

Abstract
Background:
A positive association between circulating C-reactive protein (CRP) and colorectal cancer (CRC) survival was reported in observational studies, which are susceptible to unmeasured confounding and reverse causality. We used a Mendelian randomization approach to evaluate the association between genetically-predicted CRP concentrations and CRC-specific survival.
Methods:
We used individual-level data for 16,918 eligible CRC cases of European ancestry from 15 studies within the International Survival Analysis of Colorectal Cancer Consortium. We calculated a genetic risk score based on 52 CRP-associated genetic variants identified from genome-wide association studies. Due to the non-collapsibility of hazard ratios from Cox proportional hazards models, we used the additive hazards model to calculate hazard differences (HD) and 95% confidence intervals (CI) for the association between genetically-predicted CRP concentrations and CRC-specific survival, overall and by stage at diagnosis and tumor location. Analyses were adjusted for age at diagnosis, sex, body mass index, genotyping platform, study, and principal components.
Results:
Of the 5,395 (32%) deaths accrued over up to 10 years of follow-up, 3,808 (23%) were due to CRC. Genetically-predicted CRP concentration was not associated with CRC-specific survival (HD= −1.15, 95% CI: −2.76 to 0.47 per 100,000 person-years, P =0.16). Similarly, no associations were observed in subgroup analyses by stage at diagnosis or tumor location.
Conclusions:
Despite adequate power to detect moderate associations, our results did not support a causal effect of circulating CRP concentrations on CRC-specific survival.
Impact:
Future research evaluating genetically-determined levels of other circulating inflammatory biomarkers (i.e. interleukin-6) with CRC survival outcomes is needed.
Keywords: C-reactive protein, genetic variants, colorectal cancer survival, Mendelian randomization
INTRODUCTION
Chronic inflammation plays an important role in colorectal cancer (CRC) development and progression.(1) Elevated level of inflammation after CRC diagnosis may lead to increased expression of proinflammatory mediators and promote tumor growth and progression.(2)
C-reactive protein (CRP) is an abundant acute-phase protein produced mainly by hepatocytes in response to pro-inflammatory cytokines.(3) Observational studies of CRC outcomes have reported positive associations between pre-diagnostic and pre-operative concentrations of CRP and larger tumor size, metastases, and survival.(4–8) These studies, however, may have been subject to bias as most were unadjusted or insufficiently adjusted for potential confounders and factors related to inflammation and survival, such as adiposity, use of non-steroidal anti-inflammatory drugs (NSAIDs), and smoking. Furthermore, disease progression itself could lead to enhanced tumor-associated inflammation and elevated concentrations of circulating pro-inflammatory markers. Thus, reverse causation is also a potential source of bias.
Most studies of CRP and CRC only had a single measurement of CRP, which may not represent lifelong levels of chronic inflammation. Mendelian randomization utilizes inherited germline genetic markers known to be associated with the risk factor of interest, in this case circulating CRP concentrations. These genetic variants can serve as non-modifiable markers of long-term susceptibility to chronic inflammation. Because of the natural random assortment of alleles during gamete formation, genetic variants are not affected by environmental factors that occur after conception and are non-modifiable by disease progression.(9) Over the last of 15 years, genome-wide association studies (GWAS) have accumulated robust evidence on genetic variants associated with various inflammatory biomarkers, including CRP.(10,11) “Mendelian randomization” has become a common approach for observational studies of inflammatory biomarkers in association with cancer risk, providing a way to minimize reverse causality and residual confounding. However, Mendelian randomization studies of inflammatory biomarkers and cancer survival are scarce.(12)
In this study, we aimed to test the association of genetically predicted concentrations of CRP with CRC-specific survival using a Mendelian randomization approach. As a secondary aim, we evaluated stage- and tumor site-specific associations between genetically predicted circulating CRP concentration and CRC survival. To achieve this, we used the existing data on germline genetic variants and epidemiological and clinical factors from the International Survival Analysis in Colorectal Cancer Consortium (ISACC).
MATERIALS AND METHODS
Study sample
We included individuals diagnosed with incident, invasive CRC from ISACC, a consortium consisting of clinical trials, case-control, and cohort studies from North America, Europe, and Australia. Of the 26,282 eligible ISACC participants who had GWAS and survival data available (Figure 1), we excluded individuals whose GWAS data didn’t pass QC (n= 1,154), whose epidemiologic data was not available (n=217), and those with non-European ancestry (n=1,200) for this analysis. Further exclusion of studies and individuals without data on CRC-specific survival outcome (n=6,793) resulted in a total of 16,918 subjects included in this analyses from the following fifteen studies: Colon Cancer Family Registry (CCFR) (13), Cancer Prevention Study-II (CPS-II) (14), German Darmkrebs: Chancen der Verhütung Durch Screening (DACHS) (15), Diet Activity and Lifestyle Study (DALS) (16), Early Detection Research Network (EDRN) (17), European Prospective Investigation into Cancer (EPIC) (18), Health Professionals Follow-up Study (HPFS) (19), Melbourne Collaborative Cohort Study (MCCS) (20), Nurses’ Health Study (NHS) (21), North Central Cancer Treatment Group (NCCTG) N9741 randomized trial (ClinicalTrials.gov, Identifier: NCT00003594) (22), Physician’s Health Study (PHS) (23), Prostate, Lung, Colorectal, and Ovarian Study (PLCO) (24), UK Biobank (UKB) (25), VITamins And Lifestyle Study (VITAL) (26), and Women’s Health Initiative (WHI).(27) Study-specific details are summarized in Supplementary Table 1. All studies were approved by their respective Institutional Review Boards and participants provided written informed consent.
Figure 1.
