

Graphical abstract
Keywords: SNP, Drug response, Breast cancer, Prognosis
Abstract
Detecting SNPs associated with drug efficacy or toxicity is helpful to facilitate personalized medicine. Previous studies usually find SNPs associated with clinical outcome only in patients received a specific treatment. However, without information from patients without drug treatment, it is possible that the detected SNPs are associated with patients’ clinical outcome even without drug treatment. Here we aimed to detect drug response SNPs based on data from patients with and without drug treatment through combing the cox proportional-hazards model and pairwise Kaplan-Meier survival analysis. A pipeline named Detection of Drug Response SNPs (DDRS) was built and applied to TCGA breast cancer data including 363 patients with doxorubicin treatment and 321 patients without any drug treatment. We identified 548 doxorubicin associated SNPs. Drug response score derived from these SNPs were associated with drug-resistant level (indicated by IC50) of breast cancer cell lines. Enrichment analyses showed that these SNPs were enriched in active epigenetic regulation markers (e.g., H3K27ac). Compared with random genes, the cis-eQTL genes of these SNPs had a shorter protein–protein interaction distance to doxorubicin associated genes. In addition, linear discriminant analysis showed that the eQTL gene expression levels could be used to predict clinical outcome for patients with doxorubicin treatment (AUC = 0.738). Specifically, we identified rs2817101 as a drug response SNP for doxorubicin treatment. Higher expression level of its cis-eQTL gene GSTA1 is associated with poorer survival. This approach can also be applied to identify new drug associated SNPs in other cancers.
1. Introduction
Cancer is a group of diseases involving uncontrolled growth and spread of abnormal cells. Cancer incidence and mortality are rapidly growing. Much work is ongoing to achieve better treatment of this group of diseases. However, as an extremely heterogeneous condition, it is estimated that any particular class of drugs is ineffective in about 75% of cancer patients [1]. Therefore, how to realize personalized medicine (i.e., selecting optional therapy according to patients’ personal profile) remains a key challenge.
It is reported that genetics account for 20%–95% of variability in drug disposition and effects [2]. Single nucleotide polymorphisms (SNPs) account for about 80% of the overall genomic heterogeneity [3]. Moreover, a number of pharmacogenomics studies have demonstrated that SNPs could influence the efficacy and side effects of drugs [4]. For anti-cancer drugs (e.g., irinotecan, mercaptopurine, 5-flurouracil, and tamoxifen), several SNPs have been reported to be associated with drug efficacy or toxicity [5], [6], [7]. These early studies generally focused on SNPs within pre-specified genes of interest, which might miss other potentially significant polymorphisms. Recently, with the advances in high throughput technologies, genome-wide association study (GWAS) provides a hypothesis-free approach to identify novel SNPs that are responsible for drug response. For example, Khan et al. [8] reported that the common SNP rs8113308 mapped to 19q13.41 was associated with reduced survival among endocrine treated breast cancer patients. Cairns et al. [9] identified a SNP in CSMD1 associated with breast cancer–free interval in a phase III randomized trial of anastrozole versus exemestane. Generally, these studies usually tested the association between SNPs and drug responses (e.g., recurrence-free survival) only in patients received a specific treatment. However, without information from patients without drug treatment, this design cannot discriminate drug response SNPs from clinical outcome associated SNPs. That’s, it is possible that the detected SNPs are associated with patients’ clinical outcome even without drug treatment.
