

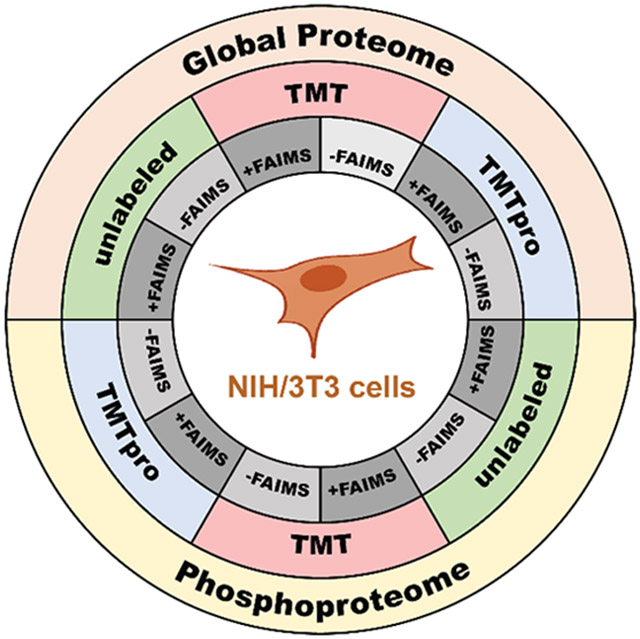
Abstract
Targeted mass spectrometry-based assays typically rely on previously acquired large data sets for peptide target selection. Such repositories are widely available for unlabeled peptides. However, they are less common for isobaric tagged peptides. Here we have assembled two series of six data sets originating from a mouse embryonic fibroblast cell line (NIH/3T3). One series is of peptides derived from a tryptic digest of a whole cell proteome and a second from enriched phosphopeptides. These data sets encompass three labeling approaches (unlabeled, TMT11-labeled, and TMTpro16-labeled) and two data acquisition strategies (ion trap MS2 with and without FAIMS-based gas phase separation). We identified a total of 1 509 526 peptide-spectrum matches which covered 11 482 proteins from the whole cell proteome tryptic digest, and 188 849 phosphopeptides from the phosphopeptide enrichment. The data sets were of similar depth, and while overlap across data sets was modest, protein overlap was high, thus reinforcing the comprehensiveness of these data sets. The data also supported FAIMS as a means to increase data set depth. These data sets provide a rich resource of peptides that may be used as starting points for targeted assays. Future data sets may be compiled for any genome-sequenced organism using the technologies and strategies highlighted herein. The data have been deposited in the ProteomeXchange Consortium with data set identifier PXD024298.
Keywords: targeted proteomics, 3T3, FAIMS, eclipse, TMT, TMTpro
Graphical Abstract
INTRODUCTION
Many targeted mass spectrometry-based assays are established using large previously collected data sets;1 however, such collections are uncommon for isobaric tagged peptides, particularly for studies using ion trap-based MS2 analysis. Here we aim to assemble several peptide data sets from the NIH/3T3 mouse embryonic fibroblast cell line. This cell line was generated from random-bred Swiss mouse embryos, and is commonly used in genetics and molecular biology.2 NIH/3T3 cells are often used to study transforming viruses and cellular oncogenes.3 Whereas HeLa cervical carcinoma cells and many other transformed cell lines grow uncontrolled in culture, 3T3 cells were spontaneously immortalized but exhibit very high sensitivity to contact inhibition.4 Indeed, many of the viral ras oncogenes and their cellular counterparts were discovered by means of the NIH/3T3 transformation assay, in which transformed clones of growing cells can be easily distinguished from background, growth-arrested cells.5,6 NIH/3T3 cells remain a powerful tool in many fields of biology, as over 30 000 publications using this cell line have been made public to date, and as such, we have selected this cell line as the basis for our data sets.
