

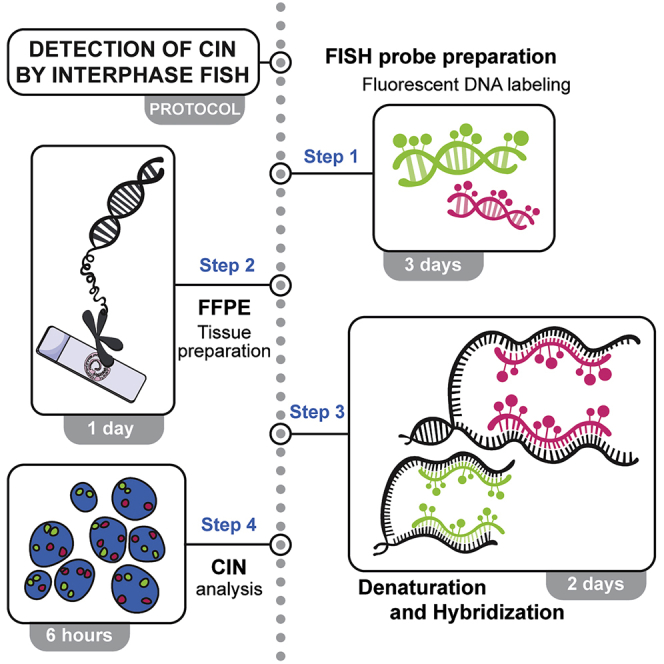
Summary
Chromosomal instability (CIN), a type of genomic instability, favors changes in chromosome number and structure and it is associated with the progression and initiation of multiple diseases, including cancer. Therefore, CIN identification and analysis represents a useful tool for cancer diagnosis and treatment. Here, we report an optimized molecular cytogenetic protocol to detect CIN in formalin-fixed, paraffin-embedded mouse and human tissues, using fluorescent in situ hybridization to visualize and quantify chromosomal alterations such as amplifications, deletions, and translocations.
For complete information on the generation and use of this protocol, please refer to Brandt et al. (2018).
Subject areas: Cell Biology, Single Cell, Microscopy, Molecular Biology, In Situ Hybridization
Graphical abstract
Highlights
-
•
A protocol for single-cell CIN analysis in paraffin-embedded tissues
-
•
Design of FISH probes for CIN detection
-
•
Identification, visualization, and quantification of CIN by FISH
Chromosomal instability (CIN), a type of genomic instability, favors changes in chromosome number and structure and it is associated with the progression and initiation of multiple diseases, including cancer. Therefore, CIN identification and analysis represents a useful tool for cancer diagnosis and treatment. Here, we report an optimized molecular-cytogenetic protocol to detect CIN in formalin-fixed, paraffin-embedded mouse and human tissues, using fluorescent in situ hybridization to visualize and quantify chromosomal alterations such as amplifications, deletions, and translocations.
Before you begin
Fluorescence in situ hybridization (FISH) analysis
FISH probes are short fluorescently-labeled DNA fragments complementary to a specific region of interest, and produced from template plasmids that contain large genomic inserts. Though, there are plenty of commercially available FISH probes used for human sample analysis, the list of FISH probes for mouse samples is very restricted, making it necessary to design and prepare new customized probes.
Probes can be produced using various cloning vectors such as: cosmids, fosmids, PI bacterial artificial chromosomes (BACs) and filamentous phage artificial chromosomes (PACs), wich major difference is the length of the probe. Cosmid and fosmids have a size of ∼40 kb, BACs size ranges between ∼150–350 kb and the size of PACs is ∼300 kb. However, the most commonly employed are BACs and PACs since they yield brighter FISH signals. Importantly, probes smaller than cosmids are associated with weak fluorescence.
Clones can be located using:
-
•
UCSC Human Genome Browser: (http://genome.ucsc.edu)
-
•
NCBI Clone Finder database (http://www.ncbi.nlm.nih.gov/mapview/mvhome/clonehome.html)
Moreover, clones can be obtained from:
-
•
BACPAC Resources Center (https://bacpacresources.org/)
-
•
Source Bioscience (https://www.sourcebioscience.com/life-science-research/clones/genomic-clones/).
The protocol bellow describes the specific steps to find BACs for a particular locus using the UCSC Human Genome Browser, and thenafter, FISH-probe design.
BAC clone selection
Timing: 1 h
-
1.
Go to Genome Browser webpage (https://genome.ucsc.edu/cgi-bin/hgGateway) and select mouse as a model organism (top left mouse brown icon).
-
2.
Chose a region of interest by typing a gene name or the locus position in the search window.
-
3.
In order to visualize BACs information, select the BACs libraries of choice in the Clone Ends Track Setting window and select clone ends (Figure 1A).
-
4.Make your BAC selection (Figure 1B).
-
a.Order the selected BAC clones.
-
a.
Figure 1.

BAC clone selection for the preparation of FISH probes using the UCSD genome browser
Screenshot of the Genome Browser Display.
(A) (1). Once you have selected your region of interest you need to adjust tracks: to visualize BACs information make sure that you have activated the “clone ends” display option to “Full”. Information from different BAC libraries can be activated from the clone ends track settings. (2). Different companies provide clones from different libraries.
