

Abstract
The growing quantity of public and private datasets focused on small molecules screened against biological targets or whole organisms provides a wealth of drug discovery relevant data. This is matched by the availability of machine learning algorithms such as Support Vector Machines (SVM) and Deep Neural Networks (DNN) that are computationally expensive to perform on very large datasets with thousands of molecular descriptors. Quantum computer (QC) algorithms have been proposed to offer an approach to accelerate quantum machine learning over classical computer (CC) algorithms, however with significant limitations. In the case of cheminformatics, which is widely used in drug discovery, one of the challenges to overcome is the need for compression of large numbers of molecular descriptors for use on a QC. Here we show how to achieve compression with datasets using hundreds of molecules (SARS-CoV-2) to hundreds of thousands of molecules (whole cell screening datasets for plague and M. tuberculosis) with SVM and the data re-uploading classifier (a DNN equivalent algorithm) on a QC benchmarked against CC and hybrid approaches. This study illustrates the steps needed in order to be “quantum computer ready” in order to apply quantum computing to drug discovery and to provide the foundation for this field to build on.
INTRODUCTION
The rapidly growing public and private datasets that are focused on small molecules screened against known biological targets or whole organisms 1 provides a wealth of data to facilitate drug discovery. Increasingly, this is used to create machine learning models 2 which can be used for enabling target-based design 3-5, predict on- or off-target effects and create scoring functions 6, 7. The pharmaceutical industry and academic laboratories are increasingly using and exploring machine learning applications in drug discovery to mine and model their data generated from years of high throughput screening 8. This is allowing rapid identification of molecules for neglected diseases such as Ebola 9 and Chagas disease 10, presenting lead compounds which can then be moved rapidly into in vivo models 11-13 and this approach can be more widely applied in cheminformatics. Recent examples have illustrated the speed with which the machine learning combined with in vitro testing continuum can generate new leads compared to traditional efforts 14. The availability of thousands of structure-activity datasets, (some of which in turn contain data for hundreds of thousands of molecules screened against a single target or organism 2, 15), presents a computational challenge with machine learning methods such as classifiers like support vector machines (SVM) on a classical computer (CC) 16.
Recently the potential of quantum machine learning has been illustrated using two methods such as a variational quantum circuit and a quantum kernel estimator 17. In addition, new machine learning methods for quantum computing continue to be developed 18, 19 which creates opportunities to expand the possible applications of machine learning to drug discovery and toxicology. Like many emerging technologies, the quantum computer (QC) has been proposed as likely to transform early-stage pharmaceutical research and development as well as providing a potential solution for datasets that would be intractable if performed on a CC 20, 21. One area of interest in early drug discovery is in cheminformatics for virtual screening and optimization, where small molecules are frequently described by fingerprint descriptors which can lead to tens of thousands of vectors called multiple fingerprint features (MFF) 22. While this may be important for many aspects of applying machine learning to chemistry 23, it also creates significant challenges when using these massive numbers of descriptors on a QC. This is mainly because we do not have enough resources and techniques to represent compounds with such large descriptors on QCs as the system will collapse.
Herein we describe how we have applied multiple approaches to compress the descriptors for QC while also demonstrating applications to drug discovery datasets on a range of scales that would be broadly applicable for drug discovery. We also describe hybrid approaches that merge QC with CC for machine learning applied to these datasets curated from public sources. These include 132 small molecule inhibitors of SARS-CoV-2 in Vero cells 24, 18,886 inhibitors of Mycobacterium tuberculosis 25 as well as several larger datasets for inhibitors of Krabbe disease (β-Galactocerebrosidase, Pubchem Assay 1159614, 44,809 compounds), Cathepsin B (Pubchem Assay 453, 63,331 compounds), Plague (Yersinia pestis, Pubchem Assay 898, 139,861 compounds), a second much larger dataset for M. tuberculosis (293,937 compounds) 26 and hERG (306,587 compounds) 15. All these datasets were curated and prepared using Assay Central 25 (See Methods) and the models can be applied for predicting new molecules for these targets or diseases.
METHODS
Experimental Procedures
Data curation
Our proprietary Assay Central software 27, 28 is a framework for curating high-quality datasets and building Bayesian machine learning models. Each dataset was subjected to the same standardization processes (i.e. removing salts, metal complexes and mixtures) prior to building models. Duplicate parent compounds with finite concentration activities are merged into a single entry. Classification models such as these require a defined threshold of bioactivity. The SARS-CoV-2 model used a threshold of 6.65 μM; Cathepsin B used >20% inhibition; the three M. tuberculosis models used the thresholds as described; the Krabbe model used actives defined by the authors; plague used a threshold at ≥50% inhibition; the hERGcentral dataset was >50% inhibition; The large M. tuberculosis model used a cut off where MIC90 or IC90 < 10 mg/ml or 10mM and a selectivity index (SI) greater than 10 was used (where SI = MIC90 or IC90/CC50) 26. The curated files that were output from Assay Central were then employed for quantum machine learning.
