

Abstract
Background
Reversible enzymatic methylation of mammalian mRNA is widespread and serves crucial regulatory functions, but little is known to what degree chemical alkylators mediate overlapping modifications and whether cells distinguish aberrant from canonical methylations.
Methods
Here we use quantitative mass spectrometry to determine the fate of chemically induced methylbases in the mRNA of human cells. Concomitant alteration in the mRNA binding proteome was analyzed by SILAC mass spectrometry.
Results
MMS induced prominent direct mRNA methylations that were chemically identical to endogenous methylbases. Transient loss of 40S ribosomal proteins from isolated mRNA suggests that aberrant methylbases mediate arrested translational initiation and potentially also no-go decay of the affected mRNA. Four proteins (ASCC3, YTHDC2, TRIM25 and GEMIN5) displayed increased mRNA binding after MMS treatment. ASCC3 is a binding partner of the DNA/RNA demethylase ALKBH3 and was recently shown to promote disassembly of collided ribosomes as part of the ribosome quality control (RQC) trigger complex. We find that ASCC3-deficient cells display delayed removal of MMS-induced 1-methyladenosine (m1A) and 3-methylcytosine (m3C) from mRNA and impaired formation of MMS-induced P-bodies.
Conclusions
Our findings conform to a model in which ASCC3-mediated disassembly of collided ribosomes allows demethylation of aberrant m1A and m3C by ALKBH3. Our findings constitute first evidence of selective sanitation of aberrant mRNA methylbases over their endogenous counterparts and warrant further studies on RNA-mediated effects of chemical alkylators commonly used in the clinic.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12967-021-02948-6.
Keywords: Alkylating agents, ALKBH3, ASCC3, Epitranscriptome, 1-Methyladenosine, 3-Methylcytosine, 7-Methylguanosine, No-go decay, P-bodies, Ribosome quality control
Background
Enzymatic modification of nitrogen bases in mRNA regulates processing, transport, lifetime and translation of mRNA molecules. In turn, this influences cell differentiation, stress responses, immunity, cognition and cancer development ([1–3] and references therein). To date, 11 endogenous base modifications have been described in mammalian mRNA. These are 7-methylguanosine (m7G, residing in the mRNA cap structure [4] as well as internally [5–7]), pseudouridine (Ψ) [8], inosine (I) [9], N4-acetylcytidine (ac4C) [10], 5-methylcytidine (m5C) [11], 5-hydroxymethylcytidine (hm5C) [12], 3-methylcytidine (m3C) [13], 1-methyladenosine (m1A) [14, 15], 6-methyladenosine (m6A) [16, 17] and its derivatives 6-hydroxymethyladenosine (hm6A) and 6-formyladenosine (f6A) [18]. Moreover, in silico analysis recently suggested a large number of potential 5-methyluridine (m5U) modification sites in mRNA [19]. Additional methylations at the ribose moieties further contribute to this complexity [2, 20]. Enzyme “writers” that are able to establish each of the base modifications have been described, and enzymes that catalyze their removal (“erasers”) have been described for the methylated bases except internal m7G. Proteins that specifically bind to and dictate the functional outcomes (“readers”) of the base modifications in mRNA are much less characterized, and are primarily confined to m6A, m1A and the m7G mRNA cap [21–27].
Notably, at least three of the endogenous mRNA modifications, m1A and m3C and m7G, may also be introduced in nucleic acids by direct non-enzymatic methylation by endogenous and environmental methylating agents, including tobacco-specific nitrosamines and agents frequently used as cytostatic drugs in cancer treatment [28, 29]. Although chemical methylation of human mRNA has previously not been specifically addressed, treatment of total RNA from different species with methyl methanesulfonate (MMS) or 1-methyl-nitrosourea (MNU) resulted in prominent formation of m7G as well as several fold higher relative levels of m1A and m3C in total RNA than in DNA [28]. m1A, m3C and m7G all introduce a positive charge in addition to the methyl group. This can dramatically affect RNA–protein interactions and secondary structures through electrostatic effects, and may interfere with mRNA–tRNA interactions [29–32]. A crucial question then is whether these aberrant methylations are recognized and processed differentially compared to their endogenous counterparts. We previously demonstrated that the human AlkB-homolog ALKBH3 is able to demethylate m1A and m3C in RNA as well as in DNA with single-strand substrate preference [33, 34]. Another member of the same family, ALKBH1, may contribute to demethylation of certain structured m1A sites in mRNA [35]. ALKBH3 is able to restore RNA function by reactivating chemically methylated RNA bacteriophages and tRNA and has been shown to act as an eraser of endogenous m1A in mRNA in vivo [15, 33]. Thousands of reversible m1A sites have been identified in mRNA from ALKBH3 knockout cells [15], whereas hundreds of sites have been identified in wild type cells [36]. Whether ALKBH3 shares a similar function in mRNA demethylation of endogenous m3C [13], remains unknown. The YTH-domain containing proteins YTHDF1-3 and YTHDC1, but not YTHDC2, have been reported to be m1A reader proteins [26, 37]. Presently, no readers are known for m3C or internal m7G in mRNA and no studies have attempted to monitor alterations in the human mRNA binding proteome after treatment with methylating agents.
To begin addressing these issues, we separately quantified endogenous and MMS-induced methylbases in HeLa mRNA after MMS treatment and monitored concomitant alterations in the mRNA binding proteome. Immediately after MMS treatment, we observed markedly increased levels of m1A, m3C and m7G in mRNA. This was accompanied by reduced binding of protein members of the ribosomal 40S subunit, whereas binding of 60S proteins was essentially unaffected. The selective 40S loss could be explained by ribosomal collisions at MMS-induced methylbases or secondary structures, thereby activating the ribosomal quality control (RQC) pathway (reviewed in [38]), GIGYF2/4EHP-dependent sequestration of the mRNA cap and blocked recruitment of 43S preinitiation complexes (PICs) to affected mRNAs [39, 40].
ASCC3 was among the very few proteins that increased binding to mRNA after MMS treatment. ASCC3 is a DNA helicase that associates with ALKBH3 to facilitate unwinding and efficient demethylation of m1A and m3C in DNA [41, 42]. In addition, it was recently identified to bind stalled ribosomes as part of the RQC trigger (RQT) complex that facilitates ribosomal splitting [43]. Our results suggest that cells exploit these properties of ASCC3 to allow ALKBH3-mediated demethylation of aberrant m1A and m3C in the coding region of mRNA, whereas canonical methylbases remain unaffected. In addition, ASCC3 has a role in P-body formation, potentially by promoting ribosomal detachment from mRNAs harboring difficult-to-repair lesions and assembly of such transcripts in P-bodies for further repair or degradation. Although the molecular details of such a mechanism remain to be elucidated, our results provide first evidence of selective removal of aberrant methylbases from the human mRNA pool and may have significant implications to modulate the effects of cytotoxic alkylating drugs.
Methods
Chemicals, plasmids and antibodies
Complete protease inhibitor cocktail was from Roche Inc. Dulbecco’s modified Eagle medium (DMEM) minus l-arginine and l-lysine, dialyzed fetal calf serum (dFCS) and trypsin were from Thermo Scientific. Stably isotope labeled l-arginine and l-lysine were from Cambridge Isotope Laboratories. All other chemicals were from Sigma Aldrich. Full-length ASCC3 with a C-terminal HA-tag and CFP-DCP1A have been described previously [44, 45].
Primary antibodies and dilutions: ALKBH3 (Santa Cruz, sc-376520, IF: 1/100, WB: 1/2000), ASCC3 (Atlas, HPA001439, IF: 1/50), β-actin (Abcam, ab8226, IB: 1/2000), CELF1 (Santa Cruz, SC-20003, IB: 1/1000, IF: 1/200), EIF2S1 (Atlas, HPA064885, IB: 1/1000), EIF2S1-pS51 [Abcam, ab32157, IB: 1/500. Note that in the canonical human EIF2S1 sequence, this serine is at position 52 (UniProtKB-P05192)], HA-tag (Santa Cruz, SC-805, IF: 1/100). Histone H3 (Abcam, ab1791, IB: 1/2000), HNRNPA1 (Santa Cruz Biotechnology, SC-32301, IB: 1/3000, IF: 1/200), RPS10 (Atlas, HPA048084, IB: 1:125), RPL18 (Atlas, HPA046572, IB: 1:250), SERBP1 (Sigma, WH0026135M1, IB: 1/1000, IF: 1/50), SND1 (Sigma, HPA002632, IB: 1/1000, IF: 1/50), TIA1 (Abcam, ab196382, IF: 1/100), HRP-conjugated swine anti-rabbit (P0399) and rabbit anti-mouse (P0260) secondary antibodies were from Dako Chemicals. IRDye® 680RD Goat anti-Mouse IgG and IRDye® 680RD Goat anti-Rabbit IgG secondary antibodies were from LiCor Biotechnology. Secondary antibodies for confocal microscopy were from Life Technologies; Alexa Fluor® 488 rabbit anti-mouse (#A-11059) and donkey anti-rabbit (#A-21206), Alexa Fluor® 532 goat anti-mouse (#A-11002) and goat anti-rabbit (#A-11009), Alexa Fluor® 647 donkey anti-goat (#A-21477) and goat anti-rabbit (#A-21244).
Cell culture
HeLa S3 cells were from ATCC. PC-3 WT and ASCC3 knockout cells were from the Washington University Genome Engineering and iPSC Center (GEiC) and the knockout cells generated as described [42]. Primary stocks were employed subsequent to amplification [41]. Cells were grown in 5% CO2 at 37 °C in DMEM supplemented with 10% fetal calf serum, 0.1 mg/ml gentamicin, 2.5 µg/ml amphotericin B and 0.03% l-glutamine. All cell lines were mycoplasma free as determined by LC–MS/MS peptide analysis. For Stable Isotope Labeling with Amino acids in cell culture (SILAC) experiments, HeLa cells were grown as above, but using DMEM minus l-arginine and l-lysine, and dFCS. For labeling of light, medium and heavy HeLa cells, l-lysine-12C6/l-arginine-12C6, l-lysine-2H4/l-arginine-13C6 and l-lysine-15N213C6/l-arginine-15N413C6 were added to the media, respectively. To increase sensitivity of the MS analysis, we reduced the number of target peptides resulting from arginine-to-proline conversion by reducing the amount of l-arginine (and l-lysine) in the growth media as previously described [46, 47]. Cells were transfected with Fugene HD® (Roche) according to the manufacturer’s instructions and analyzed 24 h later. CRISPR–Cas9-mediated editing of ASCC3 in PC-3 was described previously [42].
Cell cycle analysis and resazurin reduction assay
Flow cytometric analysis was performed as previously described [48]. Briefly, cells were fixed in methanol (70%), RNase treated (100 µg/ml in PBS, 37 °C, 30 min) and propidium iodide stained (50 µg/ml in PBS, 37 °C, 30 min) prior to analysis by FACS flow cytometry (FASCAria, BD Bioscience). 24 h after MMS treatment, 20 µl 2.5 mM resazurin, PBS was added to each well of a 96-well plate containing 100 µl cell growth medium/well and incubated further for 2 h. Resorufin fluorescence was subsequently determined using a microplate reader (BMG Labtech Fluostar Omega, Oslo, Norway, excitation 544 nm, emission 590 nm).