(B) You can change the display: zoom in or zoom out, display additional genomic features, etc. (3). Select the appropriate item from search results.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Bacterial cells from E. coli | BACPAC Resources Center | |
| Chemicals, peptides, and recombinant proteins | ||
| Glycerol | Sigma | G-6279 |
| Glucose | Sigma | G8769 |
| Tris | Sigma | T1503 |
| EDTA | Merck | 1084520250 |
| NaOH | Merck | 106462 |
| SDS | Bio-Rad | 1610302 |
| Potassium acetate | Sigma | P1190 |
| Isopropanol | Sigma | I9516 |
| Ethanol | Sigma | 51976 |
| RNAse | QIAGEN | 19101 |
| Sodium acetate | Sigma | S7899 |
| Glacial acetic acid | Sigma | A6283 |
| Agarose | Conda | 8010.22 |
| Xylene | AnalaR NORMAPURE | 28975.291 |
| Green-dUTP | Abbott Molecular | 02N32-050 |
| Red-dUTP | Abbott Molecular | 02N34-050 |
| HCL | Fisher Chemical | 10467640 |
| PBS | Sigma | D8537 |
| Wash Buffer | Agilent Technologies | G9401A;G9402A |
| Proteinase K | Dako | S3020 |
| Vysis LSI/WCP hybridization buffer | Vysis | 30-804826 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | H-1200 |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories | H-1400 |
| Buffer TE | Invitrogen | AM9849 |
| Sspe 20× buffer | Gibco | 15591-043 |
| N-Lauroylsarcosine sodium salt solution | Sigma | L-7414 |
| Sodium borohydride | Sigma | 452882 |
| BSA | Sigma | A9418 |
| Post-Hybridization Wash Buffer | Vitro-Master diagnostica | MAD-FS0105-1 |
| Critical commercial assays | ||
| Nick Translation Kit | Roche | 10976776001 |
| SureTag DNA Labeling Kit | Agilent Technologies | 5190-3399 |
| SureTag DNA Labeling Kit Purification Columns | Agilent Technologies | 5190-3391 |
| Dako’s Histology FISH Accessory Kit | Dako | K5799 |
| Vysis IntelliFISH Universal FFPE Tissue Pretreatment and Wash Reagents | Vysis | 08N85-005 |
| Vysis IntelliFISH Hybridization Buffer | Vysis | 08N87-001 |
| DNeasy Blood & Tissue Kits | QIAGEN | Cat#69506 |
| Oligo aCGH/Chip-on-chip Hybridization Kit | Agilent Technologies | 5188-5220 |
| SurePrint G3 Mouse Genome CGH Microarray Kit, 4×180K | Agilent Technologies | G4839A |
| SurePrint G3 Human CGH Microarray Kit, 4×180K | Agilent Technologies | G4449A |
| SurePrint G3 Human CGH Microarray Kit, 8×60K | Agilent Technologies | G4450A |
| Pretreatment Kit | Vitro-Master diagnostica | MAD-FISH-PTK |
| Experimental models: organisms/strains | ||
| Mouse: Mouse: MCRS1lox/lox: Mcrs1tm1.1Ndj | Dr. Nabil Djouder, (Fawal et al., 2015) Spanish National Cancer Research Center (CNIO) | J:232965 |
| Mouse: mTORlox/lox: B6.129S4-Mtor tm1.2Koz/J | (Risson et al., 2009) | J:011009 |
| Mouse: p53 lox/lox: B6.129P2-Trp53tm1Brn/J | (Marino et al., 2000) | J:61961 |
| Mouse: Villin-CreERT2 : Tg(Vil-cre/ERT2)23Syr | (el Marjou et al., 2004) | J:92295 |
| Software and algorithms | ||
| CytoVision | Leica Microsystems | N/A |
| Other | ||
| Mouse Cot-1 DNA | Invitrogen | Cat#18440-016 |
| Human Cot-1 DNA | Invitrogen | Cat#15279-011 |
| Positively charged glass slides | Menzel-Glaser | N/A |
| ThermoBrite instrument | Leica | N/A |
| Leica DM5500B fluorescence microscope | Leica | N/A |
| Rubber cement | Marabu | 29010010000 |
| 25×50 mm Cover slips | Menzel-Glaser | 15747592 |
Materials and equipment
GTE buffer
The GTE buffer contains 50 mM Glucose, 25 mM TrisHCI pH 8.0, and 10 mM EDTA pH 8.0; It has to be filter sterilized.
Storage conditions: It can be kept at 4°C for several days.
| Reagent | Final concentration | Amount |
|---|---|---|
| Glucose 1 M | 50 mM | 5 mL |
| TrisHCl 1 M pH 8.0 | 25 mM | 2.5 mL |
| EDTA 0.5 M pH 8.0 | 10 mM | 2 mL |
| Milli-Q Water | n/a | 90.5 mL |
| Total | n/a | 100 mL |
Lysis solution
The lysis solution contains 200 mM NaOH, 1% SDS buffer; It has to be filter sterilized. Storage conditions: It can be kept between 20°C–25°C until its use.
Note: The lysis solution has to be made freshly every time that it would be necessary.
| Reagent | Final concentration | Amount |
|---|---|---|
| NaOH 10 M | 200 mM | 0.2 mL |
| 20% SDS buffer | 1% (w/v) | 0.5 mL |
| Milli-Q Water | n/a | 9.3 mL |
| Total | n/a | 10 mL |
Neutralization solution
The neutralization solution contains 7.5 M Potassium acetate at pH 5.5.
Storage conditions: It can be stored at 4°C for several weeks.
| Reagent | Final concentration | Amount |
|---|---|---|
| Potassium Acetate | 5 M | 60 mL |
| Glacial acetic acid | n/a | 11.5 mL |
| Milli-Q Water | n/a | 28.5 mL |
| Total | n/a | 100 mL |
Nick translation mix
The Nick Translation mix should be prepared on ice and with reduced light exposure; the enzyme should be the last component added to the mix.
Note: The Nick Translation mix has to be prepared freshly every time it would be needed.