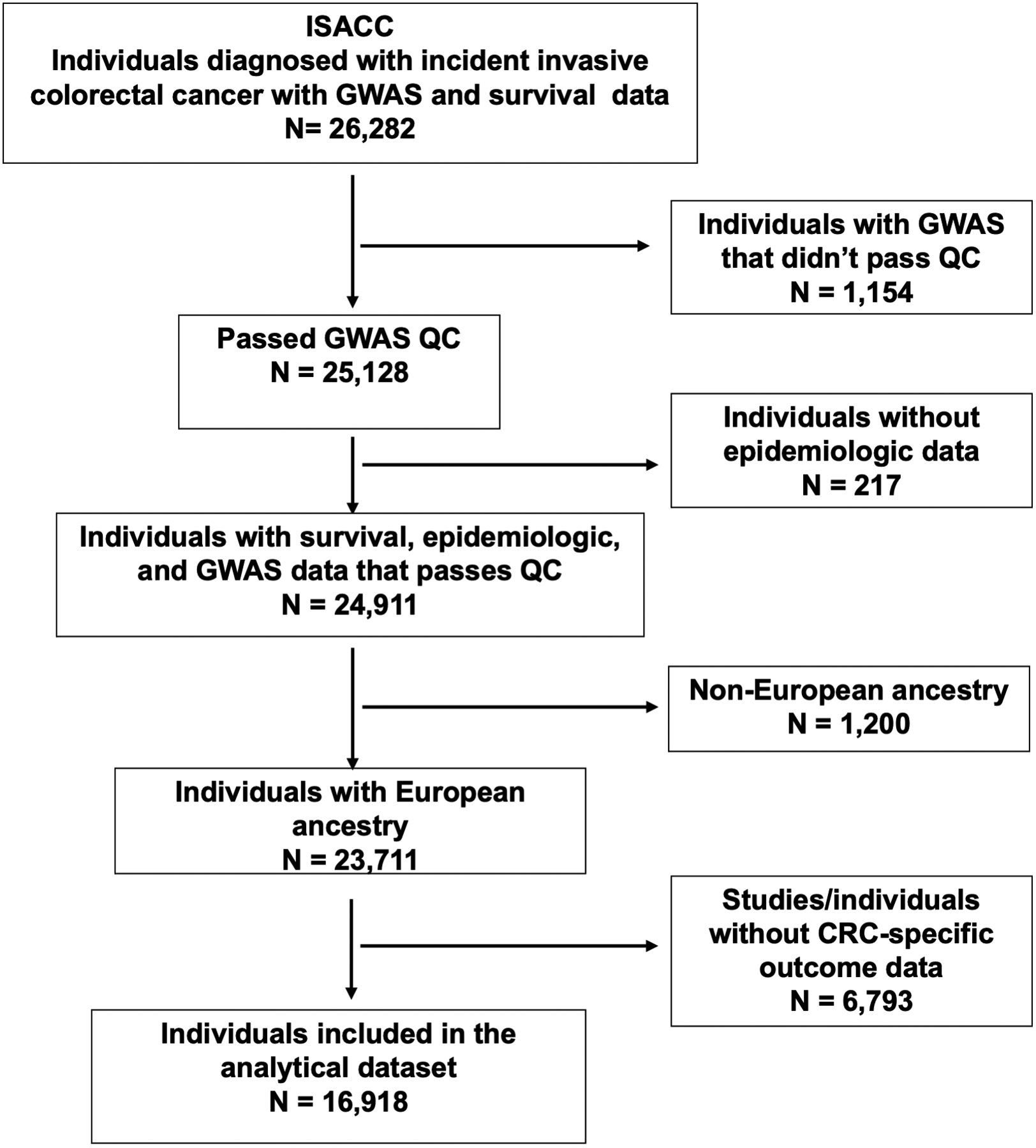
Study sample diagram. Of the 26,282 eligible ISACC participants with both GWAS and survival data, we further excluded individuals based on GWAS QC, genetic ancestry, availability of epidemiologic data and disease-specific survival outcomes, leaving a total of 16,918 subjects included in the analysis.
Ascertainment of environmental variables and survival outcomes
Demographic and epidemiologic factors were collected using self- or interviewer-administered questionnaires at enrollment according to study-specific protocols. A multistep data harmonization process was conducted centrally to define epidemiologic and clinicopathological variables in the same way across studies, as described previously.(28) Information on cancer diagnosis, such as age at diagnosis, tumor location (proximal, distal colon, or rectum) and stage at diagnosis (local: American Joint Committee of Cancer [AJCC] stage I; regional: AJCC stage II/III; or distant: AJCC stage IV), was obtained from cancer registries and/or medical records.
All study participants were followed for vital status. Date and cause of death were ascertained through linkages to the National Death Index or cancer registries (CCFR, CPSII, DACHS, DALS, EPIC, MCCS, UKB, VITAL) or via active follow-up with dates/cause of death verified by the review of death certificates and/or medical records (HPFS, NHS, PHS, PLCO, WHI, N9741). Time to event was defined as days between CRC diagnosis and death, last date of contact, or the end of study follow-up. To evaluate 10-year CRC-specific survival, we censored cases at 10 years from the date of CRC diagnosis. Cases who died from causes other than CRC within 10 years from diagnosis were censored at the time of death. We used the International Classification of Diseases-9 (ICD-9) or ICD-10 (depending on year of linkage) to define CRC-specific deaths (ICD-9: 153.0–153.4, 153.6–153.9, or 154.0–154.1; ICD-10: C18.0–20.0 or C26.0).
Genotyping, quality control (QC), and imputation
Details of genotyping and QC methods have been reported previously.(29–33) Briefly, genomic DNA was extracted from blood or buccal samples using conventional methods. Genotyping was performed using multiple platforms (Supplementary Table 1). All genotype data underwent standardized QC procedures including the exclusion of samples and SNPs with low call rates (<98%), chromosomal anomalies, samples with discrepancies in self-reported and genetically-determined sex, and SNPs out of Hardy–Weinberg Equilibrium. To investigate population structure, we used Plink (v1.9) to conduct principal components analysis (PCA). We restricted our analytic sample to participants with estimated European ancestry based on the PCA due to the low numbers of participants of other ancestries (detailed in Supplementary Methods). We imputed genotypes to infer unobserved genotypes and increase the density of genetic variants. All samples were first phased using SHAPEIT2 (34) and imputed to the Haplotype Reference Consortium (HRC) panel (35) using the University of Michigan Imputation Server.(36)
Selection of instrumental variables
The Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) is the largest GWAS of circulating CRP concentrations to date, analyzing 204,402 individuals of European descent.(11) It reported 48 lead genetic variants from the HapMap GWAS and four additional variants from the 1000 Genome GWAS that were associated with CRP at the genome-wide statistical significance (P<5×10−8). Together these 52 SNPs explained 6.5% of the variance in circulating CRP.(11)
We also searched for, but did not identify additional variants from the NHGRI-EBI GWAS Catalog (37) (downloaded on 03/09/20) that met the following criteria: 1) association with CRP at a genome-wide statistical significance level (P<5×10–8); 2) study population of European ancestry; 3) not in LD (R2<0.3) with previously selected SNPs; and 4) available information on effect sizes and standard errors.
We included the 52 variants as instrumental variables in our Mendelian randomization analyses. The imputation quality (r2) of all 52 CRP-associated SNPs in our data was greater than 0.8. We then calculated a 52-SNP genetic risk score (GRS) (38) by taking the sum of the number of risk (CRP-increasing) alleles for each of the 52 genetic variants weighted by the β coefficients reported by the CHARGE study.(11) The β coefficients represent the change in the natural-log-transformed CRP per copy increment in the risk allele (Table 1).
Table 1.