MMS treatment, UV cross-linking and oligo(dT) capture
Oligo(dT) capture of mRNA was performed as previously described [49] with minor modifications. For each condition, 2 × (for western analysis) or 4 × (for MS analysis) 15 cm cell culture dishes were used, and the volumes of buffers were equally reduced according to cell growth area compared to the original protocol. MMS was added to the growth medium at a final concentration of 1 mM, plates covered with Parafilm and cells incubated further for 1 h. After a brief wash with pre-warmed PBS, new medium was added, and the cells incubated further for various times prior to analysis. After medium removal, culture plates (60–80% confluence) were placed on ice, washed 2 × with ice cold PBS prior to UVC irradiation (254 nm) in a UV Stratalinker® 2400. To reduce the amount of starting material in the SILAC approach, we first optimized the published protocol [49] with respect to the UV dose. A reduced dose (25 mJ/cm2 UVC) mediated the same extent of cross-linking as higher dosages while also reducing the tendency of beads sticking to the surfaces of plastic tubes and pipette tips, thus improving capture of mRNA binding proteins (Additional file 1: Figure S1A). Comparison of oligo(dT)-captured samples from UVC-irradiated versus non-irradiated cells by SDS-PAGE and SimplyBlue™ staining confirmed the specificity of the protocol (Additional file 1: Figure S1B). After UV cross-linking at 25 mJ/cm2, cells were harvested, added lysis buffer (20 mM Tris–HCl pH 7.5, 0.5 M LiCl, 1% lithium dodecyl sulphate (LDS, w/v), 5 mM EDTA, 5 mM DTT, 1 × Complete protease inhibitor cocktail) and homogenized by passing the lysates several times through a syringe with different needles (2 × 23G, 2 × 25G and 3 × 27G) and the homogenates snap-frozen in liquid N2. After thawing, samples were clarified by centrifugation at 12,000×g and protein concentration measured by NanoDrop spectrophotometry. For SILAC experiments, equal amounts of protein from control (PBS), noUV and 0 h MMS extract, or control, 4 h MMS and 15 h MMS extract were mixed and subjected to oligo(dT) capture. Poly(A)-containing RNAs and cross-linked proteins were captured using oligo(dT)25 magnetic beads (New England Biolabs). Beads were then washed 3 × with 4.5 ml buffer 1 (20 mM Tris–HCl pH 7.5, 0.5 M LiCl, 0.5% LDS (w/v), 5 mM EDTA, 1 mM DTT) and then 1 × with 4.5 ml buffer 2 [20 mM Tris–HCl pH 7.5, 0.2 M LiCl, 0.5% NP-40 (v/v), 1 mM EDTA, 1 mM DTT]. To avoid LiCl/LDS precipitation, steps including lysis buffer and buffer 1/2 were performed at room temperature (RT). Beads were eluted with elution buffer (20 mM Tris–HCl pH 7.5, 1 mM EDTA) for 3.5 min at 70 °C. The entire oligo(dT) capture procedure was then repeated 2 × with the same extract and magnetic beads. Eluates were pooled and treated with RNase T1 and RNase A (Sigma), concentrated using Amicon Ultra 0.5 ml Filters (3 kDa cut-off, Millipore) and separated by PAGE as described below.
Electrophoresis and western analysis
Proteins were denatured in 1 × LDS sample buffer (Invitrogen) at 70 °C for 10 min and then electrophoresed in 10% Novex Bis–Tris gels using MOPS running buffer. Gels were then either stained (Simply Blue™ Safe Stain, Invitrogen) or electroblotted onto PVDF-membranes (XCell II™ Blot Module, Immobilon®-PSQ) according to the manufacturer’s instructions. Blots were blocked in PBST (phosphate-buffered saline, 0.1% Tween 20), 5% fat-free dry milk for 1 h, and then incubated for 1 h with primary antibody in PBST, 1% fat-free dry-milk. After 3 × 5 min washes in PBST, membranes were incubated for 1 h in secondary-HRP-conjugated antibody (1/5000) in PBST, 1% fat-free dry-milk (for antibodies recognizing phosphorylated proteins PBST was replaced by TBST). Subsequent to 3 × 5 min washes in PBST/TBST, membranes were developed using SuperSignal West Femto (Pierce) and visualized using a Kodak Image Station 4000R and images analyzed using Kodak molecular imaging software 4.0. When IRDye® secondary antibodies were used, the membranes were visualized using Odyssey Imager (LiCor) and images analyzed using Image Studio 3.1.
Protein mass spectrometry and data analysis
To analyze overall proteome changes subsequent to MMS treatment, 100 µg of each of the mixed SILAC extracts (pre-oligo(dT) capture) were precipitated using chloroform/methanol [50] and separated by LDS-PAGE as described above in parallel with the mRNA-binding protein (mRBP) samples. Each total protein lane was cut into 5 slices, while each of the two mRBP enriched lanes were cut into 3 slices. Proteins in each slice were then reduced and alkylated prior to in-gel trypsination as described [51]. Peptides were resuspended in 0.1% formic acid and analyzed by LC–MS/MS using an EASY-nLC 1000 UHPLC system (Thermo Scientific/Proxeon) coupled to an LTQ-Orbitrap Elite hybrid mass spectrometer operating in positive ion- and data dependent acquisition mode. Peptides were injected onto a C-18 trap column (Acclaim PepMap100, 75 μm i. d. × 2 cm, C18, 5 μm, 100 Å, Thermo Scientific) and further separated on a C-18 analytical column (Acclaim PepMap100, 75 μm i. d. × 50 cm, C18, 3 μm, 100 Å, Thermo Scientific) using either 120 min (oligo(dT) eluates) or 180 min (total protein) multi-step gradients from 2 to 40% CH3CN in 0.1% formic acid at a flow rate of 250 nl/min. The following parameters were used for the LTQ-Orbitrap Elite hybrid mass spectrometer: electrospray voltage 2.2 kV, CID fragmentation with normalized collision energy 35, automatic gain control (AGC) target value of 1E6 for Orbitrap MS1 and 1E3 for MS2 scans. Each MS1 scan (m/z 400–16,000) was acquired at a resolution of 120,000 FWHM, followed by 20 MS2 scans triggered for intensities above 500, at a maximum ion injection time of 200 ms for MS1, and 50 ms (total protein) or 120 ms (oligo(dT) eluates) for MS2 scans.
Raw files from the three independent biological replicates were analyzed in Proteome Discoverer 2.0 (Thermo Scientific) using Mascot version 2.2.06 with Uniprot database from Jan 2014 and Sequest HT bundled with Proteome Discoverer version 2.0.0.802 using Uniprot database from Apr 2015 for Homo sapiens [52]. The following search parameters were used: enzyme specified as trypsin with a maximum of two missed cleavages; precursor mass tolerance 10 ppm and fragment mass tolerance 0.6 Da. Carbamidomethyl (C) was set as fixed modification, while N-terminal acetylation, methionine oxidation, phosphorylation (S, T, Y) and isotope labeled l-lysines and l-arginines were set as dynamic modifications. The Percolator tool was used for peptide validation using a cutoff value of 0.01 for false discovery rate (FDR), and thus only peptides with a high confidence were used for final protein identification and quantification. Statistical analysis was performed using Perseus 1.5 [53]. SILAC ratios were normalized by protein median and log2 transformed. Proteins with a ratio for noUV/PBS and not showing an enrichment of at least twofold (non-normalized log2 noUV/PBS ≤ 1) in at least one biological replicate were filtered out, and proteins with an inconsistent directionality of SILAC ratios in label swap experiments were also excluded. The list was further filtered to only contain proteins quantified with at least six out of nine ratios (0 h, 4 h and 15 h). The three time points (0 h, 4 h and 15 h) were subjected to one-way ANOVA analysis using Benjamini–Hochberg FDR correction [54]. As an altered protein ratio in the oligo(dT) eluates might be caused either by altered mRNA binding of the protein or altered protein amount in the input extracts, we performed a pairwise t-test between oligo(dT) eluates and input extracts for each time point. Correcting for multiple testing was performed as above. ANOVA and t-test results were only considered significant if corrected p-value < 0.05 and median log2 SILAC-ratio/t-test difference less than − 0.5 or more than 0.5.
LC–MS/MS quantification of methylated nucleosides
RNA samples were digested by incubation with 0.2 U nuclease P1 (Sigma, N8630), 20 U benzonase (Santa Cruz Biotech, SC-391121B) and 0.1 U alkaline phosphatase (Sigma, P5931) in a buffer containing NH4Ac pH 6.0 and 1 mM MgCl2 at 40 °C for 40 min. The hydrolysates were added three volumes of ice-cold acetonitrile and centrifuged at 16,000×g for 30 min at 4 °C. Supernatants were dried and dissolved in 50 µl water for LC–MS/MS analysis of methylated nucleosides. A portion of each sample was diluted for LC–MS/MS analysis of unmodified nucleosides. Chromatographic separation was performed using an Agilent 1290 Infinity II UHPLC system with an ZORBAX RRHD Eclipse Plus C18 150 × 2.1 mm ID (1.8 μm) column protected with an ZORBAX RRHD Eclipse Plus C18 5 × 2.1 mm ID (1.8 µm) guard column (Agilent). The mobile phase consisted of water and methanol (both added 0.1% formic acid) run at 0.3 ml/min for methylated nucleosides starting with a 6-min gradient of 5–90% methanol, followed by 3 min re-equilibration with 5% methanol, and for unmodified nucleosides maintained isocratically with 20% methanol. Mass spectrometry detection was performed using an Agilent 6495 Triple Quadrupole system operating in positive electrospray ionization mode, monitoring the mass transitions 282.1/150.1 (m1A, m6A), 285.1/153.1 (D3-m1A, D3-m6A), 258.1/126.1 (m3C, m5C), 261.1/129.1 (D3-m3C, D3-m5C), 298.1/166.1 (m7G, m6G), 301.1/169.1 (D3-m7G, D3-m6G), 268.1/136.1 (A), 244.1/112.1 (C), 284.1/152.1 (G), 245.1/113.1 (U), 266.1/150.1 (m1dA), 252.1/136.1 (dA), 228.1/112.1 (dC), 268.1/152.1 (dG), and 243.1/127.1 (dT). Quantification was performed by comparison with pure nucleoside standards.
Confocal microscopy and image analysis
Cells were examined using a Zeiss LSM 510 or a Leica SP8 laser scanning microscope equipped with a Plan-Apochromat 63 × /1.4 oil immersion objective. DAPI was excited at 405 nm and detected at 420–480 nm. CFP was excited at 458 nm and detected using a 545 secondary beam splitter combined with LP 475. Alexa Fluor® 488 was excited at 488 nm and detected at 505–530 nm. Alexa Fluor® 532 and 555 were excited at 543 nm and detected at 560–615 nm. Alexa Fluor® 647 was excited at 633 nm and detected above 650 nm. Optical section thickness was kept constant for all channels and a pinhole diameter of 1 Airy unit was used for the 633 nm excitation wavelength to keep high signal-to-noise ratio in all channels.