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× Nick Translation buffer | 1× | 5 uL |
| 0.1 mM dNTP mix (dATP/dGTP/dCTP) | 0.02 mM | 10 uL |
| 0.1 mM dTTP | 0.01 mM | 5 uL |
| 0.1 mM Fluorescence labeled dUTP | 0.01 mM | 5 uL |
| Extracted DNA | 20 ug/mL | 1 ug |
| Nick Tranlation enzyme (DNAse I) | n/a | 10 uL |
| Nuclease-free water | n/a | Up to 50 uL |
| Total | n/a | 50 uL |
Step-by-step method details
BAC DNA isolation
Note: Stab cultures are typically shipped at 20°C–25°C and should be placed at 4°C upon arrival. The culture has few weeks of finite life at 4°C, thus, a glycerol stock should be prepared for long term storage (described below). The addition of glycerol stabilizes the frozen bacteria, preventing cell membrane damage keeping the cells alive. A glycerol stock of bacteria can be stored stably at −80°C for many years. To recover the bacteria from the glycerol stock, open the tube and use a sterile loop to scrape the frozen bacteria off from the top, and grow the bacteria from 12–16 h using the appropriate medium. Importantly, do not let the glycerol stock unthaw.
The protocol below describes a rapid alkaline lysis miniprep method. The method works very well producing high quality DNA in terms of concentration and purity. However, commercial alternatives such as Nucleobond Xtra BAC kit (Macherey-Nagel), BACMAX purification kit (Cambio) or QIAGEN Large-construct kit (QIAGEN) are also available.
-
1.
Streak a small amount of E. coli onto a selective plate and incubate from 12–16 h at 37°C to obtain single colonies.
-
2.
Pick a single colony and inoculate into 10 mL of lysogeny broth (LB) medium containing the selective antibiotic in a 50 mL falcon tube.
CRITICAL: Culture poor aeration may contribute to produce a low DNA yield. The optimal culture volume to air volume ratio is 1:4 or less.
-
3.
Grow with vigorous shaking at 37°C in a shaking incubator for 12–16 h.
Note: Freeze each plasmid mixing 800 μl of bacteria culture and 200 μl of sterile glycerol. Store at −80°C.
-
4.
Harvest the bacterial cells by centrifuging at 1792 g (4,000 rpm) for 10 min. The temperature or the spin is not critical at this stage. Carefully discard the supernatant.
-
5.
Resuspend the bacterial pellet in 300 μL of GTE buffer and transfer it to a 2.2 mL centrifuge tube.
-
6.
Add 600 μL of freshly made lysis solution, mix gently by inverting, and incubate on ice for 5 min.
-
7.Slowly add 500 μL of neutralization buffer drop by drop, mix by inverting the tube four to six times (do not vortex), and incubate on ice for 10 min. A thick white precipitate of bacterial DNA and protein will form.
-
a.Centrifuge at 11200 g (10,000 rpm) for 10 min at 4°C.
-
a.
-
8.Transfer the supernatant using a P1000 pipette to a new 2.2 mL centrifuge tube.
-
a.Centrifuge at 11200 g (10,000 rpm) for 5 min at 4°C.
-
a.
-
9.Transfer the supernatant using a P1000 pipette to a new 1.5 mL centrifuge tube containing 700 μL of ice-cold isopropanol trying to avoid any white precipitate material.
-
a.Mix by inverting the tube a few times.
-
b.Centrifuge at 21952 g (14,000 rpm) for 20 min at 4°C.
-
a.
-
10.Discard the supernatant and wash the DNA pellet with 0.5 mL of 70% ethanol.
-
a.Mix by inverting the tube several times
-
b.Centrifuge at 21,952 g (14,000 rpm) for 5 min at 4°C.
-
a.
-
11.
Remove as much supernatant as possible with a pipette and air-dry the pellet at 20°C–25°C for 10 min.
-
12.
Once the pellet turns from white to translucent in appearance, dissolve the DNA into 100 μL of nuclease-free water by gentle occasional tapping of the tube.
-
13.
Add 1 μL of RNAse (100 ug/mL) and incubate for 30 min at 37°C.
-
14.Precipitate the DNA in a mix of 1/10 vol 3 M sodium acetate and 2.5 vol of 100% ethanol for 60 min at −80°C.
-
a.Centrifuge at 21,952 g (14,000 rpm) for 15 min at 4°C.
-
a.
-
15.Discard the supernatant and wash the DNA pellet with 0.5 mL of 70% ethanol
-
a.Centrifuge at 21,952 g (14,000 rpm) for 5 min at 4°C.
-
a.
-
16.
Discard the supernatant and dissolve the DNA into 100 μL of nuclease-free water.
-
17.
Check the DNA integrity by running a 1% agarose gel (Figure 2).
Note: The agarose gel electrophoresis allows us to confirm the efficiency of the Nick translation reaction by analyzing the product size distribution.
Note: The quality of the probe is vital for a successful FISH, and this is in turn strictly related to the quality of the DNA template. Make sure to use good quality, purified DNA, free of RNA, protein or other contaminants. The purity of the preparation should be determined using classical gel electrophoresis.
-
18.
Store at −20°C.
Note: The average yield of the DNA should be approximately 4–8 μg. Commercial alkaline lysis/column purifications kits can be used as an alternative. However, in our hands the alkaline lysis miniprep method described above produces high yields of high-quality DNA needed for the Nick translation protocol.
Figure 2.

Exemplary analytical check of DNA integrity after miniprep BAC clones purification
(A) Representative images of 5 ul of each precipitated BAC analyzed on a 1% agarose gel. The red square represents the sheared and DNA smear low yield DNA extractions. (B) Suitable DNA for labeling protocol. Arrows indicate not sheared BAC DNA.
