

Abstract
A mechanistic understanding of Aβ aggregation and high-resolution structures of Aβ fibrils and oligomers are vital to elucidating relevant details of neurodegeneration in Alzheimer’s disease, which will facilitate the rational design of diagnostic and therapeutic protocols. The most detailed and reproducible insights into structure and kinetics have been achieved using Aβ peptides produced by recombinant expression, which results in an additional methionine at the N-terminus. While the length of the C-terminus is well established to have a profound impact on the peptide’s aggregation propensity, structure, and neurotoxicity, the impact of the N-terminal methionine on the aggregation pathways and structure is unclear. For this reason, we have developed a protocol to produce recombinant Aβ1-42, sans the N-terminal methionine, using an N-terminal SUMO-Aβ1-42 fusion protein in reasonable yield, with which we compared aggregation kinetics with AβM01-42 containing the additional methionine residue. The data revealed that Aβ1-42 and AβM01-42 aggregate with similar rates and by the same mechanism, in which the generation of new aggregates is dominated by secondary nucleation of monomers on fibrils surface. We also recorded MAS NMR spectra that demonstrated that excellent spectral resolution is maintained with both AβM01-42 and Aβ1-42 and that the chemical shifts are virtually identical in dipolar recoupling experiments that provide information about rigid residues. Collectively, these results indicate that the structure of the fibril core is unaffected by N-terminal methionine. This is consistent with the recent structures of AβM01-42 where the M0 is located at the terminus of a disordered 14 amino acid N-terminal tail.
INTRODUCTION
Amyloid fibril formation is associated with over 40 diseases1 including Alzheimer’s disease,2 Parkinson’s disease,3-6 type II diabetes7, 8, and dialysis-related amyloidosis.9, 10 Of these, Alzheimer’s disease (AD) is the most prevalent, currently affecting an estimated 5.5 million Americans according to the Alzheimer’s Association. By 2050 the patient population is expected to grow to 16 million people, providing an imperative to develop diagnostic and therapeutic protocols to address AD. Multiple lines of evidence indicate that the aggregation of β-amyloid (Aβ) peptide is integral to the disease pathogenicity. Aβ peptides are generated by the cleavage of the amyloid precursor protein (APP) by the membrane associated β- and γ-secretases,11, 12 yielding a variety of Aβ with different lengths, the two most common being Aβ1-40 (APP 672-711) and Aβ1-42 (APP 672-713). The former is more abundant and the latter is significantly more amyloidogenic and neurotoxic.13, 14 The difference between the two isoforms is the addition of I41 and A42 (APP 712-713), which dramatically alters the aggregation kinetics,15 neurotoxicity,16 and structure.17-19 Furthermore, it has recently been demonstrated that AβM01-40 and AβM01-42 do not cross seed one another, while each one is highly efficient in self-seeding, suggesting as well that fibrils of Aβ40 and Aβ42 possess different structures.17, 20
A more detailed understanding of Aβ in its various aggregated forms is vital to determining its pathogenicity and structure, both of which are necessary for rational design and development of diagnostic and therapeutic tools. To that end, substantial efforts have been devoted to understanding the details associated with the mechanism of aggregation as well as aggregate structure and how these features relate to toxicity 1, 21-23. Aβ fibrils are sparingly soluble and do not diffract to high resolution, rendering conventional tools for biological structure determination such as solution NMR spectroscopy and X-ray diffraction of limited utility in characterizing fibril samples. Fortunately, magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy has proven to be a powerful technique to elucidate the structural details of amyloid fibrils on an atomic level, including backbone conformations, supramolecular organization, and registry of inter-strand arrangements of amyloid fibrils.24-26
Because of their relatively short length, Aβ1-40 and Aβ1-42 peptides used in MAS NMR spectroscopy and in many other biophysical studies have historically been prepared via peptide synthesis. In the case of NMR experiments, this permits specific labeling of individual residues that was necessary because of the broad linewidths (2-4 ppm) observed in early spectra.27-29 However, the recent report that recombinant Aβ1-42 aggregates significantly faster and is more neurotoxic when compared with synthetic Aβ1-4216 has stimulated significant interest in developing recombinant methods for preparation. In particular, recombinant Aβ is more neurotoxic in cultured rat primary cortical neurons, and more toxic in vivo, as demonstrated by enhanced induction of abnormal phosphorylation of tau leading to tau aggregation into neurofibrillary tangles in brains of transgenic mice. 16 Recombinant Aβ1-42 is therefore likely more relevant to the pathogenicity of AD than is synthetic protein.16 The origin of the differences between synthetic and recombinant Aβ are not completely understood, but could be due to racemization and/or coupling errors that occur during synthesis leading to a large number of similar sequences. Such impurities are very difficult to separate. In contrast, recombinant expression in for example E. coli provides excellent fidelity with respect to both sequence homogeneity and chirality. Finally, for NMR structure experiments it is desirable to prepare uniformly 13C,15N labeled Aβ samples and the most cost effective approach to producing such samples is biosynthesis (vide infra). Thus, it is critical to have a robust biosynthetic method of preparation of Aβ for the many biophysical and structural studies of this important peptide.
Over the past decade, MAS NMR has become the method of choice to characterize amyloids including PI3-SH3,30, 31 β2-microglobulin,9, 32-34 α-synuclein3, 35-47 and Aβ.17, 19, 20, 48-52 To obtain an atomic level structure, it is necessary to complete spectral assignments of all residues and assemble an extensive set of distance constraints (>5 per residue). This typically requires a set of chemically identical peptides with different isotopic labeling to generate 2D and 3D correlation spectra combined with a series of dipolar recoupling experiments to generate inter- and intra-molecular constraints. Uniform 13C/15N labeling of proteins is easily performed when expressed recombinantly, and partial labeling is achieved with schemes utilizing 15NH4Cl as the source of 15N and 1,6-13C2-glucose, 1,3-13C2-glycerol, 2-13C1-glycerol, or REDOX labeling using 13C acetate53 as the 13C source. All of these approaches are feasible and have been found to yield inter- and intra-molecular constraints, and minimize dipolar truncation allowing for long distance correlation experiments.