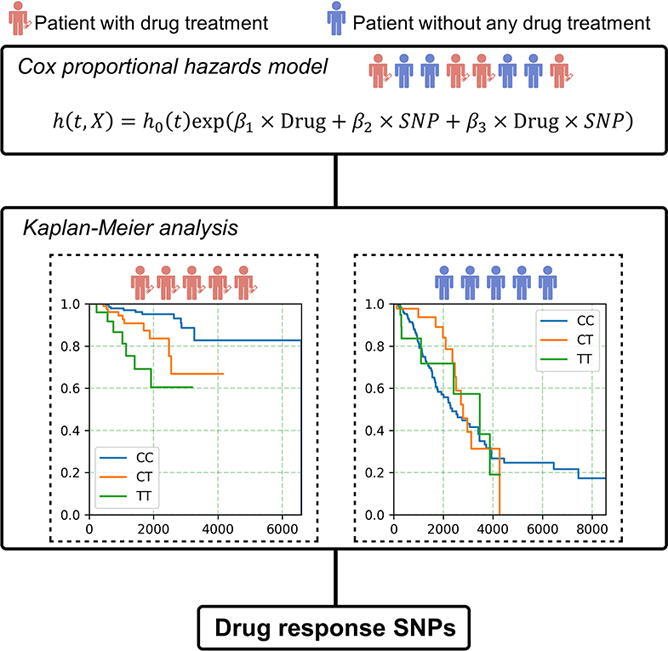
In this study, we aimed to find drug response SNPs based on data from patients with and without drug treatment. A pipeline named Detection of Drug Response SNPs (DDRS) was built through combing the cox proportional-hazards model and pairwise Kaplan-Meier survival analysis. Briefly, first, we fit a Cox’s proportional hazards model including three variates, including SNP, drug treatment status, and an interaction term between SNP and drug treatment in data including patients with and without drug treatment. SNPs with significant drug treatment and interaction term were remained. Second, Kaplan-Meier (KM) survival analysis were performed in patients with different genotypes to further generate the final set of SNPs. SNPs with significant KM results in patients without drug treatment were removed. To test the performance of DDRS, data of breast cancer patients from The Cancer Genome Project (TCGA) project were analyzed.
2. Methods
2.1. Pipeline of DDRS
The outline of DDRS is shown in Fig. 1. The input data of the study population included the genotype and patient’s survival information. Specifically, patients without drug treatment were also included. We used the patients’ survival status to indicate drug response since overall survival is believed as the primary end point to evaluate the outcome of any drug [10]. Overall survival has also been used as the endpoint to find drug response and toxicity loci in previous studies [11], [12]. First, a Cox’s proportional hazards model was built to detect significant SNP-drug interactions (P < 0.05 for ).
| (1) |
Fig. 1.

An overview of the approach. Step 1: We performed SNP × drug interaction analysis in all patients (including patients with drug treatment and patients without any drug treatment). SNPs with significant drug term (P < 0.05 for) and significant interaction term (P < 0.05 for) were remained. Step 2: For SNPs obtained from the first step, we performed Kaplan-Meier (KM) analysis in subjects with different genotypes to select SNPs associated with drug response in patients with drug treatment. KM analysis in patients without any drug treatment was also performed to remove the SNPs associated with survival but this association was not related to drug treatment. Step 3: We performed univariate cox proportional hazards analysis for each drug response SNPs get from the first two steps. The coefficients for all SNPs was used to calculate drug response score (DRS) in another population to test the performance of drug response prediction.
We only focused on drugs with significant effect on survival (P < 0.05 for) and significant interaction terms (P < 0.05 for ). Second, we performed KM survival analysis in patients with and without drug treatment separately to further select the SNPs related to drug response. For KM analysis, only subgroups with more than 20 patients (at least 5 patients with dead events) were remained for analysis. SNPs with FDR-adjusted P < 0.05 in the KM analysis of patients with drug treatment were selected. In addition, SNPs with P < 0.05 in the KM analysis of patients without drug treatment were removed to rule out the associations unrelated to drug treatment. Finally, to test the generalization ability of the selected SNP sets in other populations, we calculated the correlation between each SNP and survival in one population:
| (2) |
and constructed a drug response score (DRS) for each individual in another dataset as follows:
| (3) |
where represents the coefficient of SNPi in Eq. (2) and represents the tested allele’s copies of SNPi in Eq. (2). The correlation between S and drug response were then calculated to test whether the selected SNP sets could be used for survival prediction.
2.2. Genotype and clinical data collection processing
Clinical information, drug treatment information, germline DNA genotypes for TCGA breast cancer samples (N = 1096) were obtained from the Genomic Data Commons Data Portal (https://cancergenome.nih.gov/, GDC portal). The genotyping platform for all patients was the Affymetrix 6.0 array. Genotypes with score < 0.1 are considered to be highly confident (Broad institute, BIRDSUITE software) and 928,706 SNPs were retained in the study.
2.3. DRS and drug-resistant levels in cell lines
We constructed DRSs for 45 cancer cell lines (Table S1) to test whether DRS can be used to predict drug response. Gene expression and drug response data for these cell lines were obtained from the Genomics of Drug Sensitivity in Cancer (GDSC) database [13]. We used IC50 (half maximal inhibitory concentration) for cell apoptosis to indicate the drug-resistant level. Corresponding genotype data for all cell lines were obtained from the Gene Expression Omnibus (GEO) database (GSE34211 and GSE41308). We used R package crlmm [14] for genotype calling.