Sample multiplexing strategies in mass spectrometry-based quantitative analyses, such as tandem mass tags (TMT) and isobaric tags for relative and absolute quantitation (iTRAQ) exhibit many advantages for whole proteome profiling.7,8 Tandem mass tags (TMT) are robust and versatile reagents for sample multiplexing.9 Currently, two sets of commercial amine-reactive TMT reagents are available: (1) the original TMT6/10/11 set (“TMT11”) and (2) the more recently released TMTpro16/18.10,11 These two reagent sets are structurally different, but the labeling chemistry is identical. The tags consist of a reporter ion group, a mass normalization group, and an amine reactive group. The distribution of heavy isotopes in the structure of TMT tags varies across the reporter and normalization groups but sum to the same mass for labels in the multiplex set. These reagents label lysine residues and peptide N-termini, with TMT11 adding 229.163 Da and TMTpro16 adding 304.207 Da per tag. Here, we assemble and compare data sets comprised of unlabeled, TMT11-labeled, and TMTpro16-labeled peptides, including data sets of Fe2+-NTA enriched phosphopeptides.
In addition to different labeling approaches, we also investigate two data acquisition strategies, namely: standard ion trap-based MS2 with collision-induced dissociation (CID)-fragmentation and the same method, but coupled with high-field asymmetric-waveform ion-mobility spectrometry (FAIMS).12 Ion trap MS2 analysis with CID-fragmentation was selected due to its use in synchronous precursor selection (SPS)-MS3 and real-time database searching (RTS)-MS3 workflows.13,14 FAIMS separates ionized molecules in the gas phase by their mobility in a high or low electric field.15-17 A given ion traverses the electrodes at a specific characteristic value of a tunable DC voltage (the compensation voltage, or CV), and alternating CVs can rapidly separate different types of ions.18 FAIMS adds an orthogonal dimension of online gas phase-fractionation to a traditional LC-MS workflow. This facilitates the separation of coeluting isobaric species, including typically non-tryptic singly charged contaminant ions. As such, FAIMS can minimize instrument downtime preventing contaminating ions and salts from entering the instrument as they deposit on the easily accessible central electrode. It also reduces the coisolation and cofragmentation of peptides and can significantly improve the depth and accuracy of multiplex proteomic analyses.19,20 We selected three commonly used CVs (−40, −60, and −80 V) for our FAIMS-based analyses.21,22
In all, we assemble two series (whole cell proteome tryptic digest and Fe2+-NTA enriched phosphopeptides) of six data sets originating from the NIH/3T3 cell line, encompassing three labeling approaches, each with two different data acquisition strategies, as described above. We also compare peptide and protein identifications across data sets as well as their overlap. In addition, we investigate several peptide properties in efforts to characterize those phosphopeptides that are identified solely in a single data set. These data sets provide the community with a rich resource of peptides that may be used as starting points for further datamining and the development of targeted assays.
METHODS
Materials
Tandem mass tag (TMT11 and TMTpro16) isobaric reagents, trypsin, and the High-Select Fe2+-NTA Phosphopeptide Enrichment Kit were from ThermoFisher Scientific (Rockford, IL). Water and organic solvents were from J.T. Baker (Center Valley, PA). Gibco TrypLE Express (Cat# 12-604-021), Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) was from LifeTechnologies (Waltham, MA). Antimycotic solution (Cat# A5955) was from Sigma-Aldrich (St. Louis, MO). Trypsin was purchased from Pierce Biotechnology (Rockford, IL) and Lys-C from Wako Chemicals (Richmond, VA). Calyculin A (Cat# 9902S) was purchased from Cell Signaling Technology (Danvers, MA). Orthovanadate (Cat# P0758) was from New England BioLabs (Ipswich, MA). tC18 solid-phase extraction (SPE) Sep-Pak columns and Oasis HLB μElution plate (Cat # 186001828BA) were from Waters (Milford, MA). Unless otherwise noted, all other chemicals were from Pierce Biotechnology (Rockford, IL).