Currently several methods exist to produce recombinant Aβ containing an N-terminal methionine. Its small size and its propensity to aggregate permit direct expression resulting in the formation of inclusion bodies. This approach has been widely used to prepare Aβ due to the ease of production and purification.54 Although this method is effective, it leaves an N-terminal methionine (M0) residue whose impact on the aggregation of Aβ and the fibril structure is as yet unknown. Peptides cleaved just before M671 have to our knowledge not been observed in human samples, even though in APP, M671 precedes D672, numbered D1 in the released Aβ peptides, many variants with extended N-termini are found in blood and CSF 55, 56 Furthermore, while N-terminal extension of Aβ42 with 5-40 residues from APP has been found to retard aggregation in a manner proportional to the length of the extension57 the effect of the N-terminal methionine in AβM01-42 on the aggregation mechanism and rate remains to be found.
AβM01-40, AβM01-42, as well as the N-terminally extended variants all aggregate by the same general mechanism, which relies on at least three microscopic steps: primary nucleation of monomers in solution, elongation by monomer addition at fibril ends and secondary nucleation of monomers on fibril surfaces.15, 57, 58 The difference between the Aβ length variants lies in the rate constants of these three processes and one could imagine that the N-terminal methionine may have an impact on some rate constants or produce structurally different Aβ fibrils from those produced sans the N-terminal methionine. One can avoid the presence of the N-terminal methionine by utilizing an N-terminal fusion protein, and several methods have been reported which utilize various fusion proteins in recombinant expression of Aβ.59 These include glutathione-S-transferase,60 ubiquitin,61 intestinal fatty acid binding protein (IFABP)62, hen-egg lysozyme63 and NANP.64 Although these preparations were used previously, they were typically for Aβ1-40 and the yields reported make it challenging to generate sufficient material to fill an MAS NMR rotor (~30 mgs). Recently a preparation using SUMO (small ubiquitin-like modifier) as an N-terminal fusion protein was reported, which possessed excellent protein overexpression yields in rich media.65, 66
Here, we chose to utilize this construct to overexpress the SUMO-Aβ1-42 fusion protein in isotopically enriched minimal media and have modified the purification to produce sufficient quantities of Aβ monomers for kinetic experiments and subsequently for fibrils for MAS NMR spectra. Measurements of the aggregation kinetics revealed only very small differences between AβM01-42 and Aβ1-42, all rate constants are within a factor of 2.5, and this along with the ability of AβM01-42 and Aβ1-42 to cross seed each other, suggest that both the mechanism of aggregation and fibril structure are highly similar. For more detailed structural comparison, we compared the MAS NMR spectra of AβM01-42 with Aβ1-42 and found few chemical shift differences among residues in the fibril core. Thus, the presence of an additional methionine on the N-terminus has little impact on the aggregation rate, or fibril core structure as ascertained by MAS NMR. More explicitly, these findings indicate that the mechanism of aggregation is likely similar, and that the microscopic structure of the amyloid core is identical.
EXPERIMENTAL
Preparation of SUMO-Hydroxlyase.
The expression and purification of His6-tagged SUMO-1 protease (ULP-1-His6) has been described in detail previously,67 and is summarized below. The plasmid coding for ULP-1-His6 was kindly provided by Dr. Jens Tyedmers (University Heidelberg, Germany). ULP-1-His6 was overexpressed in E. coli BL21 (DE3) by inoculating 100 mL of LB medium by a single colony and was grown overnight at 30°C. 5 mL of the overnight culture was added to 1 L of LB broth and grown at 30°C until an OD600=1.0 was reached. Subsequently the temperature was lowered to 20°C followed by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) one hour after temperature change. Expression were conducted over night and cells were subsequently harvested and stored at −80°C. The pellet was subjected to two freeze-thaw cycles (frozen in liquid N2 and thawed in a 45°C water bath) and resuspended in a buffer containing 40 mM HEPES/KOH pH 7.4, 150 mM KCl and 20 mM β-mercaptoethanol (buffer A), followed by addition of 10 mg lysozyme, 0.5 mg of DNAse, and an EDTA-free Complete® tablet. The pellet was sonicated with a tip sonicator (Branson 450 sonifier) 50 % duty cycle, 2 min followed by 2 min rest (2x) and pelleted by centrifugation for 20 min at 20.000g. The supernatant was added to 5 mL of Ni-NTA resin (GE Healthcare) and subsequently washed with 150 ml buffer A and eluted with buffer B (buffer A + 250 mM imadizole). Fractions containing protein were pooled. The eluted protein was placed into 10 kDa snakeskin dialysis tube and left to dialyze for 2 days at 4°C in 40 mM HEPES/KOH pH 7.4, 100 mM KCl, and 10 mM β-mercaptoethanol with 3 buffer changes every 12 h, 4 L each. Following dialysis, the sample was concentrated using a 10 kDa centrifugal concentrator to a final volume of 2 mL with an equal volume of glycerol added, and 100 μL aliquots were stored at −80°C until used.
Preparation of Aβ1-42.
DNA coding for SUMO and Aβ was extracted from commercial vectors and prepared by de novo gene synthesis, respectively, and overlapped via PCR to generate a chimeric construct of SUMO fused with Aβ1-42 containing the gateway handles (e.g. attR1 and attR2). The DNA coding for SUMO-Aβ1-42 was inserted into a pDEST17 vector containing an N-terminal His6 tag via gateway cloning and transformed into BL21 Star (DE3) pLysS cells (Invitrogen), individual colonies were sent for DNA sequencing, and appropriate colonies were cultured overnight in 5 mL of LB broth (100 μg/mL ampicillin and 38 μg/mL of chloramphenicol), with glycerol stocks prepared the following morning. 200 μL of the glycerol stock was plated onto LB agar containing 100 μg/mL ampicillin and 38 μg/mL of chloramphenicol and grown overnight at 37°C. Several single colonies were used to inoculate 50 mL LB medium containing 100 μg/mL ampicillin and 38 μg/mL of chloramphenicol and cultured over night at 37°C. Subsequently, 1 ml of each over-night culture was used to inoculate 500 mL of M9 medium consisting of 2 g/L glucose, 1 g/L NH4Cl, 2 mM MgSO4, 100 μM CaCl2, 10 mg/L thiamine, 10 mg/L of FeSO4, 100 μg/mL ampicillin, and 38 μg/mL of chloramphenicol. Cells were grown at 37°C until an OD600 of 0.8 was reached, at which time overexpression was induced with IPTG. Cells were allowed to express at 37°C for four hours, and were subsequently harvested by centrifugation and stored at −80°C. Typically, a final OD600 of 1.8-2.0 was reached prior to pelleting.