Fish probe preparation: DNA labeling
Note: Probes can be labeled by direct incorporation of nucleotides conjugated to fluorochromes (i.e., fluorescein iso-thiocyanate (FITC) or tetramethyl rhodamine). Generally, deoxyuridine 5′-triphosphate (dUTP) is conjugated directly to the fluorochrome and used in the place of dTTP in a labeling reaction. Labeling approximately 1 μg of extracted DNA is enough for ten FISH experiments when using a commercially available kit for labeling DNA with modified dNTPs (i.e., Roche or ThermoFisher Nick Translation Kit).
-
19.Set-up the Nick translation reaction in a final volume of 50 μL in a tube on ice.
-
a.Vortex and centrifuge the tube for 10 s.
-
b.Incubate the tube for 90 min at 16°C.
-
a.
-
20.
Place the reaction on ice and determine the product size distribution by running 5 μL of the mix in a 1% agarose gel.
Figure 3.

Exemplary Analytical Check of Probe Size Determination by 1% Agarose Electrophoresis
As the incubation time increases the size distribution shifts to smaller probe fragments ((A) 60 min digestion; (B) 120 min digestion). Image A represents an optimal digestion experiment whereas image B is over-digested. Red squares indicate the bulk of the fragments.
Note: The structure and net charge of the different fluorophores affect the migration pattern on the gel. The unincorporated dUTP appears on the gel as brighter areas. Unincorporated Green_dUTP appears at the bottom of the run and Orange_dUTP appears around the 500 bp region overlapping with the probe DNA smear.
-
21.
If the size of the bulk of the fragments is larger than 500 bp incubate the mix for another 30 min at 16°C, if not, inactivate DNaseI by incubating the mix at 75°C for 10 min.
-
22.
Purify the DNA with a PCR purification kit (i.e., QIAGEN), and elute in 60 μL of nuclease-free water.
-
23.
Precipitate the DNA in a mix of 1/10 vol 3 M sodium acetate (pH 5.2), 2.5 vol of 100% ethanol, and 1 μg of Cot-1 DNA.
Note: Mouse or human Cot-1 DNA is a mixture of highly repetitive DNA sequences, use to block non-specific hybridization of FISH probes.
-
24.Incubate from 12-16 h at −20°C.
-
a.Centrifuge at 21,952 g (14,000 rpm) for 15 min at 4°C.
-
a.
-
25.
Wash pellet with 500 μL 70% ethanol and allow to air dry.
-
26.
Resuspend the pellet in 10 μL of nuclease free-water.
-
27.
Store at −20°C.
Note: The DNA is stable in water for long periods (up to 1–2 years) if stored at −20°C until use.
Formalin-fixed paraffin-embedded (FFPE) tissue section preparation
Note: Time dependents on the number of blocks and the number of cuts required.
In Brandt et al., FFPE of intestinal tissues obtained from MCRS1(lox/lox)Int, mTOR(lox/lox)Int and p55(lox/lox)Int mice were used (Brandt et al., 2018). Importantly, these steps describe a general protocol for FFPE mouse and human tissue preparation. Thus any FFPE tissue derived from any organism (human/mouse/etc) can be used.
-
28.
Cut FFPE tissue sections of 4–6 μm and mounted them on a poly-L-lysine-coated or positively-charged glass slide.
Note: Use gloves at all times when handling specimens. All reagents should be handled as if hazardous.
Pretreatment of FFPE tissue sections
Note: This step describes the protocol to pre-treat FFPE tissue sections. Formalin fixation introduces a macromolecular network which significantly reduces the access of FISH probes to target DNA. For that reason, the slides need to be pre-treated to diminish the extracellular matrix of proteins which potentially limit the accessibility of the probe to the cells, preventing tissue autofluorescence. Only tissues preserved in neutral-buffered formalin and paraffin-embedded are suitable for use. Other fixatives are not suitable. The use of tissue exposed to acid decalcification is not recommended. EDTA as decalcifier has been reported to preserve the DNA better for FISH techniques.
-
29.
Melt the paraffin by incubating the slides at 65°C for 12–16 h.
-
30.
Deparaffinize the tissue slides in a xylene bath and incubate for 3 min at 65°C.
-
31.Change the xylene bath and repeat once.
-
a.Tap gently on a paper towel to draw off the excess of liquid.
-
a.
-
32.
Place the slides in 100% ethanol for 3 min at 20°C–25°C.
-
33.Change the ethanol bath and repeat once.
-
a.Tap off the excess of liquid.
-
a.
-
34.Place the slides in 80% ethanol for 3 min at 20°C–25°C.
-
a.Tap off the excess of liquid.
-
a.
-
35.
Place the slides in 70% ethanol for 3 min at 20°C–25°C.
-
36.
Heat the water bath and the Pre-Treatment (10 mM citrate buffer, pH 6.0) solution at 95°C–99°C in a plastic Coplin jar.
Note: The Pre-Treatment Solution is designed for single use application only.
-
37.
Immerse the deparaffinized sections into 0.2 N HCL for 20 min at 20°C–25°C.
-
38.
Incubate the slides into the pre-heated Pre-Treatment solution for 20 min at 95°C–99°C.
-
39.
Allow the slides to cool in the Pre-Treatment Solution for 15 min at 20°C–25°C.
-
40.Wash the slides in a Coplin jar with 1× PBS and soak the slides for 3 min at 20°C–25°C.
-
a.Replace 1× PBS and soak the sections for another 3 min.
-
a.
-
41.
Dry the slides by touching gently the bottom edge and wiping the underside with a paper towel.
Alternatives: Commercially available kits include Dako’s Histology FISH Accessory Kit, or Vysis IntelliFISH Universal FFPE Tissue Pretreatment and Wash Reagents, and can be used for the pre-treatment protocol.