Cell Growth and Harvesting
Methods of cell growth and propagation followed techniques utilized previously.23,24 In brief, NIH/3T3 mouse embryonic fibroblast cells were propagated in DMEM supplemented with 10% FBS and 1× antibiotic antimycotic solution. For total protein, cells were seeded in three 175 cm2 cell culture flasks. Upon achieving ~90% confluency, the growth media was aspirated, and the cells were washed once with phosphate-buffered saline (PBS). The cells were dislodged with Gibco TrypLE Express Enzyme without phenol red, harvested by trituration following the addition of PBS, and pelleted by centrifugation at 1000g for 5 min at 4 °C. The supernatant was removed, and the cells were washed twice with PBS. Two milliliters of 200 mM EPPS, 8 M urea, pH 8.5 and 2% sodium dodecyl sulfate (SDS) supplemented with 1× Pierce Protease Inhibitors, Mini was added to the cell pellet. For phosphoproteome analysis, cells were seeded in 15 cm cell culture plates. When cells reached ~90% confluency, the growth media was aspirated, and each plate was washed once with 10 mL 20 mM HEPES pH 7.0–7.6, 120 mM NaCl. Cells were then incubated with 1 mM pervanadate and 25 nM calyculin A in 20 mM HEPES pH 7.0–7.6, 120 mM NaCl for 1 h in a cell culture incubator at 37 °C. Pervanadate was prepared fresh from sodium orthovanadate as described previously.25 Cells were harvested by scraping and trituration and collected in 50 mL conical tubes. Cells were pelleted by centrifugation at 1000g for 5 min at 4 °C, the supernatant was removed, and the cells were washed once with PBS. Cell pellets were frozen in liquid nitrogen and stored at −80 °C until further processing. The seeding and treatment were repeated twice for a total of forty-five 15 cm cell culture plates. A 3.3 mL aliquot of 200 mM EPPS, 8 M urea, pH 8.5 supplemented with 1× Pierce Protease, Mini was added to each cell pellet.
Cell Lysis and Protein Digestion
Cells were homogenized by 12 passes through a 22-gauge (1.25 in. long) needle. The homogenate was sedimented by centrifugation at 21 000g for 20 min and the supernatant was transferred to a new tube. Protein concentrations were determined using the bicinchoninic acid (BCA) assay. Approximately 3 mg protein were processed for total proteomics analysis and 60 mg protein for phosphoproteomics analysis. Proteins were subjected to disulfide bond reduction with 5 mM tris (2-carboxyethyl) phosphine (room temperature, 15 min) and alkylation with 10 mM iodoacetamide (room temperature, 30 min in the dark). Excess iodoacetamide was quenched with 10 mM dithiotreitol (room temperature, 15 min in the dark). Methanol–chloroform precipitation was performed prior to protease digestion. In brief, 4-parts neat methanol were added to each sample and vortexed, 1-part chloroform was added to the sample and vortexed, and 3-parts water was added to the sample and vortexed. The sample was centrifuged at 14 000 rpm for 2 min at room temperature and subsequently washed once with 100% methanol. Samples were resuspended in 200 mM EPPS, pH 8.5 and digested at room temperature for 14 h with Lys-C protease at a 50:1 protein-to-protease ratio. Trypsin was then added at a 100:1 protein-to-protease ratio and the reaction was incubated for 6 h at 37 °C.
Fe2+-NTA Phosphopeptide Enrichment
Phosphopeptides were enriched with the High-Select Fe2+-NTA Phosphopeptide Enrichment Kit according to the manufacturer’s specifications using approximately 2.5 mg protein digest per enrichment column. We have shown previously the efficiency of the High-Select Fe-NTA Phosphopeptide Enrichment Kit for phosphopeptide analysis.26 We began with a total of 60 mg of LysC/trypsin-digested peptides, which we divided into 30 individual spin columns (2 mg of protein per spin column). We followed the manufacturer’s instructions with the exception that we eluted the phosphopeptides into a tube containing 100 μL 10% formic acid (FA) (for a final FA concentration of 1%) to neutralize the pH of the elution buffer. Our yield was ~900 μg phosphopeptides (with ~98% enrichment), which we divided into three aliquots for the unlabeled, TMT-labeled, and TMTpro-labeled analysis.