Running our algorithms on QC
We used IBM’s ibmq_rochester for executing our algorithms. Figure S1 shows the architecture of ibmq_rochester. Colors represent error probabilities for controlled-NOTs and readout on qubits 29. This architecture has 53 qubits linked in the network. These 53 qubits are assembled and connected following the property of hexagonal lattices which is advantageous when it comes to minimizing unwanted interactions (Figure S1) 29. The error in reporting the accuracies can be ±3% depending on the time of day the code is run due to their recalibration. The QC is recalibrated once a day at unknown times. The algorithm was run with shots=2048.
Descriptor Compression for Quantum Machine Learning of SARS-CoV-2 data
As the extended connectivity fingerprint diameter 6 (ECFP6) has been widely applied in cheminformatics 27 we used the Morgan Fingerprint (with radius 3), which is equivalent to the ECFP6 fingerprint in RDkit 30. The Morgan Fingerprint generates binary numbers whose default size is 2048 bits. While this is acceptable for use on a CC, it is not acceptable for use on a QC as 2048 bits exceeds the maximum capacity of current state-of-technology gate-based QCs. Apart from this, having a relatively big descriptor size, around 1000, can also be a challenge. This is because with the increase in size/usage of qubits on the QC the network, to represent descriptors, accuracy fails because of decoherence noise introduced in the qubit system 31. Depending on the architecture of QC being used, it can happen that not all qubits are linked with each other. Having no linkage or communication between every qubit adds further noise to this system. We attempted two approaches to solve this utilizing QC alone or a hybrid where part of the code is run on the QC.
We propose four methods (utilizing QC alone or a hybrid where part of the code is run on the QC) to encode the 2048 descriptor features. Method 1 used Principal Component Analysis (PCA) that is widely applied in data compression 32. Method 2 used a common dimension reduction technique of Linear Discriminant Analysis (LDA) which also considers the target class along with the predictors 33. In this we simply take a projection of points into some other hyperplane. In Method 3, we designed an algorithm where first we divide the 2048 molecule fingerprint bits into ‘x’ groups, such that, each group has ‘k’ bits. This means that k should divide 2048 completely leaving out with 0 as the remainder. These ‘k’ bits are then converted into base 10 or decimal value. This process is repeated until all the groups are converted to decimal. Method 4 uses an algorithm where we keep track of positions of 1 in the whole array.
Another approach we have applied is a hybrid approach in which the QC is used to perform part of the calculation while performing the remainder on a CC. This removes the storage limitation on QC as this is provided by the CC, hence leaving the processing to the QC. A data re-uploading classifier was used as described further below 34. Here, unlike SVM where we were reducing to 2-3 dimensions, we can reduce to 3-10 dimensions to consider the qubit architecture. For our purpose, we reduced to 6 dimensions using the Method 1 and Method 3 for SVM. In this approach, we load data into a single qubit by performing a simple unitary operation U (x1, x2, x3) where x1, x2, x3 are the coordinates of the point. When dimensions are greater than three, we can have U (x1, x2, x3, x4, x5, x6) to (U (x1, x2, x3) U (x4, x5, x6)). Using this approach, we can therefore store a datapoint which has 6 dimensions. We can have a similar unitary U (θ1, θ2, θ3) for rotation of datapoint in bloch sphere (Figure 1). Here we make a two-qubit connected layer to introduce non-linearity in our network.
Figure 1.

A high-level abstraction of how data is stored in the bloch sphere. The two layers - A and B, with U(xA) denoting unitary applied on input vector, U(θA) representing the unitary applied to rotate the vector in the bloch sphere.
The hybrid algorithm helps in introducing non-linearity to the model which makes use of the Adam stochastic gradient optimizer 35. We tuned different parameters using an iterative process for the following: Optimizer (Adagrad and Adam), Epochs, batch-size and the number of hidden layers.
The CC details on which the algorithms were executed:
The computational server used was a Supermicro EATX DDR4 LGA 2011, Intel Computer CPU 2.1 8 BX80660E52620V4, Crucial 64GB Kit (16GBx4) DDR4 2133 (PC42133) DR x4 288 Pin Server Memory CT4K16G4RFD4213 / CT4C16G4RFD4213, 1-5 x EVGA GeForce GTX 1080 Ti FOUNDERS EDITION GAMING, 11GB GDDR5X, Intel 730 SERIES 2.5Inch Solid State Drive SSDSC2BP480G410, WD Gold 4TB Datacenter Hard Disk Drive 7200 RPM Class SATA 6 Gb/s 128MB Cache 3.5 Inch WD4002FYYZ, Supermicro 920 Watt 4U Server.