Association between 52 SNPs and circulating CRP concentrations identified in Ligthart et al. (11) and between SNPs-and CRC-specific survival associations in ISACC
| rs | chr: pos* | Count/ Alternative allele | Count allele freq | Ligthart et al. SNP-CRP associations | ISACC SNP-survival associations | ||||
|---|---|---|---|---|---|---|---|---|---|
| beta** | se | P | HD*** (per 100,000 person year) | se | P | ||||
| rs2293476 | 1:40036847 | C/G | 0.23 | 0.030 | 0.004 | 8.27E-13 | 0.124 | 3.44E-06 | 0.72 |
| rs1805096 | 1:66102257 | G/A | 0.62 | 0.104 | 0.004 | 2.17E-183 | −0.121 | 2.99E-06 | 0.68 |
| rs4129267 | 1:154426264 | C/T | 0.61 | 0.088 | 0.004 | 1.20E-129 | −0.474 | 2.91E-06 | 0.10 |
| rs2794520 | 1:159678816 | C/T | 0.66 | 0.182 | 0.004 | 4.17E-523 | −0.049 | 3.17E-06 | 0.88 |
| rs10925027 | 1:247612562 | T/C | 0.40 | 0.036 | 0.004 | 4.25E-21 | −0.759 | 2.92E-06 | 0.01 |
| rs1260326 | 2:27730940 | T/C | 0.41 | 0.073 | 0.004 | 2.72E-92 | 0.278 | 2.95E-06 | 0.35 |
| rs13409371 | 2:113838145 | A/G | 0.39 | 0.048 | 0.004 | 5.07E-36 | −0.209 | 2.91E-06 | 0.47 |
| rs13233571 | 7:72971231 | C/T | 0.88 | 0.057 | 0.005 | 2.95E-25 | −0.180 | 4.22E-06 | 0.67 |
| rs4841132 | 8:9183596 | G/A | 0.92 | 0.065 | 0.006 | 2.00E-25 | 0.442 | 5.40E-06 | 0.41 |
| rs10778215 | 12:103537266 | T/A | 0.52 | 0.033 | 0.004 | 1.86E-20 | −0.222 | 2.87E-06 | 0.44 |
| rs7310409 | 12:121424861 | G/A | 0.60 | 0.137 | 0.004 | 2.54E-299 | 0.105 | 2.88E-06 | 0.71 |
| rs340005 | 15:60878030 | A/G | 0.63 | 0.030 | 0.004 | 1.01E-15 | 0.352 | 2.92E-06 | 0.23 |
| rs10521222 | 16:51158710 | C/T | 0.95 | 0.104 | 0.011 | 2.06E-22 | −1.450 | 6.99E-06 | 0.04 |
| rs2852151 | 18:12841176 | A/G | 0.40 | 0.025 | 0.004 | 1.36E-11 | −0.002 | 2.94E-06 | 0.99 |
| rs4420638 | 19:45422946 | A/G | 0.83 | 0.229 | 0.006 | 1.23E-305 | −0.425 | 4.16E-06 | 0.31 |
| rs1800961 | 20:43042364 | C/T | 0.97 | 0.112 | 0.011 | 4.63E-23 | −0.742 | 8.51E-06 | 0.38 |
| rs469772 | 1:91530305 | C/T | 0.81 | 0.031 | 0.005 | 5.54E-12 | 0.242 | 3.60E-06 | 0.50 |
| rs12995480 | 2:629881 | C/T | 0.83 | 0.031 | 0.005 | 1.24E-10 | 0.329 | 3.92E-06 | 0.40 |
| rs4246598 | 2:88438050 | A/C | 0.46 | 0.022 | 0.004 | 5.11E-10 | −0.200 | 2.89E-06 | 0.49 |
| rs9284725 | 2:102744854 | C/A | 0.24 | 0.027 | 0.004 | 7.34E-11 | −0.434 | 3.36E-06 | 0.20 |
| rs1441169 | 2:214033530 | A/G | 0.47 | 0.025 | 0.004 | 2.27E-11 | −0.130 | 2.81E-06 | 0.64 |
| rs2352975 | 3:49891885 | C/T | 0.31 | 0.025 | 0.004 | 6.43E-10 | 0.161 | 3.27E-06 | 0.62 |
| rs17658229 | 5:172191052 | C/T | 0.04 | 0.056 | 0.010 | 5.50E-09 | −0.274 | 6.67E-06 | 0.68 |
| rs9271608 | 6:32591588 | G/A | 0.17 | 0.042 | 0.005 | 2.33E-17 | 0.094 | 4.15E-06 | 0.82 |
| rs12202641 | 6:116314634 | C/T | 0.60 | 0.023 | 0.004 | 3.00E-10 | 0.187 | 2.94E-06 | 0.53 |
| rs1490384 | 6:126851160 | C/T | 0.49 | 0.025 | 0.004 | 2.65E-12 | 0.175 | 2.83E-06 | 0.54 |
| rs9385532 | 6:130371227 | C/T | 0.66 | 0.026 | 0.004 | 1.90E-11 | −0.403 | 3.25E-06 | 0.21 |
| rs1880241 | 7:22759469 | A/G | 0.51 | 0.028 | 0.004 | 8.41E-14 | −0.313 | 2.90E-06 | 0.28 |
| rs2710804 | 7:36084529 | C/T | 0.37 | 0.021 | 0.004 | 1.30E-08 | 0.298 | 2.91E-06 | 0.31 |
| rs2064009 | 8:117007850 | T/C | 0.58 | 0.027 | 0.004 | 2.28E-14 | −0.697 | 3.03E-06 | 0.02 |
| rs2891677 | 8:126344208 | T/C | 0.54 | 0.020 | 0.004 | 1.59E-08 | 0.212 | 3.00E-06 | 0.48 |
| rs643434 | 9:136142355 | A/G | 0.37 | 0.023 | 0.004 | 1.02E-09 | −0.041 | 3.05E-06 | 0.89 |
| rs1051338 | 10:91007360 | G/T | 0.30 | 0.024 | 0.004 | 2.27E-09 | 0.514 | 3.14E-06 | 0.10 |
| rs10832027 | 11:13357183 | A/G | 0.67 | 0.026 | 0.004 | 4.43E-12 | −0.394 | 2.96E-06 | 0.18 |
| rs10838687 | 11:47312892 | T/G | 0.79 | 0.031 | 0.004 | 9.12E-13 | 0.016 | 3.39E-06 | 0.96 |
| rs1582763 | 11:60021948 | G/A | 0.63 | 0.022 | 0.004 | 2.37E-09 | −0.083 | 2.97E-06 | 0.78 |
| rs7121935 | 11:72496148 | G/A | 0.62 | 0.022 | 0.004 | 5.28E-09 | 0.090 | 3.04E-06 | 0.77 |
| rs11108056 | 12:95855385 | C/G | 0.58 | 0.028 | 0.004 | 5.42E-14 | 0.318 | 3.02E-06 | 0.29 |
| rs2239222 | 14:73011885 | G/A | 0.37 | 0.035 | 0.004 | 9.87E-20 | 0.415 | 3.15E-06 | 0.19 |
| rs4774590 | 15:51745277 | G/A | 0.62 | 0.022 | 0.004 | 2.71E-08 | 0.110 | 3.07E-06 | 0.72 |
| rs1558902 | 16:53803574 | A/T | 0.40 | 0.034 | 0.004 | 5.20E-20 | 0.030 | 2.84E-06 | 0.92 |
| rs178810 | 17:16097430 | T/C | 0.56 | 0.020 | 0.004 | 2.95E-08 | −0.060 | 2.83E-06 | 0.83 |
| rs10512597 | 17:72699833 | C/T | 0.80 | 0.037 | 0.005 | 4.44E-14 | −0.048 | 3.87E-06 | 0.90 |
| rs4092465 | 18:55080437 | G/A | 0.62 | 0.027 | 0.004 | 3.11E-10 | −0.154 | 3.11E-06 | 0.62 |
| rs12960928 | 18:57897803 | C/T | 0.26 | 0.024 | 0.004 | 1.91E-09 | −0.296 | 3.34E-06 | 0.38 |
| rs2315008 | 20:62343956 | G/T | 0.68 | 0.023 | 0.004 | 5.36E-10 | 0.118 | 3.11E-06 | 0.70 |
| rs2836878 | 21:40465534 | G/A | 0.73 | 0.043 | 0.004 | 7.71E-26 | 0.289 | 3.10E-06 | 0.35 |
| rs6001193 | 22:39074737 | A/G | 0.63 | 0.028 | 0.004 | 6.53E-14 | −0.678 | 3.10E-06 | 0.03 |
| rs75460349 | 1:27180088 | A/C | 0.98 | 0.086 | 0.014 | 4.50E-10 | 0.477 | 9.43E-06 | 0.61 |
| rs1514895 | 3:170705693 | G/A | 0.30 | 0.027 | 0.004 | 2.70E-09 | 0.002 | 3.26E-06 | 0.99 |
| rs112635299 | 14:94838142 | G/T | 0.98 | 0.107 | 0.017 | 2.10E-10 | −2.150 | 1.22E-05 | 0.08 |
| rs1189402 | 15:53728154 | A/G | 0.63 | 0.025 | 0.004 | 3.90E-09 | 0.474 | 3.20E-06 | 0.14 |
Chromosome: position, hg19
beta, SNP-sepcific coefficients for association with circulating concentrations of CRP obtained from Ligthart et al, per unit increase in natural log transformed CRP (mg/L)
hazards difference for CRC-specific survival per unit increase in the count allele based on additive hazards model
Abbreviations: ISACC: the International Survival Analysis in Colorectal Cancer Consortium; HD: hazards difference; se: standard error
Statistical Analysis
The genetic variants selected as an instrumental variable in a Mendelian randomization analysis need to meet three assumptions: (1) they are robustly associated with the exposure (“relevance”), (2) they do not share a common cause with the outcome (“exchangeability”), and (3) they affect the outcome only through the exposure (“exclusion restriction”).