Tandem Mass Tag Labeling
The classic TMT reagents (0.8 mg) were dissolved in anhydrous acetonitrile (40 μL) of which 20 μL was added to the peptides (200 μg) with 60 μL of acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). The TMT11 reagent has the identical structure and labeling properties as TMT10, but has a mass difference of 0.06 Da. Peptides labeled with the TMT11 reagent are indistinguishable at the MS1 stage from those labeled with TMT10 during routine TMT analysis. TMTpro reagents (0.8 mg) were dissolved in anhydrous acetonitrile (40 μL) of which 28 μL was added to the peptides (200 μg) with 52 μL of acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). A greater amount of labeling reagent were used in TMTpro16 compared to TMT11 reactions due to the increased size of the TMTpro16 molecule and the need to ensure >95% labeling efficiency.11 Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.3% (v/v). The sample was vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (SPE, Sep-Pak).
Off-Line Basic pH Reversed-Phase (BPRP) Fractionation
We fractionated the peptide samples using BPRP HPLC.27 We used an Agilent 1200 pump equipped with a degasser and a UV detector. Peptides were subjected to a 50 min linear gradient from 5% to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 at a flow rate of 0.6 mL/min over an Agilent 300 Extend C18 column (3.5 μm particles, 4.6 mm ID and 250 mm in length). The peptide mixture was fractionated into a total of 96 fractions, which were consolidated into 24 superfractions (in a checkerboard-like pattern) for the whole proteome digest28 and into 12 fractions (eluted across rows, but collected down the each column) for the phosphopeptide-enriched fractions. Samples were subsequently acidified with 1% formic acid and vacuum centrifuged to near dryness. Each consolidated fraction was desalted using an Oasis HLB μElution plate,29 dried again via vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid Chromatography and Tandem Mass Spectrometry
Mass spectrometric data were collected on an Orbitrap Eclipse mass spectrometer coupled to a Proxeon NanoLC-1200 UHPLC. The 100 μm capillary column was packed with 35 cm of Accucore 150 resin (2.6 μm, 150 Å; ThermoFisher Scientific). Data were acquired for 75 min per fraction for the whole proteome samples and 90 min for the phosphopeptide fractions. The scan sequence began with an MS1 spectrum: Orbitrap analysis, resolution 120 000, 350–1400 Th, automatic gain control (AGC) target 5 × 105, maximum injection time 100 ms. MS2 analysis consisted of CID with multistage activation (MSA), quadrupole ion trap analysis, AGC 1 × 104, NCE (normalized collision energy) 35, q-value 0.25, isolation window 0.4 Th, maximum injection time set to “auto” and TopSpeed set at 3 s. Advanced peak detection (APD) was turned on. For data acquisition that included FAIMS, the dispersion voltage (DV) was held constant at 5000 V, the compensation voltages (CVs) were set at −40, −60, and −80 V, and TopSpeed parameter was set to 1 s. No MS3 scans were acquired for these data sets.
Data Analysis
Spectra were converted to mzXML via MSconvert.30 Database searching included all entries from the mouse UniProt Database (downloaded: November 2019). The database was concatenated with one composed of all protein sequences for that database in the reversed order. Searches were performed using a 50-ppm precursor ion tolerance and the product ion tolerance was set to 0.9 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Comet searches and linear discriminant analysis.31,32 TMT tags on lysine residues and peptide N-termini (+229.16 Da for TMT11 and +304.207 Da for TMTpro16 where appropriate), as well as carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications. Oxidation of methionine residues (+15.995 Da) and phosphorylation (i.e., for the Fe2+-NTA enriched samples) on serine, threonine, and tyrosine (+79.966 Da) were set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR).33,34 PSM filtering was performed using a linear discriminant analysis, as described previously32 and then assembled further to a final protein-level FDR of 1%.34 Data analysis and visualization was performed in Microsoft Excel or R Bioconductor35 with the “peptides”, “dplyr”, “DT”, and “shiny” packages.