Quantum SVM:
The initial algorithm we have compared is an SVM implemented using the Qiskit library 36 which uses Least-Square SVM (LS-SVM) 37. Qiskit chooses Mij =xi xj as the ansatz where Mij is the kernel matrix and xi xj are the datapoints in the dataset 38. When working with QC we first formulate the ansatz and try to minimize it. This ansatz formulates the SVM hyperplane which divides the datasets. We have chosen the ansatz such that we get a line as an output. The main reason we selected linear fit and not polynomial fit because we wanted to avoid the scenario of non-convergence kernels. This ansatz equation determines the shape the hyperplane will take. We made use of already available QSVM (Quantum SVM) code available in the Qiskit library. For the dataset, the SVM’s depth was set to 2. Entanglement was set to ‘full’ and the skip_qobj_validation parameter for quantum instance was set to False.
Data re-uploading classifier:
The MFF molecular descriptor 22 was also used with the four methods to achieve better accuracy rates. The data re-uploading classifier is very similar to a deep neural network (DNN) 34 ,implemented using the existing library in the Pennylane tool 39, where a single qubit represents a neural network layer. A DNN consists of 2 or more hidden layers. So, to replicate the DNN properties and introduce non-linearity, we make use of 2 qubits as an analogy to 2-layer Neural networks. With the above MFF descriptor we get 71,375 vectors. Sandfort et al. 22 postulated that this leads to overfitting and the same results were observed with MFF followed by our data reduction algorithm. For all datasets in this study, the data re-uploading classifier had hyperparameters set to – Training set size: (70% of dataset), Test set size: (30% of dataset), train accuracy rate: 61.2%, test accuracy rate: 61%, number of layers: 4, batch-size: 32, epochs: 10, optimizer: Adam, learning rate: 0.6, cross fold validation: 5.
RESULTS
Descriptor Compression for Quantum Machine Learning of SARS-CoV-2 data
Within the QC, a quantum algorithm is used to solve the direct product 37 to solve for matrix operations and using it to then calculate the M(the kernel matrix)/. Then a quantum algorithm can be used to transform into waveforms to solve the system of linear equations 38. This approach solves the complete SVM on a QC. While SVM generally provides promising results, it takes considerable time to solve linear equations to solve for Kernel Matrix using feature maps (1.24 minute for each datapoint) and hence, any time advantage for QC is likely not achievable. It should be noted that the connection to the qubit architecture also plays an instrumental role in driving the accuracy rates which may vary slightly for the best accuracy value. The accuracy rates reported are therefore a best value achieved on the QC or QC simulator.
In order to reduce the molecular descriptor features such that they can be represented and stored in our limited number of qubits (53 qubits for the ibmq-rochester, Fig. S1). We have initially demonstrated the application of QC with a SARS-CoV-2 Vero cell inhibition dataset consisting of 132 compounds of which 66 were found to be active with the IC50 activity threshold of 6.65 μM which was selected using our Assay Central software 24. We reduced the molecular descriptor dimensions from 2048 descriptor features to 2-3 using multiple techniques. Method 1 (i.e., PCA) resulted in accuracy rates on a CC with 1GPU of 37% using the kernel algorithm = RBF (Radial Basis Function) whereas on a QC accuracy was 33% (N= 3). Method 2 (i.e., LDA) resulted in accuracy rates of 40% using a kernel algorithm on a CC while on QC this was 39% (N = 3). An example of a molecule compressed using Method 3 is shown in Figure 2. Method 3 resulted in a greatly improved accuracy rate of 61% on a CC using kernel algorithm=‘poly’ and 59.6% on a QC (n=3).
Figure 2.