We first verified the “relevance” assumption by evaluating the association between GRS and post-diagnosis circulating CRP concentrations in a subset of CRC cases from Seattle CCFR (n=285) whose CRP leves were measured in between one to three years after diagnosis to rule out active treatment effects.(39) We estimated the proportion of variance (R2) explained by the 52 genetic variants and calculated the F statistic, a measure of instrument strength, based on R2, the sample size (n), and the number of instruments (k) as described in the formula: . A strong instrumental variable is defined as having F≥10.(40)
For the second “exchangeability” assumption, we examined several epidemiologic and clinicopathological factors that may confound the CRP- survival association, including smoking, body mass index (BMI), NSAID use, tumor location, and stage at diagnosis. Each was assessed for association with the GRS. BMI was statistically significantly associated with the GRS and therefore it was included as an additional adjustment variable in the following Mendelian randomization analysis. No other variables were statistically significantly associated with GRS.
The “exclusion restriction” assumption was assessed in a series of sensitivity analyses. We used MR- Egger regression to assess the horizontal pleiotropic effect. The test of a non-zero intercept indicates whether there are averaged pleiotropic effects.(41) We also restricted the instrumental variable to rs2794520 in the CRP gene to minimize the probability of horizontal pleiotropy. This variant itself explained 1.4% of the variance in circulating CRP.(11)
We performed the Mendelian randomization analyses using a two-stage regression approach.(42) Additive hazards model offers a flexible alternative for modeling associations on the hazard scale: a hazard difference (HD), unlike the hazard ratio (HR) from the Cox proportional hazards model, is a collapsible effect measure over strata of unmeasured and unknown confounders. (42,43) We used additive hazards models to calculate HD and 95% confidence intervals (CI) for the associations between CRP-associated GRS and CRC-specific survival. The R package “timereg” was used for fitting additive hazards models.(44)
We also evaluated the association between genetically determined CRP circulating concentration and CRC-specific survival using the inverse-variance weighted (IVW) method (45), MR-Egger regression (41), and the estimator from the weighted median approach (46) based on summary statistics on SNP-specific associations with CRC survival. In secondary analyses, we evaluated the associations between genetically predicted concentrations of CRP and CRC-specific survival according to tumor stage and location.
In the sensitivity analyses, Cox proportional hazards models were used for hypothesis testing. We also compared results with and without adjustment of BMI in addition to age at diagnosis, sex, genotyping platform, study, and the first nine principal components. All analyses were conducted using R version 3.6.0.
Statistical power
Currently, there is no available power calculation tool for survival outcomes in Mendelian randomization analysis, we first took a conservative approach treating CRC-specific survival as a binary outcome and used the methods described by Burgess. (47) With a total of 16,916 CRC cases and 23% CRC-specific deaths occurring over up to 10 years follow-up, we have more than 85% power to detect an OR of 1.25 for the association between CRP and CRC-specific survival at a significance level of 0.05, assuming 5.9% variance of CRP explained by the genetic variance.
In addition, we ran a simulation using the additive hazards model for power calculation. With the number of CRC cases and 3,808 CRC deaths accrued over a 10-year follow-up, the population-averaged hazard was estimated to be 3808/(16918*10) =0.023 per person*year. We have at least 83% power to detect a 25% difference in hazard (HD=0.0058) for every 1 SD increase of CRP assuming 5.9% of the variance of CRP was explained by GRS. The R code for the simulation is included in the Supplementary Materials.
RESULTS
We included 16,918 eligible CRC cases from ISACC in this study (Figure 1). Study participants were diagnosed at a median of 67 years of age, and 49.7% were female. Over the maximum 10-year follow up, there were 5,395 (32%) deaths accrued with 3,808 (23%) due to CRC. Study-specific summaries are shown in Supplementary Table 1. SNP-specific associations with circulating CRP concentrations and CRC-specific survival are summarized in Table 1.
In evaluating the “relevance” assumption, we observed strong associations between the GRS and circulating CRP concentrations in a subset of the study participants (n= 285). A one-unit increase in GRS was associated with a 1.22-unit increase in the natural-log-transformed CRP (95% CI: 0.65–1.80, P= 4.33×10−5) and explained 5.9% of the variance of the natural-log-transformed CRP concentrations. The estimated F statistic was 20.2, indicating a strong instrumental variable.
Among the 16,918 participants from ISACC, the distribution of the CRP-associated GRS calculated based on individual-level data is shown in Supplementary Figure 2. Based on additive hazards model, we observed that one unit increase in GRS was associated with 1.15 fewer deaths due to CRC per 100,000 patients each year (HD= −1.15, 95% CI: −2.76 to 0.47 per 100,000 person-year, Table 2). However, it didn’t reach statistical significance (P= 0.16). No associations between quartiles of GRS and CRC-specific survival were observed (Table 2). Results based on IVW, MR-Egger, and weighted median approaches using summary statistics were consistent with those based on individual GRS data (Table 2). Sensitivity analyses using Cox proportional hazards models for hypothesis testing showed similar null associations between GRS and CRC-specific survival (HR= 0.90, 95% CI= 0.79 to 1.02, P= 0.10, Table 2).
Table 2.
Association between genetically determined CRP concentrations and CRC-specific survival
| Additive hazards model | Cox proportional hazards model | ||||||
|---|---|---|---|---|---|---|---|
| HD (95% CI) per 100,000 person-year | P | HR (95% CI) | P | ||||
| Using individual-level data | |||||||
| 52-SNP GRS | |||||||
| Continuous* | −1.15 | (−2.76, 0.47) | 0.16 | 0.90 | (0.79, 1.02) | 0.10 | |
| By quartiles | |||||||
| Q1 (2.05,3.06] | 1.00 | Ref | 1.00 | Ref | |||
| Q2 (3.06,3.24] | 0.31 | (−0.87, 1.49) | 0.61 | 1.02 | (0.93, 1.12) | 0.67 | |
| Q3 (3.24,3.41] | −0.52 | (−1.71, 0.68) | 0.40 | 0.96 | (0.88, 1.05) | 0.41 | |
| Q4 (3.41,4.08] | −0.73 | (−1.87, 0.41) | 0.21 | 0.93 | (0.85, 1.02) | 0.14 | |
| Using summary statistics | |||||||
| IVW | −1.12 | (−2.72, 0.48) | 0.17 | 0.90 | (0.79, 1.02) | 0.10 | |
| MR-Egger | −1.29 | (−3.68, 1.11) | 0.29 | 0.88 | (0.72, 1.06) | 0.18 | |
| Weighted median | −0.77 | (−3.02, 1.47) | 0.50 | 0.93 | (0.77, 1.11) | 0.40 | |
Per one-unit increment in GRS;
All models adjusted for age at diagnosis, sex, body mass index, genotyping platform, study and principal components.