2.4. Functional annotation of the selected SNPs
We annotated the epigenetic regulatory features for SNPs using data from the following resources: 1) we downloaded the 8 types of ChIP-seq data from the GEO database (GSE85158 [15]) for 11 breast cancer cells. Sequencing reads were mapped to the human genome reference (hg19) using Bowtie2 [16] with default settings. MACS2 [17] was used to call peaks (-g hs -q 0.05 -n --keep-dup all). 2) we downloaded the predicted enhancer region for BRCA from CistromeCancer [18]. The genome coordinates were converted from hg38 to hg19 using liftover [19]. Next, we performed 10,000 permutation tests by using random SNP sets generated by SNPsnap [20] with default settings. To calculate the enrichment p-value, we simply count the number of sets with annotated SNPs as or more extreme than our selected SNP set, and divide that number by the total number (10,000). The enrichment score (ES) was calculated as follows:
where is the number of annotated SNPs from our selected SNP set, and is the number of annotated SNPs in the ith random SNP set.
2.5. eQTL analysis
Gene expression data normalized using RSEM [21] and segmented copy number variation data were downloaded from Genome Data Analysis Center database (GDAC, http://gdac.broadinstitute.org/). Absolute gene copy numbers were calculated from segmented copy number files by ABSOLUTE [22], and then used as covariate to adjust the gene expression data [23]. To remove the effect of population structure on gene expression, we used smartpca in the EIGENSOFT [24] program to perform principal component (PC) analysis, and selected the top 10 PCs from genome-wide genotype data as covariates. To remove the hidden batch effects and other potential confounders in the gene expression data, we also used the Probabilistic Estimation of Expression Residuals [25] (PEER) method to select the first 15 PEER factors as covariates. Age, pathologic stage and race were also used as covariates. EQTL analysis was performed using matrix eQTL [26]. SNPs with false discovery rates (FDR) < 0.05 were defined as eQTL genes. Cis-eQTL genes were defined if the SNP was located within 1 Mb from the gene transcriptional start site (TSS).
2.6. Extraction of drug target genes and protein–protein interactions (PPIs)
We collected doxorubicin targets genes from the DrugBank database [27] and Therapeutic Target Database [28]. We also collected doxorubicin related enzymes, carriers and transporters from DrugBank. Genes directly interacted with doxorubicin (confidence score over 0.7) were collected from STITCH database [29]. To get PPIs, we downloaded the human interactome published by Cheng et al. [30], which collected 243,603 PPIs of 16,677 unique proteins. Next, we performed 10,000 permutation tests by randomly chose same number of genes from 16,677 proteins. To calculate the permutation p-value, we simply compared the mean shortest PPI distances from eQTL genes to doxorubicin and from randomly chosen genes to doxorubicin. We count the number of randomly chosen gene sets with shorter mean PPI distances than our eQTL gene set, and divide that number by the total permutation number (10,000) [30].
2.7. Doxorubicin resistance prediction model
We constructed multigene classifiers using these eQTL genes and Linear Discriminant Analysis (LDA) in GSE20194, which has 230 patients received 6 months of preoperative chemotherapy including doxorubicin. 182 patients were categorized as residual invasive cancer (RD) and 48 patients were categorized as pathological complete response (pCR, no residual invasive cancer in the breast or lymph nodes). We performed 1000 times repeated 1-fold cross-validation. The classifier performance on the validation data were assessed by using the area under the receiver operating characteristic curve (ROC-AUC).
2.8. Pathway enrichment and pathway activity inference
The eQTL genes were ranked according to the product of the β from eQTL analysis and the β from Cox analysis (i.e., βcox * βeQTL). For genes with more than one significant SNPs, the results with the maximum absolute value was remained. Pathway enrichment analysis for these genes with pre-ranked values was then performed using GSEA [31]. Positive pathways enriched at P < 0.05 were recognized as doxorubicin-resistant/risk pathways and negative pathways enriched at P < 0.05 were recognized as doxorubicin-sensitive/protective pathways. The pathways’ activity score (PAS) were calculated with diffrank [32].