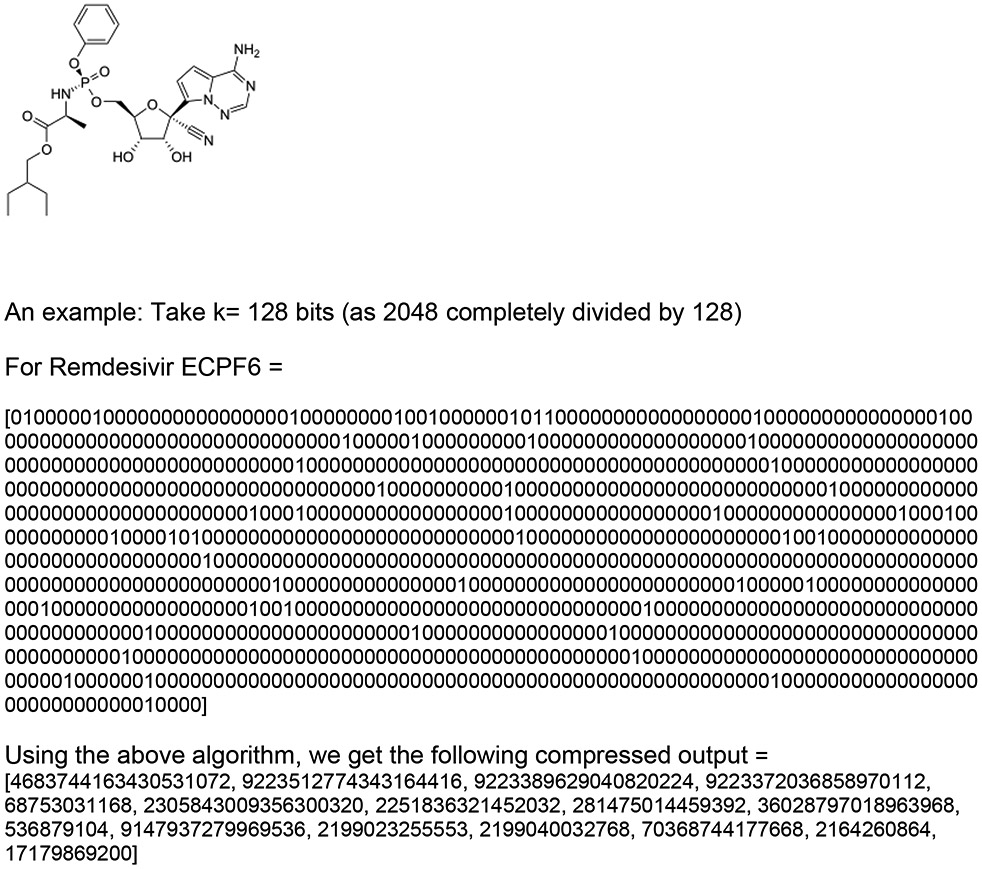
An example of a molecule and its compression using Method 3. 2D representation of the antiviral Remdesivir, and an illustration of the ECFP6 descriptor for Remdesivir and the compressed output.
Applying Method 4 using the same example of Remdesivir, we get two values [−21086,−502690]- where −21086 is storing the positions of 1’s and −502690 is storing the positions of 0’s. Using this approach, we get two dimensions that can be easily fed into the model. All the algorithms discussed above try to retain most of the information from the molecular descriptors as much as possible. This resulted in accuracy rates on the CC of 59% using kernel algorithm=‘sigmoid’ and on the QC of 59.25% (with n=2).
These four methods were implemented on the QC and then tried as combinations and the accuracy was compared with the CC (Table 1). The most promising results were obtained when combining Method 1 and Method 3 (k=128). The improved results for k=128 are due to the increased spread (more variance) as compared to other k values when combined with PCA (Figure 3). This makes it easier to obtain a hyperplane separating the two classes (active or not active). The Kernel matrix obtained from running the SVM model on a QC is shown in Figure 4.
Table 1.
The accuracy rates achieved with different combinations of methods. Method 1 (PCA), Method 2 (LDA), Method 3 and Method 4 using SVM on the classical and the quantum computer.
| Combination | Accuracy on QC (%) | Accuracy on CC (%) |
|---|---|---|
| PCA + Method 3(k=64) | <30 | 47 |
| PCA + Method 3(k=128) | 62.9 | 65 |
| PCA + Method 3(k=256) | 59.25 | 60 |
| PCA + Method 3(k=512) | 41 | 44 |
| LDA + Method 3(k=64) | 31 | 36 |
| LDA + Method 3(k=128) | <30 | 35 |
| LDA + Method 3(k=256) | <30 | 31 |
| LDA + Method 3(k=512) | 32 | 59 |
| PCA + Method 4(2) | 59.25 | 60 |
| LDA + Method 4(2) | 57 | 57 |
Figure 3.

The spread of the data for the SARS-CoV-2 dataset after applying Method 1 and Method 3. “x1” and “x2” are the top two features respectively (for all the compounds) obtained after applying these two methods.
Figure 4.

Kernel Matrix obtained using PCA and Method 3 for the SARS-CoV-2 dataset.