Abbreviations: CI: confidence interval; GRS: genetic risk score; HD: hazard difference; HR: hazard ratio; IVW: inverse-variance weighted; MR: Mendelian randomization.
We further evaluated this association by stage at diagnosis and tumor location, and found no evidence of statistically significant association in these subgroup analyses using Cox proportional hazards models, whereas the additive hazards model did not converge due to limited number of events in subgroups. (Table 3). Among inidividuals diagnosed with colon cancer, we observed a boarderline significant association: one unit increase in GRS was associated with improved CRC-specific survival (HR=0.87, 95% CI: 0.75 to 1.00, P= 0.06, Table 3).
Table 3.
Association between genetically determined CRP concentrations and CRC-specific survival, by subgroups
| 52-SNP GRS | Total | Events* | Cox proportional hazards model | |||
|---|---|---|---|---|---|---|
| HR (95% CI)** | P | |||||
| All | 16,918 | 3,808 | 0.90 | (0.79, 1.02) | 0.10 | |
| By stage at diagnosisⱡ | ||||||
| Local | 3,341 | 142 | 0.50 | (0.24, 1.02) | 0.06 | |
| Regional | 6,420 | 1,177 | 0.92 | (0.73, 1.17) | 0.51 | |
| Distant | 1,845 | 1,387 | 0.97 | (0.75, 1.24) | 0.79 | |
| By tumor location║ | ||||||
| Colon | 12,000 | 2,791 | 0.87 | (0.75, 1.00) | 0.06 | |
| Proximal | 6,205 | 1,365 | 0.86 | (0.69, 1.07) | 0.18 | |
| Distal | 4,879 | 932 | 0.98 | (0.75, 1.29) | 0.91 | |
| Rectum | 4,729 | 974 | 1.02 | (0.79, 1.33) | 0.85 | |
Death events due to CRC within up to 10-year follow-up
HRs represent per one-unit increase in GRS, and were adjusted for age at diagnosis, sex, body mass index, genotyping platform, study, and principal components; Additive models do not converge in subgroup analysis.
Stage at diagnosis was defined using SEER summary stage (local: AJCC stage I; regional: stage II-IIl; distant: stage IV)
Proximal colon was defined as from the cecum through transverse colon; distal colon was from the splenic flexure to sigmoid colon; rectum included the rectosigmoid junction and rectum. Abbreviations: CI: confidence interval; GRS: genetic risk score; HR: hazard ratio; AJCC: American joint committee on cancer.
We plotted the SNP-specific associations with CRC-specific survival against coefficients of SNP-CRP associations (Figure 2). After conducting MR-Egger regression analysis, we found that the intercept was not statistically significantly different from zero (β0 = 1.28×10−7, 95% CI = −1.23×10−6 to 1.48×10−6, P= 0.85) when using additive hazards models. This suggested no horizontal pleiotropic effect. The MR-Egger regression using Cox proportional hazards estimates (Figure 2B) yielded similar results compared to the one using additive hazards models (Figure 2A). We then restricted the instrumental variable to rs2794520 in the CRP gene and repeated the Mendelian randomization analysis. A null association with CRC survival was observed (additive hazards model: HD= −0.049 per 100,000 person-year, P= 0.88; Cox proportional hazards model: HR= 0.99, 95% CI: 0.94–1.04, P=0.60).
Figure 2.
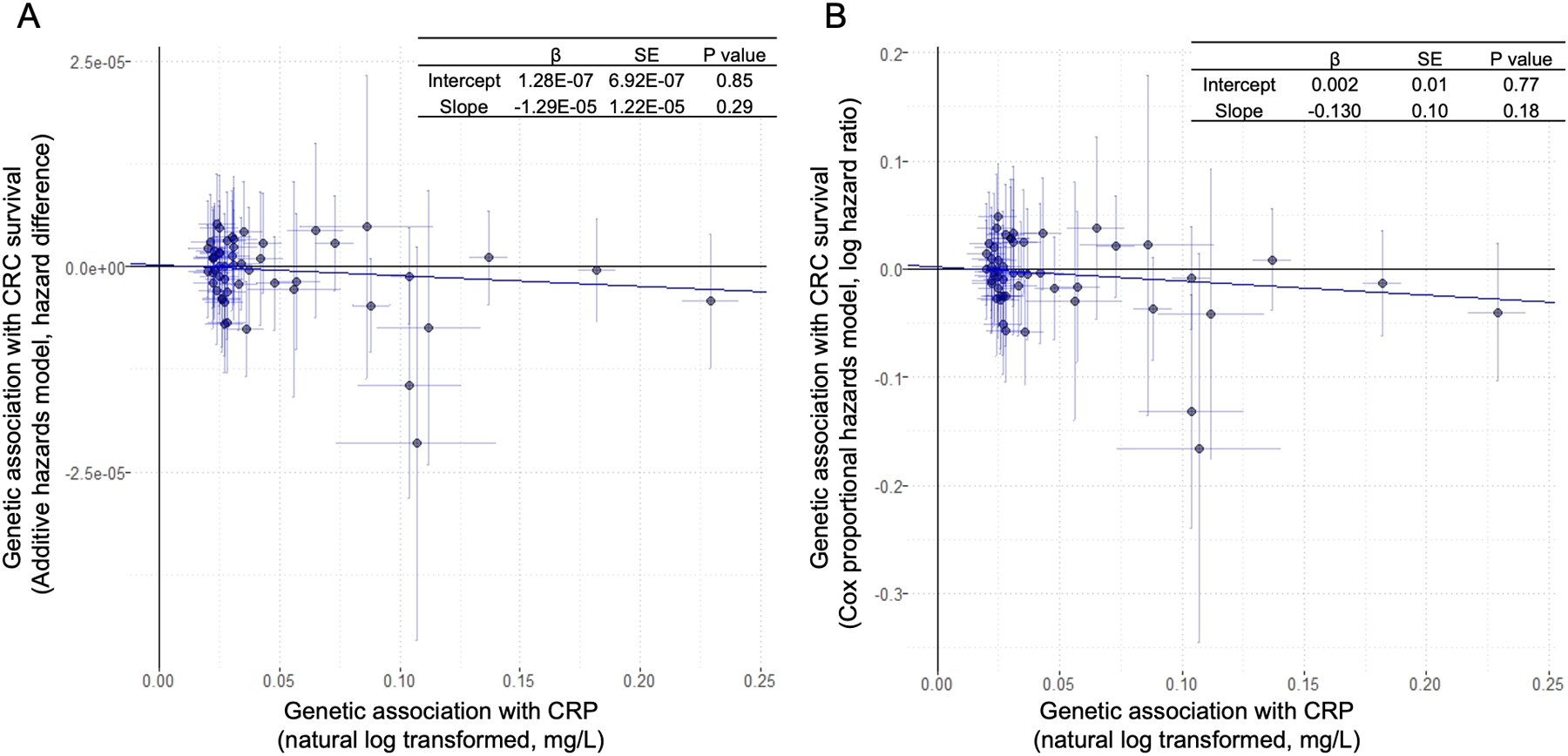
Scatter plot of SNP-specific associations with CRC survival against coefficients of SNP-CRP associations among CRC cases from ISACC using A) additive hazards models and B) Cox proportional hazards models. The slope of the regression line provides an estimate of the association between genetically predicted circulating concentration of CRP and CRC survival; the intercept is an estimate of the average pleiotropic effect across all the genetic variants.