Cells were grown on no. 1.5 glass coverslips (Marienfeld) in phenol red-free growth media and washed with ice-cold PBS before fixation in 4% paraformaldehyde for 15 min at RT and permeabilization with 0.1% Triton-X 100, PBST for 15 min at RT. Cells were then washed 3× with PBST and blocked in 1% newborn goat serum (NGS), PBST for 45 min at RT. Staining was performed overnight at 4 °C with the primary antibodies at 20 µg/ml in 0.5% NGS in PBST. Coverslips were washed 4× in PBST and stained with Alexa Fluor® secondary antibodies (1:400 in PBST) for 2 h at room temperature. Coverslips were mounted using Vectashield® mounting medium with DAPI or SlowFade Antifade Diamond mounting medium without DAPI (Thermo Scientific). Since nuclear staining was not feasible when three differentially stained proteins were analyzed, nuclei and cytoplasm were confidently identified by intermittent differential interference contrast (DIC). Images were analyzed in Imaris, version 8.2.0. The sample size [10] to estimate the number of P-bodies per cell, as well as the size of the P-bodies in wt vs. ASCC3 knockdown cells was chosen by estimating a mean reduction in difference by 50%, a standard deviation of 40% of the mean, a statistical power of the test of 80%, and a type 1 error of 0.05. The sample size to determine the fraction of cells positive for P-bodies (> 25) was chosen by roughly estimating 60% for the wt and 20% for the knockdown cells, based on previous observations, a power of 80% and a type-1 error of 0.05 [55].
Results
MMS mediates direct induction of methylbases that are also present endogenously in human mRNA
Several studies have quantified levels of various methylated RNA bases subsequent to treatment with alkylating agents ([28, 56] and references therein). However, to the best of our knowledge, no studies have specifically monitored chemical methylation in human mRNA. To determine a relevant and non-lethal dose of MMS, HeLa cells were first treated for 1 h with increasing doses of MMS and cell viability analyzed 24 h later. Based on the data, we chose to treat cells with 1 mM MMS for 1 h, mediating a relative survival of 90% (Additional file 2: Figure S2A) but with a clear S-phase delay around 4 h after MMS treatment (Additional file 2: Figure S2B). 24 h after treatment, cells had returned to essentially normal asynchronous distribution. To quantify the levels of methylated RNA bases, total RNA was extracted from non-treated and MMS-treated HeLa cells. mRNA was enriched by oligo(dT) capture from the same samples and global modification levels in total- and mRNA were quantified by LC–MS/MS. Although oligo(dT) captures both mRNAs and other polyadenylated RNA species like long non-coding RNAs (lncRNAs), about 90% generally constitutes protein-coding RNA [57].
To individually monitor endogenous (enzymatic) and MMS-induced (non-enzymatic) methylation, cells were cultured for > 10 generations in medium containing a 1:1 mixture of non-labelled and deuterated methionine prior to MMS treatment. Within cells, methionine is converted to S-adenosyl methionine (SAM), the only known methyl donor for enzymatic RNA methylation. Thus, enzymatic methylation alone will mediate essentially equal amounts of light and heavy methylated ribonucleosides whereas non-enzymatic MMS-induced methylation will mediate increased CH3/CD3 ratios in the methylated ribonucleosides. Mass chromatograms from total- and mRNA analyses are illustrated in Fig. 1A, B. The relative quantities of each modification in untreated and MMS-treated cells are illustrated in Fig. 1C, D. Indeed, essentially identical levels of CH3 and CD3 were observed for all five methylnucleosides quantified in total RNA in the absence of MMS treatment (Fig. 1A, upper panel). MMS treatment resulted in markedly increased CH3/CD3 ratios in m1A, m3C and m7G, whereas the CH3/CD3 ratios in m5C and m6A remained essentially unchanged (Fig. 1A, lower panel), consistent with the notion that MMS does not induce these lesions [28].
Fig. 1.
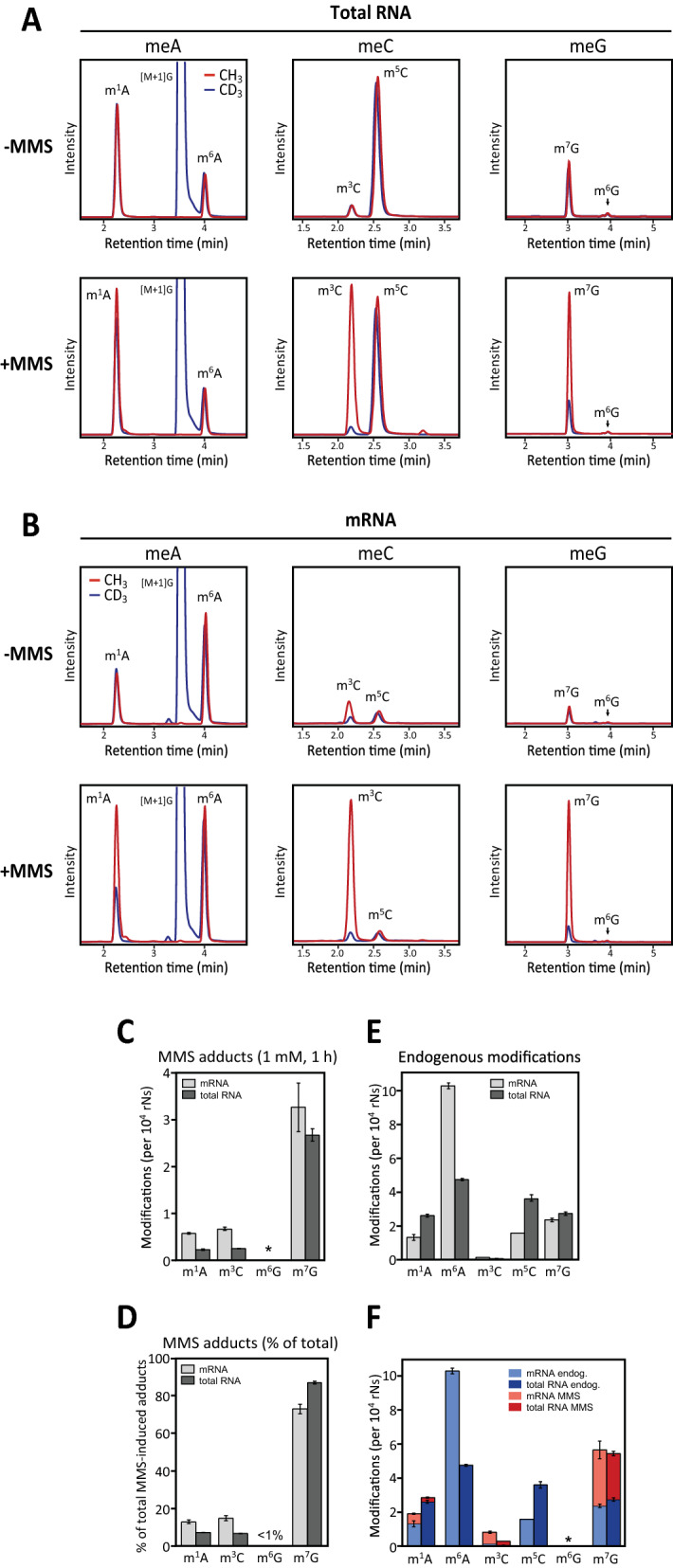
Endogenous and MMS-induced base methylations in total- and mRNA in HeLa cells. A Mass chromatograms of methylated bases in total RNA isolated from non-treated (upper panels) or MMS-treated (lower panels) HeLa cells after 1 h exposure of 1 mM MMS. Red graphs represent fraction of the bases containing light (CH3) methyl and blue graphs represent fraction containing deuterated (CD3) methyl in each modified nucleoside. Increased CH3/CD3 ratios after MMS treatment are mediated by non-enzymatic, MMS-mediated methylation (CH3). B Similar as in A, but with mRNA. C Concentrations of MMS-induced methyl adducts in total- and mRNA. D Relative distribution of MMS-induced methyl adducts in total- and mRNA. E Endogenous levels of methylated bases in total- and mRNA in untreated cells. F Total number of various base methylations in total- and mRNA with the relative amounts of endogenous and MMS-induced base methylations as indicated by differential coloring. Each bar in C-F represents the mean of three biological replicates with SDs as indicated
In poly(A)RNA from untreated cells (Fig. 1E), m6A was found to be the dominant endogenous base methylation, in agreement with previous reports [58, 59]. Moreover, substantial amounts of m1A, m7G, m5C and m3C were also detected. Whereas m6A was twofold enriched upon oligo(dT) capture, m1A was correspondingly depleted. This verifies the successful enrichment of mRNA, since m1A is more abundant in the rRNA and tRNA pools than in mRNA [60, 61], whereas m6A is most abundant in mRNA [62]. Nearly identical levels of deuterated and non-deuterated methyl groups were observed for all the methylbases except m3C, which displayed a markedly higher CH3/CD3 ratio (Fig. 1B, upper panel). The cause of this is unknown, but could indicate a kinetic isotope effect or, more likely, isotope exchange catalyzed by the m3C methyltransferase METTL8 [13] or a yet unidentified m3C methyltransferase. Notably, SAM-independent hydrogen isotope exchange has previously been reported for tRNA m5U methyltransferase [63].
MMS treatment mediated markedly increased CH3/CD3 ratios in m1A, m3C and m7G, (Fig. 1B, lower panel). Levels of the different MMS-adducts (per 104 unmodified ribonucleosides) in total- and mRNA are shown in Fig. 1C and conform well to those previously reported in total RNA subsequent to MMS treatment [28, 56]. The levels of endogenous methylations in untreated cells are shown in Fig. 1E, F illustrates the sum of each endogenous and MMS-induced methylation in total- and mRNA. MMS induced higher levels of the methyl adducts in mRNA compared to total RNA. This was especially evident for m1A and m3C, which were induced at more than twofold higher density in mRNA relative to total RNA (Fig. 1C).
We then monitored reversal of m1A, m3C and m7G in total- and mRNA at various time points after 1 h MMS treatment (Fig. 2). Upon MMS removal, a gradual reduction of all three lesions in mRNA followed essentially first order kinetics. Notably, whereas m1A and m3C returned to near pre-treatment levels after 24 h (~ 10% MMS-induced lesions remaining), about 30% residual MMS-induced m7G adducts were still present. m1A and m3C are both substrates for ALKBH3, but no human RNA m7G demethylase is known. Thus, removal of MMS-induced m7G is likely mediated by mRNA degradation only, whereas removal of m1A and m3C may be mediated by combined mRNA degradation and ALKBH3-mediated demethylation. Moreover, removal of all three methylbases was confined to the MMS-induced lesions, whereas the corresponding endogenous methylbases remained essentially unaffected. This strongly suggests that the cells harbor mechanisms to specifically recognize and remove aberrant methylbases from mRNA.