Another approach we have applied is that of the hybrid approach (i.e., QC performs part of the calculation and CC the remainder) to remove the storage limitation on QC. We reduced the datapoint to 6 dimensions using the Method 1 and Method 3 for SVM. We loaded data into a single qubit by performing a simple unitary operation U (x1, x2, x3) where x1, x2, x3 are the coordinates of the point. When dimensions are greater than three, we can have U (x1, x2, x3, x4, x5, x6) to (U (x1, x2, x3) U (x4, x5, x6)). We have a similar unitary U (θ1, θ2, θ3) for rotation of datapoint in bloch sphere (Figure 4) to make a two-qubit connected layer. For the same SARS-CoV-2 dataset, we obtained accuracy rates of 61%.
Applications of Quantum Machine Learning to M. tuberculosis datasets
The same hybrid algorithm was then implemented on M. tuberculosis datasets, which were representative of high throughput screening data (i.e., tens of thousands of compounds). For all these datasets, we applied Method 1 and Method 3 for reducing the dimension and feeding it into our data re-uploading classifier as well as for plotting and visualization purposes. We also worked with the MFF descriptors 22 which generated a 71,375-descriptor vector. Datasets for in vitro inhibitors of M. tuberculosis had three variants, namely with a cutoff at of 100nM, 1μM and 10 μM 25. Figure 5 shows a plot for the 100nM cutoff M. tuberculosis dataset when we reduced it into 2 dimensions. For these three datasets the data re-uploading classifier was run and then compared with the results obtained with CC (Table 2, Table S1). In Table 2 we see that the accuracy obtained on a QC is closer to that obtained on a CC with a slight time advantage over CC, such that there is a trade-off of accuracy and speed with these datasets. All of the above reported data are implemented on a QC (ibmq_rochester).
Figure 5.

A 2D plot representing the spread of data for the M. tuberculosis (100nM) dataset. This takes the important features using dimension = 2 instead of 6. “x1” and “x2” are the top two features respectively (for all the compounds) that have been calculated using Method 1 and Method 3.
Table 2.
Comparing accuracy and run time results for M. tuberculosis inhibition datasets (18,886 compounds) 25 using data re-uploading classifier on CC with 3 GPU’s versus QC with 5-fold cross-validation using SVM.
| Dataset threshold (number of actives) |
Time on CC (min) |
CC Accuracy (%) |
Time on QC (min) |
QC Accuracy (%) |
|---|---|---|---|---|
| 100 nM (645) | 125 | 97.1 | 104 | 90.5 |
| 1 mM (2351) | 144 | 90.4 | 101 | 81.4 |
| 10 mM (7762) | 229 | 75.6 | 153 | 54.9 |
Applications of Quantum Machine Learning to Large in vitro Datasets
The data re-uploading classifier algorithm 34 was implemented with the ECFP6 molecular descriptor, and tested with five considerably larger drug discovery datasets ranging from 44,000 – 293,000 molecules on a QC simulator (Table 3). Running them on a QC with the transfers of data was the major overhead here for such large datasets. We find that the results obtained are very comparable with a slight time advantage for the QC simulator over CC. The linearity of calculation time on a CC with dataset size was apparent and this plateaued for the QC simulator (Figure 6).
Table 3.
Comparing large scale drug discovery datasets on a Quantum Computer Simulator and CC with 5 GPU’s for data re-uploading classifier using the ECFP6 descriptor obtained from Method 1 and Method 2. Algorithms on QC Simulator were run for 10 epochs with 5 fold cross validation unless noted. ND = not determined.
| QC | CC | |||||
|---|---|---|---|---|---|---|
| Dataset target |
Total compounds (active) |
Training accuracy (%) |
Testing accuracy (%) |
Time on QC (min/epoch) |
Training accuracy (%) |
Time on CC (min) |
| Cathepsin B | 63,331 (75) |
99.4 | 99.1 | 35 (10 epochs) |
99.8 | 341.25 |
| Krabbe disease | 44,809 (63) |
91.2 | 92.4 | 31 (10 epochs) |
99.9 | 497.91 |
| Plague | 139,861 (223) |
92.9 | 93.1 | 47 (10 epochs) |
99.8 | 958.59 |
| M. tuberculosis | 293,937 (6104) |
90.4 | 91.3 | 71 (10 epochs) |
97.9 | 2329.4 |
| hERG | 306,587 (233) |
82.7 | 82.5 | 313 (ran for 5 epochs) |
ND | ND |
Figure 6.

Comparing dataset size with run time for Quantum Computer (QC) simulator and Classical Computer (CC). Linear or non-linear (semi-log) fit lines for the data generated. These show the likely relationship of CC and QM model size versus calculation time is on a linear and semi-log scale, respectively. The dotted lines represent the 99% confidence bands of these lines to highlight the likelihood of this relationship.
