figure S1D).
Fig. 1.

The hypoxia mimic CoCl2 induces mitochondrial oxidative stress and mitophagy in HeLa cells. The cells were treated with 500 μM CoCl2 in combination with 25 μM Z-VAD-FMK for 24 h unless otherwise indicated. Additionally, the cells were pretreated with 10 mM N-Acetyl Cysteine for 2 h before treatment with CoCl2 wherever indicated. A. HeLa cells were treated with 500 μM CoCl2 alone for the indicated time points. The whole cell lysates were subjected to immunoblotting to analyze the expression of HIF-1α, LC-3B, cleaved caspase-3 and GAPDH (as a loading control). B. The mitochondrial respiration measurements were performed on Seahorse Extracellular Flux analyzer. Changes in the oxygen consumption rate (OCR) were measured by sequential injections of Oligomycin, FCCP and equimolar concentration of Rotenone + Antimycin A. The OCR values were normalized by protein concentration per well determined by Bradford assay at the end of experiment. C. The extent of mitochondrial fragmentation and mitochondrial ROS levels were determined by live cell imaging dyes Mitotracker Green and MitoSOX red using a confocal microscope and represented as fold change relative to control. D. Evaluation of functional mitophagy by confocal microscopy-based Mitokeima assay. The extent of mitophagy was represented as mitophagy index. E. Alternatively, the extent of mitophagy was determined by mitokeima-based FACS assay and expressed as percent cells showing the red mitokeima fluorescence. F. The control and CoCl2 treated whole cell extracts were analyzed for the expression of the indicated mitochondrial proteins with GAPDH as a loading control. The blots are representative of three independent experiments. The data is expressed as mean + SEM of three independent experiments. Scale bar: 10 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
As mitochondria are the principal source of cellular ROS and CoCl2 is reported to cause oxidative stress in cells [34], we next evaluated the extent of mitochondrial ROS upon CoCl2 treatment. By using MitoSOX Red superoxide indicator dye (Fig. 1C) and ELITE Mitochondrial ROS assay (Supplementary Figure S1E), we found that CoCl2 induces significant (p < 0.001) ROS accumulation in mitochondria and caused extensive fragmentation of the mitochondrial network. Interestingly, the antioxidant N-Acetyl Cysteine (NAC) protected the cells from the damaging effects of CoCl2 by preventing buildup of ROS and aided in maintenance of the normal mitochondrial architecture (Fig. 1C). These observations indicate that CoCl2 causes oxidative damage to mitochondria leading to fragmentation and hampering respiratory capacity even in presence of normal levels of oxygen.
Next, we investigated the fate of these dysfunctional oxidation-damaged mitochondria. Typically, damaged mitochondria are engulfed by autophagosomes that later on fuse with lysosomes to form mitophagolysosomes. Indeed, in response to CoCl2-induced oxidative stress, some of the fragmented mitochondria colocalized with the autophagosomal marker LC-3B and with the lysosomal marker LAMP-1 (Supplementary Figure S1F, S1G). Labeling mitochondria with Keima - Mitokeima, a fluorescence reporter probe for studying active mitophagy flux, confirmed that these observations reflect functional mitophagy (Fig. 1D and E). The end result was that over time, the levels of several mitochondrial proteins such as VDAC1, TOM20, MFN1, MFN2 and COX4, localizing to different subcompartments, decreased in response to CoCl2 treatment (Fig. 1F, Supplementary Figure S1H). Of note, the degradation of mitofusins MFN1 and MFN2 points towards mitochondrial fragmentation, as discussed above. OMM proteins VDAC1, TOM20 and the inner membrane protein COX4 also exhibited degradation. Taken together, these observations suggest that oxidation-damaged mitochondria undergo fragmentation which are then eliminated by mitophagy.
2.2. Genuine hypoxia induces mitochondrial oxidative stress and fragmentation in HeLa cells
In order to validate the applicability of CoCl2 as a mitochondria-damaging agent, we compared the effects of CoCl2 on the mitochondrial morphology and physiology (as demonstrated above in Fig. 1) with those of genuine hypoxia (1% Oxygen). First, we confirmed the sustained stabilization of HIF-1α in response to hypoxia as early as 6 h and up to 36 h (Fig. 2A). Here too, the mitochondrial network showed extensive fragmentation and perinuclear aggregation (Fig. 2B). As a testimony, exposure to the hypoxic conditions caused a significant (p < 0.01) decrease in the extent of OCR (Fig. 2C), suggesting that cells are responding to the hypoxic conditions by switching their energy production from OXPHOS to anaerobic glycolytic mode. Similar to CoCl2 treatment, the mitochondrial fragmentation was associated with an increase in the ROS levels as assessed by the mitochondrial ROS indicator dye MitoSOX Red (Fig. 2D). It is noteworthy that treatment of cells exposed to hypoxia with the mitochondria-targeted antioxidant Mitoquinone (MitoQ) led to a significant decrease in the HIF-1α protein levels, suggesting that mitochondria-derived ROS play a key role in stabilization of the HIF-1α protein under hypoxic conditions (Fig. 2E). Apparently, oxidative stress constitutes an integral part of the hypoxic stress response whether generated by CoCl2 or by genuine hypoxia.
Fig. 2.

Genuine hypoxia causes mitochondrial oxidative stress leading to their ubiquitination. HeLa cells were exposed to 1% Oxygen for 24 h (Hypoxia) or 21% Oxygen (Normoxia). A. The cell lysates corresponding to the indicated time points following hypoxia were analyzed for detection of HIF-1α and GAPDH by western blotting. B. The cells were immunostained with HSP60 antibody followed by image acquisition on a confocal microscope and the percent mitochondrial fragmentation was determined by evaluating a minimum of 100 cells across 10 different microscopic fields. C. The Oxygen Consumption Rate (OCR) was determined by Seahorse Extracellular Flux analyzer using the MitoStress test kit. D. The cells were stained with live cell imaging dyes Mitotracker Green and MitoSOX red and observed under a confocal microscope. The MitoSOX Red staining intensity was expressed in terms of fold change relative to the control. At least 100 cells across 10 different microscopic fields per condition were evaluated. E. The cells were exposed to normoxia and hypoxia alone or in combination with 1 μM Mitoquinone Q (MitoQ) or vehicle control (VC) and whole cell extracts were subjected to immunoblotting for the detection of HIF-1α, PHD2 and Actin (loading control). F. The cells were stained with MitoTracker Red followed by immunostaining with LC-3B antibody. G. The whole cell lysates of HeLa cells exposed to Normoxia (N) and Hypoxia (H) were subjected to immunoblotting for detection of the indicated mitochondrial proteins. H. The isolated mitochondrial fractions from control and CoCl2 treated cells (left) and normoxic and hypoxic cells (right) were subjected to western blotting for evaluating the level of total ubiquitin and ATP5a (loading control). I. Wild type HeLa cells were stained with Mitotracker Red followed by immunostaining with Ubiquitin and LC-3B antibodies. The numbered circles mark their corresponding position in the individual channels and in the merged image to indicate the extent of their overlap. Scale bar for microscopy images: 10 μm. The blots are representative of three experiments. The data is expressed as mean + SEM of three experiments. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, we demonstrated mitophagy, as a fate of these oxidatively damaged mitochondria, by colocalization between mitochondria and the autophagosome marker LC-3B (Fig. 2F). Furthermore, we assessed the levels of key mitochondrial proteins and documented a dramatic decrease in levels of OMM proteins such as TOM20 and MFN2 as well as of an inner membrane protein, TIM23 (Fig. 2G). Induction of mitophagy was further supported by conversion of LC-3B, a marker of general autophagy (Fig. 2G). In the case of membrane-depolarized mitochondria, ubiquitination typically marks them for mitophagy [27], therefore we wished to test whether hypoxia also leads to mitochondrial ubiquitination. To this end, we isolated mitochondria from HeLa cells following an earlier protocol [16] by sequential application of differential centrifugation followed by density gradient ultracentrifugation (Supplementary Figure S2A). Interestingly, mitochondria isolated from cells exposed to either CoCl2 or to hypoxia exhibited increased ubiquitination as compared to mitochondria purified from cells growing under normoxia (Fig. 2H). Mitochondrial ubiquitination in response to hypoxia was further confirmed by immunofluorescence microscopy, where fragmented mitochondria exhibited partial colocalization with ubiquitin and LC-3B foci (Fig. 2I). All these observations suggest that the aberrant effects of CoCl2 on the mitochondrial organization and function coincide to a large extent with those of genuine hypoxia. We speculated that mitochondrial ubiquitination may be involved in the mitophagy of oxidation damaged mitochondria.