Fig. 2.

Kinetics of removal of endogenous and MMS-induced m1A, m3C and m7G from total-and mRNA subsequent to 1 h exposure of HeLa cells to 1 mM MMS. Grey curves: endogenous (CD3) modifications; Black curves: aberrant, MMS-induced (CH3) + endogenous (CH3) modifications. Grey rectangle: period of MMS treatment. Dashed line: Baseline MMS-induced lesions
MMS-induced changes in the mRNA binding proteome
To identify proteins differentially binding to mRNA at various time points after MMS treatment, we adapted a previously published UV cross-linking protocol [49] (Additional file 3: Figure S3A) combined with a triple-SILAC mass spectrometry (MS) approach [64] (Additional file 3: Figure S3B). By combining two time-course experiments using a common control (PBS treatment), a four time-point profile of the changes was achieved: (i) prior to MMS treatment (PBS), (ii) immediately after 1 h MMS treatment (0 h), (iii) 4 h and (iv) 15 h after end of MMS treatment (Additional file 3: Figure S3B). We also included a control that was neither MMS-treated nor UVC cross-linked (no UV). After covalent cross-linking by UVC irradiation at an optimized dose of 25 mJ/cm2 (Additional file 1: Figures S1A,B), cells were lysed and equal amounts of protein extract from the three cell populations were mixed (no UV/PBS/0 h and PBS/4 h MMS/15 h MMS). mRNA was enriched using oligo(dT) magnetic beads and mRNA binding proteins as well as proteins in the extracts were quantified using Orbitrap Elite LC–MS/MS. After filtering of three biological replicates, 336 proteins were quantified with at least six out of nine ratios (Additional file 4: Table S1). All of these proteins have previously been reported to be RNA-binding proteins (RBPs) [65, 66], underscoring the specificity of our experimental approach. In a previous study by Boucas et al. of proteins differentially binding to mouse embryonic fibroblast (MEF) mRNA subsequent to etoposide treatment [67], 184 proteins were quantified in both non-treated and treated cells. Out of these, 127 were also identified in our dataset (Additional file 3: Figure S3C), suggesting that the mRNA surveillance machinery is highly conserved between the species.
Figure 3A-C show the log2 SILAC ratios at the various time points after MMS treatment plotted against the corresponding ANOVA p-value. Differentially expressed proteins (Benjamini Hochberg corrected p-values < 0.05, absolute median log2 SILAC-ratio > 0.5) observed in at least one time point are highlighted in red in the plots and provided in Table 1 together with additional selected proteins. The log2 SILAC ratios revealed very few and small alterations, and nearly all the differentially bound proteins showed reduced binding to mRNA after MMS treatment. Furthermore, very few RBPs showed significantly altered log2 ratios except immediately after 1 h MMS treatment (0 h). As changes in the SILAC ratios could be due to either altered mRNA binding or alterations of the protein levels in the input extracts induced by MMS treatment, we subjected the SILAC ratios of the input extracts to a one-way ANOVA analysis. None of the oligo(dT)-enriched proteins showed significantly altered amounts in the input extracts (Additional file 4: Table S1). We also performed pairwise t-tests between oligo(dT)-enriched samples and the corresponding input extracts, shown in Fig. 3D–F. Here, 6 proteins at 0 h (Fig. 3D), while no proteins at 4 h (Fig. 3E) and 15 h (Fig. 3F) were significantly altered between oligo(dT) eluates and input extracts. Thus, MMS treatment mediated rapid and transient loss of a small subset of RBPs from the enriched mRNA.
Fig. 3.

Temporal changes in the mRNA binding proteome after MMS treatment. A–C ANOVA p-values (−log10) plotted against median SILAC ratios (log2) at 0 h (A), 4 h (B) or 15 h (C). D–F Volcano plots showing t-test p-values (−log10) versus median SILAC ratios (log2) of oligo(dT)/input extract at 0 h (D), 4 h (E) and 15 h (F). Significantly altered proteins after Benjamini Hochberg FDR correction (< 0.05) are shown in red. G-I Verification of SILAC data by western analysis. HeLa cells were treated as indicated with either PBS (control) or MMS for 1 h, lysed and subjected to oligo(dT) enrichment. G SND1 and SERBP1 show reduced binding to m RNA, while HNRNPA1 does not alter its binding. H Input of extracts utilized for oligo(dT) enrichment in G. Note that the protein levels in lanes 1–3 remain the same. I Reduced mRNA binding is not caused by cross-linking bias. As in G, but MMS-treated cells were irradiated with either 25 mJ/cm2 (standard) or 100 mJ/cm2 (4 × standard dose). Note that increasing the UVC dose does not lead to increased cross-linking (lane 3 vs. lane 4)
Table 1.
Proteins displaying most affected binding to mRNA subsequent to MMS treatment
| Gene | Protein name | Anova | t-test significance | Median log2 SILAC ratios |
||||
|---|---|---|---|---|---|---|---|---|
| 0 h | 4 h | 15 h | 0 h | 4 h | 15 h | |||
| Significant differential binding | ||||||||
| ZC3HAV1 | Zinc-finger CCCH-type antiviral protein 1 | + | + | − 0.51 | − 0.52 | − 0.19 | ||
| SND1 | Staphylococcal nuclease domain-containing protein 1 | + | + | − 1.03 | − 0.04 | − 0.22 | ||
| HDLBP | Vigilin | + | − 0.65 | − 0.11 | − 0.16 | |||
| ZC3H8 | Zinc finger CCCH domain-containing protein 8 | + | − 0.77 | − 0.23 | 0.27 | |||
| SERBP1 | Plasminogen activator inhibitor 1 RNA-binding protein | + | + | − 1.10 | − 0.05 | − 0.06 | ||
| NSUN2 | tRNA (Cytosine(34)-C(5))-methyltransferase | + | + | − 0.19 | 0.15 | 0.59 | ||
| RPSA | 40S ribosomal protein SA | + | + | − 0.58 | − 0.05 | 0.05 | ||
| RPS3 | 40S ribosomal protein 3 | + | + | − 0.93 | − 0.08 | 0.00 | ||
| RPS10 | 40S ribosomal protein 10 | + | − 0.74 | 0.03 | 0.07 | |||
| RPS14 | 40S ribosomal protein 14 | + | + | − 0.91 | − 0.07 | − 0.05 | ||
| RPS26P11 | Putative 40S ribosomal protein S26-like 1 | + | − 0.81 | − 0.08 | 0.05 | |||
| Sub-significantly increased binding | ||||||||
| ASCC3 | Activating signal cointegrator 1 complex subunit 3 | 0.72 | − 0.02 | 0.21 | ||||
| YTHDC2 | Probable ATP-dependent RNA helicase YTHDC2 | 0.56 | 0.03 | − 0.01 | ||||
| TRIM25 | E3 ubiquitin/ISG15 ligase TRIM25 | 0.80 | 0.05 | 0.06 | ||||
| GEMIN5 | Gem-associated protein 5 | 0.62 | 0.78 | 0.50 | ||||
To further validate our data, the levels of selected proteins were also monitored by Western blot analysis. Here, SND1 and SERBP1 displayed reduced binding to mRNA immediately after MMS treatment, whereas no change was observed for HNRNPA1 (Fig. 3G). The levels of all three proteins remained equal in the input extracts (Fig. 3H). These results are entirely in agreement with the SILAC data. We also tested whether the reduction in RBPs after MMS treatment was due to formation of higher order complexes shielding them from UVC-induced cross-linking, by increasing the UVC dose from 25 to 100 mJ/cm2. As seen in Fig. 3I (lanes 2–4), increasing the dosage did not increase the amount of RBPs. Thus, there was no evidence of bias caused by differential RBP cross-linking after stress.
The low number of proteins with significantly altered mRNA binding after MMS treatment was somewhat surprising. A potential explanation could be that MMS likely mediates very low stoichiometry in the modification at individual sequence sites. It might then be assumed that proteins displaying differential mRNA binding immediately after MMS treatment represent proteins that directly interact with the methylated bases and have little dependence on sequence context, or that belong to RNA metabolic events directly triggered by such recognition.
Reduced mRNA binding of 40S ribosomal subunits supports blocked 5ʹ-entry of 43S preinitiation complexes
Of the proteins displaying significantly reduced mRNA binding immediately after MMS treatment, several were subunits of the small (40S) ribosomal subunit (Table 1). In addition, the binding of all except one of the other 40S subunits in our dataset was reduced immediately after MMS treatment, although non-significantly according to our criteria (Additional file 4: Table S1). These 40S subunits displayed the same binding pattern during the time course of the experiment, returning to control levels at 4 h after MMS (Fig. 4A left panel, Additional file 4: Table S1), strongly supporting that MMS mediates transient loss of the 40S subunit from the captured mRNA. Conversely, none of the eight large ribosomal subunits in our dataset displayed significantly altered mRNA binding after MMS (Fig. 4A right panel, Additional file 4: Table S1). There is currently no experimental evidence suggesting that 40S ribosomal subunits may be lost from intact ribosomes while retaining the 60S subunit attached to the mRNA. Thus, it is more likely that the MMS treatment mediates reduced loading and accumulation of 43S preinitiation complexes (PICs) at the 5ʹ-UTR. Several forms of cell stress result in an integrated stress response mediated by phosphorylation of the guanine exchange factor EIF2S1 [68], thereby inhibiting formation of 43S PICs. To test this, we quantified phosphorylated EIF2S1 in untreated HeLa cells and at varying time points after 1 h MMS exposure. As shown in Fig. 4B, MMS-treatment mediated negligible change in EIF2S1 phosphorylation within 15 h after MMS treatment, rendering reduced PIC accumulation via this pathway less likely. Notably, two recent studies identified an alternative pathway to block translation initiation and that is independent of EIF2S1 phosphorylation. Instead, ribosomal collisions trigger recruitment of GIGYF2 and 4EHP, which sequester the mRNA cap and blocks recruitment of 43S PICs [39, 40]. To investigate this, we treated the cells with cycloheximide prior to MMS treatment. Cycloheximide arrests ribosomal movement after one translocation step [69] and would thus obstruct ribosomal collisions. Whereas MMS treatment alone mediated a marked loss in the ratio of RPS10 to RPL18, the loss was reversed to varying degrees by cycloheximide prior to the MMS treatment (Fig. 4C, right panel). This supports that the MMS-induced lesions promote RQC-associated arrest of translation initiation at affected mRNAs, thereby mediating the reduced 40S/60S ratio. Potentially, cycloheximide treatment could also block RQC-associated no-go decay (NGD). In yeast, endonucleolytic cleavage occurs at persistently collided ribosomes [70]. This would lead to selective loss of the 5’-UTR-containing fragment and accumulated 43S PICs during oligo(dT) enrichment. However, in human cells mRNA degradation is apparently less associated with RQC than in yeast [71].
Fig. 4.