3. Result
3.1. Identification of doxorubicin response SNPs in breast cancer
We applied this approach to TCGA breast cancer data to detect doxorubicin response SNPs. 363 patients with doxorubicin treatment and 321 patients without any drug treatment was included. Detailed characteristics of the patients are provided in Table 1. Using the cox’s proportional hazards model, we identified 8020 SNPs that might be interacting with doxorubicin treatment. Next, KM analysis in patients with doxorubicin treatment showed that subjects with different genotypes of 619 SNPs had significant different survival status (FDR P < 0.05). In addition, using KM analysis in patients without any drug treatment, we further ruled out the SNP-survival associations not related to drug treatment and 71 SNPs were removed. Therefore, after analyzing with the DDRS pipeline, a total of 548 doxorubicin associated SNPs were identified with the detailed information summarized in Supplementary Table S2.
Table 1.
Pre-treatment characteristics of the patients.
| Patient with doxorubicin treatment | Patients without any drug treatment | All patients | |
|---|---|---|---|
| Number | 363 | 321 | 684 |
| Age | |||
| <=50 | 173 (48%) | 71 (22%) | 244 (36%) |
| >50 | 189 (52%) | 240 (77%) | 429 (64%) |
| Mean (SD) | 52 (10) | 62 (14) | 56 (13) |
| Unknown | 1 | 10 | 11 |
| Nodal status | |||
| Positive | 238 (66%) | 152 (48%) | 390 (57%) |
| Negative | 122 (33%) | 158 (49%) | 280 (41%) |
| Unknown | 3 (1%) | 11 (3%) | 14 (2%) |
| T stage | |||
| 1 | 73 (21%) | 79 (25%) | 152 (22%) |
| 2 | 233 (64%) | 177 (55%) | 410 (60%) |
| 3 | 52 (14%) | 41 (13%) | 93 (14%) |
| 4 | 5 (1%) | 22 (7%) | 27 (4%) |
| Unknown | 0 (0%) | 2 (0%) | 2 (0%) |
| Pathologic stage | |||
| 1 | 36 (10%) | 59 (18%) | 95 (14%) |
| 2 | 213 (59%) | 174 (54%) | 387 (57%) |
| 3 | 108 (30%) | 67 (21%) | 175 (26%) |
| 4 | 2 (0%) | 9 (3%) | 11 (1%) |
| Unknown | 4 (1%) | 12 (4%) | 16 (2%) |
| ER Status | |||
| Positive | 244 (67%) | 225 (70%) | 469 (69%) |
| Negative | 106 (29%) | 79 (25%) | 185 (27%) |
| Unknown | 13 (4%) | 17 (5%) | 30 (4%) |
| PR Status | |||
| Positive | 209 (58%) | 188 (59%) | 397 (58%) |
| Negative | 139 (38%) | 115 (36%) | 254 (37%) |
| Unknown | 15 (4%) | 18 (5%) | 33 (5%) |
| HER2 Status | |||
| Positive | 45 (12%) | 61 (19%) | 106 (15%) |
| Negative | 207 (57%) | 144 (45%) | 351 (51%) |
| Unknown | 111 (31%) | 116 (36%) | 227 (34%) |
3.2. DRS derived from selected SNP are associated with drug resistant level
We calculated the DRS in the rest 269 TCGA breast cancer patients with other drug treatment. As shown in Fig. 2A, compared with other patients, the basal like patients had the highest DRS. We also calculated doxorubicin-associated DRS in breast cancer cell lines (Fig. 2B). These cells were divided into doxorubicin-resistant group (IC50 higher than median value) and doxorubicin-sensitive group (IC50 lower than median value). The DRS in the doxorubicin-resistant group was significantly higher than the doxorubicin-sensitive group (P = 0.044).
Fig. 2.

A: Boxplot of DRS of patients in different molecular subtypes. Patients were stratified into groups of Luminal A, Luminal B, HER2 and Basal like by the ER, PR and HER2 markers. B: Boxplot of DRS between doxorubicin-resistant cells (High IC50) and doxorubicin-sensitive (Low IC50) cells. C: Enrichment analysis heatmap plot of those significant SNPs’ epigenetic annotation. Enrichment score (ES) are color-coded from light to dark.
3.3. The selected SNPs are enriched in active regulatory epigenetic markers
According to the genomic region annotation results, over 94% SNPs are located in the intergenic or intronic region. We further examined whether these doxorubicin-associated SNPs were associated with active epigenetic regulation using ChIP-seq data from 11 breast cancer cell lines (Table S1, Fig. 2C). The results showed that 34% SNPs are located in active epigenetic mark regions of at least one breast cancer cell lines. Further analysis showed that these SNPs were significantly enriched in histone epigenetic regions associated with active enhancer (H3K27ac [33], H3K4me1 [34] and H4K8ac [35]), active gene transcription (H3K36me3 [36], H3K9ac [33] and H2BK120ub1 [37]) (Fig. 2C) and also in predicted BRCA enhancer regions (P = 0.0008, 10,000 permutations; 1.46-fold).