2.3. CoCl2-induced mitophagy occurs independent of Parkin or ubiquitin-binding autophagy adaptor proteins
In order to analyze the nature of mitochondrial ubiquitination, we evaluated the isolated mitochondrial fractions with antibodies for specific ubiquitin linkages (Supplementary Figure S2B). In addition to the overall increase in total polyubiquitin conjugates associated with the mitochondrial fractions of CoCl2-treated cells (Fig. 2H), there was a predominant increase in Lysine 48-linked (K48) polyubiquitin, whereas Lysine 63-linked (K63) polyubiquitin increased to a relatively lesser extent. Immunoblotting of isolated mitochondria revealed increased recruitment of proteasomal subunits at the mitochondria from the CoCl2-treated cells consistent with the elevated levels of K48-linked polyubiquitin (Supplementary Figure S2C). More generally, mass spectrometry analysis of isolated mitochondria from CoCl2-treated cells identified several components of the UPS: UBA1 (E1 ubiquitin-activating enzyme), UBE2S (an E2 ubiquitin-conjugating enzyme), several E3 ubiquitin ligases (HUWE1, MARCH5, UBR4, TRIM25), USP33 (a deubiquitinating enzyme), AAA ATPase/p97 (required for protein extraction during the MAD pathway), UBXN4/UBXD2 (an auxiliary factor for p97) and FKBP8 (which docks proteasome on the OMM and also harbors LIR motifs) (Supplementary Figure S3). This analysis also confirmed numerous proteasome subunits indicating proteasome recruitment to the oxidation-damaged mitochondria (Supplementary Figure S3). All these results suggest that oxidation damaged mitochondria associate with multiple components of the UPS, undergo extensive ubiquitination primarily of K48-linkages, and possibly activate proteasomal degradation of certain mitochondrial targets.
Typically, HeLa cells do not express detectable levels of Parkin [ [35] and Supplementary Figure S4A]. Nevertheless, since Parkin is central to mitophagy of depolarized mitochondria, we overexpressed Parkin (Supplementary Figure S4A) and observed that in stark contrast to CCCP treatment, CoCl2 failed to induce translocation of Parkin to the damaged mitochondria (Supplementary Figure S4B). Furthermore, neither by immunoblotting nor by immunofluorescence could we detect PINK1 stabilization or the presence of phospho-Ubiquitin (pS65 Ub) following CoCl2 treatment (Supplementary Figure S4C, S4D). By contrast, as a positive control, CCCP treatment did stabilize PINK1 leading to ubiquitin phosphorylation at mitochondria (Supplementary Figure S4C and S4D). These results emphasized that the PINK1-Parkin pathway is dispensable for CoCl2-induced mitochondrial ubiquitination and mitophagy.
In order to elucidate whether ubiquitination is a cause or a consequence of mitochondrial ROS, we evaluated whether antioxidants reduce the extent of mitochondrial ubiquitination. Indeed, NAC prevented CoCl2-induced total ubiquitination as well as both K48 and K63-linked polyubiquitin linkages detected on isolated mitochondria (Fig. 3A). Furthermore, by coimmunofluorescence microscopy, we showed that upon CoCl2-induced oxidative damage, ubiquitin foci colocalize with the fragmented mitochondria. However, treatment with the antioxidant NAC not only maintained the organization of mitochondrial network but also suppressed the mitochondrial ubiquitination (Fig. 3B). Taken together, mitochondrial ubiquitination appears to be a direct consequence of oxidative stress.
Fig. 3.

Oxidative stress causes extensive mitochondrial ubiquitination but mitophagy occurs independent of ubiquitin-binding autophagy receptors. HeLa cells were treated with 500 μM CoCl2 along with 25 μM Z-VAD-FMK for 24 h. A. The cells were either treated with CoCl2 alone or in combination with NAC. The isolated mitochondrial fractions were analyzed for the extent of total ubiquitin, K48 and K63-linked polyubiquitin by immunoblotting. B. The cells were stained with Mitotracker Red followed by immunostaining with LC-3B and Ubiquitin antibodies. Panels on the right display co-localization between mitochondria, ubiquitin and LC-3B by fluorescence intensity line measurement (yellow line). Scale bar: 10 μm. C. The whole cell extracts of CoCl2 treated or control HeLa WT and 5KO cells were analyzed for HIF-1α, MFN2 and tubulin (loading control) by immunoblotting. Alternatively, isolated mitochondrial fractions from these cells were subjected to western blotting for detection of total ubiquitination and HSP60 (loading control). D. Control or CoCl2 treated WT and 5KO HeLa cells stably expressing Mitokeima were analyzed by FACS for mitophagy quantification (ns: not significant). Data is shown as mean + SEM of three experiments. E. Control, CoCl2 and CCCP (10 μM for 6 h) treated HeLa cells overexpressing Parkin were immunostained with TOM20 and OPTN or NDP52 antibodies. Scale bar: 10 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, we asked if following CoCl2 treatment, the polyubiquitin chains on the damaged mitochondria participate in mitophagy by engaging with the ubiquitin-binding autophagy receptor proteins similar to PINK1-Parkin pathway. To address this question, we used a Penta KO HeLa line (5KO) devoid of the five conserved ubiquitin-binding autophagy receptors (adaptors): p62, NDP52, Optineurin (OPTN), NBR1, and TAX1BP1 (Supplementary Figure S5A). The extent of mitochondrial ubiquitination following CoCl2-treatment in the wild type versus 5KO cells was comparable, as was MFN2 turnover (Fig. 3C). In agreement with this observation, the extent of mitochondrial fragmentation and MFN2 turnover was similar in WT and 5KO cells upon exposure to hypoxia (Supplementary Figure S5B and S5C). Moreover, wild type cells exhibited an active flux of general autophagy upon exposure to hypoxia, as evident from reduced levels (marked degradation) of the autophagy adapter proteins and increased conversion of LC-3BI to LC-3BII. On the other hand, the 5KO cells exhibited compromised autophagic flux evident from decreased LC-3BII levels in response to hypoxia (Supplementary Figure S5C), suggesting that general autophagy induced during hypoxic stress may be ubiquitin-dependent. Nevertheless, the 5KO cells did not show any defect in mitophagy upon CoCl2 treatment as evaluated by microscopy or flow cytometry (Fig. 3D and Supplementary Figure S5D). Moreover, by immunofluorescence, we confirmed that two key ubiquitin-binding mitophagy receptors NDP52 and OPTN were not recruited at the damaged mitochondria in response to CoCl2 even though they were actively recruited following CCCP treatment (Fig. 3E). These observations indicate that despite extensive mitochondrial ubiquitination, CoCl2-induced mitophagy occurs independent of the five-known ubiquitin-binding autophagy adaptor proteins.