Selective loss of 40S subunits from MMS-treated mRNA. A Five out of ten 40S subunits showed statistically significant transiently reduced mRNA binding after MMS treatment, whereas additional four 40S subunits demonstrated very similar, although sub-significantly altered binding pattern (left panel). Such transiently reduced binding was not observed with the 60S subunits (right panel). B 1 h MMS-treatment mediated negligible change in the level of phosphorylated (S52) EIF2S1 within 15 h after treatment (normalized to actin). C Western analysis (left panel) of mRNA-binding proteins after oligo(dT) capture confirmed a marked decrease in the ratio of bound 40S (RPS10) versus 60S (RPL18) subunits in agreement with the SILAC results. Notably, treatment with cycloheximide (10 μg/ml) for 15 min prior to the MMS treatment (CHX) to decrease ribosomal collision, partially or fully restored the RPS10/RPS18 ratio. The right panel shows RPS10/RPL18 ratios bound to mRNA in three independent experiments (given as three individual graphs) after western blotting and probing with IRDye® secondary antibodies and demonstrating the same trend (Spearman rank correlation = 1 between the three series)
The most downregulated protein immediately after MMS treatment was SERBP1, a multifunctional RNA binding protein associated with the 40S ribosomal subunit and actively translating ribosomes [72]. This agrees with a transiently reduced recruitment of 43S PICs. ZC3HAV1 (ZAP), and SND 1 also displayed significantly reduced binding immediately after MMS treatment (Table 1). ZC3HAV1has been shown to target mRNA translation by interfering with the translation initiation complex [73] whereas SND1 is a multifunctional protein that recently was demonstrated to be a bona fide m6A-binding protein [74]. It is a known binder of metadherin (MTDH), a dsRNA binder and proposed oncogene overexpressed in many cancers [75]. MTDH displayed a similar mRNA binding pattern as SND1 but was quantified in only in only two out of three experiments (1.7- and 2.2-fold reduced binding immediately after MMS treatment) and thus reported as non-significant. This also holds true for the de-ubiquitinase USP10, which displayed 1.8- and 2.7-fold reduced binding immediately after treatment. Recent PAR-CLIP analyses demonstrated that USP10 is enriched at the CDS and that it rescues mono-ubiquitinated 40S subunits subsequent to ribosomal collision and splitting, from otherwise programmed lysosomal degradation [76].
MMS mediates increased mRNA binding of proteins associated with methylated RNA bases
ANOVA analysis identified that the m5C methyltransferase NSUN2 differentially bound to mRNA after MMS treatment, displaying 1.5-fold increased binding after 15 h (Table 1). NSUN2-mediated deposition of m5C on mRNA is induced by stress in plants [77], whereas in mammals NSUN2 modulates protein synthesis subsequent to stress by forming m5C in tRNAs [78, 79]. Since m5C is not directly induced by MMS (Fig. 1), it is tempting to speculate that the increased binding reflects increased NSUN2 targeting to mRNA as part of an overall stress response.
Using our stringent criteria, no proteins displayed statistically significant increased binding to mRNA immediately after MMS treatment. However, manual inspection of the dataset identified four proteins that were readily identified immediately after the treatment, but that were barely detectable at later time points or in the control. This led to large variances in their SILAC-ratios (Additional file 4: Table S1) contributing to making their p-values non-significant [80]. These were gem-associated protein 5 (GEMIN5), probable ATP-dependent RNA helicase and m6A reader YTHDC2, the E3 ligase/ISG15 ligase TRIM25 and the activating signal cointegrator 1 complex (ASCC) subunit 3 (ASCC3) (Table 1). GEMIN5 is a 169 kDa multidomain protein that is part of the SMN complex that is essential for spliceosome formation. In addition, GEMIN5 has a key role in reprogramming cellular translation. It can bind to the m7G cap and the large ribosomal subunit and is able to mediate global translational repression while enhancing translation of mRNAs harboring thermodynamically stable secondary structure motifs [81]. To what degree GEMIN5 recognizes internal m7G or secondary mRNA structures induced by methylation remains, however, to be investigated. The RNA-dependent E3 ubiquitin ligase TRIM25 plays an important role in innate immunity, but the mechanisms underlying this are still poorly understood ([82] and references therein). Very recently, TRIM25 was shown to ubiquitinylate the m6A readers IGF2BP1/2/3 and mediate their degradation. This was promoted by binding of the IGF2BPs to m6A-modified circular RNA circNDUFB2 and was dependent on the RNA-binding motif in TRIM25 [83]. To what extent methylated ribonucleosides modulates RNA binding and E3 ligase activity of TRIM25 itself is unknown, but such studies are warranted given known strategies used by viruses to modulate their RNA methylation pattern to avoid host immune responses [84]. The 3ʹ–5ʹ RNA helicase YTHDC2 is an m6A reader that is essential for male and female fertility in mice [85]. It has also been found to associate with the 40S ribosomal subunit as well with XRN1, which has a role in degradation of polyadenylated NGD fragments [86].
The increased binding of ASCC3 after MMS treatment was especially interesting. In addition to its role in the ASCC complex [87], ASCC3 was very recently also shown to be part of the human RQT complex, which recognizes stalled ribosomes during mRNA translation to induce subunit dissociation and facilitate RQC [43]. In the nucleus, ASCC3 binds directly to ALKBH3 and stimulates DNA demethylation of m3C and m1A via its DNA helicase activity. Knockdown of either ALKBH3 or ASCC3 significantly increased sensitivity towards MMS [41]. Since we previously demonstrated that ALKBH3 demethylates RNA with about the same efficiency as ssDNA [33], we speculated whether ASCC3 could mediate a similar stimulatory effect upon demethylation of RNA as of DNA, e.g. by recruiting ALKBH3 or alleviate steric hindrance to demethylation by stalled ribosomes.
ASCC3 contributes to m1A and m3C sanitation from mRNA following MMS treatment
To investigate whether ASCC3 influences removal of aberrant methylbases from the mRNA pool, WT and ASCC3-deficient PC-3 prostate cancer cells (Fig. 5A) were subjected to MMS treatment for 1 h and the levels of m1A, m3C and m7G in mRNA were quantified in at various time points. In agreement with the analyses in HeLa cells (Fig. 1), MMS-treatment did not mediate a significant change in the enzymatically induced methylbases (CD3), whereas a strong induction of chemical methylation (CH3) was observed. Notably, significantly delayed removal of m1A (4 h and 12 h, Fig. 5B) and m3C (4 h Fig. 5C) was observed after MMS treatment of the ASCC3-deficient cells compared to controls, whereas no such delay was observed for m7G (Fig. 5D). This supports a function of ASCC3 in promoting selective removal of aberrant m1A and m3C from the mRNA pool and suggests that ASCC3 promotes discrimination between the MMS-induced and endogenous methylations.
Fig. 5.
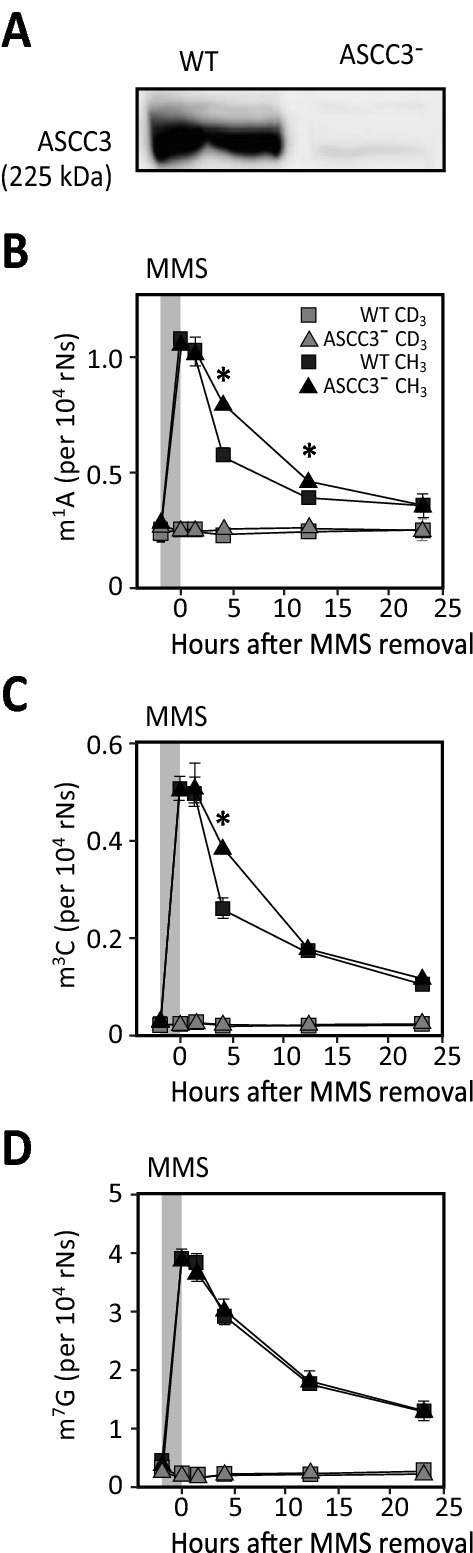
Knockdown of ASCC3 mediates delayed removal of aberrant m1A and m3C, but not m7G, from the mRNA pool. PC-3 prostate cancer cells were subjected to stable lentiviral knockdown of ASCC3. Knockdown and wild type (WT) cells were treated with 1 mM MMS for 1 h (light grey field) and mRNA extracted at various time points. Each data point represents the mean of three biological replicates with SDs as indicated. Removal of MMS-induced (CH3) m1A (A) and m3C (B) was significantly reduced in the ASCC3-deficient cells compared to WT at 4 h and 12 h (m1A) and 4 h (m3C), respectively (* t-test, p < 0.001), whereas no such effect was observed for removal of m7G (C)
ASCC3 promotes assembly of MMS-induced, ALKBH3-containing P-bodies
To further study a potential role of ASCC3 in recognition- and processing of methylated mRNA we investigated to what degree ASCC3 associated with known cellular ribonucleoprotein structures. We recently demonstrated that ASCC3 predominantly localizes to nuclei both in untreated and MMS-treated cells [42]. To avoid potential leakage of cytoplasmic factors caused by the nuclear permeabilization- and washing steps in the previous study, we reduced the concentration of Triton X-100 from 0.2 to 0.1% and omitted the use of NP-40. This resulted in a clear cytoplasmic staining of endogenous ASCC3 in HeLa cells, forming a dense pattern of diffuse foci throughout the cytoplasm and fewer foci in the nucleus (Fig. 6A, upper panel). A similar, but less dense pattern of ALKBH3-staining was observed throughout the cells, but we could not detect evident focal co-localization of ASCC3 and ALKBH3. Treatment of the cells with 5 mM MMS for 1 h mediated little change in distribution of either ASCC3 or ALKBH3 (Fig. 6A, middle panel). To investigate whether MMS-treatment mediated re-localization to stress-inducible cytoplasmic RNP compartments, we used antibodies against the stress granule (SG) marker TIA1, and transfection with a vector encoding the P-body marker mRNA-decapping enzyme 1A (DCP1A). MMS treatment did not induce stress granules in the cells (data not shown), in agreement with previous findings [88]. Conversely, large and diffuse P-bodies were present in the nucleus both without and after 5 mM MMS-treatment compared to smaller and more sharply defined P-bodies in the cytoplasm, especially after 5 mM MMS treatment. Aggregation of large, sharply defined cytoplasmic P-bodies were observed after an elevated (10 mM) dose of MMS (Fig. 6A, bottom panel). These MMS-induced P-bodies were generally enriched in ALKBH3 (Fig. 6B), whereas ASCC3 was often enriched around the P-bodies. This was most evident from x/y cross-sectional images as illustrated in Fig. 6B (bottom panel).