Cell pellets were thawed and resuspended in 10 mM Tris pH 8.0, 1 mM EDTA (buffer C) and sonicated (Branson Sonifier 450) on ice for 2 minutes and pelleted for 10 minutes at 15,000g. The supernatant was removed, the pellet was resuspended in buffer C and sonicated three additional times, for times of 30 seconds, 20 seconds and 15 seconds, respectively, each sonication was followed by centrifugation for five minutes. Subsequently the remaining pellet was dissolved in lysis buffer (buffer C + 8 M urea) and sonicated for 2 minutes followed by 2 minutes rest (3x), followed by centrifugation at 15,000g for 15 minutes. The supernatant was decanted and subsequently diluted with buffer C by a factor of 4 to yield 2 M urea and added to 30 mL of DEAE cellulose. The protein was allowed to bind for 30-60 min, and the resin was subsequently loaded into a column. The column was then washed with 50 mL of buffer C + 2 M urea with increasing amounts of NaCl: 10 mM (2x), 25 mM (2x), 75 mM (1x), 200 mM (4x), and 500 mM (1x). We found that SUMO-Aβ1-42 elutes at 200 mM NaCl. The eluate was concentrated by a factor of four in a centrifugal concentrator (3.5 kDa cutoff), followed by dilution by a factor of four with buffer C, to yield 0.5 M urea in buffer C. A representative yield of the SUMO-Aβ fusion protein is ~100 mg/L M9 minimal medium. ULP-1 was added immediately after dilution and the fusion protein allowed to cleave for at least two hours at room temperature, followed by lyophylization. The lyophilized material was dissolved in 6 M GuHCl and subjected to size exclusion chormatography (GE Superdex 26/600 75pg) and eluted with buffer containing 20 mM sodium phosphate pH 8.0, 0.2 mM EDTA to isolate monomeric Aβ1-42. The monomer solution contained residual SUMO domain and was therefore lyophylized, dissolved in 6 M GuHCl and subjected to size exclusion chromatography one more time. Fibrils were formed by seeding with AβM01-42.
Aggregation kinetics by thioflavin T fluorescence.
The monomer from above was lyophilized, dissolved in 6 M GuHCl and subjected to a third round of size exclusion chormatography on a gel filtration column (GE Superdex 10/300 75pg) in 2 buffer containing 20 mM sodium phosphate pH 8.0, 0.2 mM EDTA to isolate pure monomer. The monomer was diluted with the same buffer to prepare a series of samples with peptide concentration ranging from 1 to 6 μM. All samples contained 10 μM thioflavin T. The experiment was initiated by placing the 96-well plate at 37°C in a plate reader (Fluostar Omega, Optima or Galaxy from BMG Labtech, Offenburg, Germany). The ThT fluorescence was measured through the bottom of the plate every 60 or 120 seconds (with excitation filter 440 nm, and emission filter 480 nm). The data were analyzed globally using the AmyloFit interface.68 with the equations described in references 58 and 68
MAS NMR spectroscopy.
Fully formed (mature) fibrils were packed into a 3.2 mm Bruker rotor using a home-built centrifugal packing tool. Spectra were recorded on a Bruker 800 MHz AVANCE III spectrometer equipped with a 3.2 mm triple channel HCN Bruker probe (Bruker Biospin, Billerica, MA). Spectra were recorded at ωr/2π = 20 kHz and regulated to ±10 Hz using a Bruker spinning frequency controller. All experiments were conducted at 277 K. Spectra were processed using TopSpin 3.1, and analyzed in Sparky.69
RESULTS AND DISCUSSION
Preparation of Aβ1-42.
The SUMO fusion protein was placed N-terminally of Aβ1-42 to eliminate the M0 residue present when directly expressed in E. coli. Most of the SUMO-Aβ fusion protein is in the supernatant following lysis, and that it can be effectively separated using DEAE cellulose resin (data not shown). The SUMO fusion protein is cleaved by SUMO-1 protease (ULP-1-His6, Figure 1) which recognizes and cleaves SUMO based on SUMO’s tertiary structure,67 necessitating that SUMO be refolded following lysis. We found that SUMO-Aβ has a propensity to aggregate in the absence of a denaturant and therefore the SUMO-Aβ fusion protein was cleaved with 0.5 M urea present, the highest concentration of urea that would permit reasonable cleavage yields (data not shown).
Figure 1.
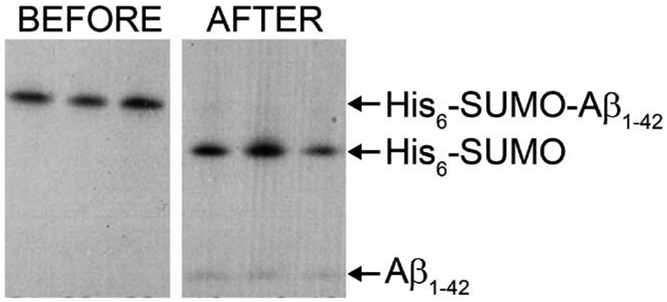
SDS-PAGE gel showing the cleavage with ULP-1-His6. The cleavage is complete within 24 hours with 0.5 M urea present. The gel was stained using Coomassie Brilliant Blue. The three lanes in each case are repetitions of the same sample.
Following cleavage, Aβ1-42 was separated from SUMO using size exclusion chromatography in an identical manner to the purification employed before preparing AβM01-42 fibrils.17, 54 The yield of Aβ1-42 in M9 minimal medium was comparable with other protocols found in the literature and was about 20 mg/liter prior to gel filtration, which was sufficient to produce enough amyloid fibrils for characterization by MAS NMR spectroscopy. With further optimization of the purification we anticipate an increase in the yield of fusion protein and are currently exploring additional purification options to enhance the yield of Aβ.