2.4. Mitochondrial ROS-induced HIF-1α drives BNIP3/NIX-mediated mitophagy
If ubiquitin-binding autophagy receptors are not required for hypoxia-driven mitophagy, what other factors participate? It has been long known that HIF-1α stabilization during hypoxia drives the expression of two structurally related but distinct proapoptotic BCL-2 family proteins, BNIP3 and NIX, which localize to the OMM [36]. We confirmed that the expression of BNIP3 and NIX proteins was indeed induced by both hypoxia and CoCl2 in our experimental system (Fig. 4A). By real-time PCR analysis, we confirmed that their upregulation was primarily at the transcriptional level (Fig. 4B). By immunofluorescence, we further confirmed that upon induction, BNIP3 and NIX localized to the damaged mitochondria (Fig. 4C and D). Interestingly, the antioxidant NAC diminished HIF-1α protein levels and the expression of its downstream targets BNIP3 and NIX, in-line with their expression primarily driven by HIF-1α transcription factor (Fig. 4E).
Fig. 4.

BNIP3 and NIX proteins drive mitophagy of oxidatively damaged mitochondria. A. Whole cell extracts of CoCl2 (500 μM CoCl2 for 6 h) and hypoxia-treated (1% O2 for 24 h) WT HeLa cells were analyzed for the expression of HIF-1α, BNIP3, NIX, GAPDH and Tubulin proteins by immunoblotting. B. The expression of BNIP3 and NIX transcripts in response to CoCl2 treatment (500 μM CoCl2 for 6 h) was analyzed by SYBR Green real time PCR relative to GAPDH and expressed as 2−ΔCt. WT HeLa cells treated with 500 μM CoCl2 plus 25 μM Z-VAD-FMK for 16 h were stained with Mitotracker Red followed by immunostaining with BNIP3 (C) and NIX (D). E. Following the indicated treatments for 24 h, the whole cell extracts of WT HeLa cells were analyzed for HIF-1α, BNIP3, NIX and Tubulin proteins by western blotting. F. The mitochondrial bioenergetics were studied by Seahorse flux analyzer. G. The whole cell extracts of control and CoCl2-treated WT and DRP1 KO HeLa cells were analyzed for DRP1, HIF-1α, MFN2, TOM20, BNIP3, NIX, total ubiquitin and GAPDH proteins (loading control) by immunoblotting. H. The control and CoCl2-treated WT and DRP1 KO HeLa cells were stained with MitoSOX red to evaluate the extent of ROS generated in response to CoCl2-induced oxidative stress and the MitoSOX red staining intensity was expressed as fold change relative to the control. Scale bar for microscopy images: 10 μm. Data represented as Mean + SEM of three experiments. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To identify the extent by which mitochondrial ROS contribute to HIF-1α stabilization in response to CoCl2, we treated cells with the mitochondria-targeted antioxidant Mitoquinone Q (MitoQ). MitoQ prevented HIF-1α protein stabilization and the subsequent expression of its downstream targets BNIP3 and NIX (Fig. 4E). Since the effects of MitoQ and NAC were similar, we concluded that mitochondria-derived ROS are instrumental in CoCl2-induced mitochondrial aberrations. Consistent with its ability to protect mitochondria from oxidative damage, NAC also significantly restored the mitochondrial respiratory capacity which includes the basal level of respiration, active ATP production, and maximal respiratory capacity (Fig. 4F).
2.5. Mitochondrial ROS accumulation and ubiquitination occurs upstream of fragmentation
Mitochondrial oxidative stress has been shown to cause mitochondrial fragmentation by regulating the key fusion-fission proteins MFN2 and DRP1 [37]. Since, depletion of MFN2 is a robust phenomenon during CoCl2-induced mitophagy (Fig. 1, Fig. 2, Fig. 3), we wished to evaluate whether removal of mitofusins contributes to mitochondria fragmentation and mitophagy, or is itself a consequence of mitochondrial damage. To do so, we utilized a DRP1 knockout HeLa cell line (DRP1 KO) resulting in mitochondrial elongation and enlargement due to unrestricted mitochondrial fusion [8]. We observed that in response to CoCl2 treatment, the degradation of MFN2 and TOM20 occurred to the same extent in the wild type and in DRP1 KO HeLa cells (Fig. 4G). Likewise, induction of polyubiquitination by CoCl2 was comparable in the two cell lines (Fig. 4G). Moreover, the HIF-1α-driven expression of BNIP3 and NIX proteins was also seemingly unaffected independent of DRP1 status (Fig. 4G). All these observations point to mitochondrial ubiquitination, turnover of mitochondrial proteins, and induction of mitophagy receptors as a direct response to hypoxia or oxidative stress rather than a consequence of mitochondrial fragmentation.
We then sought to test the contribution of the resulting mitochondrial ROS to mitochondrial fragmentation. By using the mitochondrial ROS indicator dye MitoSOX Red, we observed that accumulation of mitochondrial ROS occurs to similar extent in both wild type and the DRP1 KO HeLa cell lines, suggesting that ROS generation occurs upstream and independent of DRP1-dependent mitochondrial fragmentation (Fig. 4H). By immunofluorescence microscopy, we also found that mitochondrial ubiquitination upon CoCl2 treatment was roughly equivalent in both WT and DRP1-null HeLa cells (Fig. 5A). All the above observations indicate that ROS generation and ubiquitination events occur independently of DRP1-dependent mitochondrial fragmentation.
Fig. 5.

Mitochondrial ROS generation and ubiquitination occurs independent of DRP1-dependent fragmentation. HeLa WT and DRP1 KO cells were either treated with 500 μM CoCl2 plus 25 μM Z-VAD-FMK for 24 h or left untreated (control). A. The cells were stained with Mitotracker Red dye followed by immunostaining with ubiquitin and LC-3B antibodies. Right panels indicate semiquantitative co-localization analysis between mitochondria, ubiquitin and LC-3B by fluorescence intensity line measurement (blue line). Scale bar: 10 μm. B. The whole cell extracts of WT and BNIP3-NIX double knockout (DKO) HeLa cells were analyzed for HIF-1α, NIX, BNIP3, MFN2, TOM20 and Tubulin proteins by immunoblotting. C. The mitophagy analysis was performed by mitokeima FACS analysis of WT, DRP1 KO and DKO cells. D. The enriched mitochondrial fractions from WT, DRP1 KO and DKO cells were analyzed for total ubiquitin levels by immunoblotting with HSP60 being the loading control. The data is representative of three experiments and expressed as mean + SEM. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.6. DRP1-dependent mitochondrial fragmentation and BNIP3/NIX proteins are critical for CoCl2-induced mitophagy
With the information that BNIP3 and NIX are induced following hypoxia-associated oxidative stress and recruited to mitochondria regardless of fragmentation status, we wished to test whether they are required for mitophagy. To address this question, we generated a BNIP3-NIX double knockout (DKO) HeLa cell line by the CRISPR-Cas9 methodology (Fig. 5B). We then stably expressed the mitophagy reporter fluorescence probe Mitokeima by retroviral transduction of wild type, DRP1 KO, and DKO HeLa cell lines to monitor the mitophagy flux. By FACS analysis, we deduced that the DRP1 KO cells had a drastic (p < 0.01) decrease in the extent of mitophagy. The BNIP3-NIX DKO cells also exhibited a modest but significant (p < 0.05) reduction in the extent of mitophagy in comparison to wild-type cells (Fig. 5C). We conclude that DRP1-dependent mitochondrial fragmentation and BNIP3-NIX-mediated clearance of the damaged mitochondria are critical for CoCl2-induced mitophagy.