Data Access
RAW files will be made available upon request. The data have been deposited in the ProteomeXchange Consortium via the PRIDE36 partner repository with the data set identifier PXD024298.
RESULTS AND DISCUSSION
Six Data Sets Were Collected Encompassing Three Labeling and Two Data Acquisition Strategies to Investigate Proteolytically Digested Peptides from a Whole Cell Proteome
NIH/3T3 cells were processed using the SL-TMT protocol37 (Figure 1). Following proteolytic digestion, the peptide pool was divided equally into three aliquots. One aliquot remained unlabeled, the second aliquot was labeled with a mix of TMT11 reagents, and the third aliquot was labeled with a mix of isobaric labels of the TMTpro16 reagent. We noted that as only collision-induced dissociation (CID)—not higher energy-collisional dissociation (HCD)—was used for peptide fragmentation, peptide fragment ions generally retained the isobaric tag on lysine residues and peptide N termini. These three sets of peptides were each separated into 96 fractions using basic-pH reversed-phase (BPRP) chromatography and concatenated down to 24 superfractions.28 Each superfraction was analyzed over a 75 min gradient on an Orbitrap Eclipse mass spectrometer. We estimated 1 μg of peptide was loaded on-column for non-FAIMS analysis, and 2 μg of peptide were loaded for FAIMS-based analysis to compensate for sample loss as ions traverse the FAIMS interface.21 In total, we analyzed 144 samples, that together comprised six separate data sets of unlabeled, TMT11-labeled, and TMTpro16-labeled samples, each of which were analyzed with and without FAIMS.
Figure 1.

Workflow for whole proteome tryptic digest data sets. A mouse embryonic cell line (NIH/3T3) was harvested, lysed, and processed further using the SL-TMT protocol.37 The tryptic peptide pool was divided into three aliquots: (1) unlabeled, (2) labeled with TMT11 reagents, and (3) labeled with TMTpro16 reagents. Each aliquot of peptides was fractionated into 96 fractions using basic-pH reversed-phase (BPRP) chromatography and concatenated into 24 superfractions. Each superfraction was analyzed on an Orbitrap Eclipse mass spectrometer with and without FAIMS.
Similar Numbers of Unique Peptides and Proteins Were Identified among the Six Whole Proteome Tryptic Digest Data Sets Regardless of Tag or Data Acquisition Strategy
A total of 1 509 526 peptides were matched to a mouse tryptic database at a global peptide false discovery rate (FDR) of less than 1%. Of these peptides, 247 630 were unique in sequence. These peptides corresponded to 11 482 proteins in total across the six data sets at a 1% protein FDR (Figure 2A). We sought to determine if the analytical depths of the six data sets were comparable. We noted that the average number of unique peptides per fraction was approximately 122 000 with a slight increase (~5%) in the number of peptides in data sets acquired with FAIMS. However, overall, no single data set matched substantially more peptides. We observed a similar trend when proteins were inferred from the peptide matches. The number of proteins was slightly higher for all three peptide populations (i.e., unlabeled, TMT11-labeled, and TMTpro16-labeled) when samples were analyzed with FAIMS, although this value was again less than 10%. It follows that the average number of peptides per protein ranges from 11.5 to 12.5 for all six data sets. Together, these data sets comprise a large resource comparing mouse peptides using multiple labeling chemistries (unlabeled, TMT11, and TMTpro16) and data acquisition strategies (i.e., with and without FAIMS). Overall, we conclude that the depths of these data sets are similar considering the number of identified peptides and proteins.
Figure 2.

Overview of the whole proteome tryptic digest data sets. (A) Table of total and unique peptides, as well as proteins for all six whole proteome tryptic digest data sets. Proteins identified with single peptides are included in the tally. Venn diagrams illustrating the overlap of the unlabeled, TMT11, and TMTpro16 data sets at the peptide level (B) without using FAIMS and (C) with FAIMS, and at the protein level (D) without FAIMS and (E) with FAIMS.