Aggregation kinetics of Aβ1-42.
Measuring the aggregation kinetics, which are typically monitored by thioflavin T fluorescence, followed by global fitting of the data68 can elucidate the mechanistic details of aggregation. Recently, the aggregation of AβM01-42 was shown to proceed through a secondary nucleation step, suggesting a positive feedback loop between monomers and already formed fibrils.58 Here we examined the kinetics and concentration dependence of Aβ1-42 aggregation to evaluate mechanistic similarities or differences between AβM01-42 and Aβ1-42. Examples of aggregation kinetics data for Aβ1-42 are shown in Figure 2. The experiment was repeated three times with similar results. In comparison with previous data for AβM01-42, we find that the removal of M0 retains the shape of individual aggregation traces (Figure 2A) and produces a highly similar concentration dependence of the half time for fibril formation (Figure 2B), implying that fibrillization of both species (AβM01-42/Aβ1-42) occurs by a similar mechanism.70 Fitting of a power function to the concentration dependence of the half time yields an exponent of −1.3, in agreement with earlier findings in reference 58. The AmyloFit interface68 was used to fit various kinetic models in the form of master equations to the ThT fluorescence data for fibril formation versus time for all peptide concentrations globally. While models including only primary nucleation and elongation, or primary nucleation, elongation and fragmentation failed to fit the data, as previously found for AβM01-42,58 a model including primary nucleation, elongation and secondary nucleation produced acceptable global fits to the data as shown in Figure 2A. This analysis further revealed that all rate constants are within a factor of 2.5 when the two peptides are compared, and there is a slightly higher dominance of the secondary pathway in the shorter peptide. Still the difference between the two peptides is so small that the main finding is that there is no major change in mechanism or rate constants. For the product rate constant k+k2, we obtain a value of 1 x 1011 M−3s−2 for Aβ1-42 (vs. 4 x 1010 M−3s−2 for AβM01-42)58 while we obtain similar values for the combined rate constant k+kn for both peptides (900 M−2s−2). As a comparison, removing two residues at the C-terminus leads to a 1800-fold reduction in k+kn and a 44-fold reduction in k+k2.15 However, the minor difference in aggregation kinetics between Aβ1-42 and AβM01-42 does not provide information about the atomic level structure of the two fibrils.
Figure 2.

Kinetics monitored by thioflavin T at 37°C. A. Examples of aggregation data at 11 peptide concentrations in quadruplicate together with a global fit k+kn =900 M−2s−2; k+k2 = 1·1011 M−3s−2). B. Half time of aggregation versus peptide concentration combined from three experiments of the type shown in panel A, plus a fitted power function with exponent −1.3.
Fibrils of Aβ1-42 and AβM01-42 have rigid and dynamic regions.
To compare the structures of AβM01-42 and Aβ1-42, we therefore recorded MAS NMR spectra to determine what impact, if any, the N-terminal methionine has on the microscopic structure of Aβ42 fibrils. Amyloid fibrils of AβM01-42 have a rigid amyloid core that sequesters residues Q15-A42 into parallel in register beta sheets while residues M0-H14 are less ordered.17 Overall, removal of the M0 did not result in striking change in the rigid regions of these fibrils.
We collected 1D 13C and 15N cross-polarization spectra that were recorded at ω0H/2π=800 MHz. We note that the 13C cross polarized spectrum (Figure 3A) of Aβ1-42 is slightly more well resolved than AβM01-42, possibly due to a more compact core structure, but is otherwise comparable with respect to chemical shifts. Similarly, the amide region of the 15N cross polarization spectrum of Aβ1-42 (Figure 3C) is similar to that of AβM01-42. However, there are some differences in the side chains. The lysine signal for Aβ1-42 fibrils consists of two lines with one more intense than the other and the arginine resonances are more intense than they are in AβM01-42 fibrils. In the case of the lysine signals we assigned the more intense component to K28 and the latter to K16 that is difficult to observe in AβM01-42 in 2D and 3D spectra.17 Similar statements are applicable to the R5 resonances in AβM01-42 where we observed that the Nℇ and Nη1 and Nη2 are attenuated indicating that R5 is more dynamic on a ns-μs time-scale in AβM01-42. Finally, the 15N-His resonances at ~180 ppm are broadened probably due to exchange and dynamics on a ns-μs time-scale. We note that the histidine 15N tensors are large71 and therefore sensitive to both to the dynamics and protonation state of the histidine side-chain. To visualize any dynamic regions of these amyloid fibrils, we recorded a 1D INEPT spectrum (Figure 3B), which appears to have more intense signals than observed previously, due to the absence of the N-terminal methionine, which had a very efficient transfer based on J-couplings. The increase in spectral intensity in the 13C aromatic region is probably due to F4 (vide infra).
Figure 3:

1D MAS NMR spectra of Aβ1-42 (red) and AβM01-42 (black) recorded at ω0H/2π= 800 MHz at 277 K and ωr/2π =20 kHz with 83 kHz 1H decoupling during acquisition. (a) 13C cross polarization 1D spectrum recorded with 512 transients. (b) 13C-INEPT spectrum recorded with 1024 transients. (c) 15N cross polarization 1D spectrum recorded with 512 transients.
The fibrils of AβM01-42 are structurally heterogeneous - the N-terminal 15 residues (M0-H14) are flexible and show only weak resonances in dipolar recoupling experiments, while the C-terminal core (Q15-A42) forms a rigid uniform structure. Initial studies suggested that fibrils of Aβ1-42 may have both a slightly expanded rigid region, particularly with respect to the protein side chains. Moreover, removal of the first methionine residue appears to result in greater mobility in the N-terminal tail as evidenced by the increase in the chemical shifts and intensity of the J-coupled spectra. Thus, we will use homo and heteronuclear correlation spectroscopy to determine the differences in both the amyloid core and mobile N-terminal tail between Aβ1-42 and AβM01-42.
The rigid core structures of Aβ1-42 and AβM01-42 fibrils are identical.