-
42.Digest the pre-treated tissue by covering it with 40 μL of ready to use pepsin.
-
a.Incubate samples for 30 min at 37°C.
-
a.
-
43.Place the slides in 70% ethanol for 3 min at 20°C–25°C.
-
a.Change ethanol bath and repeat once.
-
b.Tap off the excess of liquid.
-
a.
-
44.Place the slides in 80% ethanol for 3 min at 20°C–-25°C.
-
a.Tap off the excess of liquid.
-
a.
-
45.
Place the slides in 96% ethanol for 3 min at 20°C–25°C.
-
46.
Air-dry the slides from 2 to 5 min.
-
47.
Place the slide on a 45°C–50°C slide warmer to allow the remaining ethanol to evaporate.
Probe hybridization
Note: This step describes the protocol to perform the FISH assay using a ThermoBrite instrument that automates the denaturation and hybridization steps in the FISH protocol. Refer to the ThermoBrite Operations Manual for instructions on instrument use.
Alternatives: A heated plate can be used for the denaturation steps, and a humidified chamber or a plastic slide storage box containing moist tissue paper, incubated at 37°C can be used for the hybridization steps.
-
48.
Moisten humidity cards with water and place in the card slots of the ThermoBrite instrument. Ensure that the surface of the ThermoBrite instrument is clean and free of debris.
-
49.Set the following conditions:
-
a.Denaturation temperature (Melt Temp): 75°CDenaturation time (Melt Time): 3 min
-
b.Hybridization temperature (Hyb Temp): 37°CHybridization time (Hyb Time): 16 h
-
a.
-
50.Thaw labeled probe and Vysis IntelliFISH hybridization buffer (08N87-001) at 20°C–25°C.
-
a.Prepare the probe mixture with 1 μL of labeled probe, 3 μL of nuclease-free water and 7 μL of hybridization buffer.
-
b.Vortex the mix and centrifuge using a micro-centrifuge for 2–3 s at maximum speed.
-
a.
-
51.
Apply 10 μL of the probe mixture to the center of each tissue section, and immediately apply a coverslip. Ensure that no air bubbles are present in the probe mixture prior to applying the coverslip (Figure 4).
Note: The volume of the probe mixture, and the size of the coverslip can be changed according to the tissue size. An easy way to get rid of bubbles is to gently roll the tip of a pencil on the coverslip.
-
52.
Seal the coverslip with rubber cement (Figure 4).
Note: Make sure that the rubber cement straddles the edge of the coverslip.
-
53.Place the slides on the ThermoBrite instrument and begin the denaturation-hybridization program.
-
a.Hybridize the slides incubating them from 12–16 h.
-
a.
Figure 4.

Representative images of step-by-step coverslip mounting for FISH probe hybridization
From left to right, 10 μL of probe mixture is applied to the slide with the mounted tissue, immediately, an appropriated size coverslip is applied. Once no bubbles are present, the coverslip is sealed with rubber cement. Ensuring a proper sealing is key to avoid evaporation. After hybridization using the ThermoBrite denaturation-hybridization program (not shown), the rubber cement seal is carefully removed by pulling one of the corners of the seal with forceps.
Slide washes
Note: This step describes the protocol to perform the slide washes. Importantly the post-hybridization washes can significantly affect the intensity and stability of the FISH signal. When the conditions are inappropriate, for instance by using inadequate temperature or time of washing, background autofluorescence, unspecific signals or loss of the signals can appear.
Note: 30 min before you start the slide washes pre-warm the wash solution
-
54.
Pre-warm the wash solution (0.4× SSC/0.3% NP-40) in a Coplin jar at 75°C in a water bath for at least 30 min prior to use.
-
55.
Prepare a second container of 2× SSC/0.1% NP-40 at 20°C–25°C.
Note: The wash solutions are for single use application only.
-
56.
Remove the rubber cement seal with forceps by gently pulling one of the corners of the seal (Figure 4).
-
57.
Remove the coverslip by soaking the slides in a Coplin jar with 2× SSC at 20°C–25°C.
Note: If the cover slip does not come out, it can be removed by tapping gently the slide in the Coplin jar. If the coverslip is still stuck to the slide, slide the blade of a scalpel under one corner of the slide and lift it gently before immersing the slide into the Coplin jar. This may need to be repeated several times if the coverslip remains stuck.
-
58.Immediately place the slide into the 0.4× SSC/0.3% NP-40 wash solution at 73 ± 1°C.
-
a.Agitate the slide for 5 s.
-
b.Repeat with each slide.
-
a.
Note: Do not wash more than four slides in each Coplin jar.
-
59.
Wash the slide/s for 2 min into the 0.4× SSC/0.3% NP-40 wash solution at 73 ± 1°C.
-
60.Place the slide/s in the 2× SSC/0.1% NP-40 wash solution at 20°C–25°C.
-
a.Agitate the slide for 5 s and let them stand for 5 min.
-
a.
-
61.
Air dry slide in the dark.
-
62.
Apply to every slide 10 μL of counterstain dye (0.5 vol 4′,6-diamidino-2-phenylindole DAPI II/0.5 vol antifade) and mount the slides with a 25×50 mm coverslip.
Selecting and enumerating cells for FISH analysis
Note: This step describes how to select and enumerate cells within the selected target area, for fish quantification and analysis.
-
63.Analyze the FISH signals at 1000× magnification using the appropriate filters.
-
a.Select a representative area of the tissue with of good nuclear distribution (i.e., where individual nuclei can be distinguished (Figure 5).
-
b.Using a 100× objective and prescribed filters enumerate and record the number of signals within each nucleus. Focus up and down to find all of the signal’s present in the nucleus.
-
c.Move to another representative area for enumeration until 200–300 tumor cells have been enumerated.