Having found that the loss of ubiquitin-independent mitophagy receptors, BNIP3 and NIX, diminishes the extent of CoCl2-induced mitophagy, we next asked whether the absence of these receptors also affect the turnover of OMM proteins. To our surprise, CoCl2-induced turnover of MFN2 and TOM20 was unaffected in the BNIP3-NIX DKO (Fig. 5B). We could reproduce these results with genuine hypoxia to a large extent. As expected, the DKO cells showed compromised mitophagy in response to hypoxia as evident from sustained levels of intramitochondrial proteins COX2, COX4 and TIM23. However, hypoxia-induced MFN2 degradation was consistent in the WT and DKO cells (Supplementary Figure S6A). The DKO cells however exhibited a normal flux of general autophagy evident from degradation ubiquitin-binding autophagy receptors TAX1BP1, NDP52, and OPTN (Supplementary Figure S6A). Interestingly, consistent with MFN2 degradation, the extent of hypoxia-induced mitochondrial fragmentation was also similar in the WT and DKO cells (Supplementary Figure S6B). The levels of mitochondrial ROS were higher in the DKO cells compared to the WT cells, consistent with the fact that BNIP3-NIX are important in clearance of damaged mitochondria during hypoxia to limit ROS generation (Supplementary Figure S6B). These results suggest that mitochondrial fragmentation and ROS accumulation occurs upstream of BNIP3-NIX-dependent mitophagy step. A possible explanation was that mitochondrial ubiquitination underlies the turnover of key OMM proteins even if the mitophagy step itself appears to be driven by ubiquitin-independent mitophagy receptors. Indeed, mitochondria isolated from DRP1 KO or BNIP3-NIX DKO cells showed similar or even greater ubiquitination than those from wild-type cells (Fig. 5D). We conclude that mitochondrial ubiquitination occurs independent of mitochondrial fragmentation or of BNIP3-NIX-dependent mitophagy, and likely serves for a distinct role.
2.7. Proteasome function at the damaged mitochondria is critical for CoCl2-induced mitophagy
Given that ubiquitination is an early event in CoCl2-induced mitophagy and is predominantly K48-linked, we speculated that the 26 S proteasome plays an important role in this process. The specific 26 S proteasome inhibitor Velcade blocked the CoCl2-induced degradation of key OMM proteins such as MFN2 and TOM20 (Fig. 6A). By contrast, the DRP1 inhibitor mDivi-1 or the lysosomal inhibitor Chloroquine did not prevent degradation of MFN2 and TOM20, indicating that their degradation occurs independent of mitochondrial fragmentation or of mitophagy. Nevertheless, addition of Velcade to CoCl2-treated cells did not alter the ratio of LC-3BII to LC-3BI, indicating that Velcade did not affect the flux of general autophagy in CoCl2-treated cells (Fig. 6A). Conversely, the lysosome inhibitor Chloroquine did potently suppress general autophagy, yet did not significantly stabilize OMM proteins such as MFN2 or TOM20 (Fig. 6A), corroborating the conclusion that their turnover by the UPS is largely independent of bulk mitochondria elimination by mitophagy.
Fig. 6.

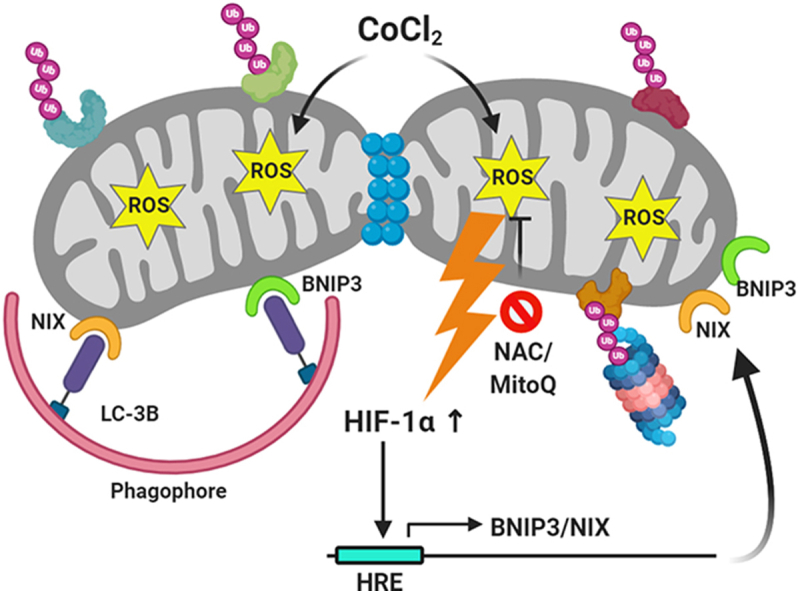
Proteasome inhibition blocks CoCl2-induced mitophagy. HeLa WT cells were treated with 500 μM CoCl2 plus 25 μM Z-VAD-FMK either alone or in combination with NAC (10 mM; pretreatment for 2 h), mDivi-1 (20 μM; throughout 24 h), Velcade (500 nM; for the last 12 h) and Chloroquine (50 μM; for the last 6 h). A. The whole cell extracts were analyzed by immunoblotting for the expression of HIF-1α, TOM20, MFN2, DRP1, LC-3B and GAPDH (loading control). The normalized expression of TOM20 and MFN2 relative to GAPDH is indicated. Comparing the ratio of LC-3BII/LC-3BI indicates that CoCl2 exhibits an active flux of autophagy with reference to the lysosomal (autophagy) inhibitor Chloroquine. B. The analysis of functional mitophagy was performed by Mitokeima FACS assay. C. Mitokeima analysis by microscopy and the extent of mitophagy is indicated as mitophagy index. The data is represented as mean + SEM of three experiments. D. The proposed model of oxidative stress-induced mitophagy depicts ROS-mediated stabilization of HIF-1α, mitochondrial ubiquitination followed by fragmentation and subsequent expression of ubiquitin-independent mitophagy receptors BNIP3 and NIX.
Although inhibition of proteasome did not appear to significantly affect general autophagy in CoCl2-treated cells, stabilization of MFN2 would be expected to affect mitochondria fragmentation and potentially interfere with efficiency of mitophagy. Therefore, we tested the effect of proteasome blockage on mitophagy and revealed that Velcade significantly (p < 0.05) reduced the extent of CoCl2-induced mitophagy (Fig. 6B and C). Although proteasome inhibition prevented MFN2 degradation, we could not detect a decrease in the extent of mitochondrial fragmentation, possibly because proteasome inhibition itself causes mitochondrial fragmentation and aggregation [16,25]. To summarize, mitochondrial ubiquitination and proteasome-dependent turnover of OMM proteins constitutes a critical event upstream of ubiquitin-independent mitophagy receptors.
3. Discussion
As the primary consumers of molecular oxygen, mitochondria are both producers as well as targets of ROS generated due to hypoxia [2,4]. The resulting oxidative damage associated with hypoxia causes mitochondrial dysfunction which underlies many pathophysiological conditions in humans. In the present study we used genuine hypoxia or Cobalt Chloride (CoCl2) as a hypoxia mimic to induce mitochondrial oxidative damage and to study their fate. One outcome was that oxidatively damaged mitochondria were removed by mitophagy, which can be negated to a large extent by antioxidants.
The transcription factor HIF-1α is critical for metabolic adaptation to hypoxia by altering downstream gene expression. Rapid stabilization of the HIF-1α protein during hypoxia has been studied in great detail [38]. Cobalt Chloride (CoCl2) has been widely used as a hypoxia mimic as it also induces rapid stabilization of HIF-1α protein [39,40]. Several mechanisms have been proposed for the induction of HIF-1α by CoCl2, most of which point towards ROS-dependent stabilization of HIF-1α [34]. ROS is known to directly inhibit Prolyl Hydroxylase Domain (PHD) enzymes by chelating and oxidizing Fe2+ bound to the active site of PHD to Fe3+ [[41], [42], [43]]. The high levels of mitochondrial ROS production during hypoxia or in presence of CoCl2 thus may potently inhibit PHD activity [41], leading to HIF-1α stabilization. Indeed, in the present study, we found evidence for ROS-dependent stabilization of HIF-1α, whether by hypoxia or CoCl2, and that antioxidants can overturn HIF-1α stabilization even at low oxygen concentrations. This finding places mitochondria-generated ROS as an early event in hypoxia/CoCl2-induced mitophagy. For this reason, antioxidants such as NAC improve mitochondrial function by counteracting ROS-induced mitochondrial damage in several models of human pathophysiological conditions [44,45]. By protecting against mitochondria-generated ROS, we demonstrate in the current study that antioxidants also mitigate the extent of mitochondrial stress, mitophagy, and restored mitochondrial bioenergetics. Consistent with our observations, NAC has been shown to prevent mitochondrial dysfunction and oxidative stress in Alzheimer's disease patient fibroblasts [46] and in animal models of acute kidney damage [47].