Peptide Overlap Was Minimal, but Most Identified Proteins Were Common among the Six Data Sets Regardless of Label or Data Acquisition Strategy
Once we established that no difference was apparent in the number of peptides or proteins identified across the six data sets, we compared the overlap across the data sets. We first illustrated the overlap among data sets at the peptide level using an upset plot (Figure S1A). This plot showed that 29 346 peptides were matched in all six data sets, which is approximately 12% of the total number of peptides across data sets (i.e., of the 247 630 total unique peptides). However, this plot also depicted a total of 68 807 peptides identified in only one data set, specifically, 10 183 from the unlabeled data, 16 010 from the unlabeled with FAIMS data set, 7319 from the TMT11 data set, 14 635 from the TMT11 with FAIMS data set, 6700 from the TMTpro16 data set, and 13 960 from the TMTpro16 with FAIMS data set. These data supported further that the highest number of data set-unique peptides were obtained from the FAIMS data sets. Instrument methods with FAIMS were designed to alternate across three compensation voltage (CVs), specifically −40, −60, and −80 V, for sequential scans in efforts to select a diverse population of ions. Liquid chromatography-based fractionation combined with the advantages of gas-phase separation allowed us to substantially increase the depth of peptide identification for each labeling approach as FAIMS reduced the chemical noise and enhanced the dynamic range of quantification.32,33 These findings were also illustrated in a series of Venn diagrams. Comparing the three labeling approaches with data acquired without FAIMS revealed that 56 239 peptides (30%) were common among all three data sets, while 39–44% were common between any two data sets (Figure 2B). However, we observed slightly less overlap in the data sets analyzed with FAIMS among the differentially labeled data sets. Specifically, 50 155 peptides (23%) were common in all three data sets, while 30–37% were common between any two data sets (Figure 2C). The discrepancy between the number of overlapping peptides acquired with and without FAIMS supported again our previous observations that greater peptide depth and diversity was obtained when using FAIMS regardless of labeling strategy.
We next investigated if the lack of overlap persisted in a similar analysis of the proteins identified in each data set. We again illustrated the protein level overlap using an upset plot (Figure S1B). This plot showed that 8410 proteins (of 11 482 total) were represented in all six data sets, which is over 72% of the total proteins across the six data sets. In fact, the highest number of proteins identified in only a single data set was 34. These data suggested that although the peptide overlap was modest, those findings did not translate to the protein level as we identified many of the same proteins in all six data sets. We also illustrated these findings in a series of Venn diagrams. Comparing the three labeling approaches without FAIMS revealed that 9081 proteins (82%) were common in all three differentially labeled data sets, while that value approached 85% considering overlap in at least two data sets (Figure 2D). Likewise, with data acquired using FAIMS, 9837 proteins (89%) were common in all differentially labeled data sets, while only slightly more (~93%) were common when including those between any two labeling approaches (Figure 2E). The high overlap at the protein level reinforced further the comprehensiveness of these data sets.
To emphasize further the advantage of FAIMS, we performed pairwise comparisons of the number of peptides and proteins for each labeling approach between data acquired with and without FAIMS using a series of Venn diagrams. The peptide overlap ranged between 44% and 51% for unlabeled (Figure S2A), TMT11-labeled (Figure S2B), and TMTpro16-labeled (Figure S2C) peptides, supporting the data presented in the corresponding upset plot (Figure S1A). As expected, the protein level data again demonstrated high overlap. Specifically, the protein overlap ranged between 87% and 90% for unlabeled (Figure S2D), TMT11-labeled (Figure S2E), and TMTpro16-labeled (Figure S2F) data sets. We noted that for all comparisons between FAIMS and non-FAIMS data sets, two to three times more data set-unique peptides were identified in the FAIMS data sets. Coincidently, the number of data set-unique proteins in the FAIMS data sets were also two to three times higher than in the non-FAIMS data sets. These data support further the use of FAIMS for increasing data set depth.