-
a.
Figure 5.

Nuclei recognition by FISH: Selecting and enumerating cells within the selected target area
(A–C) Exemplary of three representative areas of tissue with a good nuclear distribution. Individual nuclei can be distinguished and show a strong FISH signal together with weak or no fluorescence background. Only the tumor nuclei with intact nuclear membrane, that do not overlap are used for the analysis. Images were captured using a Leica DM5500B fluorescence microscope with a 100× objective running the Cytovision v7.4 software. (A–C) White dashed line highlights single nuclei that meet the described criteria; scale bars, 10 μm.
We performed FISH imaging on a Leica DM 5500B fluorescence microscope equipped with a 100× oil-immersion objective, Leica DM DAPI, Aqua, Green and Orange fluorescence filter cubes and a CCD camera (Photometrics SenSys camera) connected to a PC running the Zytovision image analysis system (Applied Imaging Ltd., UK) with focus motor and Z stack software.
Note: The background should not contain particles that interfere with enumeration. In some cases, fluorescent haze or glow may be noticeable outside of the nuclei, but it is acceptable as long as the fluorescent haze/glow does not cover the nuclei and interfere with the enumeration.
Note: Probe signal intensity: Analyze signal intensity on single color filters. The signals should be bright, distinct, and easily evaluable. Signals should be compact, round or oval shapes. Rubbish generally appears to be brighter and shinier compared to real signals, and background appear fuzzy and indistinct compared to real signal. Overly diffuse signals should be avoided.
Note: FISH signal analysis can be done manually or automatized by fluorescence image acquisition scanning systems. For manual analysis two experienced scientists or technicians should count the signals of each individual nucleus from 200–300 cells directly on the microscope using the appropriate filters. As an alternative, if the scientists or technicians have not enough expertise in microscopy analysis, then representative images can be captured to complete 200–300 cells. In this case, FISH signals can be counted based on the captured images.
CIN analysis
Note: average time for an experienced observer to visualize one FISH probe slide.
The CIN score (CS) is a metric devised to quantify the alterations (copy number changes) detected in a sample, that would lead to chromosomal heterogeneity and could drive different cancer patterns from initiation, progression, evolution and drug resistance patterns. Depending on the size of the genomic alteration it can be classified as: structural CIN if it involves translocations, inversions, deletions and amplifications that range from genes to complete chromosome arms (Geigl et al., 2008); or as numerical CIN if it involves whole chromosomes gain or losses.
In order to quantify these alterations, there are many available techniques, with their own pros and cons (Lepage et al., 2019). Based in our research paper (Brandt et al., 2018), we have detailed here how to measure the CS by FISH.
To perform manual FISH analysis, visualize, count and annotate every FISH signal of each nucleus under the microscope using its specific filter. Computes a summary index of the FISH signals based on all examined cells. Alternative FISH analysis can be done counting signals on captured images. Different capture platforms are available; some examples are Cytovision (Leica) or Metafer (MetaSystems). This step describes the protocol to perform the FISH analysis on captured images.
-
64.
Take representative images from several tissue areas that account for at least 200–300 nuclei signals. Select only intact nuclei that are not folded, overlapped or obstructed by debris (as described in the FISH protocol section) (Figure 6).
-
65.
Evaluate and annotate the total number of signals of each nucleus for any of the probes designed before to start with the following one to determine the final 200–300 nuclei count.
Note: CS is used to describe the aneuploidies within a given sample. When calculated for a single chromosome is known as CS, and for several stained chromosomes as CSC (CIN score combined).
-
66.
Calculate the value for each chromosome probe [e.g., CS for chromosomes 1, 2, or 3 (CS1, CS2, or CS3, respectively)] of a given sample, applying the following formula:
Where:
Figure 6.

Pepsin treatment calibration
Representative pictures were serial sections of FFPE tissues were incubated at different time points with ready to use pepsin to test the correct time for digestion. Incubations were performed at (A) 30 min, (B) 45 min, (C) 60 min and (D) 120 min (only Dapi channel are shown) before probe hybridization. The total time of wash buffer incubation and hybridization was equal for all conditions. Images were captured using a Leica DM5500B fluorescence microscope with a 100× objective running the Cytovision v7.4 software. (A–D) scale bars, 10 μm.
accounts for the number of nuclei analyzed.
: are the observed signals for a specific chromosome ().
: are the value that corresponds to the expected signals for the given chromosome (). The ep value is usually equal to 2, that is the number of signals for a wild type cell (2n).
-
67.
Sum each of the individual values to obtain the corresponding for a given sample, as follows:
Note: Alternatively, an overall mean can be calculated for each sample by summing the CSC for each nucleus analyzed withing a sample and dividing by the total number of nuclei evaluated:
Note: As a reference, indicates a wild type nucleus has been analyzed. By the contrary, a value higher than 0 indicates gains/losses of chromosomes and hence, a specific degree of chromosomal instability. The higher the value, the greater the instability.
Expected outcomes
As described, the CS is a metric devised to quantify chromosomal alterations in the samples through the use of two probes hybridized on each nucleus analyzed. We assessed CS aneuploidies of chromosomes 11 and 2, and described that CIN may be a driver for inflammation-induced tumorigenesis (Brandt et al., 2018). Figures 7 illustrates FISH probe hybridization of chromosomes to visualize frequently occuring chromosomal abnormalities and its corresponding FISH signaling pattern. Figure 8 illustrates probe hybridization of chromosomes 11 and 2 used in (Brandt et al., 2018), and frequent scenarios during CIN quantification.
Figure 7.

Schematic representation of chromosomal abnormalities visualized after FISH assay
(A) Schematic representation at genomic level of a control diploid cell containing a pair of chromosomes, and its corresponding normal FISH signal pattern is illustrated in the second column (Top).