To further explore the similarities and differences between the fibril core of Aβ1-42 and AβM01-42 we recorded 13C-13C RFDR72 and 13C-15N ZF-TEDOR73 (Figure 4). We collected these spectra using short dipolar mixing times and these spectra report on one and two bond correlations. To probe for longer-range correlations we also collected a 13C-13C DARR74 spectrum with a longer mixing time (Figure 5). We have recently published a list of assignments for AβM01-4217 and used spectra recorded on that sample as a baseline to compare with the Aβ1-42 fibrils discussed herein. In the Aβ1-42 spectra we observed a single set of chemical shifts indicating that only one fibril form is present. Polymorphism were not observed for the fibril core, consistent with our previous observations.17 Furthermore the chemical shifts were virtually identical in Aβ1-42 and AβM01-42 indicating that the fibril core is unchanged by the presence of the N-terminal methionine. However, we observed subtle differences between the two fibrils; we found that 1) some signals were much stronger in Aβ1-42 than in AβM01-42, 2) minor chemical shift perturbations and 3) a small number of additional peaks in the spectra of Aβ1-42 fibrils. The resonance signals in Aβ1-42 that most prominently exhibited increased intensity were A21, E22 and D23 (Figure 4). These peaks were considerably weaker in spectra of AβM01-42. Interestingly, these residues reside in the ‘toxic corner’ of the recently published structure of AβM01-42 that showed increased flexibility for these residues compared to the rest of the fibril core. However, it is not clear if these intensity variations are due to the presence or absence of the N-terminal methionine or if these occur for some other reason such as minor variations in pH or ionic strength during fibril growth.
Figure 4:

(A) 1.6 ms mixing RFDR spectra of Aβ1-42 (red) and AβM01-42 (black) recorded at 800 MHz, ωr/2π=20 kHz, VT gas regulated to 277 K with 83 kHz CW 1H decoupling during evolution, and 83 kHz TPPM 1H decoupling during acquisition. (B) 1.6 ms mixing ZF-TEDOR spectra of Aβ1-42 (red) and AβM01-42 (black) recorded at 800 MHz, ωr/2π=20 kHz, VT gas regulated to 277K with 83 kHz TPPM during acquisition.
Figure 5.

2D 13C-13C-T DARR of Aβ1-42 (red) and AβM01-42 (blue) recorded at T= 277K, ω0H/2π= 800 MHz, ωr/2π= 20 kHz, and τmix(DARR) = 100 ms. A 83 kHz 1H decoupling field was applied during acquisition.
Furthermore, most resonances of the fibril core show no or only minor chemical shift perturbations (CSP) between the AβM01-42 and Aβ1-42 and the results of the chemical shift analysis are summarized pictorially in Figure 6. The most prominent difference is at K16 and amounts to ~0.37 ppm and V36 and which is 0.23 ppm. K16 is the residue that is directly adjacent to the beginning of the disordered region in AβM01-42 fibrils (Figure 6B, red). Because the other differences were smaller than 0.15 ppm, which is less than the error in our experiments, we conclude that M0 does not have a significant effect on the structure of the Aβ1-42 core.
Figure 6:

(A) 13C chemical shift perturbations (CSP) between AβM01-42 and Aβ1-42 plotted as a function of residue position. The only residues where the shifts differ significantly are K16 and V36. (B) Structure of AβM01-42 illustrating the positions of K16 and V36 and other residues with 0.1<CSP<0.2.
Interestingly, there are several peaks present in the 13C-13C RFDR and 13C-15N ZF-TEDOR (Figure 4), as well as 13C-13C DARR spectra (Figure 5) that do not originate from chemical shift perturbations or increased rigidity. Most likely, these resonances belong to residues of the N-terminal tail in a second, more rigid conformation as evidenced by a second resonance in the 3C-13C RFDR and 13C-15N ZF-TEDOR, as well as 13C-13C DARR spectra at 56-62 ppm most likely belonging to S8. This observation is in agreement with studies on Aβ1-42 by Meier and co-workers that show polymorphism in the absence of zinc during fibrillization.19
The N-terminal tail of Aβ1-42.
Having determined that the N-terminal methionine does not significantly impact the fibril core, we next sought to determine what difference, if any, would be observed on the N-terminus, which is known to be flexible. We recorded a 2D INEPT-13C-13C TOBSY75 (Figure 5) spectrum. Leaving aside the obvious difference that results from the additional methionine in AβM01-42, the most apparent differences were for the aromatic resonances of F4, which were more intense in spectra of Aβ1-42 fibers than in AβM01-42 fibrils. (Figure 7, see also Figure 3). As indicated in the INEPT spectra (Figure 3), the INEPT-TOBSY spectra demonstrate that Aβ1-42 has several flexible residues, and that some, but not all of these residues are distinct from those in AβM01-42. Overall this suggests that M0 may change the mobility of the flexible N-terminus by predisposing this region to sample a variety of transient interactions with the amyloid core that are non-specifically driven by the hydrophobicity of the methionine side chain. When the methionine is removed, the energetically preferred interaction point between the flexible tail and the core uses F4 as a tether point, leading to a small number of additional peaks in the dipolar–selective spectra.
Figure 7.

2D 13C-13C-TOBSY of Aβ1-42 (red) and AβM01-42 (blue) recorded at T= 277K, ω0H/2π= 800 MHz, ωr/2π= 20 kHz, and τmix(TOBSY) = 9.6 ms. A 83 kHz 1H decoupling field was applied during acquisition.
Conclusions
We report a new preparation method to produce and purify Aβ1-42 in sufficient yield to produce samples for MAS NMR spectroscopy. Monomers isolated by size exclusion chromatography can be incubated to yield fibrils of a monomorphic fibril core that exhibits excellent resolution and permitted us to determine a structure whose core is identical to that of AβM01-42. In particular, by comparing the MAS NMR spectra of Aβ1-42 with and without an N-terminal methionine, we determined that this residue has little impact on the structure of the fibril’s core, as the chemical shifts are nearly identical between the two different species. Yet, despite this striking structural conservation, we found that the deletion of the N-terminal methionine residue changes these fibers in subtle ways. Aβ1-42 fibers are more rigid than AβM01-42 fibers, which is seen as a small increase in the number of interactions (a handful of new peaks become visible) and a change in the dynamics of some regions of the core (increased peak intensities, particularly in the “toxic corner” of the fibril). Because Aβ1-42 and AβM01-42 follow the same aggregation mechanism with highly similar rate constants and because these dynamic differences are minor, structural studies that use an AβM01-42 should be generalizable to Aβ1-42. However, investigations of the role of the disordered regions and their possible interactions should consider M0 as a variable.