3.4. The eQTL gene expression levels could be used to predict clinical outcome
We identified 958 cis-eQTL genes for these 548 doxorubicin associated SNPs. Compared with random selected genes, these genes had significantly shorter mean PPI distances to doxorubicin target genes, doxorubicin related enzymes/carriers/transporters, and doxorubicin-interacting genes (Fig. 3A, P < 0.001). Next, we estimated these genes’ doxorubicin-response predictive power. We used all the genes’ expression as features to train a LDA classifier to distinguish patients from RD and pCR. The ROC-AUC was 0.738 (CI: 0.736–0.741) (Fig. 3B).
Fig. 3.

A: Violin plot of mean shortest PPI distances to doxorubicin target. Red bars represent the mean PPI distance of the cis-eQTL genes to doxorubicin target genes, doxorubicin related enzymes, doxorubicin related enzymes/carriers/transporters, and doxorubicin-interacting genes. Blue bars represent the mean shortest PPI distance of 10,000 groups of randomly selected genes. B: ROC curve for the predictive performance of the LDA genomic pCR predictor with these eQTL genes as features. C: Volcano plot of univariate cox proportional hazards results of eQTL genes. Horizontal axis showed the univariate cox proportional hazards confidences and vertical axis showed the negative log of the P values. Doxorubicin-resistant genes are shown on upper and doxorubicin-sensitive genes are shown on below. D: Volcano plot of univariate cox proportional hazards results of PAS of enriched pathways. Horizontal axis shows the univariate cox proportional hazards confidences and vertical axis showed the negative log of the P values. Positive pathways are shown on upper and negative pathways are shown on below. E: Univariate cox proportional hazards confidences of 6 positive pathways had significant risk PAS and 7 negative pathways had significant protective PAS. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.5. The direction of the association between gene expression and clinical outcome is consistent with the product of βcox and βeQTL of SNPs
We further used the product of βcox and βeQTL to classify these genes into two categories: doxorubicin-resistant/risk (product with positive sign) and doxorubicin-sensitive/protective (product with negative sign). 25 genes with different signs when referring to different SNPs were excluded from subsequent analysis. Using data from GSE25055, we performed univariate cox proportional hazards analysis to test the association between these genes and patients’ survival. As shown in Fig. 3C, the directions of cox analysis results were mostly consistent with our definition of doxorubicin-resistant/sensitive genes (Chi-square test: P = 1.93 × 10−10).
We performed GSEA pre-ranked pathway enrichment of these eQTL genes and identified 11 positive pathways and 52 negative pathways. Similar to single gene analysis, we defined the positive pathways as doxorubicin-resistant/risk and negative pathways as doxorubicin-sensitive/protective. We calculated the PAS for these pathways and performed univariate cox proportional hazards analysis using data from GSE25055. As shown in Fig. 3D, the directions of cox analysis results were also mostly consistent with our definition of doxorubicin-resistant/risk and sensitive/protective pathways (Chi-square test: P = 0.039). Specifically, we illustrate the pathways with the same direction in both enrichment and PAS cox analyses (Fig. 3E). For example, two immune related pathways, including ‘regulation of immune effector process (GO: 0002697)’ and ‘Leukocyte cell adhesion (GO: 0007159)’, were doxorubicin-resistant/risk pathways whose PAS were significant risk factors to patients’ survival. The pathway ‘response to steroid hormone (GO: 0048545)’ was a doxorubicin-sensitive/protective pathway whose PAS was significant protective factors to patients’ survival.
3.6. High expression of GSTA1 is a risk factor to doxorubicin treatment
We identified rs2817101 and its cis-eQTL gene GSTA1, which had the highest product of βcox and βeQTL. Cox proportional hazards analysis showed that both doxorubicin treatment and the interaction term (rs2817101 × doxorubicin) were significant risk factors to breast cancer patients (Fig. 4A). Patients received doxorubicin treatment with TT allele of rs2817101 suffered from poorer prognosis than those with CC alleles (Fig. 4B). Meanwhile, in patients received no treatment, no difference was detected between subjects with different genotypes of rs2817101 (Fig. 4B). We further performed two multivariate cox’s proportional hazards model in patients with and without drug treatment separately. The following covariates were used as confounding factors: age, pathologic stage, histological subtypes, lymph nodes status, ER, PR and HER2 status. We found that the rs2817101 was an independent prognostic factor (HR = 0.90, 95% CI 0.41–1.38; P = 2.85 × 10−4) (Fig. 4C) for patients received doxorubicin treatment. In contrast, for patients without doxorubicin treatment, there was no survival difference for subjects with different rs2817101 genotypes.