Ample evidence suggests that pervasive ubiquitination of depolarized mitochondria by Parkin leads to their autophagic clearance mediated by conserved ubiquitin-binding autophagy receptors [28,35]. Although hypoxia-associated oxidation-damaged mitochondria were extensively ubiquitinated, we found that oxidation-induced mitophagy did not rely on five conserved ubiquitin autophagy receptors that are critical during PINK1-Parkin mitophagy [28]. Indeed, the well-studied PINK1-Parkin pathway for the ubiquitin-dependent removal of membrane-depolarized mitochondria appears to be dispensable for mitophagy induced by hypoxia or CoCl2. We did not find evidence for ubiquitin phosphorylation [pS65-Ub is a hallmark of PINK1 action on OMM proteins] [48,49], indicating the existence of a distinct mechanism to eliminate oxidation-damaged mitochondria. Mitochondria ubiquitination is an early event in hypoxia-induced mitophagy and is predominantly K48-linked. K48-linkages have been classically implicated as a signal for proteasome-mediated degradation of target proteins [19,50], whereas K63-linked polyubiquitin chains are important in the endosomal-lysosomal pathway [51]. Recruitment of proteasome and other components of the UPS pathway to the stressed mitochondria in this study (including UBA1, UBE2S, HUWE1, MARCH5, UBR4, TRIM25, USP33, VCP/p97, UBXN4/UBXD2, and FKBP8) provides some insight into what drives mitochondria ubiquitination, fragmentation, and clearance. For instance, UBA1 is the E1 ubiquitin-activating enzyme, UBE2S is one of the E2 ubiquitin-conjugating enzymes which catalyzes the formation of K48-linked polyubiquitin chains, HUWE1 is an E3 ubiquitin ligase known to be critical for PINK1-Parkin independent mitophagy [52] and independently has been shown to ubiquitinate MFN2 thus contributing to mitochondrial fragmentation [53]. MARCH5 is the E3 ligase that localizes to the OMM, is known to ubiquitinate several OMM proteins including TOM20 [17] and MFN2 [54]. VCP/p97 is a AAA ATPase that catalyzes extraction of OMM proteins during MAD for proteolytic destruction [55] including even MFN2 [56], and FKBP8 plays an important role in stress-induced Parkin-independent mitophagy [57,58]. The potential functions of UBR4, TRIM25, USP33, and UBXN4 in mitophagy are yet to be elucidated. Turnover of key OMM proteins such as MFN2 promotes fragmentation of the mitochondrial network in response to hypoxia, which also requires the dynamin-like GTPase DRP1 to complete the fission process. Interestingly, the ubiquitin signals on mitochondria were brought back to basal levels upon exposure to NAC, suggesting that hypoxia-triggered mitochondrial ubiquitination occurs as a response to oxidative stress.
In parallel to mitochondria ubiquitination and fragmentation, HIF-1α stabilization also induced BNIP3 and NIX that are sufficient to drive mitophagy. The receptor-mediated pathway is an alternative mechanism for mitophagy that does not require ubiquitin-binding autophagy adapters [26]. We show that upon exposure to CoCl2, stabilized HIF-1α induced the expression of OMM proteins BNIP3 and NIX. These two proteins harbor an LC-3-interacting region (LIR), through which they directly bind to LC3 and thereby participate in Receptor-mediated mitophagy [59]. Notably, antioxidant NAC and MitoQ-mediated suppression of HIF-1α levels also diminished the expression of BNIP3 and NIX. Interestingly, BNIP3 and NIX deficiency have been linked to defective mitophagy leading to excessive ROS production [60,61], suggesting that ROS generating defective mitochondria requires BNIP3/NIX for their mitophagy. We found that indeed BNIP3 and NIX play crucial roles in CoCl2-induced mitophagy but the extent of mitochondrial ubiquitination in BNIP3-NIX DKO increased probably due to a block in mitophagy and possibly due to compromised autophagy in general [59]. BNIP3/NIX double knockout strain led to a marked – but not complete – deficiency of CoCl2-induced mitophagy, hinting possibly at involvement of other ubiquitin-independent mitophagy receptors such as FUNDC1 [62] or FKBP8 [57] among others which can function in a Parkin-independent manner. More importantly, the degradation of MFN2 and TOM20 still occurred in the BNIP3-NIX DKO cells, pointing towards an upstream role of UPS during BNIP3/NIX-dependent mitophagy.
Mitochondrial fragmentation is a prerequisite for efficient clearance of damaged mitochondria by mitophagy [14]. The UPS tightly regulates mitochondrial fusion-fission dynamics by controlling the relative levels of the proteins involved in mitochondrial fusion (such as MFN1/2) or fission (such as DRP1, FIS-1) [12,37,63]. Moreover, CoCl2 has been shown to induce mitochondrial fragmentation in a DRP1-dependent manner [64]. In this study we show that the OMM profusion protein MFN2 is readily degraded in response to oxidative stress along with the mitochondrial protein import receptor TOM20. In addition to MFN2 degradation, the recruitment of mitochondrial fission factor DRP1 to OMM drives fragmentation of the mitochondrial network. Indeed, we show that in addition to the MFN2 degradation, DRP1-dependent mitochondrial fragmentation is critical for CoCl2-induced mitophagy. Moreover, in absence of Parkin, DRP1 is regarded as a critical factor for maintaining the integrity of mitochondria [65]. Interestingly, mitochondrial oxidative stress and ubiquitination occur upstream of DRP1-dependent mitochondrial fragmentation. Overall this study dissects oxidation-induced mitophagy into two sequential steps: an initial ubiquitin-proteasome-dependent turnover of OMM proteins that promotes fragmentation of mitochondria, and a subsequent ubiquitin-independent engulfment of damaged mitochondria entailing BNIP3 and NIX.
As evidence of a link between the UPS and mitophagy, proteasome inhibition blocked the CoCl2-induced mitophagy and also restored the levels of mitochondrial proteins. However, blockade of autophagy or mitochondrial fragmentation did not prevent the degradation of the OMM proteins. This unilateral relationship suggested that proteasome-mediated processing of mitochondrial membrane proteins may be an early step critical for mitophagy. To summarize, our findings reveal that both UPS and autophagy systems are induced in the event of mitochondrial oxidative stress and cooperate to mediate the safe disposal of damaged or dysfunctional mitochondria (Fig. 6D). Antioxidants, therefore, offer great potential in the management of oxidative stress-associated mitopathies in humans.
Declaration of competing interest
All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We sincerely thank Prof. Richard Youle and his team for kindly providing us the penta knockout (5KO) and DRP1 knockout HeLa cell lines, and Dr. Michael Lassarou for the mitokeima plasmid. We thank Dr. Nitsan Dahan and all members of the Microscopy and sorting units of the LS&E institute at the Technion for painstakingly aiding fluorescence microscopy and FACS. We thank all members of the Smoler Proteomics Center at the Technion for sample handling, and members of the Glickman lab for critical comments. This work was supported by a Technion Integrated Cancer Center (TICC) fellowship to PS, grant 755/19 from the Israel Science Foundation for studying ubiquitination at mitochondria (MHG), and a grant 76251-12-9/19 (ZN3457) with S. Dennerlein from the Ministry of Science and Culture of Lower Saxony for research on mitochondria (MHG).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102047.