DISCUSSION
In this large Mendelian randomization study, we did not find evidence of an association between genetically predicted CRP circulating concentration and CRC-specific survival in a cohort of individuals diagnosed with incident invasive CRC and followed up for survival. No associations were observed in subgroups defined by tumor stage at diagnosis and location. Our findings do not support a causal relationship between circulating CRP and CRC-specific survival.
Previous studies of CRP and CRC incidence and survival do not provide convincing evidence of causation. For CRC risk, meta-analyses of prediagnostic circulating CRP concentrations showed that one unit change in natural logarithm CRP was associated with a 12% increased risk of developing CRC.(48) Conversely, we showed in a large multi-consortium Mendelian randomization study with more than 30,400 cases and 22,800 controls no association between genetically determined CRP concentrations and CRC risk.(49) For CRC-specific survival, results from observational studies of circulating CRP concentration were inconsistent. Some studies observed that circulating CRP concentration measured before surgery was not statistically significantly associated with survival after multivariable adjustment.(7,8) Other studies observed that elevated concentrations of pre-operative (4–6,50) and post-treatment (51,52) CRP were associated with worse CRC survival outcomes. However, the CRP measures in these studies were crude. Several of these studies used CRP ≥10mg/L as the cut-off to dichotomize circulating CRP concentrations.(4,5,50) Elevated CRP concentrations ≥10mg/L are likely driven by acute inflammatory conditions other than chronic inflammation. Similarly, in our recent study, circulating concentration of CRP was no longer associated with CRC survival after we excluded CRC cases who had post-treatment CRP>10mg/L.(39)
In this study, we used genetic variants as proxies of circulating CRP concentrations that can help address potential biases due to residual confounding and reverse causality, but existing evidence on CRC survival outcomes is limited. Slattery et al. evaluated four tag SNPs in the CRP gene in relation to CRC survival among 1,574 cases, however, none were statistically significantly associated with CRC-specific survival within 5 years after diagnosis.(53) Another study with 421 CRC cases of East Asian ancestry showed that two SNPs from the CRP gene were associated with CRC survival: rs3093059 was associated with disease-free survival, whereas rs1205 was associated with CRC-specific survival.(54) Although these two variants were not included in our study, we evaluated rs2794520, at CRP locus that is in high LD with these two SNPs. The allele frequencies of these SNPs are twice as common in the East Asian population (ASN) compared with the European population (EUR): rs3093059 (ASN: 0.14; EUR: 0.07), rs2794520 and rs1205 (ASN: 0.60; EUR: 0.31). This could partially explain the different study findings.
There are some limitations when interpreting our study results. First, the restriction of our study sample to individuals diagnosed with CRC by design could be a potential source of selection bias (also known as collider bias) particularly if CRP is causally associated with increased risk of developing CRC. By conditioning on the collider- CRC risk (selecting only CRC cases into the study sample), it can induce an association between genetic variants and risk factors of CRC. However, evidence from our previous Mendelian randomization study suggests that CRP is not causally associated with CRC risk.(49) To further address this potential selection bias, we evaluated the associations between the genetic variants with both potential confounders of CRP and CRC survival associations and common risk factors of CRC risk. BMI was identified as the only variable being statistically significantly associated with the GRS for CRP in our study sample and was adjusted for in all analyses. However, as BMI is an inheritable trait that shares some genetic suscpetibilites with CRP, we also assessed whether there was potential bias due to BMI adjustment (55,56) and compared main analysis with and without adjustment of BMI (Supplementary Table 2). We observed minimal changes due to BMI adjustment. Second, the 52-SNP GRS for CRP explained only less than 6% of the variance of the natural-log-transformed CRP concentrations. The null results of our study cannot rule out a weaker causal effect of CRP on CRC-specific survival. Third, the genetic variants shown to be robustly associated with circulating CRP were identified from a GWAS based on study sample from the general population. The SNP-CRP associations may be different in a sample of CRC cases. Although we evaluated the “relevance” assumption in a subset of our study sample and observed a strong association between the CRP-associated GRS and post-diagnostic circulating CRP concentrations among CRC cases, the small sample size limited the statistical power to evaluate SNP-specific associations with CRP among CRC cases. In addition, our subgroup analyses had insufficient statistical power even though our main analysis was well powered. The limited number of events in subgroups also led to convergence issues when using the additive hazards model. Lastly, since the study sample was limited to individuals with European ancestry, our findings may not be generalizable to other racial/ethnicity groups.
Our study also has many strengths. This is the first study that evaluates circulating biomarkers in relation to CRC survival using a Mendelian randomization approach. Our large sample size possessed adequate statistical power to detect associations with moderate effect sizes. Also, the well-characterized study sample with individual-level genotype data and detailed information on epidemiologic and clinic factors allowed us to compare study results with those based on summary statistics, to evaluate the “exchangeability” assumptions, and to conduct subgroup analysis by stage at diagnosis and tumor location, however we weren’t able to account for several clinical prognostic factors for CRC survival, such as treatement, due to data availability. A subset of study participants had data on both genotypes and circulating CRP concentrations allowing us to evaluate the “relevance” assumption. By carefully examining the three assumptions, our Mendelian randomization study is less susceptible to confounding and reverse causality compared with observational studies.
In summary, our study did not find evidence of an association between genetically predicted circulating CRP concentration and CRC-specific survival, overall or in subgroups defined by stage at diagnosis or tumor location. Future research should be conducted to determine if other circulating inflammatory biomarkers, such as interleukin 6, are associated with CRC survival outcomes to better understand chronic inflammation and disease progression among CRC patients.
Supplementary Material
ACKNOWLEDGEMENTS
ISACC: The authors would like to thank all those at the ISACC Coordinating Center for helping bring together the data and people that made this project possible. This research was funded by in part through National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (R01 CA176272) and the NIH/NCI Cancer Center Support Grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685.
CCFR: The Colon CFR graciously thanks the generous contributions of their 42,505 study participants, dedication of study staff, and the financial support from the U.S. National Cancer Institute, without which this important registry would not exist.
CCFR (www.coloncfr.org) is supported in part by funding from the National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). The CCFR Set-1 (Illumina 1M/1M-Duo) and Set-2 (Illumina Omni1-Quad) scans were supported by NIH awards U01 CA122839 and R01 CA143247 (to GC). The CCFR Set-3 (Affymetrix Axiom CORECT Set array) was supported by NIH award U19 CA148107 and R01 CA81488 (to SBG). The CCFR Set-4 (Illumina OncoArray 600K SNP array) was supported by NIH award U19 CA148107 (to SBG) and by the Center for Inherited Disease Research (CIDR), which is funded by the NIH to the Johns Hopkins University, contract number HHSN268201200008I. The content of this manuscript does not necessarily reflect the views or policies of the NCI, NIH or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government, any cancer registry, or the CCFR.
SCCFR: The authors would like to thank the study participants and staff of the Seattle Colon Cancer Family Registry and the Hormones and Colon Cancer study (CORE Studies).
[OFCCR ARCTIC]: Additional funding for the OFCCR/ARCTIC was through award GL201-043 from the Ontario Research Fund (to BWZ), award 112746 from the Canadian Institutes of Health Research (to TJH), through a Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society (to SG), and through generous support from the Ontario Ministry of Research and Innovation.
[SCCFR (Illumina HumanCytoSNP (300k)): The SCCFR Illumina HumanCytoSNP array was supported through NCI award R01 CA076366 (to PAN).