Fig. 6.

MMS-induced P-bodies in HeLa are enriched in ALKBH3 and localized adjacent to ASCC3. A HeLa cells transfected with P-body marker CFP-DCP1A were treated for 1 h as indicated, fixed, and double stained with anti-ASCC3 (red) and anti-ALKBH3 (green). B Enlarged images from white squares in A showing overlap of DCP1A and ALKBH3, and accumulation of ASCC3 at the P-body periphery (upper panel, corresponding to section 1). The lower panel shows z-stacks of section 2 demonstrating that ASCC3 accumulates around MMS-induced P-bodies whereas ALKBH3 accumulates inside the same P-bodies (lower panel)
In parental PC-3 prostate cancer cells, a similar staining pattern of ASCC3 and ALKBH3 was observed as in HeLa, except that more distinct cytoplasmic P-bodies were present in the untreated PC-3 cells (Fig. 7A, upper panel). In PC-3 cells treated with 2 mM MMS, ALKBH3 accumulated within P-bodies whereas little ASCC3 was seen in the interior volume of the cytoplasmic P-bodies compared to the surrounding volume, similar to MMS-treated HeLa cells (Fig. 7A, middle and bottom panels). In the ASCC3-deficient cells the ASCC3 strongly reduced and limited to distinct small foci of which many co-localized with ALKBH3 (Fig. 7B, upper panel). This verified efficient depletion of ASCC3 as well as specificity of the antibody. MMS-treatment of the ASCC3-deficient PC-3 cells mediated redistribution of ALKBH3 into fewer and enlarged foci, similar to that observed in HeLa, and of which several overlapped with ASCC3 (Fig. 7B, bottom panel). Strikingly, we found a marked reduction in the fraction of ASCC3-deficient cells positive for P-bodies, and the number and size of P-bodies present in these cells (Fig. 7B,C). This strongly suggests that ASCC3 is involved in the formation of MMS-induced P-bodies but does not constitute a prominent part of the P-body itself.
Fig. 7.

ASCC3 promotes formation of P-bodies in PC-3 cells. PC-3 cells transfected with P-body marker CFP-DCP1A were MMS-treated for 1 h as indicated, fixed, and double stained with anti-ASCC3 (red) and anti-ALKBH3 (green). A PC-3 cells proficient in ASCC3 contain P-bodies both in the absence (upper panel) and presence (middle panel) of MMS-treatment. The bottom panel shows enlarged z-stack demonstrating co-localization of DCP1A and ALKBH3, whereas the P-body interior contains less ASCC3 than the volume immediately surrounding the same P-body. B Formation of P-bodies was severely impeded in ASCC3 knockdown PC-3 cells both in the presence and absence of MMS. C Quantitative analysis of P-bodies in WT and ASCC3 knockdown (Kd) PC-3 after 2 mM MMS treatment demonstrated that significantly fewer ASCC3-deficient cells contained P-bodies compared to WT. In the cells that contained P-bodies, these were also significantly fewer and smaller than in the parental cells
Discussion
Here we show that the methylating agent MMS induces direct formation of m1A, m3C and m7G in human poly(A)-enriched RNA at levels considerably exceeding those in total RNA, while the overall levels of the corresponding endogenous modifications remain essentially unaffected.
Somewhat surprisingly, the aberrant methylbases induced by MMS did not mediate prominent skewing of the mRNA binding proteome. Thermodynamic dissociation constants (KD) so far measured for proteins able to bind modified mRNAs suggest that the modifications mediate modestly (2- to 20-fold) altered mRNA binding [1]. Moreover, binding of individual proteins is also affected by local sequence context, and thus altered binding of specific RBPs will likely affect only small subsets of mRNAs. Translating ribosomes constitutes an exception to this by offering a means of universal quality control of coding RNA, and ribosomal collisions signal potentially defective mRNA. RQC has been shown to be activated in response to MMS and oxidizing agents in yeast, and mutants defective in RQC components recover poorly subsequent to treatment [89]. Recent studies in yeast [70] and mammalian cells [90] demonstrated that the 40S–40S interface at persistently stalled and collided ribosomes are detected by ZNF598 (Hel2 in yeast), which ubiquitinylates the leading 40S subunit and mediates recruitment of the ASCC complex containing ASCC3 (Slh1 in yeast). The helicase activity of ASCC3 is essential for splitting of the leading ribosome [90]. Very likely, this increases accessibility of ALKBH3 to demethylate translation-blocking m1A and m3C buried within the decoding center of the leading ribosome. Independent of its ubiquitin ligase activity, ZNF598 also mediates formation of a ZNF598-GIGYF2-4EHP complex at collided ribosomes. Here, 4EHP (EIF4E2) outcompetes binding of eIF4E to the 5’-cap of affected transcripts, thereby inhibiting translation initiation by blocking access of 43S PICs [39, 40]. Since multiple PICs are bound at the 5’-UTR [91–93] this would lead to selective loss of 40S subunits after MMS treatment (Fig. 4D). In yeast, Hel2 and Slh1 were both crucial for RQC as well as for degradation of affected transcripts by NGD. Endonucleolytic cleavage occurred in the region spanning the leading stalled ribosome and the colliding ribosome [70], and the 5ʹ- and 3ʹ-fragments are degraded by the exosome (stimulated by Dom34:Hbs1) and the 5ʹ–3ʹ-exonuclease Xrn1, respectively [94]. In human cells, activation of the RQC pathway appears to be less accompanied by mRNA degradation compared to yeast [71]. Nevertheless, it cannot be ruled out that NGD contributes to the observed loss of 40S subunits after MMS-treatment, since mRNA cleavage would result in loss of the 5’-UTR during oligo(dT)-mediated capture and loss of associated 43 PICs.
Several lines of evidence support a function of ASCC3 in promoting removal of MMS-induced m1A and m3C from the mRNA pool: (i) ASCC3 displays transiently increased mRNA binding subsequent to MMS-treatment, (ii) knockdown of ASCC3 mediates delayed removal of m1A and m3C, but not m7G, from the mRNA pool after MMS treatment, (iii) ASCC3 promotes formation of MMS-induced P-bodies, which are prominent sites of mRNA turnover and (iv) ASCC3 and its yeast homolog Slh1 both couple ribosome arrest to RQC-mediated ribosome stripping and nascent polypeptide degradation [90, 95]. Within the translated pool of mRNA, ASCC3 might serve a dual function in the sanitation of aberrant methylbases: When ribosomes stall at m1A or m3C it mediates ribosomal splitting and recruits ALKBH3 to catalyze direct demethylation and allow restored translation. When these substrates are located in less accessible secondary structures, prolonged ribosomal stalling may promote transfer of the aberrantly methylated mRNA to P-bodies, similar to the postulated role of YTHDF2 in the transfer of m6A-containing mRNA from the translatable pool to RNA decay sites [21]. P-bodies are formed by phase separation largely driven by interaction between proteins rich in lysine-containing disordered regions, and RNA [96]. It is tempting to speculate that ASCC3-mediated stripping of ribosomes, which are highly structured ribonucleoproteins with an overall negative surface [97], is an important step to induce phase separation and formation of P-bodies. This is also supported by studies showing that P-bodies are virtually devoid of ribosomal proteins [98].
The precise contribution of ASCC3/ALKBH3-mediated demethylation versus depletion of m1A and m3C by RNA degradation cannot be precisely determined from the present study. Nevertheless, some rough estimates can be made from the kinetics of depletion of MMS-induced m1A, m3C and m7G. Mammalian mRNA, which constitutes the bulk of the poly(A)-RNA pool, has an average half-life of 9 h [99]. Thus, the bulk of mRNA harboring aberrant methylations should be degraded by canonical mRNA turnover alone within 24 h after MMS treatment. In addition to the canonical turnover, translated transcripts harboring aberrant methylbases will be subject to ASCC3/ALKBH3-mediated demethylation and potentially also NGD. At early time points (4 h) ASCC3-mediated removal, most likely by ALKBH3-catalyzed demethylation, is responsible for about one-third of the MMS-induced m1A and m3C lesions (Fig. 5B, C). Based on the virtually identical removal of mRNA containing aberrant m7G at early time points after treatment in the ASCC3-proficient cells compared to the removal of m1A and m3C in the ASCC3-deficient cells (4 h, Fig. 5B–D), aberrant m7G is likely removed by canonical RNA turnover and potentially also NGD, since m7G may modulate mRNA secondary structure [6] and base pairing of m7G with G is often observed in double stranded RNAs [32]. It cannot be excluded, however, that mammalian cells also express a demethylase that contributes to internal m7G removal. Interestingly, a recent study found that internal m7G is enriched at the 5ʹ-UTR and AG-rich sequences in unstressed cells, whereas increased m7G deposition was observed in the CDS and 3ʹ-UTR after H2O2 and heat shock treatments [7]. Increased m7G in the 3ʹ-UTR was accompanied by increased translation efficiency of a minigene reporter and the authors suggest that stress-induced m7G might be involved in the signaling pathways for specific stress responses [7].
Based on accumulated experimental evidence and our data, we propose a working model (Fig. 8) in which MMS induces aberrant methylbases that inhibit translation and potentially affect other mRNA functions. In the translated mRNA, aberrant methylbases in the coding region mediate ribosomal stalling and collision. ASCC3 recruits ALKBH3 to the arrested ribosomes, promotes splitting of the first arrested ribosome and renders m1A and m3C accessible for oxidative demethylation by ALKBH3. mRNAs harboring persistent translational blocks may then accumulate in P-bodies. Prior to accumulation in P-bodies, aberrantly methylated mRNAs and mRNA fragments are stripped of ribosomal subunits, likely via an ASCC3-initiaded RQC pathway involving degradation of partial nascent polypeptides and recycling of ribosomal subunits (analogous to the function of Slh1 in yeast [95]). P-bodies contain the endonucleolytic activities necessary for degradation of the mRNA as well as RNA helicases that may relieve secondary structures and potentially allow accessibility to repairable modifications [100]. After repair, the mRNAs can then be routed back to the translatable pool of mRNAs.
Fig. 8.