Appendix A. Supplementary data
The article contains the following Supplementary data:
Material and methods
Cell culture
HeLa cells (Wild type, Penta knockout, and DRP1 knockout cell lines) kindly provided by Prof. Richard Youle (NINDS, NIH, USA) were maintained in DMEM supplemented with 10% Fetal bovine serum (FBS), 2 mM Glutamine, 1 mM Sodium Pyruvate and 1% antibiotics (Penicillin and Streptomycin). pCHAC-mitoKeima retroviral construct was a kind gift from Dr. Michael Lassarou (Monash University, Australia; Addgene plasmid repository #72342). The retroviral plasmid pBMN-Parkin for overexpression of untagged human Parkin was a gift from Michael Lazarou (Addgene plasmid repository #89299). The BNIP3-NIX double knockout (DKO) HeLa cell line was generated by CRISPR-Cas9 approach. We used the following targeting sequence to design guide RNAs: BNIP3: GGAGAGAAAAACAGCTCAC and NIX: CAGGACAGAGTAGTTCCAG. The guide RNA oligonucleotides were annealed and ligated to BpiI digested pSp-Cas9-BB-2A-Puro (PX459) V 2.0 vector (a gift from Feng Zhang; Addgene Plasmid #62988). Equal amounts of BNIP3 and NIX CRISPR plasmids were transfected together using X-treme Gene HP transfection reagent (Roche) into wild type HeLa cells followed by selection with Puromycine (2 μg/ml). The individual clones obtained after selection were expanded and exposed to 1% hypoxia for 24 h to screen them for the expression of BNIP3 and NIX by immunoblotting. The HeLa cell lines (WT, penta KO, DRP1 KO, and DKO) stably expressing mitochondria-targeted keima (Mitokeima) were generated by retroviral transduction.
Antibodies, reagents, and inhibitors
HIF-1α (ab179483), LC-3B (ab51520) and ATP5a (ab14748) antibodies were obtained from Abcam. Cleaved caspase-3 (Cat#9664) and HSP60 (Cat#12165) antibodies were purchased from Cell Signaling. LAMP-1 (sc-20011), VDAC1 (sc-8828), TOM20 (sc-17764), MFN-1 (sc-166644), MFN-2 (sc-100560), DRP1 (sc-271583), BNIP3 (sc-56167), NIX (sc-166332), Ubiquitin (sc-8017) and Calnexin (sc-11397) antibodies were bought from Santacruz Biotechnology. Ubiquitin antibody from Dako (Cat#Z0458). Following antibodies were obtained from Milipore: Ubiquitin Lysine48-linkage specific (Cat#05–1307), Ubiquitin Lysine63-linkage specific (Cat#1308) and Phospho-Ubiquitin specific (Ser65) (Cat#ABS 1513-I). GAPDH antibody was from Sigma (Cat#G9545). COX-4 antibody (A kind gift from Sven Dennerlein, University of Gottingen, Germany). Antibodies specific to proteasome subunits Alpha 6 and Rpn2 were kindly provided by Dr. Rasmus Hartmann-Petersen, University of Copenhagen. The fluorescence secondary antibodies were purchased from Molecular Probes, Invitrogen. The live-cell dyes MitoTracker Green FM (M7514) and MitoSOX Red (M36008) were obtained from Molecular Probes, Invitrogen. The mitochondrial ROS level was alternatively measured by using the ELITE Mitochondrial ROS activity assay kit (eEnzyme, Cat#CA-R933). The staining procedure was performed as per the recommendations of the manufacturer. For each experiment, a stock of 50 mM CoCl2 was freshly prepared and filter sterilized. All the chemicals were purchased from Sigma unless otherwise mentioned.
Measurement of mitochondrial bioenergetics
The mitochondrial respiration measurements were performed using the Extracellular Seahorse Flux Analyzer Assay (Agilent) according to the manufacturer's protocol. Briefly, 10,000 cells were seeded per well in the 8-well culture miniplate (except the wells at either end which serve as blank). After the treatment period, the cells were washed twice with XFp assay medium and then incubated in a non-CO2 incubator at 37 °C for 1 h. Meanwhile, the overnight hydrated sensor cartridge was loaded with the inhibitors: 1 μM Oligomycin A (Port A), 1 μM FCCP (Port B), and 0.5 μM of each of Rotenone + Antimycin A (Port C) and equilibrated followed by insertion of the culture miniplate. At the end of the assay, the OCR values for each well were normalized relative to the corresponding protein amount determined by Bradford assay.
Immunofluorescence, live-cell imaging and confocal microscopy
Following the treatment period, the cells were fixed using warm 4% Paraformaldehyde for 20 min at RT and then permeabilized with 1% Triton X-100 for 5 min at RT. Blocking was done with 5% BSA and then incubated with primary antibodies either at room temperature for 1 h or at 4 °C overnight. The coverslips were washed with PBS thrice and then incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature in dark. The coverslips were again washed with PBS thrice and finally mounted with Fluoromount G (Thermo) on glass slides and stored at −20 °C until image acquisition. The colocalization analysis was performed by using the Profile analysis tool in the ZEN lite (V 3.0) software (Zeiss) by fluorescence intensity line measurement approach.
For live-cell imaging, the cells were grown in glass-bottom (confocal) plates. To assess the extent of mitochondrial oxidative stress, the cells were stained with 5 μM MitoSOX red for 15 min at 37 °C.
Analysis of mitochondrial ROS levels
Evaluation of mitochondrial ROS levels was performed by using mitochondrial superoxide indicator dye MitoSOX Red as described earlier [64]. The cells growing in live-cell imaging plates following the treatment were stained with 200 nM MitoTracker Green FM and 5 μM MitoSOX Red (Invitrogen) for 30 min at 37 °C. After the staining procedure, the cells were washed with PBS and were maintained in a Live-cell imaging solution (Molecular probes) supplemented with 25 mM Glucose. All the imaging was performed on LSM710 fluorescence confocal microscope (Zeiss) using the Zen software. At least 100 cells across 10 random microscopic fields were analyzed for quantification of MitoSOX Red fluorescence intensity. The data analysis was performed using the ImageJ software (NIH).
Mitochondria isolation
Briefly, cells were harvested by trypsinization and washed twice with cold PBS followed by resuspending them in ice-cold HM buffer (250 mM Sucrose, 1 mM EDTA, 1 mM EGTA, 20 mM HEPES pH 7.4 containing 1 mM N-Ethylmaleimide and protease inhibitor cocktail). The cells were homogenized in a precooled Dounce homogenizer with ~30 strokes on ice. The homogenate was centrifuged at 1000×g for 10 min at 4 °C and the resulting supernatant was transferred to a fresh tube and was further centrifuged at 10,000×g for 20 min at 4 °C. The supernatant served as cytosolic fraction whereas the pellet represented Crude mitochondria. The crude mitochondrial pellet was resuspended in a small volume of ice-cold HM buffer and layered on top of a continuous gradient of 17.5% Opti-Prep (Sigma) solution (total 7 ml). It was then subjected to ultracentrifugation in a fixed angle rotor at 270,000×g for 3 h at 4 °C. The resulting gradient was carefully subdivided into 14 fractions each of 0.5 ml which was then diluted twice with ice-cold HM buffer. The tubes were centrifuged at 13,000×g for 20 min at 4 °C. The supernatant was discarded and pellets were resuspended in a small volume of HM buffer, protein concentration was estimated by Bradford assay and stored at −80 °C.