DISCUSSION
We have discussed four approaches and their combination for compression of the molecular descriptors, followed by calculation of the machine learning model on a QC. We found that the results were optimal when combining Method 3 and Method 1, most likely due to the increased spread of data after employing these techniques. Further, we applied both the QC and hybrid approach to train our model. With the current QC hardware available, the option of choosing the hybrid approach for drug discovery is likely optimal. When dealing with bigger in vitro structure-activity datasets on the order of tens of thousands of molecules, we found that the data communication overhead between computer and cloud-based QC was much larger than the actual time taken for a circuit to execute on QC. Plotting the time versus size of a dataset for CC vs QC simulator also suggests the likely potential speed of model building on a QC with larger datasets if we are able to replicate the same settings on a QC (Figure 6). Now that we have optimized these steps for machine learning we have demonstrated that QC can handle ‘very large’ drug discovery datasets on the order of hundreds of thousands of molecules. At the time of this work data for SARS-CoV-2 available for machine learning was in the hundreds of molecules and now it is likely in the low thousands of molecules 24 and yet such datasets clearly do not require the performance of QC. However, the larger high-throughput screening datasets for other targets and diseases that have been amassing in public databases like PubChem 40 and ChEMBL 1 present significant challenges for SVM and deep neural network as well as other computationally intensive tools. QC is therefore a viable approach to overcoming some of these limitations and allowing practical computing times. With thousands of such datasets now readily available, being able to curate and update them quickly will be important as new screening data are added. Obviously, as we start to see DNA encoded libraries with sizes in the millions to possibly billions of molecules being used for generating high throughput screening data, then we will likely need QC for machine learning with algorithms that are compute intensive in order to see results in a reasonable time. This study demonstrates the non-linear scaling of compute time on a QC with multiple independent datasets of different sizes, compared with the linearity observed on a CC. As quantum machine learning develops, the accessibility of QC will increase for drug discovery cheminformatics applications as we have demonstrated here. Future studies to evaluate these and other quantum machine learning models will also need to involve prospective prediction and experimental validation in order to provide convincing evidence of their value for drug discovery.
Supplementary Material
Acknowledgments
Professor Daniel D. Stancil (NC State) is kindly acknowledged for his helpful guidance and on quantum computing and overall support of this project. Access to the IBM Q Network was obtained through the IBM Q Hub at NC State. We gratefully acknowledge our many colleagues and collaborators who assisted in curating these datasets used as examples for this study as well as Mr. Valery Tkachenko for hardware support and Dr. Alex M. Clark for Assay Central support. We kindly acknowledge NIH funding to develop the software from R44GM122196-02A1 “Centralized assay datasets for modelling support of small drug discovery organizations” from NIGMS and NIEHS for 1R43ES031038-01 “MegaTox for analyzing and visualizing data across different screening systems”. “Research reported in this publication was supported by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number R43ES031038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.” V.O.G. was supported by FAPESP funding: 2019/25407-2.
Abbreviations
- CC
Classical computer
- DNN
Deep Neural Networks
- ECFP6
Extended connectivity fingerprint radius 6
- LDA
Linear Discriminant Analysis
- MFF
multiple fingerprint features
- PCA
Principal Component Analysis
- QC
Quantum computer
- SVM
Support Vector Machines
Footnotes
Competing interests
S.E., K.M.Z., D.H.F., E.M., and T.R.L. work for Collaborations Pharmaceuticals, Inc. K.B. and V.O.G. have no conflicts of interest.
Data and Software Availability
All datasets and software are available from the author upon written request.
Supporting Information available
Supporting information is available online including a supplemental figure describing the IBM Rochester architecture map and a supplemental Table describing the large tuberculosis dataset model confusion tables.