Acknowledgement
This work was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institute of Health under grants EB-001960 and EB-002026 (to RGG), NS087557 (to SLL), and the Swedish Research Council (VR) and a European research council (ERC) Advanced Grant (to SSL). R.S. is funded by a DFG research fellowship (SI2105/1-1).
REFERENCES
- [1].Chiti F, and Dobson C (2006) Protein misfolding, functional amyloid, and human disease., Annu Rev Biochem 75, 333–366. [DOI] [PubMed] [Google Scholar]
- [2].Dobson CM (2006) Protein aggregation and its consequences for human disease, Protein Peptide Lett 13, 219–227. [DOI] [PubMed] [Google Scholar]
- [3].Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, Woods WS, Lipton AS, George JM, and Rienstra CM (2011) Structured Regions of alpha-Synuclein Fibrils Include the Early-Onset Parkinson's Disease Mutation Sites, J Mol Biol 411, 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, and Baldus M (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR, Proc. Natl. Acad. Sci. U.S.A 102, 15871–15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Olanow CW, and Brundin P (2013) Parkinson's Disease and Alpha Synuclein: Is Parkinson's Disease a Prion-Like Disorder?, Movement Disord 28, 31–40. [DOI] [PubMed] [Google Scholar]
- [6].Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, and Riek R (2008) The fold of alpha-synuclein fibrils, Proc. Natl. Acad. Sci. U.S.A 105, 8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Marzban L, Park K, and Verchere CB (2003) Islet amyloid polypeptide and type 2 diabetes, Exp Gerontol 38, 347–351. [DOI] [PubMed] [Google Scholar]
- [8].Westermark P, Andersson A, and Westermark GT (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus, Physiol Rev 91, 795–826. [DOI] [PubMed] [Google Scholar]
- [9].Debelouchina GT, Platt GW, Bayro MJ, Radford SE, and Griffin RG (2010) Intermolecular Alignment in beta(2)-Microglobulin Amyloid Fibrils, J Am Chem Soc 132, 17077–17079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Debelouchina GT, Platt GW, Bayro MJ, Radford SE, and Griffin RG (2010) Magic Angle Spinning NMR Analysis of beta(2)-Microglobulin Amyloid Fibrils in Two Distinct Morphologies, J Am Chem Soc 132, 10414–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, and Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein, Nature 391, 387–390. [DOI] [PubMed] [Google Scholar]
- [12].Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller L, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, and Citron M (1999) beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE, Science 286, 735–741. [DOI] [PubMed] [Google Scholar]
- [13].Jarrett JT, Berger EP, and Lansbury PT (1993) The Carboxy Terminus of the Beta-Amyloid Protein Is Critical for the Seeding of Amyloid Formation - Implications for the Pathogenesis of Alzheimers-Disease, Biochemistry-Us 32, 4693–4697. [DOI] [PubMed] [Google Scholar]
- [14].Jarrett JT, Berger EP, and Lansbury PT Jr. (1993) The C-terminus of the beta protein is critical in amyloidogenesis, Ann N Y Acad Sci 695, 144–148. [DOI] [PubMed] [Google Scholar]
- [15].Meisl G, Yang XT, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SIA, Dobson CM, Linse S, and Knowles TPJ (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the A beta 40 and A beta 42 peptides, Proc. Natl. Acad. Sci. U.S.A 111, 9384–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Finder VH, Vodopivec I, Nitsch RM, and Glockshuber R (2010) The Recombinant Amyloid-beta Peptide A beta 1-42 Aggregates Faster and Is More Neurotoxic than Synthetic A beta 1-42, J Mol Biol 396, 9–18. [DOI] [PubMed] [Google Scholar]
- [17].Colvin MT, Silvers R, Frohm B, Su Y, Linse S, and Griffin RG (2015) High Resolution Structural Characterization of Aβ42 Amyloid Fibrils by MAS NMR, J. Am. Chem. Soc 137, 7509–7518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, and Griffin RG (2016) Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils, J Am Chem Soc 138, 9663–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Guüntert P, Meier BH, and Riek R (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril, Proc. Natl. Acad. Sci. U.S.A 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cukalevski R, Yang X, Meisl G, Weininger U, Bernfur K, Birgitta Frohm, Knowles TPJ, and Linse S (2015) The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation, Chemical Science 6, 4215–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aguzzi A, and O’Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives, Nat. Rev. Drug Discov 9, 237–248. [DOI] [PubMed] [Google Scholar]
- [22].Haass C, and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide, Nat. Rev. Mol. Cell Biol 8, 101–112. [DOI] [PubMed] [Google Scholar]
- [23].Koo EH, Lansbury PTJ, and Kelly JW (1999) Amyloid diseases: abnormal protein aggregation in neurodegeneration, Proc. Natl. Acad. Sci. U.S.A 96, 9989–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tycko R (2011) Solid-State NMR Studies of Amyloid Fibril Structure, Annu Rev Phys Chem 62, 279–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Debelouchina GT, Bayro MJ, Fitzpatrick AW, Ladizhansky V, Colvin MT, Caporini MA, Jaroniec CP, Bajaj VS, Rosay M, MacPhee CE, Vendruscolo M, Maas WE, Dobson CM, and Griffin RG (2013) Higher Order Amyloid Fibril Structure by MAS NMR and DNP Spectroscopy, J Am Chem Soc 135, 19237–19247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tanzi RE, and Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective, Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- [27].Lansbury PT, Costa PR, Griffiths JM, Simon EJ, Auger M, Halverson KJ, Kocisko DA, Hendsch ZS, Ashburn TT, Spencer RGS, Tidor B, and Griffin RG (1995) Structural model for the [beta]-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a C-terminal peptide, Nature structural & molecular biology 2, 990–998. [DOI] [PubMed] [Google Scholar]