Protein extraction and western blotting
The cells were harvested by trypsinization and washed twice with PBS. The cell pellet was resuspended in ice-cold cell lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40 containing protease inhibitor cocktail) followed by vigorous pipetting and brief vortexing before incubating on ice for 15 min. The cell lysates were centrifuged at 13,000×g for 15 min at 4 °C. The supernatants were transferred to fresh tubes and protein estimation was performed by Bradford assay. Equal amounts of whole-cell lysates were subjected to SDS PAGE followed by immunoblotting for the detection of desired proteins. The blots were incubated with specific primary antibodies either at room temperature for 1 h or overnight at 4 °C. The blots were then incubated with the respective secondary antibodies for 1 h at room temperature. The signals were acquired using a chemiluminescence detection system (GE).
Quantification of mitophagy
To monitor mitophagy, we used Mitokeima (mitochondria-targeted Keima), a ratiometric, pH-sensitive fluorescent protein that is resistant to lysosomal proteases. As Keima is characteristically excited at 561 nm under acidic conditions and at 488 nm under neutral pH environments, a high ratio of 561/488 nm fluorescence represents the presence of mitochondria in acidic lysosomes (mitochondria actively undergoing mitophagy). The extent of mitophagy was assessed by confocal microscopy and indicated as mitophagy index [determined as High (561/488) ratio area/total mitochondrial area] [66]. Ratio images were created by the Ratio Plus plugin of ImageJ (NIH). High ratio areas were segmented and quantified by analyzing the particle plugin of ImageJ.
For quantitation of mitophagy by FACS [28], the cells following treatment were harvested by trypsinization and resuspended in FACS buffer (145 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 10 mM HEPES, 10 mM glucose, 0.1% BSA). Measurements of lysosomal Mitokeima were made using dual-excitation ratiometric pH measurements at 488 (pH 7) and 561 (pH 4) nm lasers with 620/29 nm and 614/20 nm emission filters, respectively. A control mitokeima gate was drawn aound the untreated population of wild type cells which was used to determine the lysosomal mitokeima population in the rest of the samples for that specific experiment. The control gate was set with about 1% population positive for lysosomal mitokeima. A vertical shift in the cell population above the control gate was considered as positive for mitophagy (gate P4) and those below the gate were classified as not undergoing mitophagy (gate P3). Minimum 10,000 events were recorded for data analysis. Data acquisition was performed on Cytek Aurora Spectral analyzer flow cytometer.
RNA extraction and real-time qPCR analysis
Total RNA was extracted using TRIzol reagent (Thermo) according to the manufacturer’s protocol. Reverse transcription was performed using qPCRBIO cDNA synthesis kit (PCR Biosystems PB30.11-02), and real-time PCR using qPCRBIO fast qPCR SyGreen blue mix (PCR Biosystems PB20.16–51). The following primers were used: BNIP3 FW: 5'-CAGGGCTCCTGGGTAGAACT-3', RV: 5' CTACTCCGTCCAGACTCATGC 3'. NIX FW: 5′-TGCGGACTGGGTATCAGACTG-3′, RV: 5′-GGGTGTCTGAAGTGGAACTCCT-3′. GAPDH FW: 5’-TGCACCACCAACTGCTTAG-3’, RV: 5’-GATGCAGGGATGATGTTC-3’. The BNIP3 and NIX transcript expression was determined relative to GAPDH as housekeeping and expressed as fold-change 2-ΔCt.
Statistical analysis
All the statistical data was analyzed using the GraphPad Prism (V 5.0) software. Data from at least 3 experiments was analyzed by one-way ANOVA or t test to determine the statistical significance. All error bars represents mean + standard error mean (SEM).
Appendix B. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Vafai S.B., Mootha V.K. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491(7424):374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- 2.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pomatto L.C., Davies K.J. The role of declining adaptive homeostasis in ageing. J. Physiol. 2017;595(24):7275–7309. doi: 10.1113/JP275072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solaini G., Baracca A., Lenaz G., Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta Bioenerg. 2010;1797(6–7):1171–1177. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Semenza G.L., Jiang B.-H., Leung S.W., Passantino R., Concordet J.-P., Maire P. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996;271(51):32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 6.Iyer N.V., Kotch L.E., Agani F., Leung S.W., Laughner E., Wenger R.H. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12(2):149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenza G., editor. Cold Spring Harbor Symposia on Quantitative Biology. Cold Spring Harbor Laboratory Press; 2011. Regulation of metabolism by hypoxia-inducible factor 1. [DOI] [PubMed] [Google Scholar]
- 8.Youle R.J., Van Der Bliek A.M. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nargund A.M., Pellegrino M.W., Fiorese C.J., Baker B.M., Haynes C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker M.J., Tatsuta T., Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harbor perspectives in biology. 2011;3(7):a007559. doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clague M.J., Heride C., Urbé S. The demographics of the ubiquitin system. Trends Cell Biol. 2015;25(7):417–426. doi: 10.1016/j.tcb.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Livnat-Levanon N., Glickman M.H. Ubiquitin–proteasome system and mitochondria—reciprocity. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2011;1809(2):80–87. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Sulkshane P., Ram J., Glickman M.H. Ubiquitination of intramitochondrial proteins: implications for metabolic adaptability. Biomolecules. 2020;10(11):1559. doi: 10.3390/biom10111559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burman J.L., Pickles S., Wang C., Sekine S., Vargas J.N.S., Zhang Z. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. JCB (J. Cell Biol.) 2017;216(10):3231–3247. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pickles S., Vigié P., Youle R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018;28(4):R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulkshane P., Duek I., Ram J., Thakur A., Reis N., Ziv T. Inhibition of proteasome reveals basal mitochondrial ubiquitination. J. Proteonomics. 2020;229:103949. doi: 10.1016/j.jprot.2020.103949. [DOI] [PubMed] [Google Scholar]
- 17.Phu L., Rose C.M., Tea J.S., Wall C.E., Verschueren E., Cheung T.K. Dynamic regulation of mitochondrial import by the ubiquitin system. Mol. Cell. 2020;77(5):1107–1123. doi: 10.1016/j.molcel.2020.02.012. e10. [DOI] [PubMed] [Google Scholar]
- 18.Escobar-Henriques M., Altin S., Brave F. Interplay between the ubiquitin proteasome system and mitochondria for protein homeostasis. Curr. Issues Mol. Biol. 2019;35:35–58. doi: 10.21775/cimb.035.035. [DOI] [PubMed] [Google Scholar]
- 19.Glickman M.H., Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82(2) doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 20.Korolchuk V.I., Menzies F.M., Rubinsztein D.C. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy. 2009;5(6):862–863. doi: 10.4161/auto.8840. [DOI] [PubMed] [Google Scholar]
- 21.Ding W.-X., Ni H.-M., Gao W., Yoshimori T., Stolz D.B., Ron D. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007;171(2):513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatenay-Lapointe M., Shadel G.S. Stressed-out mitochondria get MAD. Cell metabolism. 2010;12(6):559–560. doi: 10.1016/j.cmet.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heo J.-M., Livnat-Levanon N., Taylor E.B., Jones K.T., Dephoure N., Ring J. A stress-responsive system for mitochondrial protein degradation. Mol. Cell. 2010;40(3):465–480. doi: 10.1016/j.molcel.2010.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao P.-C., Wolken D.M.A., Serrano E., Srivastava P., Pon L.A. Mitochondria-associated degradation pathway (MAD) function beyond the outer membrane. Cell Rep. 2020;32(2):107902. doi: 10.1016/j.celrep.2020.107902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livnat-Levanon N., Kevei E., Kleifeld O., Krutauz D., Segref A., Rinaldi T. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014;7(5):1371–1380. doi: 10.1016/j.celrep.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 26.Palikaras K., Lionaki E., Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018;20(9):1013–1022. doi: 10.1038/s41556-018-0176-2. [DOI] [PubMed] [Google Scholar]