References
- 1.Gaulton A; Hersey A; Nowotka M; Bento AP; Chambers J; Mendez D; Mutowo P; Atkinson F; Bellis LJ; Cibrian-Uhalte E; Davies M; Dedman N; Karlsson A; Magarinos MP; Overington JP; Papadatos G; Smit I; Leach AR, The ChEMBL database in 2017. Nucleic Acids Res 2017, 45, D945–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lane TR; Foil DH; Minerali E; Urbina F; Zorn KM; Ekins S, Bioactivity Comparison across Multiple Machine Learning Algorithms Using over 5000 Datasets for Drug Discovery. Mol Pharm 2021, 18, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosc N; Atkinson F; Felix E; Gaulton A; Hersey A; Leach AR, Large scale comparison of QSAR and conformal prediction methods and their applications in drug discovery. J Cheminform 2019, 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nogueira MS; Koch O, The Development of Target-Specific Machine Learning Models as Scoring Functions for Docking-Based Target Prediction. J Chem Inf Model 2019, 59, 1238–1252. [DOI] [PubMed] [Google Scholar]
- 5.Imrie F; Bradley AR; van der Schaar M; Deane CM, Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J Chem Inf Model 2018, 58, 2319–2330. [DOI] [PubMed] [Google Scholar]
- 6.Shaikh N; Sharma M; Garg P, An improved approach for predicting drug-target interaction: proteochemometrics to molecular docking. Mol Biosyst 2016, 12, 1006–14. [DOI] [PubMed] [Google Scholar]
- 7.Mayr A; Klambauer G; Unterthiner T; Steijaert M; Wegner JK; Ceulemans H; Clevert DA; Hochreiter S, Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem Sci 2018, 9, 5441–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekins S; Puhl AC; Zorn KM; Lane TR; Russo DP; Klein JJ; Hickey AJ; Clark AM, Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 2019, 18, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekins S; Freundlich JS; Clark AM; Anantpadma M; Davey RA; Madrid P, Machine learning models identify molecules active against the Ebola virus in vitro. F1000Res 2015, 4, 1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekins S; de Siqueira-Neto JL; McCall LI; Sarker M; Yadav M; Ponder EL; Kallel EA; Kellar D; Chen S; Arkin M; Bunin BA; McKerrow JH; Talcott C, Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl Trop Dis 2015, 9, e0003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lane TR; Massey C; Comer JE; Anantpadma M; Freundlich JS; Davey RA; Madrid PB; Ekins S, Repurposing the antimalarial pyronaridine tetraphosphate to protect against Ebola virus infection. PLoS Negl Trop Dis 2019, 13, e0007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane TR; Comer JE; Freiberg AN; Madrid PB; Ekins S, Repurposing Quinacrine Against Ebola Virus Infection In vivo. Antimicrob Agents Chemother 2019, 63, e01142–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekins S; Lingerfelt MA; Comer JE; Freiberg AN; Mirsalis JC; O'Loughlin K; Harutyunyan A; McFarlane C; Green CE; Madrid PB, Efficacy of Tilorone Dihydrochloride against Ebola Virus Infection. Antimicrob Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhavoronkov A; Ivanenkov YA; Aliper A; Veselov MS; Aladinskiy VA; Aladinskaya AV; Terentiev VA; Polykovskiy DA; Kuznetsov MD; Asadulaev A; Volkov Y; Zholus A; Shayakhmetov RR; Zhebrak A; Minaeva LI; Zagribelnyy BA; Lee LH; Soll R; Madge D; Xing L; Guo T; Aspuru-Guzik A, Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019, 37, 1038–1040. [DOI] [PubMed] [Google Scholar]
- 15.Du F; Yu H; Zou B; Babcock J; Long S; Li M, hERGCentral: a large database to store, retrieve, and analyze compound-human Ether-a-go-go related gene channel interactions to facilitate cardiotoxicity assessment in drug development. Assay Drug Dev Technol 2011, 9, 580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nalepa J; Kawulok M, Selecting training sets for support vector machines: a review. Artificial Intelligence Review 2019, 52, 857–900. [Google Scholar]
- 17.Havlicek V; Corcoles AD; Temme K; Harrow AW; Kandala A; Chow JM; Gambetta JM, Supervised learning with quantum-enhanced feature spaces. Nature 2019, 567, 209–212. [DOI] [PubMed] [Google Scholar]
- 18.Fastovets DV; Bogdanov YI; Bantysh BI; Lukichev VF Machine learning methods in quantum computing theory. https://arxiv.org/abs/1906.10175 (accessed 2021-04-09) [Google Scholar]