- [28].Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, and Tycko R (2000) Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta-sheets in Alzheimer's beta-amyloid fibrils, Proc. Natl. Acad. Sci. U.S.A 97, 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, and Tycko R (2002) A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR, Proc. Natl. Acad. Sci. U.S.A 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bayro MJ, Maly T, Birkett N, MacPhee C, Dobson CM, and Griffin RG (2010) High-resolution MAS NMR analysis of PI3-SH3 amyloid fibrils: Backbone conformation and implications for protofilament assembly and structure, Biochemistry-Us 49, 7474–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, and Griffin RG (2011) Intermolecular structure determination of amyloid fibrils with magic-angle spinning, dynamic nuclear polarization NMR, J. Am. Chem. Soc 133, 13967–13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Debelouchina GT, Platt GW, Bayro MJ, Radford SE, and Griffin RG (2010) Intermolecular Alignment in β2-Microglobulin Amyloid Fibrils, J Am Chem Soc 132, 17077–17079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sarell CJ, Woods LA, Su Y, Debelouchina GT, Ashcroft AE, Griffin RG, Stockley PG, and Radford SE (2013) Expanding the repertoire of amyloid polymorphs by co-polymerization of related protein precursors, J. Biol. Chem 288, 7327–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Su YC, Sarell CJ, Eddy MT, Debelouchina GT, Andreas LB, Pashley CL, Radford SE, and Griffin RG (2014) Secondary Structure in the Core of Amyloid Fibrils Formed from Human beta(2)m and its Truncated Variant Delta N6, J Am Chem Soc 136, 6313–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Comellas G, Lemkau LR, Zhou DHH, George JM, and Rienstra CM (2012) Structural Intermediates during alpha-Synuclein Fibrillogenesis on Phospholipid Vesicles, J Am Chem Soc 134, 5090–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gath J, Bousset L, Habenstein B, Melki R, Meier BH, and Bockmann A (2014) Yet another polymorph of alpha-synuclein: solid-state sequential assignments, Biomolecular NMR Assignments 8, 395–404. [DOI] [PubMed] [Google Scholar]
- [37].Gath J, Habenstein B, Bousset L, Melki R, Meier BH, and Bockmann A (2012) Solid-state NMR sequential assignments of alpha-synuclein, Biomolecular NMR Assignments 6, 51–55. [DOI] [PubMed] [Google Scholar]
- [38].Kloepper KD, Hartman KL, Ladror DT, and Rienstra CM (2007) Solid-state NMR spectroscopy reveals that water is nonessential to the core structure of alpha-synuclein fibrils, Journal of Physical Chemistry B 111, 13353–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kloepper KD, Woods WS, Winter KA, George JM, and Rienstra CM (2006) Preparation of alpha-synuclein fibrils for solid-state NMR: Expression, purification, and incubation of wild-type and mutant forms, Protein Expres Purif 48, 112–117. [DOI] [PubMed] [Google Scholar]
- [40].Kloepper KD, Zhou DH, Li Y, Winter KA, George JM, and Rienstra CM (2007) Temperature-dependent sensitivity enhancement of solid-state NMR spectra of alpha-synuclein fibrils, Journal of Biomolecular NMR 39, 197–211. [DOI] [PubMed] [Google Scholar]
- [41].Lemkau LR, Comellas G, Kloepper KD, Woods WS, George JM, and Rienstra CM (2012) Mutant Protein A30P alpha-Synuclein Adopts Wild-type Fibril Structure, Despite Slower Fibrillation Kinetics, J. Biol. Chem 287, 11526–11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lemkau LR, Comellas G, Lee SW, Rikardsen LK, Woods WS, George JM, and Rienstra CM (2013) Site-Specific Perturbations of Alpha-Synuclein Fibril Structure by the Parkinson's Disease Associated Mutations A53T and E46K, Plos One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Woods WS, Boettcher JM, Zhou DH, Kloepper KD, Hartman KL, Ladror DT, Qi Z, Rienstra CM, and George JM (2007) Conformation-specific binding of alpha-synuclein to novel protein partners detected by phage display and NMR spectroscopy, J. Biol. Chem 282, 34555–34567. [DOI] [PubMed] [Google Scholar]
- [44].Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, and Melki R (2013) Structural and functional characterization of two alpha-synuclein strains, Nat Commun 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lv GH, Kumar A, Giller K, Orcellet ML, Riedel D, Fernandez CO, Becker S, and Lange A (2012) Structural Comparison of Mouse and Human alpha-Synuclein Amyloid Fibrils by Solid-State NMR, J Mol Biol 420, 99–111. [DOI] [PubMed] [Google Scholar]
- [46].Mollenhauer B, Trautmann E, Otte B, Ng J, Spreer A, Lange P, Sixel-Doring F, Hakimi M, VonSattel JP, Nussbaum R, Trenkwalder C, and Schlossmacher MG (2012) alpha-Synuclein in human cerebrospinal fluid is principally derived from neurons of the central nervous system, J Neural Transm 119, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim HY, Cho MK, Kumar A, Maier E, Siebenhaar C, Becker S, Fernandez CO, Lashuel HA, Benz R, Lange A, and Zweckstetter M (2009) Structural Properties of Pore-Forming Oligomers of alpha-Synuclein, J Am Chem Soc 131, 17482–17489. [DOI] [PubMed] [Google Scholar]