[CCFR Set-1 and/or Set-2 scan (Illumina Human 1M, 1M-Duo, and/or Omni1-Quad)]: The CCFR Set-1 (Illumina 1M/1M-Duo) and Set-2 (Illumina Omni1-Quad) scans were supported by NIH awards U01 CA122839 and R01 CA143247 (to GC).
[CCFR Set-3 scan (Affymetrix Axiom CORECT Set array)]: The CCFR Set-3 (Affymetrix Axiom CORECT Set array) was supported by NIH award U19 CA148107 and R01 CA81488 (to SBG).
[CCFR Set-4 scan (Illumina OncoArray 600K SNP array)]: The CCFR Set-4 (Illumina OncoArray 600K SNP array) was supported by NIH award U19 CA148107 (to SBG) and by the Center for Inherited Disease Research (CIDR), which is funded by the NIH to the Johns Hopkins University, contract number HHSN268201200008I.
CPS-II: The authors thank the CPS-II participants and Study Management Group for their invaluable contributions to this research. The authors would also like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. This study was conducted with Institutional Review Board approval.
DACHS: We thank all participants and cooperating clinicians, and everyone who provided excellent technical assistance. This work was supported by the German Research Council (BR 1704/6-1, BR 1704/6-3, BR1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1 and BR 1704/17-1), the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A, and 01ER1505B), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and German Cancer Research Center.
DALS: National Institutes of Health (R01 CA48998 to M. L. Slattery).
EDRN: We acknowledge all contributors to the development of the resource at University of Pittsburgh School of Medicine, Department of Gastroenterology, Department of Pathology, Hepatology and Nutrition and Biomedical Informatics. This work is funded and supported by the NCI, EDRN Grant (U01 CA 84968-06).
EPIC: Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization. EPIC is financially supported by the European Commission (DGSANCO) and the International Agency for Research on Cancer. The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF), Deutsche Krebshilfe, Deutsches Krebsforschungszentrum and Federal Ministry of Education and Research (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRCItaly and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG 232997 and Nordforsk, Nordic Centre of Excellence programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPICOxford) (United Kingdom).
Harvard cohorts (HPFS, NHS, PHS): The study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the HPFS, NHS and PHS for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. HPFS is supported by the National Institutes of Health (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA137178, R01 CA151993, and R35 CA197735), NHS by the National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, R01 CA151993, and R35 CA197735) and PHS by the National Institutes of Health (R01 CA042182).
MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 509348, 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database.
PLCO: The authors thank the PLCO Cancer Screening Trial screening center investigators and the staff from Information Management Services Inc and Westat Inc. Most importantly, we thank the study participants for their contributions that made this study possible. Cancer incidence data have been provided by the District of Columbia Cancer Registry, Georgia Cancer Registry, Hawaii Cancer Registry, Minnesota Cancer Surveillance System, Missouri Cancer Registry, Nevada Central Cancer Registry, Pennsylvania Cancer Registry, Texas Cancer Registry, Virginia Cancer Registry, and Wisconsin Cancer Reporting System. All are supported in part by funds from the Center for Disease Control and Prevention, National Program for Central Registries, local states or by the National Cancer Institute, Surveillance, Epidemiology, and End Results program. The results reported here and the conclusions derived are the sole responsibility of the authors. Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Funding was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438.
UK Biobank: The authors thank all the participants and staff of UK Biobank for making such a wonderful resource available for research. This study has been conducted under Application Number 8614.
VITAL is supported by National Institutes of Health (K05 CA154337).
WHI: The authors thank the WHI investigators and staff for their dedication and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf
The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.
Conflicts of Interest:
The authors have no conflicts of interest to disclose, except that: Dr. Goldberg reports consulting/advisory role for Merck, Taiho Pharmaceutical, Merck KGaA, and Novartis, research funding from Bristol-Myers Squibb, and meeting travel reimbursement from Merck and Merck KGaA. Dr. Shi reports consulting/advisory role for Yiviva Inc and Boehringer Ingelheim Parmaceuticals, Inc (to self), stock from Johnson & Johnson, Amgen, and Merck & CO. (to self), research funds from Celgene/ Bristol-Myers Squibb and Roche/Genentech (to institution).
REFERENCE
- 1.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology 2010;138(6):2101–14 e5 doi 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 2.Klintrup K, Makinen JM, Kauppila S, Vare PO, Melkko J, Tuominen H, et al. Inflammation and prognosis in colorectal cancer. Eur J Cancer 2005;41(17):2645–54 doi 10.1016/j.ejca.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 3.Ansar W, Ghosh S. C-reactive protein and the biology of disease. Immunol Res 2013;56(1):131–42 doi 10.1007/s12026-013-8384-0. [DOI] [PubMed] [Google Scholar]
- 4.Takasu C, Shimada M, Kurita N, Iwata T, Nishioka M, Morimoto S, et al. Impact of C-reactive protein on prognosis of patients with colorectal carcinoma. Hepatogastroenterology 2013;60(123):507–11 doi 10.5754/hge11425. [DOI] [PubMed] [Google Scholar]
- 5.Crozier JE, McKee RF, McArdle CS, Angerson WJ, Anderson JH, Horgan PG, et al. The presence of a systemic inflammatory response predicts poorer survival in patients receiving adjuvant 5-FU chemotherapy following potentially curative resection for colorectal cancer. Br J Cancer 2006;94(12):1833–6 doi 10.1038/sj.bjc.6603185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koike Y, Miki C, Okugawa Y, Yokoe T, Toiyama Y, Tanaka K, et al. Preoperative C-reactive protein as a prognostic and therapeutic marker for colorectal cancer. J Surg Oncol 2008;98(7):540–4 doi 10.1002/jso.21154. [DOI] [PubMed] [Google Scholar]
- 7.Chung YC, Chang YF. Serum C-reactive protein correlates with survival in colorectal cancer patients but is not an independent prognostic indicator. Eur J Gastroenterol Hepatol 2003;15(4):369–73 doi 10.1097/00042737-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Volkova E, Willis JA, Wells JE, Robinson BA, Dachs GU, Currie MJ. Association of angiopoietin-2, C-reactive protein and markers of obesity and insulin resistance with survival outcome in colorectal cancer. Br J Cancer 2011;104(1):51–9 doi 10.1038/sj.bjc.6606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32(1):1–22 doi 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 10.Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation 2011;123(7):731–8 doi 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ligthart S, Vaez A, Vosa U, Stathopoulou MG, de Vries PS, Prins BP, et al. Genome Analyses of >200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways that Link Inflammation and Complex Disorders. Am J Hum Genet 2018;103(5):691–706 doi 10.1016/j.ajhg.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Q, Burgess S, Turman C, Bolla MK, Wang Q, Lush M, et al. Body mass index and breast cancer survival: a Mendelian randomization analysis. Int J Epidemiol 2017;46(6):1814–22 doi 10.1093/ije/dyx131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newcomb PA, Baron J, Cotterchio M, Gallinger S, Grove J, Haile R, et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev 2007;16(11):2331–43 doi 10.1158/1055-9965.EPI-07-0648. [DOI] [PubMed] [Google Scholar]