Fig. 4.

A: Multivariate cox proportional hazards results about doxorubicin, rs2817101, rs2817101 × doxorubicin (interaction term) factors in all patients. B: Pairwise Kaplan-Meier survival analysis in patients with and without drug treatment separately of rs2817101. C: Multivariate cox proportional hazards results about rs2817101, age, pathologic stage, histological subtypes, Lymph nodes status, ER, PR and HER2 status factors respectively patients with and without drug treatment. Significant P-value threshold was set at 0.05. D: Boxplot of GSTA1 expression levels (log2(TPM + 1)) based on rs2817101 genotypes. E. Kaplan-Meier survival analysis between high and low GSTA1 expression patients in GSE25055.
The SNP rs2817101 is located in the downstream of GSTA1. Subjects with the CC genotype of rs2817101 showed the lowest expression of GSTA1 (Fig. 4D). KM analysis using data from GSE25055 showed that the survival of patients with low GSTA1 expression was poorer (Fig. 4E, P = 0.046). This result indicated that patients with higher GSTA1 expression might be more resistant to doxorubicin treatment.
4. Discussion
Detecting SNPs associated with drug response is helpful to realize personalized medicine. Here we developed DDRS to detect drug response SNPs. Different from previous studies, information from the patients without drug treatment was also taken into consideration. We applied this pipeline to detect doxorubicin response SNPs using data from the TCGA database and the follow up analysis confirmed its reliability.
For these identified doxorubicin associated SNPs, we calculated the DRS in the patients with other drug treatment. The basal like patients significantly had the highest DRS. This result consistent with the known conclusion that drug-resistance is commonly observed in TNBC (Triple-Negative Breast Cancer) patients and is more common than in non-TNBC patients[38], [39].
For these identified eQTL genes, we classified and ranked these genes with the product of eQTL β * coefficient β. These genes were classified into drug-resistant/risk and drug-sensitive/protective by corresponding SNPs. The constancy of effect to drug between SNPs and genes was confirmed in GEO validation data, indicating the drug-response effect of genes are partly from the regulation of SNPs. We identified rs2817101 and its cis-eQTL gene GSTA1, which had the highest product of βcox and βeQTL, the most doxorubicin-resistant gene. Glutathione transferases (GSTs) was frequently reported to have correlation with bad prognosis and resistance against a number of different anticancer drugs [40]. It has been reported that GSTA1 could promote lung cancer cell invasion and adhesion and have effect on lung cancer cell metastasis by promoting the epithelial-mesenchymal transition [41]. A previous study reported that several polymorphisms in GST genes were associated with differences in survival for cancer patients treated with chemotherapy [42]. Our study revealed the regulation from downstream SNP rs2817101 to GSTA1 could also influenced doxorubicin-response, and rs2817101 was an independently prognostic factor to doxorubicin chemotherapy.
Except for interaction with drug related genes or other drug resistant mechanism like drug efflux or metastasis, some eQTL genes can also influence drug side effect. We also identified the activity of pathway ‘response to steroid hormone (GO: 0048545)’ was a significant protective factor to doxorubicin for it could reduce side effects of doxorubicin. Recent studies also revealed that testosterone could protects cardio myocytes against senescence caused by doxorubicin [43].
The major limitation to use DDRS is that it is hard to collect data including patients with and without drug treatment. We used the TCGA data in this study, which has the largest number of patients with drug treatment information currently. When new large-scale data is available, the results might be updated.
In summary, we presented an approach to identify drug-response SNPs and applied it to TCGA breast cancer patients. We identified a group of doxorubicin associated SNPs. We hope this method could also help to identify new drug associated SNPs in other cancers.
5. URLs
DDRS is freely available for non-commercial research institutions. Details can be obtained from https://github.com/ew314/DDRS.