(B) (Top) Schematic representation of the amplification of a specific targeted locus. Gene amplification can be observed both at genomic level and through FISH signaling pattern. (Bottom) Schematic representation of locus deletion, upon chromosome breaks. Gene deletion can be observed both at genomic level and through FISH signaling pattern.
(C) Polyploidy where the probe signal shows multiple sets of chromosomes, observed by three pair of fused genes.
Figure 8.

Probe hybridization and FISH signaling pattern
(A) Schematic representation at genomic level of probe hybridization for the detection of chromosomal aberrations in chromosomes 2 and 11.
(B) Schematic representation of fluorescent probe visualization in interphase nucleus after FISH assay, where the green signal corresponds to chromosome 2 and the red signal corresponds to 11. From top to bottom, the first nuclei represent a control diploid cell with two copies of each chromosome (two red signals, and two green signals). The second nuclei represent an abnormal signaling pattern where chromosome aneuploidy can be visualized (two green signals and four red signals).
(C) Schematic representation at genomic level of probes hybridization for the detection of chromosomal aberrations, in chromosomes 2, 9 and 19.
(D) Schematic representation of fluorescent probe visualization in interphase nucleus after FISH assay, where the yellow signal corresponds to chromosome 2, the red signal corresponds to chromosome 9, and the green signal corresponds to chromosome 19. From top to bottom, the first nuclei represent a control diploid cell with two copies of each chromosome (two yellow signals, two red signals, and two green signals). The second nuclei represent an abnormal signaling pattern where chromosome aneuploidy can be visualized (four yellow signals, three red signals two green signals and).
(E) Schematic representation at genomic level of probes hybridization for the detection of chromosomal aberrations, in chromosomes 17 and 11.
(F) Schematic representation of fluorescent probe visualization in interphase nucleus after FISH assay, where the red signal corresponds to a specific locus of chromosome 17 and the green signal corresponds to a specific locus of chromosome 11. From top to bottom, the first nuclei represent a control diploid cell with two copies of each chromosome locus (two red signals, and two green signals). The second nuclei represent an abnormal signaling pattern where the deletion of the analyzed locus can be visualized (two green signals and four red signals). (B, D, and F) Calculations for CSc score are represented in blue squares (normal signal pattern) and red squares (abnormal signal pattern), for each represented example.
Limitations
CIN refers to an ongoing rate of change that drives cell-to-cell heterogeneity rather than a static state such as aneuploidy. For that reason, it is important to assess CIN at the single cell level using techniques that are capable of capturing the full spectrum of heterogeneity and CIN existing within a given sample. In this sense, FISH allows the quantitative assessment of cell-to-cell heterogeneity within a given population analyzing karyotypic heterogeneity. Although, FISH analysis enables the labeling of specific genes, regions, or whole chromosomes, is amenable to automation and multiplexing, and can be applied to fixed samples. Its main limitation is that it only detects events involving the labeled locus/chromosomes; multiplexing depends on the availability of spectrally distinct fluorophores. Metaphase G-banding analysis may be helpful in characterizing abnormal chromosomal patterns, however only a low number of cells can be analyzed using this approach. As an alternative, single-cell high throughput sequencing could also be useful, but this approach considerably increases the cost of analysis.
Troubleshooting
Problem 1
Low DNA yield
Step: BAC DNA isolation
Potential solutions
Poor aeration of culture: The optimal culture volume to air volume ratio is 1:4. For best aeration vented or gas-permeable seal culture flasks can be used. (steps 1–5)
Incomplete neutralization: Cell debris will float to the surface. Ensure that neutralization is complete prior to centrifugation. Invert the tube an additional 2–3 times. (step 7)
Incomplete elution: Pre-warming water to 50°C prior to elution can increase the DNA yield. (step 16)
Problem 2
Low DNA quality
Step: FISH preparation: BAC DNA isolation
Potential solutions
Incomplete neutralization generates poor quality of supernatant, and results in cell debris contamination during the following steps of the protocol. Make sure the neutralization is complete prior to centrifugation. Additionally, invert the tube 2–3 times. (step 7)
RNA contamination: Ensure that RNase A has enough time to degrade RNA by allowing lysate to incubate at 20°C–25°C an additional 3–5 min (step 13)
Genomic DNA contamination: Improper handling. Genomic DNA contamination is usually caused by excessive mechanical shearing during the lysis and neutralization steps. Prolonged lysis or incomplete mixing of neutralization buffer may affect genomic DNA contamination. (steps 6 and 7)
Problem 3
Low FISH probe yield
Step: FISH preparation: BAC DNA isolation (steps 20–22 )
Potential solutions
Ensure that the length of the probe is adequate. Short fragment sizes, smaller than 200–500 bp gives low yield. Check if enough template (minimum 1 μg) was used for labeling.
DNAse I might be inhibited by contaminants such as amines. Care should be taken to ensure that all impurities which might interfere in the labeling reaction have been removed from the DNA. Phenol-chloroform DNA purification can enhance DNA purity.