Working model of cellular processing of aberrantly (MMS-) methylated mRNA. MMS-induced m1A and m3C mediate direct ribosomal stalling, whereas m7G may form translation-blocking secondary structures. Collided disomes are detected by ZNF598, which serves two functions: It stabilizes GIGYF2-4EHP to block cap-dependent 43S entry and facilitates recruitment of ASCC3/ALKBH3 to mediate ribosome splitting and removal of m1A/m3C. Repaired transcripts can then be re-routed directly to translation. Transcripts harboring persistent blocking lesions, e.g. MMS-induced secondary structures, are stripped of ribosomes by ASCC3 and accumulate in P-bodies. Here, helicase activities relieve secondary structures to promote repair by ALBH3, whereas nonrepairable mRNAs are degraded
The biological impact of ASCC3/ALKBH3-mediated removal of MMS-induced methylbases from mRNA yet remains to be investigated. Enzymatic demethylation of mRNA would be energetically favorable compared to mRNA degradation and resynthesis and could be especially important during cell stress. Interestingly, ALKBH3 was recently shown to protect against cell stress by demethylating tRNA. This rendered the tRNA susceptible to cleavage by angiogenin, thus producing tRNA-derived small RNAs (tDRs) that strengthened ribosome assembly on mRNA and prevented apoptosis triggered by cytochrome c [101]. It is tempting to speculate that excessive chemical induction of m1A and m3C in mRNA might negatively affect this defense mechanism by diverging ALKBH3 from tRNA demethylation. Depletion of either ALKBH3 or ASCC3 has previously been shown to reduce survival of several cell lines subsequent to MMS treatment [41]. However, the contribution of hampered DNA repair versus RNA repair/degradation to the increased cytotoxicity must await further studies.
Conclusion
Our study demonstrates that MMS abundantly induces methylated bases in human mRNA identical to those found endogenously. Our proteomic analyses furthermore highlight potential mechanisms that enable cells to distinguish between canonical and aberrant methylations. In translated mRNA, aberrant methylations in the coding region apparently induces RQC of the affected transcripts. ASCC3 mediates ribosomal splitting and may recruit ALKBH3 to demethylate aberrant m1A and m3C. ASCC3 also facilitates formation of P-bodies, potentially by stripping ribosomes from transcripts harboring persistent translational blocks. Our findings warrant further studies on the capacity of alkylating agents to mediate (epi)transcriptome dysregulation and the potential contribution of aberrant RNA methylation to the cytotoxic effects of alkylating drugs. These studies should include other RNA species such as tRNAs and rRNAs as well as R-loops, in which altered base methylation patterns are often associated with human cancers [102].
Supplementary Information
Additional file 1: Figure S1. Optimization of UV-C dose for RBP-RNA crosslinking.
Additional file 2: Figure S2. Effects of MMS treatment on viability and cell cycle distribution of HeLa cells.
Additional file 3: Figure S3. Experimental pipeline of the SILAC analyses.
Additional file 4: Table S1. Results from SILAC analysis of RBPs after MMS treatment.
Acknowledgements
The mass spectrometry analysis was performed at the Proteomics and Modomics Experimental Core (PROMEC), Norwegian University of Science and Technology (NTNU). The confocal imaging was performed at the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology (NTNU). CFP-DCP1A was a kind gift from Dr. Bertrand Seraphin, IGBMC, CNRS UMR 1704/INSERM U964/Université de Strasbourg, France.
Abbreviations
- ALKBH3
Alpha-ketoglutarate-dependent dioxygenase alkB homolog 3
- ASCC3
Activating signal cointegrator 1 complex subunit 3
- CDS
Coding sequence
- CFP
Cyan fluorescent protein
- DMEM
Dulbecco’s modified Eagle medium
- FACS
Fluorescence-activated cell sorting
- HA
Hemagglutinin
- LC–MS/MS
Liquid chromatography-tandem mass spectrometry
- MMS
Methyl methanesulfonate
- MNU
1-Methyl-nitrosourea
- NGD
No-go decay
- PAR-CLIP
Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation
- P-body
Processing body
- RBP
RNA binding protein
- RPL
Ribosome large (60S) subunit
- RPS
Ribosome small (40S) subunit
- RQC
Ribosome quality control
- RQT
Ribosome quality control trigger
- SAM
S-Adenosyl methionine
- SG
Stress granule
- SILAC
Stable Isotope Labeling with Amino acids in Cell culture
- UTR
Untranslated region
- YTH-domain
YT521-B homology domain
Authors’ contributions
All contributed to planning of experiments and interpretation of data. GS and KLW wrote the manuscript. LH, KLW and DMF performed protein mass spectrometry and CBV performed nucleoside mass spectrometry. AS analyzed MS raw data and performed statistical analysis. KLW, RR,TSI PAA, HE and VP performed cell culture and viability assays, microscopy and wet lab experiments. All authors read and approved the final manuscript.
Funding
The work was supported by NTNU and the Central Norway Regional Health Authority via funding of PROMEC, the Liaison Committee for Education, Research and Innovation in Central Norway (46060917), the Research Council of Norway (221538/F20), The Norwegian Cancer Society (5813920), the Svanhild and Arne Must Fund for Medical Research and the Cancer Fund at St. Olavs Hospital, Trondheim, Norway. CMIC and PROMEC are funded by the Faculty of Medicine at NTNU and Central Norway Regional Health Authority. PROMEC is a member of the National Network of Advanced Proteomics Infrastructure (NAPI), which is funded by the RCN INFRASTRUKTUR-program (295910).
Availability of data and materials
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the identifier PXD003549 [103].
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jones JD, Monroe J, Koutmou KS. A molecular-level perspective on the frequency, distribution, and consequences of messenger RNA modifications. Wiley Interdiscip Rev RNA. 2020;11(4):e1586. doi: 10.1002/wrna.1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathlin J, Le Pera L, Colombo T. A census and categorization method of epitranscriptomic marks. Int J Mol Sci. 2020;21(13):4684. doi: 10.3390/ijms21134684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai K, Cullen BR. Epigenetic and epitranscriptomic regulation of viral replication. Nat Rev Microbiol. 2020;18(10):559–570. doi: 10.1038/s41579-020-0382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shatkin AJ. Capping of eucaryotic mRNAs. Cell. 1976;9(4 PT 2):645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- 5.Chu JM, Ye TT, Ma CJ, Lan MD, Liu T, Yuan BF, et al. Existence of internal N7-methylguanosine modification in mRNA determined by differential enzyme treatment coupled with mass spectrometry analysis. ACS Chem Biol. 2018;13(12):3243–3250. doi: 10.1021/acschembio.7b00906. [DOI] [PubMed] [Google Scholar]
- 6.Zhang LS, Liu C, Ma H, Dai Q, Sun HL, Luo G, et al. Transcriptome-wide mapping of internal N(7)-methylguanosine methylome in mammalian mRNA. Mol Cell. 2019;74(6):1304–16.e8. doi: 10.1016/j.molcel.2019.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malbec L, Zhang T, Chen YS, Zhang Y, Sun BF, Shi BY, et al. Dynamic methylome of internal mRNA N(7)-methylguanosine and its regulatory role in translation. Cell Res. 2019;29(11):927–941. doi: 10.1038/s41422-019-0230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159(1):148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommer B, Kohler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67(1):11–19. doi: 10.1016/0092-8674(91)90568-J. [DOI] [PubMed] [Google Scholar]
- 10.Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175(7):1872–1886.e24. doi: 10.1016/j.cell.2018.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubin DT, Taylor RH. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975;2(10):1653–1668. doi: 10.1093/nar/2.10.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature. 2018;554(7690):123–127. doi: 10.1038/nature25434. [DOI] [PubMed] [Google Scholar]
- 13.Xu L, Liu X, Sheng N, Oo KS, Liang J, Chionh YH, et al. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J Biol Chem. 2017;292(35):14695–14703. doi: 10.1074/jbc.M117.798298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311–316. doi: 10.1038/nchembio.2040. [DOI] [PubMed] [Google Scholar]
- 16.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5' terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- 18.Fu Y, Jia G, Pang X, Wang RN, Wang X, Li CJ, et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798. doi: 10.1038/ncomms2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang J, Song B, Tang Y, Chen K, Wei Z, Meng J. m5UPred: a web server for the prediction of RNA 5-methyluridine sites from sequences. Mol Ther Nucleic Acids. 2020;22:742–747. doi: 10.1016/j.omtn.2020.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai Q, Moshitch-Moshkovitz S, Han D, Kol N, Amariglio N, Rechavi G, et al. Nm-seq maps 2'-O-methylation sites in human mRNA with base precision. Nat Methods. 2017;14(7):695–698. doi: 10.1038/nmeth.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. doi: 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai X, Wang T, Gonzalez G, Wang Y. Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal Chem. 2018;90(11):6380–6384. doi: 10.1021/acs.analchem.8b01703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levi O, Arava YS. Pseudouridine-mediated translation control of mRNA by methionine aminoacyl tRNA synthetase. Nucleic Acids Res. 2021;49(1):432–443. doi: 10.1093/nar/gkaa1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, et al. Alkylation damage in DNA and RNA–repair mechanisms and medical significance. DNA Repair. 2004;3(11):1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Yan LL, Zaher HS. How do cells cope with RNA damage and its consequences? J Biol Chem. 2019;294(41):15158–15171. doi: 10.1074/jbc.REV119.006513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, et al. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell. 2004;16(1):107–116. doi: 10.1016/j.molcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Hudson BH, Zaher HS. O6-Methylguanosine leads to position-dependent effects on ribosome speed and fidelity. RNA. 2015;21(9):1648–1659. doi: 10.1261/rna.052464.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chawla M, Oliva R, Bujnicki JM, Cavallo L. An atlas of RNA base pairs involving modified nucleobases with optimal geometries and accurate energies. Nucleic Acids Res. 2015;43(14):6714–6729. doi: 10.1093/nar/gkv606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421(6925):859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- 34.Sundheim O, Vagbo CB, Bjoras M, Sousa MM, Talstad V, Aas PA, et al. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006;25(14):3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell. 2016;167(3):816–28.e16. doi: 10.1016/j.cell.2016.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H, Rauch S, Dai Q, Cui X, Zhang Z, Nachtergaele S, et al. Evolution of a reverse transcriptase to map N(1)-methyladenosine in human messenger RNA. Nat Methods. 2019;16(12):1281–1288. doi: 10.1038/s41592-019-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo KW, Kleiner RE. YTHDF2 recognition of N(1)-methyladenosine (m(1)A)-modified RNA is associated with transcript destabilization. ACS Chem Biol. 2020;15(1):132–139. doi: 10.1021/acschembio.9b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yip MCJ, Shao S. Detecting and rescuing stalled ribosomes. Trends Biochem Sci. 2021;S0968–0004(21):00066–69. doi: 10.1016/j.tibs.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hickey KL, Dickson K, Cogan JZ, Replogle JM, Schoof M, D'Orazio KN, et al. GIGYF2 and 4EHP inhibit translation initiation of defective messenger RNAs to assist ribosome-associated quality control. Mol Cell. 2020;79(6):950–62.e6. doi: 10.1016/j.molcel.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Juszkiewicz S, Slodkowicz G, Lin Z, Freire-Pritchett P, Peak-Chew SY, Hegde RS. Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. Elife. 2020;9:e60038. doi: 10.7554/eLife.60038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, et al. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44(3):373–384. doi: 10.1016/j.molcel.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brickner JR, Soll JM, Lombardi PM, Vagbo CB, Mudge MC, Oyeniran C, et al. A ubiquitin-dependent signalling axis specific for ALKBH-mediated DNA dealkylation repair. Nature. 2017;551(7680):389–393. doi: 10.1038/nature24484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hashimoto S, Sugiyama T, Yamazaki R, Nobuta R, Inada T. Identification of a novel trigger complex that facilitates ribosome-associated quality control in mammalian cells. Sci Rep. 2020;10(1):3422. doi: 10.1038/s41598-020-60241-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol. 2004;165(1):31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Ding SC, Cho H, Chung BC, Gale M, Jr, Chanda SK, et al. A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. MBio. 2013;4(3):e00385–e413. doi: 10.1128/mBio.00385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22(9):1139–1145. doi: 10.1038/nbt1005. [DOI] [PubMed] [Google Scholar]