CRediT authorship contribution statement
Yu Rong: Conceptualization, Methodology, Software, Data curation, Writing - original draft, Writing - review & editing. Shan-Shan Dong: Formal analysis, Writing - original draft, Writing - review & editing. Wei-Xin Hu: Data curation, Validation. Yan Guo: Supervision, Writing - review & editing, Methodology. Yi-Xiao Chen: Investigation, Resources. Jia-Bin Chen: Software, Data curation. Dong-Li Zhu: Resources. Hao Chen: Visualization. Tie-Lin Yang: Resources.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
The authors thank the TCGA and Broad Institute for maintaining critical public databases and services. The normalized expression matrixes and segmented copy number variation data we used were obtained through GDAC (Genome Data Analysis Center, http://gdac.broadinstitute.org/, doi:10.7908/C11G0KM9). The controlled data of TCGA we used were obtained through authorized access in dbGaP with the accession number of phs000178.v10. p8.
Funding
This work was supported by the National Natural Science Foundation of China (81872490, 31871264), the Natural Science Basic Research Program of Shaanxi (2021JC-02), and the Fundamental Research Funds for the Central Universities.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.06.026.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Coalition PM: The personalized medicine report. Opportunity, challenges, and the future.; 2017.
- 2.Kalow W., Tang B.K., Endrenyi L. Hypothesis: comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics. 1998;8:283–289. doi: 10.1097/00008571-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Syvänen A.-C. Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat Rev Genet. 2001;2(12):930–942. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- 4.Wood A.J.J., Evans W.E., McLeod H.L. Pharmacogenomics–drug disposition, drug targets, and side effects. N Engl J Med. 2003;348(6):538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 5.Evans W.E., Hon Y.Y., Bomgaars L., Coutre S., Holdsworth M., Janco R. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19(8):2293–2301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 6.Pullarkat S.T., Stoehlmacher J., Ghaderi V., Xiong Y.-P., Ingles S.A., Sherrod A. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 2001;1(1):65–70. doi: 10.1038/sj.tpj.6500012. [DOI] [PubMed] [Google Scholar]
- 7.Iyer L., Das S., Janisch L., Wen M., Ramírez J., Karrison T. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenom J. 2002;2(1):43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 8.Khan S., Fagerholm R., Rafiq S., Tapper W., Aittomäki K., Liu J. Polymorphism at 19q13.41 predicts breast cancer survival specifically after endocrine therapy. Clin Cancer Res. 2015;21(18):4086–4096. doi: 10.1158/1078-0432.CCR-15-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns J., Ingle J.N., Dudenkov T.M., Kalari K.R., Carlson E.E., Na J. Pharmacogenomics of aromatase inhibitors in postmenopausal breast cancer and additional mechanisms of anastrozole action. JCI Insight. 2020;5 doi: 10.1172/jci.insight.137571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Driscoll J.J., Rixe O. Overall survival: still the gold standard: why overall survival remains the definitive end point in cancer clinical trials. Cancer J. 2009;15:401–405. doi: 10.1097/PPO.0b013e3181bdc2e0. [DOI] [PubMed] [Google Scholar]
- 11.Giacomini K.M., Yee S.W., Mushiroda T., Weinshilboum R.M., Ratain M.J., Kubo M. Genome-wide association studies of drug response and toxicity: an opportunity for genome medicine. Nat Rev Drug Discov. 2017;16:1. doi: 10.1038/nrd.2016.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Innocenti F., Owzar K., Cox N.L., Evans P., Kubo M., Zembutsu H. A genome-wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clin Cancer Res. 2012;18(2):577–584. doi: 10.1158/1078-0432.CCR-11-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wanjuan Y., Jorge S., Patricia G., Edelman E.J., Howard L., Simon F. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scharpf R.B., Irizarry R.A., Ritchie M.E., Carvalho B., Ruczinski I. Using the R package crlmm for genotyping and copy number estimation. J Stat Softw. 2011;40:1–32. [PMC free article] [PubMed] [Google Scholar]
- 15.Franco H.L., Nagari A., Malladi V.S., Li W., Xi Y., Richardson D. Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res. 2018;28(2):159–170. doi: 10.1101/gr.226019.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mei S, CA M, R Z, Q Q, Q W, P J, B L, X S, B W, J F: Cistrome Cancer: A Web Resource for Integrative Gene Regulation Modeling in Cancer. Cancer Research 2017, 77:e19. [DOI] [PMC free article] [PubMed]