- 19.Broughton M; Verdon G; McCourt T; Martinez AJ; Yoo JH; Isakov SV; Massey P; Niu MY; Halavati R; Peters E; Leib M; Skolik A; Streif M; Von Dollen D; McClean JR; Boixo S; Bacon D; Ho AK; Neven H; Mohseni M TensorFlow Quantum: A Software Framework for Quantum Machine Learning. https://arxiv.org/abs/2003.02989 (accessed 2021-04-09) [Google Scholar]
- 20.Langione M; Bobier J-F; Meier C; Hasenfuss S; Schulze U Will quantum computing transform biopharma R&D? https://www.bcg.com/publications/2019/quantum-computing-transform-biopharma-research-development.aspx (accessed 2021-04-09) [Google Scholar]
- 21.Schuld M, Machine learning in quantum spaces. Nature 2019, 567, 179–181. [DOI] [PubMed] [Google Scholar]
- 22.Sandfort F; Strieth-Kalthoff F; Kühnemund M; Beecks C; Glorius F, A Structure-Based Platform for Predicting Chemical Reactivity. Chem 2020, 6, 1379–1390. [Google Scholar]
- 23.Pattanaik L; Coley CW, Molecular Representation: Going Long on Fingerprints. Chem 2020, 6, 1204–1207. [Google Scholar]
- 24.Gawriljuk VO; Kyaw Zin PP; Foil DH; Bernatchez J; Beck S; Beutler N; Ricketts J; Yang L; Rogers T; Puhl AC; Zorn KM; Lane TR; Godoy AS; Oliva G; Siqueira-Neto JL; Madrid PB; Ekins S, Machine Learning Models Identify Inhibitors of SARS-CoV-2. bioRxiv 2020, 2020.June.16.154765. https://www.biorxiv.org/content/10.1101/2020.06.16.154765v1 (accessed 2021-04-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane T; Russo DP; Zorn KM; Clark AM; Korotcov A; Tkachenko V; Reynolds RC; Perryman AL; Freundlich JS; Ekins S, Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol Pharm 2018, 15, 4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ekins S; Freundlich JS; Reynolds RC, Are bigger data sets better for machine learning? Fusing single-point and dual-event dose response data for Mycobacterium tuberculosis. J Chem Inf Model 2014, 54, 2157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark AM; Dole K; Coulon-Spektor A; McNutt A; Grass G; Freundlich JS; Reynolds RC; Ekins S, Open Source Bayesian Models. 1. Application to ADME/Tox and Drug Discovery Datasets. J Chem Inf Model 2015, 55, 1231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark AM; Ekins S, Open Source Bayesian Models. 2. Mining a "Big Dataset" To Create and Validate Models with ChEMBL. J Chem Inf Model 2015, 55, 1246–60. [DOI] [PubMed] [Google Scholar]
- 29.Wootton JR Benchmarking near-term devices with quantum error correction https://arxiv.org/abs/2004.11037 (accessed 2021-04-09) [Google Scholar]
- 30.Landrum G RDkit. https://www.rdkit.org (accessed 2021-04-09) [Google Scholar]
- 31.Saki AA; Alam M; Ghosh S Study of Decoherence in Quantum Computers: A Circuit-Design Perspective https://arxiv.org/abs/1904.04323 (accessed 2021-04-09) [Google Scholar]
- 32.Paul LC; Suman AA; Sultan N, Methodological analysis of principal component analysis (PCA) method. Int J Comp Eng Management 2013, 16, 32–38. [Google Scholar]
- 33.Tharwat A; Gaber T; Ibrahim A; Aboul Ella H, Linear discriminant analysis: A detailed tutorial. AI Communications 2017, 30, 169–190. [Google Scholar]
- 34.Pérez-Salinas A; Cervera-Lierta A; Gil-Fuster E; Latorre JI, Data re-uploading for a universal quantum classifier. Quantum 2020, 4, 226. [Google Scholar]
- 35.Kingma DP; Ba J Adam: A method for stochastic optimization. https://arxiv.org/abs/1412.6980 (accessed 2021-04-09) [Google Scholar]
- 36.Anon Qiskit: An Open-source Framework for Quantum Computing. https://github.com/Qiskit/qiskit/blob/master/Qiskit.bib (accessed 2021-04-09)
- 37.Rebentrost P; Mohseni M; Lloyd S, Quantum support vector machine for big data classification. Phys Rev Lett 2014, 113, 130503. [DOI] [PubMed] [Google Scholar]
- 38.Abhijith J; Adedoyin A; Ambrosiano J; Anisimov P; Bärtschi A; Casper W; Chennupati G; Coffrin C; Djidjev H; Gunter D; Karra S; Lemons N; Lin S; Malyzhenkov A; Mascarenas D; Mniszewski S; Nadiga B; O'Malley D; Oyen D; Pakin S; Prasad L; Roberts R; Romero P; Santhi N; Sinitsyn N; Swart PJ; Wendelberger JG; Yoon B; Zamora R; Zhu W; Eidenbenz S; Coles PJ; Vuffray M; Lokhov AY Quantum algorithm implementations for beginners. https://arxiv.org/abs/1804.03719 (accessed 2021-04-09) [Google Scholar]
- 39.Anon Pennylane. https://pennylane.ai/ (accessed 2021-04-09) [Google Scholar]
- 40.Wang Y; Cheng T; Bryant SH, PubChem BioAssay: A Decade's Development toward Open High-Throughput Screening Data Sharing. SLAS Discov 2017, 22, 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.