Problem 4
FISH: Weak or no FISH signal
Step: Stated at each potential solution
Potential solutions
Ensure you obtained an adequate FISH probe yield, efficient dye incorporation and the expected probe length. You might need to optimize quality of the template, amount of the starting material, reaction temperatures and time. (step 17)
Ensure that the pre-treatment steps were made correctly; that the temperature of the pre-treatment solutions is optimal; and make sure to replace the pre-treatment solution after every batch (Figure 5). (steps 35–39)
Ensure the correct time for pepsin digestion. Importantly, the user may need to optimize their own times depending on the extent of fixation, age of archived tissue or the type of tissue to be digested. Use a start digestion time of 15 min, however increasing to 30 or 45 min incubation times can improve signal intensity or autofluorescence background in some samples (Figure 6). (step 42)
Ensure the temperature of denaturation. The denaturation temperature is set at 75°C; however, increasing it to 80°C can improve the signal strength. Time of denaturation can also be adjusted. (step 50)
Use 10 to 15 μl of probe solution and increase the volume when using larger sections. Ensure a good seal with rubber glue to prevent evaporation. Ensure that no air bubbles were trapped under the coverslip before hybridization. (steps 52 and 53)
Temperature and pH of post-hybridization washes also influence in the intensity of the FISH signals; increasing the temperature increases the stringency, and the pH determines the availability of the positive ions that counteract the repulsive negative force between the DNA backbones of both the probe and target. Ensure that the wash solutions was prepared correctly and had the proper pH and temperature. (step 55)
Prolonged storage of FISH probes can lead to substantial probe degradation. Probes must be stored at −20°C for no longer than a year.
Problem 5
FISH: High slide background
Step: Pretreatment of formalin-fixed paraffin-embedded (FFPE) tissue section
Potential solution
Ensure that the wash solutions was prepared correctly, having the proper pH and temperature. Remove the coverslip before immersing the slides in the wash solution. (steps 55–58)
High background may be due to insufficient removal of material during the pre-treatment steps. The extent of fixation, the age of archived tissue or the type of tissue can affect the results of FISH hybridization. Adjustment of digestion time can improve autofluorescence background in some samples (Figure 6). (Step 42)
High background may be due incorrect sealing off the slides with rubber cement during the pre-treatment and co-denaturation steps, since this allows the solution to evaporate and the tissue to dry out. Placing the slides in the incubator to allow the rubber cement to dry before these steps can reduce this effect. (steps 52 and 53)
Problem 6
FISH: Degraded tissue
Step: Pretreatment of formalin-fixed paraffin-embedded (FFPE) tissue section
Potential solutions
Pre-treatment steps can render the tissue softer, and it may easily fall off or get degraded. The size of the tissue gives a good indication as to the fragility of the tissue, so this should be taken into account to decide the incubation time of the pre-treatment solution. Small tissues should be incubated for 10 min (step 38–40)
Reducing pepsin enzyme treatment on the sample may also avoid this problem. (step 42)
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Nabil Djouder (Correspondence: ndjouder@cnio.es).
Further technical information should be directed to and will be fulfilled by the technical contact, Dra. Sandra Rodriguez-Perales (Correspondence: srodriguezp@cnio.es).
Materials availability
Plasmids generated in this study and materials are available upon request from the lead or technical contact; the sharing of materials described in this work could be subject to standard Material Transfer Agreements.
Data and code availability
Datasets and code generated or analyzed in this study can be found in the key resources table.
Acknowledgments
This work was funded by grants to N.D. from the State Research Agency (AEI, 10.13039/501100011033) from the Spanish Ministry of Science and Innovation (projects SAF2016-76598-R, SAF2017-92733-EXP, RTI2018-094834-B-I00, and RED2018-102723-T), cofounded by European Regional Development Fund (ERDF). S.R-P. is supported by grants from the Spanish National Research and Development Plan, Instituto de Salud Carlos III, and Spanish Ministry of Science and Innovation cofounded by ERDF (PI17/02303; DTS19/00111 and BIO2017-91272-EXP) and by the Asociación Española Contra el Cáncer (AECC). R.T.-R. was supported by a postdoctoral fellowship from the AECC. This work was developed at the CNIO funded by the Health Institute Carlos III (ISCIII) and the Spanish Ministry of Science and Innovation.
Author contributions
R.T.-R. and T.P.G. wrote the protocol with M.B. and M.M.-L.; T.P.G. made the figures and graphical abstract; R.T-R, T.P.G, S.R.-P. and N.D reviewed, edited and corrected the protocol; S.R.-P. and N.D. supervised the study, and secured funding. N.D. designed the study and was in charge of the project administration.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Raul Torres-Ruiz, Email: rtorresr@cnio.es.
Sandra Rodriguez-Perales, Email: srodriguezp@cnio.es.
Nabil Djouder, Email: ndjouder@cnio.es.
References
- Brandt M., Grazioso T.P., Fawal M.A., Tummala K.S., Torres-Ruiz R., Rodriguez-Perales S., Perna C., Djouder N. mTORC1 inactivation promotes colitis-induced colorectal cancer but protects from APC loss-dependent tumorigenesis. Cell Metab. 2018;27:118–135.e118. doi: 10.1016/j.cmet.2017.11.006. [DOI] [PubMed] [Google Scholar]
- el Marjou F., Janssen K.P., Chang B.H., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Fawal M.A., Brandt M., Djouder N. MCRS1 binds and couples rheb to amino acid-dependent mTORC1 activation. Dev. Cell. 2015;33:67–82. doi: 10.1016/j.devcel.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Geigl J.B., Obenauf A.C., Schwarzbraun T., Speicher M.R. Defining 'chromosomal instability. Trends Genet. 2008;24:64–69. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Lepage C.C., Morden C.R., Palmer M.C.L., Nachtigal M.W., McManus K.J. detecting chromosome instability in cancer: approaches to resolve cell-to-cell heterogeneity. Cancers. 2019;11:226. doi: 10.3390/cancers11020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S., Vooijs M., van Der Gulden H., Jonkers J., Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. doi: 10.1101/gad.14.8.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risson V., Mazelin L., Roceri M., Sanchez H., Moncollin V., Corneloup C., Richard-Bulteau H., Vignaud A., Baas D., Defour A. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Datasets and code generated or analyzed in this study can be found in the key resources table.