- 19.Kuhn RM, David H, James KW: The UCSC genome browser and associated tools. Briefings in Bioinformatics 2012:2. [DOI] [PMC free article] [PubMed]
- 20.Pers T.H., Timshel P., Hirschhorn J.N. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–420. doi: 10.1093/bioinformatics/btu655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q., Seo J.-H., Stranger B., McKenna A., Pe’er I., LaFramboise T. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 25.Stegle O., Parts L., Durbin R., Winn J., Regev A. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol. 2010;6(5):e1000770. doi: 10.1371/journal.pcbi.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shabalin AA: Matrix eQTL: Ultra Fast eQTL Analysis via Large MatrixOperations [R package MatrixEQTL version 2.2]. 2018, 28:1353. [DOI] [PMC free article] [PubMed]
- 27.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Research. 2017;46 doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Zhang S., Li F., Zhou Y., Zhang Y., Wang Z. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020;48:D1031–d1041. doi: 10.1093/nar/gkz981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn M., von Mering C., Campillos M., Jensen L.J., Bork P. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res. 2008;36(Database):D684–D688. doi: 10.1093/nar/gkm795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng F., Kovács I.A., Barabási A.L. Network-based prediction of drug combinations. Nat Commun. 2019;10:1197. doi: 10.1038/s41467-019-09186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X., Sun Z., Zimmermann M.T., Bugrim A., Kocher J.P. Predict drug sensitivity of cancer cells with pathway activity inference. BMC Med Genom. 2019;12:15. doi: 10.1186/s12920-018-0449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Igolkina A.A., Zinkevich A., Karandasheva K.O., Popov A.A., Selifanova M.V., Nikolaeva D. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 histone tags suggest distinct regulatory evolution of open and condensed chromatin landmarks. Cells. 2019;8(9):1034. doi: 10.3390/cells8091034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rada-Iglesias A. Is H3K4me1 at enhancers correlative or causative? Nat Genet. 2018;50(1):4–5. doi: 10.1038/s41588-017-0018-3. [DOI] [PubMed] [Google Scholar]
- 35.Qing-Lan, Li, Dan-Ya, Wang, Lin-Gao, Ju, Jie, Yao, Chuan, Gao: The hyper-activation of transcriptional enhancers in breast cancer. Clinical Epigenetics 2019. [DOI] [PMC free article] [PubMed]
- 36.Zentner G.E., Tesar P.J., Scacheri P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21(8):1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minsky N., Shema E., Field Y., Schuster M., Segal E., Oren M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat Cell Biol. 2008;10(4):483–488. doi: 10.1038/ncb1712. [DOI] [PubMed] [Google Scholar]
- 38.Wu T., Wang X., Li J., Song X., Wang Y., Wang Y. Identification of personalized chemoresistance genes in subtypes of basal-like breast cancer based on functional differences using pathway analysis. PLoS ONE. 2015;10(6):e0131183. doi: 10.1371/journal.pone.0131183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwentner L, Wolters R, Koretz K, Wischnewsky MB, Kreienberg R, Rottscholl R, W?ckel A: Triple-negative breast cancer: the impact of guideline-adherent adjuvant treatment on survival—a retrospective multi-centre cohort study. Breast Cancer Research & Treatment, 132:1073-1080. [DOI] [PubMed]
- 40.van Gisbergen M.W., Cebula M., Zhang J., Ottosson-Wadlund A., Dubois L., Lambin P. Chemical reactivity window determines prodrug efficiency toward glutathione transferase overexpressing cancer cells. Mol Pharm. 2016;13(6):2010–2025. doi: 10.1021/acs.molpharmaceut.6b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Liu F, Wang C, Wang C, Jiang Z: Glutathione S‑transferase A1 mediates nicotine‑induced lung cancer cell metastasis by promoting epithelial‑mesenchymal transition. Experimental & Therapeutic Medicine 2017, 14:1783. [DOI] [PMC free article] [PubMed]
- 42.Romero A, Martin M, Oliva B, De lT, J., Furio V, De lH, M., Garcia-Saenz JA, Moreno A, Roman JM, Diaz-Rubio E: Glutathione S-transferase P1 c.313A > G polymorphism could be useful in the prediction of doxorubicin response in breast cancer patients. Annals of Oncology Official Journal of the European Society for Medical Oncology 2012, 23:1750. [DOI] [PubMed]
- 43.Altieri P, Barisione C, Lazzarini E, Garuti A, Bezante GP, Canepa M, Spallarossa P, Tocchetti CG, Bollini S, Brunelli C, Ameri P: Testosterone Antagonizes Doxorubicin-Induced Senescence of Cardiomyocytes. J Am Heart Assoc 2016, 5. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.