- [48].Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, and Smith SO (2010) Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils, Nature structural & molecular biology 17, 561–U556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fandrich M, Schmidt M, and Grigorieff N (2011) Recent progress in understanding Alzheimer's beta-amyloid structures, Trends in Biochemical Sciences 36, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Miller Y, Ma BY, Tsai CJ, and Nussinov R (2010) Hollow core of Alzheimer's A beta(42) amyloid observed by cryoEM is relevant at physiological pH, Proc. Natl. Acad. Sci. U.S.A. 107, 14128–14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang R, Hu XY, Khant H, Ludtke SJ, Chiu W, Schmid MF, Frieden C, and Lee JM (2009) Interprotofilament interactions between Alzheimer's A beta(1-42) peptides in amyloid fibrils revealed by cryoEM, Proc. Natl. Acad. Sci. U.S.A 106, 4653–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tay WM, Huang DT, Rosenberry TL, and Paravastu AK (2013) The Alzheimer's Amyloid-beta(1-42) Peptide Forms Off-Pathway Oligomers and Fibrils That Are Distinguished Structurally by Intermolecular Organization, J Mol Biol 425, 2494–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Eddy MT, Belenky M, Sivertsen AC, Griffin RG, and Herzfeld J (2013) Selectively dispersed isotope labeling for protein structure determination by magic angle spinning NMR, Journal of Biomolecular NMR 57, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB, and Linse S (2009) A facile method for expression and purification of the Alzheimer's disease-associated amyloid beta-peptide, Febs J 276, 1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Portelius E, G B, AJ T, H Z, A W-B, and K B (2009) Identification of novel APP/Abeta isoforms in human cerebrospinal fluid, Neurodeg. Discussions 6, 87–94. [DOI] [PubMed] [Google Scholar]
- [56].Kaneko N, Yamamoto R, Sato T-A, and Tanaka K (2014) Identification and quantification of amyloid beta-related peptides in human plasma using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, Proc. Japan. Acad. Ser. B 90, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Szczepankiewicz O, Linse B, Meisl G, Thulin E, Frohm B, Frigerio CS, Colvin MT, Jacavone AC, Griffin RG, Knowles T, Walsh DM, and Linse S (2015) N-Terminal Extensions Retard A beta 42 Fibril Formation but Allow Cross-Seeding and Coaggregation with A beta 42, J Am Chem Soc 137, 14673–14685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, and Knowles TP (2013) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism, Proc. Natl. Acad. Sci. U.S.A 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang L, Yu H, Song C, Lin X, Chen B, Tan C, Cao G, and Wang Z (2009) Expression, purification, and characterization of recombinant human b-amyloid42 peptide in Escherichia coli, Protein Expres Purif 64, 55–62. [DOI] [PubMed] [Google Scholar]
- [60].Long F, Cho W, and Ishii Y (2011) Expression and purification of N-15- and C-13-isotope labeled 40-residue human Alzheimer's beta-amyloid peptide for NMR-based structural analysis, Protein Expres Purif 79, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lee EK, Hwang JH, Shin DY, Kim DI, and Yoo YJ (2005) Production of recombinant amyloid-beta peptide 42 as an ubiquitin extension, Protein Expres Purif 40, 183–189. [DOI] [PubMed] [Google Scholar]
- [62].Garai K, Crick SL, Mustafi SM, and Frieden C (2009) Expression and purification of amyloid-beta peptides from Escherichia coli, Protein Expres Purif 66, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nagata-Uchiyama M, Yaguchi M, Hirano Y, and Ueda T (2007) Expression and purification of uniformly N-15-Labeled amyloid p peptide 1-40 in Escherichia coli, Protein Peptide Lett 14, 788–792. [DOI] [PubMed] [Google Scholar]
- [64].Ovchinnikova OY, Finder VH, Vodopivec I, Nitsch RM, and Glockshuber R (2011) The Osaka FAD Mutation E22 Delta Leads to the Formation of a Previously Unknown Type of Amyloid beta Fibrils and Modulates A beta Neurotoxicity, J Mol Biol 408, 780–791. [DOI] [PubMed] [Google Scholar]
- [65].Satakarni M, and Curtis R (2011) Production of recombinant peptides as fusions with SUMO, Protein Expres Purif 78, 113–119. [DOI] [PubMed] [Google Scholar]
- [66].Weber DK, Sani MA, and Gehman JD (2014) A routine method for cloning, expressing and purifying Aβ(1–42) for structural NMR studies, Amino Acids 46, 2415–2426. [DOI] [PubMed] [Google Scholar]
- [67].Andreasson C, Fiaux J, Rampelt H, Mayer MP, and Bukau B (2008) Hsp110 is a nucleotide-activated exchange factor for Hsp70, J. Biol. Chem 283, 8877–8884. [DOI] [PubMed] [Google Scholar]
- [68].Meisl G, Kirkegaard JB, Arosio P, Michaels TCT, Vendruscolo M, Dobson CM, Linse S, and Knowles TPJ (2016) Molecular mechanisms of protein aggregation from global fitting of kinetic models, Nature Protocols 11, 252–272. [DOI] [PubMed] [Google Scholar]
- [69].Goddard TD, and Kneller DG (2008) SPARKY Version 3.115, Univ. California, San Francisco. [Google Scholar]
- [70].Cohen SIA, Vendruscolo M, Dobson CM, and Knowles TPJ (2012) From Macroscopic Measurements to Microscopic Mechanisms of Protein Aggregation, J Mol Biol 421, 160–171. [DOI] [PubMed] [Google Scholar]
- [71].Munowitz M, Bachovchin WW, Herzfeld J, Dobson CM, and Griffin RG (1982) Acid-Base and Tautomeric Equilibria in the Solid-State - N-15 Nmr-Spectroscopy of Histidine and Imidazole, J Am Chem Soc 104, 1192–1196. [Google Scholar]
- [72].Bennett AE, Ok JH, Griffin RG, and Vega S (1992) Chemical-Shift Correlation Spectroscopy in Rotating Solids - Radio Frequency-Driven Dipolar Recoupling and Longitudinal Exchange, J Chem Phys 96, 8624–8627. [Google Scholar]
- [73].Jaroniec CP, Filip C, and Griffin RG (2002) 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly C-13, N-15-labeled solids, J Am Chem Soc 124, 10728–10742. [DOI] [PubMed] [Google Scholar]
- [74].Takegoshi K, Nakamura S, and Terao T (2001) C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR, Chem Phys Lett 344, 631–637. [DOI] [PubMed] [Google Scholar]
- [75].Andronesi OC, Becker S, Seidel K, Heise H, Young HS, and Baldus M (2005) Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy, J Am Chem Soc 127, 12965–12974. [DOI] [PubMed] [Google Scholar]