- 27.Harper J.W., Ordureau A., Heo J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018;19(2):93. doi: 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- 28.Lazarou M., Sliter D.A., Kane L.A., Sarraf S.A., Wang C., Burman J.L. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank M., Duvezin-Caubet S., Koob S., Occhipinti A., Jagasia R., Petcherski A. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta Mol. Cell Res. 2012;1823(12):2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Garza-Lombó C., Pappa A., Panayiotidis M.I., Franco R. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 2020;51:105–117. doi: 10.1016/j.mito.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C., Nie P., Zhou C., Hu Y., Duan S., Gu M. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021;12(2):1–16. doi: 10.1038/s41419-021-03484-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Rita A., D'Acunzo P., Simula L., Campello S., Strappazzon F., Cecconi F. AMBRA1-mediated mitophagy counteracts oxidative stress and apoptosis induced by neurotoxicity in human neuroblastoma SH-SY5Y cells. Front. Cell. Neurosci. 2018;12:92. doi: 10.3389/fncel.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao A., Jiang J., Xie F., Chen L. Bnip3 in mitophagy: novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta. 2020;506:72–83. doi: 10.1016/j.cca.2020.02.024. [DOI] [PubMed] [Google Scholar]
- 34.Muñoz‐Sánchez J., Chánez‐Cárdenas M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019;39(4):556–570. doi: 10.1002/jat.3749. [DOI] [PubMed] [Google Scholar]
- 35.Narendra D.P., Jin S.M., Tanaka A., Suen D.-F., Gautier C.A., Shen J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1) doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sowter H.M., Ratcliffe P.J., Watson P., Greenberg A.H., Harris A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Canc. Res. 2001;61(18):6669–6673. [PubMed] [Google Scholar]
- 37.Wu S., Zhou F., Zhang Z., Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission–fusion proteins. FEBS J. 2011;278(6):941–954. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 38.Lee J.-W., Bae S.-H., Jeong J.-W., Kim S.-H., Kim K.-W. Hypoxia-inducible factor (HIF-1) α: its protein stability and biological functions. Exp. Mol. Med. 2004;36(1):1–12. doi: 10.1038/emm.2004.1. [DOI] [PubMed] [Google Scholar]
- 39.Hervouet E., Pecina P., Demont J., Vojtíšková A., Simonnet H., Houštěk J. Inhibition of cytochrome c oxidase subunit 4 precursor processing by the hypoxia mimic cobalt chloride. Biochem. Biophys. Res. Commun. 2006;344(4):1086–1093. doi: 10.1016/j.bbrc.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Zou W., Yan M., Xu W., Huo H., Sun L., Zheng Z. Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP‐1 activation. J. Neurosci. Res. 2001;64(6):646–653. doi: 10.1002/jnr.1118. [DOI] [PubMed] [Google Scholar]
- 41.Pan Y., Mansfield K.D., Bertozzi C.C., Rudenko V., Chan D.A., Giaccia A.J. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007;27(3):912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerald D., Berra E., Frapart Y.M., Chan D.A., Giaccia A.J., Mansuy D. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 43.Li Y.-N., Xi M.-M., Guo Y., Hai C.-X., Yang W.-L., Qin X.-J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014;224(2):165–174. doi: 10.1016/j.toxlet.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 44.Otte D.-M., Sommersberg B., Kudin A., Guerrero C., Albayram Ö., Filiou M.D. N-acetyl cysteine treatment rescues cognitive deficits induced by mitochondrial dysfunction in G72/G30 transgenic mice. Neuropsychopharmacology. 2011;36(11):2233–2243. doi: 10.1038/npp.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ommati M.M., Amjadinia A., Mousavi K., Azarpira N., Jamshidzadeh A., Heidari R. N-acetyl cysteine treatment mitigates biomarkers of oxidative stress in different tissues of bile duct ligated rats. Stress. 2020:1–16. doi: 10.1080/10253890.2020.1777970. [DOI] [PubMed] [Google Scholar]
- 46.Moreira P.I., Harris P.L., Zhu X., Santos M.S., Oliveira C.R., Smith M.A. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheim. Dis. 2007;12(2):195–206. doi: 10.3233/jad-2007-12210. [DOI] [PubMed] [Google Scholar]
- 47.Aparicio-Trejo O.E., Reyes-Fermín L.M., Briones-Herrera A., Tapia E., León-Contreras J.C., Hernández-Pando R. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019;130:379–396. doi: 10.1016/j.freeradbiomed.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D.G., Ritorto M.S., Hofmann K. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014;460(1):127–141. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ordureau A., Heo J.-M., Duda D.M., Paulo J.A., Olszewski J.L., Yanishevski D. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. Unit. States Am. 2015;112(21):6637–6642. doi: 10.1073/pnas.1506593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahu I., Glickman M.H. Proteasome in action: substrate degradation by the 26S proteasome. Biochem. Soc. Trans. 2021;49(2) doi: 10.1042/BST20200382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lauwers E., Jacob C., André B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. JCB (J. Cell Biol.) 2009;185(3):493–502. doi: 10.1083/jcb.200810114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Rita A., Peschiaroli A., Pasquale D., Strobbe D., Hu Z., Gruber J. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 2018;9(1):1–18. doi: 10.1038/s41467-018-05722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leboucher G.P., Tsai Y.C., Yang M., Shaw K.C., Zhou M., Veenstra T.D. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell. 2012;47(4):547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim H.-J., Nagano Y., Choi S.J., Park S.Y., Kim H., Yao T.-P. HDAC6 maintains mitochondrial connectivity under hypoxic stress by suppressing MARCH5/MITOL dependent MFN2 degradation. Biochem. Biophys. Res. Commun. 2015;464(4):1235–1240. doi: 10.1016/j.bbrc.2015.07.111. [DOI] [PubMed] [Google Scholar]
- 55.Xu S., Peng G., Wang Y., Fang S., Karbowski M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell. 2011;22(3):291–300. doi: 10.1091/mbc.E10-09-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLelland G.-L., Goiran T., Yi W., Dorval G., Chen C.X., Lauinger N.D. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife. 2018;7 doi: 10.7554/eLife.32866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhujabal Z., Birgisdottir Å.B., Sjøttem E., Brenne H.B., Øvervatn A., Habisov S. FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep. 2017;18(6):947–961. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoo S.M., Yamashita Si, Kim H., Na D., Lee H., Kim S.J. FKBP8 LIRL‐dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. Faseb. J. 2020;34(2):2944–2957. doi: 10.1096/fj.201901735R. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J., Ney P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16(7):939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chourasia A.H., Tracy K., Frankenberger C., Boland M.L., Sharifi M.N., Drake L.E. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015;16(9):1145–1163. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu X., Zheng Y., Liu M., Li Y., Ma S., Tang W. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy. 2020:1–13. doi: 10.1080/15548627.2020.1802089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012;14(2):177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 63.Heo J.-M., Rutter J. Ubiquitin-dependent mitochondrial protein degradation. Int. J. Biochem. Cell Biol. 2011;43(10):1422–1426. doi: 10.1016/j.biocel.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He Y., Gan X., Zhang L., Liu B., Zhu Z., Li T. CoCl2 induces apoptosis via a ROS-dependent pathway and Drp1-mediated mitochondria fission in periodontal ligament stem cells. Am. J. Physiol. Cell Physiol. 2018;315(3):C389–C397. doi: 10.1152/ajpcell.00248.2017. [DOI] [PubMed] [Google Scholar]
- 65.Kageyama Y., Hoshijima M., Seo K., Bedja D., Sysa‐Shah P., Andrabi S.A. Parkin‐independent mitophagy requires D rp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33(23):2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bingol B., Tea J.S., Phu L., Reichelt M., Bakalarski C.E., Song Q. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.