- 14.Calle EE, Rodriguez C, Jacobs EJ, Almon ML, Chao A, McCullough ML, et al. The American Cancer Society Cancer Prevention Study II Nutrition Cohort: rationale, study design, and baseline characteristics. Cancer 2002;94(9):2490–501 doi 10.1002/cncr.101970. [DOI] [PubMed] [Google Scholar]
- 15.Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 2011;154(1):22–30 doi 10.7326/0003-4819-154-1-201101040-00004. [DOI] [PubMed] [Google Scholar]
- 16.Slattery ML, Potter J, Caan B, Edwards S, Coates A, Ma KN, et al. Energy balance and colon cancer--beyond physical activity. Cancer Res 1997;57(1):75–80. [PubMed] [Google Scholar]
- 17.Amin W, Singh H, Dzubinski LA, Schoen RE, Parwani AV. Design and utilization of the colorectal and pancreatic neoplasm virtual biorepository: An early detection research network initiative. J Pathol Inform 2010;1:22 doi 10.4103/2153-3539.70831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riboli E, Kaaks R. The EPIC Project: rationale and study design. European Prospective Investigation into Cancer and Nutrition. Int J Epidemiol 1997;26 Suppl 1:S6–14 doi 10.1093/ije/26.suppl_1.s6. [DOI] [PubMed] [Google Scholar]
- 19.Rimm EB, Stampfer MJ, Colditz GA, Chute CG, Litin LB, Willett WC. Validity of self-reported waist and hip circumferences in men and women. Epidemiology 1990;1(6):466–73 doi 10.1097/00001648-199011000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Giles GG, English DR. The Melbourne Collaborative Cohort Study. IARC Sci Publ 2002;156:69–70. [PubMed] [Google Scholar]
- 21.Colditz GA, Manson JE, Hankinson SE. The Nurses’ Health Study: 20-year contribution to the understanding of health among women. J Womens Health 1997;6(1):49–62 doi 10.1089/jwh.1997.6.49. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004;22(1):23–30 doi 10.1200/JCO.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 23.Christen WG, Gaziano JM, Hennekens CH. Design of Physicians’ Health Study II--a randomized trial of beta-carotene, vitamins E and C, and multivitamins, in prevention of cancer, cardiovascular disease, and eye disease, and review of results of completed trials. Ann Epidemiol 2000;10(2):125–34 doi 10.1016/s1047-2797(99)00042-3. [DOI] [PubMed] [Google Scholar]
- 24.Prorok PC, Andriole GL, Bresalier RS, Buys SS, Chia D, Crawford ED, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials 2000;21(6 Suppl):273S–309S doi 10.1016/s0197-2456(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 25.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12(3):e1001779 doi 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White E, Patterson RE, Kristal AR, Thornquist M, King I, Shattuck AL, et al. VITamins And Lifestyle cohort study: study design and characteristics of supplement users. Am J Epidemiol 2004;159(1):83–93 doi 10.1093/aje/kwh010. [DOI] [PubMed] [Google Scholar]
- 27.Group TWsHIS. Design of the Women’s Health Initiative clinical trial and observational study. . Control Clin Trials 1998;19(1):61–109 doi 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 28.Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res 2012;72(8):2036–44 doi 10.1158/0008-5472.Can-11-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology 2013;144(4):799–807.e24 doi 10.1053/j.gastro.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huyghe JR, Bien SA, Harrison TA, Kang HM, Chen S, Schmit SL, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 2019;51(1):76–87 doi 10.1038/s41588-018-0286-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562(7726):203–9 doi 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmit SL, Edlund CK, Schumacher FR, Gong J, Harrison TA, Huyghe JR, et al. Novel Common Genetic Susceptibility Loci for Colorectal Cancer. J Natl Cancer Inst 2019;111(2):146–57 doi 10.1093/jnci/djy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schumacher FR, Schmit SL, Jiao S, Edlund CK, Wang H, Zhang B, et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun 2015;6:7138 doi 10.1038/ncomms8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delaneau O, Marchini J, McVean GA, Donnelly P, Lunter G, Marchini JL, et al. Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nature Communications 2014;5(1):3934 doi 10.1038/ncomms4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48(10):1279–83 doi 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet 2016;48(10):1284–7 doi 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019;47(D1):D1005–d12 doi 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med 2016;35(11):1880–906 doi 10.1002/sim.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hua X, Kratz M, Newcomb PA. Associations between Post-treatment Inflammatory Biomarkers and Survival among Stage II-III Colorectal Cancer Patients. Cancer Epidemiology Biomarkers & Prevention 2020;29(3):691- doi 10.1158/1055-9965.Epi-20-0053. [DOI] [Google Scholar]
- 40.Staiger DO, Stock JH. Instrumental variables regression with weak instruments. National Bureau of Economic Research Cambridge, Mass., USA; 1994. [Google Scholar]
- 41.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44(2):512–25 doi 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tchetgen Tchetgen EJ, Walter S, Vansteelandt S, Martinussen T, Glymour M. Instrumental variable estimation in a survival context. Epidemiology 2015;26(3):402–10 doi 10.1097/EDE.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sjolander A, Dahlqwist E, Zetterqvist J. A Note on the Noncollapsibility of Rate Differences and Rate Ratios. Epidemiology 2016;27(3):356–9 doi 10.1097/EDE.0000000000000433. [DOI] [PubMed] [Google Scholar]
- 44.Scheike T, Martinussen T, Silver J, Holst KJRpv. timereg: Flexible Regression Models for Survival Data. 2019;1(4). [Google Scholar]
- 45.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37(7):658–65 doi 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 2016;40(4):304–14 doi 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int J Epidemiol 2014;43(3):922–9 doi 10.1093/ije/dyu005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou B, Shu B, Yang J, Liu J, Xi T, Xing Y. C-reactive protein, interleukin-6 and the risk of colorectal cancer: a meta-analysis. Cancer Causes Control 2014;25(10):1397–405 doi 10.1007/s10552-014-0445-8. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Dai JY, Albanes D, Arndt V, Berndt SI, Bezieau S, et al. Mendelian randomization analysis of C-reactive protein on colorectal cancer risk. Int J Epidemiol 2019;48(3):767–80 doi 10.1093/ije/dyy244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C, Xu Q, Chen L, Luo C, Ying J, Liu J. C-reactive protein (CRP) as a prognostic factor for colorectal cancer after surgical resection of pulmonary metastases. Bull Cancer 2017;104(3):232–6 doi 10.1016/j.bulcan.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 51.Cooney RV, Chai W, Franke AA, Wilkens LR, Kolonel LN, Le Marchand L. C-reactive protein, lipid-soluble micronutrients, and survival in colorectal cancer patients. Cancer Epidemiol Biomarkers Prev 2013;22(7):1278–88 doi 10.1158/1055-9965.EPI-13-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsubara D, Arita T, Nakanishi M, Kuriu Y, Murayama Y, Kudou M, et al. The impact of postoperative inflammation on recurrence in patients with colorectal cancer. Int J Clin Oncol 2020;25(4):602–13 doi 10.1007/s10147-019-01580-1. [DOI] [PubMed] [Google Scholar]
- 53.Slattery ML, Curtin K, Poole EM, Duggan DJ, Samowitz WS, Peters U, et al. Genetic variation in C-reactive protein in relation to colon and rectal cancer risk and survival. Int J Cancer 2011;128(11):2726–34 doi 10.1002/ijc.25721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang SH, Huang CJ, Chang SC, Lin JK. Association of C-reactive protein gene polymorphisms and colorectal cancer. Ann Surg Oncol 2011;18(7):1907–15 doi 10.1245/s10434-011-1575-9. [DOI] [PubMed] [Google Scholar]
- 55.Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL, Kraft P. Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am J Hum Genet 2015;96(2):329–39 doi 10.1016/j.ajhg.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aschard H, Guillemot V, Vilhjalmsson B, Patel CJ, Skurnik D, Ye CJ, et al. Covariate selection for association screening in multiphenotype genetic studies. Nat Genet 2017;49(12):1789–95 doi 10.1038/ng.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.