- 47.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2003;2(2):173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 48.Gilljam KM, Feyzi E, Aas PA, Sousa MM, Muller R, Vagbo CB, et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J Cell Biol. 2009;186(5):645–654. doi: 10.1083/jcb.200903138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, et al. System-wide identification of RNA-binding proteins by interactome capture. Nat Protocols. 2013;8(3):491–500. doi: 10.1038/nprot.2013.020. [DOI] [PubMed] [Google Scholar]
- 50.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138(1):141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 51.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 52.UniProt C. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43(Database issue):D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012;13(Suppl 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. [Google Scholar]
- 55.Rosner B. Fundamentals of biostatistics. 5. Pacific Grove: Duxbury Press; 2000. [Google Scholar]
- 56.Tang Y, Wang Z, Li M, Zhang R, Zhang J. Simultaneous quantitation of 14 DNA alkylation adducts in human liver and kidney cells by UHPLC-MS/MS: application to profiling DNA adducts of genotoxic reagents. J Pharm Biomed Anal. 2019;166:387–397. doi: 10.1016/j.jpba.2019.01.034. [DOI] [PubMed] [Google Scholar]
- 57.Guo Y, Zhao S, Sheng Q, Guo M, Lehmann B, Pietenpol J, et al. RNAseq by total RNA library identifies additional RNAs compared to Poly(A) RNA library. Biomed Res Int. 2015;2015:862130. doi: 10.1155/2015/862130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams JM, Cory S. Modified nucleosides and bizarre 5'-termini in mouse myeloma mRNA. Nature. 1975;255(5503):28–33. doi: 10.1038/255028a0. [DOI] [PubMed] [Google Scholar]
- 59.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taoka M, Nobe Y, Yamaki Y, Sato K, Ishikawa H, Izumikawa K, et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018;46(18):9289–9298. doi: 10.1093/nar/gky811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, et al. Base-Resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68(5):993–1005.e9. doi: 10.1016/j.molcel.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 63.Santi DV, Hardy LW. Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: evidence for covalent catalysis. Biochemistry. 1987;26(26):8599–8606. doi: 10.1021/bi00400a016. [DOI] [PubMed] [Google Scholar]
- 64.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 65.Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46(5):674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 66.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 67.Boucas J, Fritz C, Schmitt A, Riabinska A, Thelen L, Peifer M, et al. Label-free protein-RNA interactome analysis identifies khsrp signaling downstream of the p38/Mk2 kinase complex as a critical modulator of cell cycle progression. PLoS ONE. 2015;10(5):e0125745. doi: 10.1371/journal.pone.0125745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11(2):113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6(3):209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ikeuchi K, Tesina P, Matsuo Y, Sugiyama T, Cheng J, Saeki Y, et al. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 2019;38(5):e100276. doi: 10.15252/embj.2018100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juszkiewicz S, Hegde RS. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol Cell. 2017;65(4):743–50.e4. doi: 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Muto A, Sugihara Y, Shibakawa M, Oshima K, Matsuda T, Nadano D. The mRNA-binding protein Serbp1 as an auxiliary protein associated with mammalian cytoplasmic ribosomes. Cell Biochem Funct. 2018;36(6):312–322. doi: 10.1002/cbf.3350. [DOI] [PubMed] [Google Scholar]
- 73.Zhu Y, Wang X, Goff SP, Gao G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012;31(21):4236–4246. doi: 10.1038/emboj.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baquero-Perez B, Antanaviciute A, Yonchev ID, Carr IM, Wilson SA, Whitehouse A. The Tudor SND1 protein is an m(6)A RNA reader essential for replication of Kaposi's sarcoma-associated herpesvirus. Elife. 2019;8:e47261. doi: 10.7554/eLife.47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dhiman G, Srivastava N, Goyal M, Rakha E, Lothion-Roy J, Mongan NP, et al. Metadherin: a therapeutic target in multiple cancers. Front Oncol. 2019;9:349. doi: 10.3389/fonc.2019.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meyer C, Garzia A, Morozov P, Molina H, Tuschl T. The G3BP1-family-USP10 deubiquitinase complex rescues ubiquitinated 40s subunits of ribosomes stalled in translation from lysosomal degradation. Mol Cell. 2020;77(6):1193–205.e5. doi: 10.1016/j.molcel.2019.12.024. [DOI] [PubMed] [Google Scholar]
- 77.Tang Y, Gao CC, Gao Y, Yang Y, Shi B, Yu JL, et al. OsNSUN2-mediated 5-methylcytosine mRNA modification enhances rice adaptation to high temperature. Dev Cell. 2020;53(3):272–86.e7. doi: 10.1016/j.devcel.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 78.Gkatza NA, Castro C, Harvey RF, Heiss M, Popis MC, Blanco S, et al. Cytosine-5 RNA methylation links protein synthesis to cell metabolism. PLoS Biol. 2019;17(6):e3000297. doi: 10.1371/journal.pbio.3000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, et al. Stem cell function and stress response are controlled by protein synthesis. Nature. 2016;534(7607):335–340. doi: 10.1038/nature18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waanders LF, Chwalek K, Monetti M, Kumar C, Lammert E, Mann M. Quantitative proteomic analysis of single pancreatic islets. Proc Natl Acad Sci USA. 2009;106(45):18902–18907. doi: 10.1073/pnas.0908351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez-Salas E, Embarc-Buh A, Francisco-Velilla R. Emerging roles of Gemin5: from snRNPs assembly to translation control. Int J Mol Sci. 2020;21(11):3638. doi: 10.3390/ijms21113868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choudhury NR, Heikel G, Michlewski G. TRIM25 and its emerging RNA-binding roles in antiviral defense. Wiley Interdiscip Rev RNA. 2020;11(4):e1588. doi: 10.1002/wrna.1588. [DOI] [PubMed] [Google Scholar]
- 83.Li B, Zhu L, Lu C, Wang C, Wang H, Jin H, et al. circNDUFB2 inhibits non-small cell lung cancer progression via destabilizing IGF2BPs and activating anti-tumor immunity. Nat Commun. 2021;12(1):295. doi: 10.1038/s41467-020-20527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sacco MT, Horner SM. Flipping the script: viral capitalization of RNA modifications. Brief Funct Genomics. 2021;20(2):86–93. doi: 10.1093/bfgp/elaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m(6)A transcripts by the 3'–>5' RNA helicase YTHDC2 Is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374–87.e12. doi: 10.1016/j.molcel.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 86.Kretschmer J, Rao H, Hackert P, Sloan KE, Hobartner C, Bohnsack MT. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5'-3' exoribonuclease XRN1. RNA. 2018;24(10):1339–1350. doi: 10.1261/rna.064238.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jung DJ, Sung HS, Goo YW, Lee HM, Park OK, Jung SY, et al. Novel transcription coactivator complex containing activating signal cointegrator 1. Mol Cell Biol. 2002;22(14):5203–5211. doi: 10.1128/MCB.22.14.5203-5211.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10(11):1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 89.Yan LL, Simms CL, McLoughlin F, Vierstra RD, Zaher HS. Oxidation and alkylation stresses activate ribosome-quality control. Nat Commun. 2019;10(1):5611. doi: 10.1038/s41467-019-13579-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Juszkiewicz S, Speldewinde SH, Wan L, Svejstrup JQ, Hegde RS. The ASC-1 complex disassembles collided ribosomes. Mol Cell. 2020;79(4):603–614.e8. doi: 10.1016/j.molcel.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gray NK, Hentze MW. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 1994;13(16):3882–3891. doi: 10.1002/j.1460-2075.1994.tb06699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berthelot K, Muldoon M, Rajkowitsch L, Hughes J, McCarthy JE. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol Microbiol. 2004;51(4):987–1001. doi: 10.1046/j.1365-2958.2003.03898.x. [DOI] [PubMed] [Google Scholar]
- 93.Shirokikh NE, Dutikova YS, Staroverova MA, Hannan RD, Preiss T. Migration of small ribosomal subunits on the 5' untranslated regions of capped messenger RNA. Int J Mol Sci. 2019;20(18):4464. doi: 10.3390/ijms20184464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Passos DO, Doma MK, Shoemaker CJ, Muhlrad D, Green R, Weissman J, et al. Analysis of Dom34 and its function in no-go decay. Mol Biol Cell. 2009;20(13):3025–3032. doi: 10.1091/mbc.e09-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sitron CS, Park JH, Brandman O. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA. 2017;23(5):798–810. doi: 10.1261/rna.060897.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ukmar-Godec T, Hutten S, Grieshop MP, Rezaei-Ghaleh N, Cima-Omori MS, Biernat J, et al. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat Commun. 2019;10(1):2909. doi: 10.1038/s41467-019-10792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Breindel L, Yu J, Burz DS, Shekhtman A. Intact ribosomes drive the formation of protein quinary structure. PLoS ONE. 2020;15(4):e0232015. doi: 10.1371/journal.pone.0232015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hubstenberger A, Courel M, Benard M, Souquere S, Ernoult-Lange M, Chouaib R, et al. P-body purification reveals the condensation of repressed mRNA regulons. Mol Cell. 2017;68(1):144–57.e5. doi: 10.1016/j.molcel.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 99.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 100.Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol Cell. 2018;69(3):517–32.e11. doi: 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 101.Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J, et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019;47(5):2533–2545. doi: 10.1093/nar/gky1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Janin M, Coll-SanMartin L, Esteller M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer. 2020;19(1):70. doi: 10.1186/s12943-020-01192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(D1):D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Optimization of UV-C dose for RBP-RNA crosslinking.
Additional file 2: Figure S2. Effects of MMS treatment on viability and cell cycle distribution of HeLa cells.
Additional file 3: Figure S3. Experimental pipeline of the SILAC analyses.
Additional file 4: Table S1. Results from SILAC analysis of RBPs after MMS treatment.
Data Availability Statement
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the identifier PXD003549 [103].