Differences in Characteristics and Physicochemical Properties of Peptides That Were Unique to Each of the Six Data Sets Were Minimal
As we noted only modest overlap in peptides matched across the six data sets, we next examined several characteristics and physicochemical properties of peptides unique to a given data set. We first examined the XCorr value, which assesses the cross-correlation between the theoretical and acquired mass spectra (Figure S3A). We noted that the median and mean of the distributions of XCorr values were similar across all data sets. We observed that the distribution of peptide lengths was slightly lower and the distribution itself tighter for data sets collected with FAIMS versus those collected without FAIMS for the same labeling approach (Figure S3B). Such a result may be expected as the constraints of the three compensation voltages adds some selectivity with regard to the ions entering the mass spectrometer. When comparing across labeling approaches, we observed a decrease in peptide length in the data sets in this order: unlabeled, TMT11, and TMTpro16. To pursue this finding further, we next examined the distribution of peptide mass (with tag, if applicable) (Figure S3C). The differences in distributions between the data sets acquired with FAIMS were smaller than those acquired without FAIMS. In addition, the overall mass distributions, as well as their means and medians were similar across the three FAIMS data sets. A slight decrease in peptide mass was observed only in the non-FAIMS data sets. The order of the data sets by descending mean peptide length was: unlabeled, TMT11 and TMTpro16. We then calculated the masses of peptides stripped of isobaric labels (if applicable) and plotted the distribution of the data set-unique peptides (Figure S3D). Overall, similar trends were observed, as we again noted a tighter distribution when using FAIMS than not. However, now we also observed a decrease in peptide mass (stripped of modifications) across labeling approaches in both the FAIMS and non-FAIMS-fractionated data set, closely mimicking the distribution of peptide length (Figure S3B). As such, we postulate that certain peptides with additional mass due to the tags fell outside the optimal mass range for the current data acquisition settings. We noted that isobaric tags added mass to peptides, such that many peptides now had the mass equivalent of two to six more amino acids. More specifically, if we estimate the average mass of an amino acid residue to be 110 Da, a single TMT11 tag on an arginine-terminating peptide will have added ~229 Da (the mass equivalent of ~2 amino acid residues), while in the extreme case, two TMTpro16 tags on a lysine-terminating peptide will have added ~608 Da (the mass equivalent of ~6 residues). These data revealed that further optimization of data acquisition parameters and investigation thereof may be needed to improve the identification of longer peptides labeled with TMT or TMTpro reagents.
In addition, we interrogated several other characteristics for data set-unique peptides, but these differences were less significant among labeling approaches. Although the distribution of isoelectric points (pI) did not differ by labeling chemistry, we observed a minor increase in pI values for FAIMS-fractionated data sets (Figure S3E). This finding has not been reported previously but may merit further investigation. We similarly noticed a slight decrease in charge state (z) distribution for the samples acquired using FAIMS compared to without, yet no difference was observed across labeling approaches (Figure S3F). Moreover, we noted no significant difference in peak width distribution for any subset of the peptides queried (Figure S3G). Likewise, the distribution of other physicochemical properties, such as the GRAVY index38 (Figure S3H), which is a measure of hydrophobicity, the aliphatic index39 (Figure S3I), which is based on the number of aliphatic residues on a peptide, and the instability index40 (Figure S3J) were similar for the data set-unique peptides across all data sets. Overall, we conclude that the starkest differences in the characteristics tested for data set-unique peptides were mass and length across labeling approaches. Moreover, the data sets acquired using FAIMS demonstrated tighter distributions that may be reflective of the selectivity attained by limiting the CV values chosen for gas phase separation.
Six Additional Data Sets Were Collected to Interrogate the Phosphoproteome of NIH/3T3 Cells Also Using Three Labeling and Two Data Acquisition Strategies
As performed for the whole cell proteome analysis, NIH/3T3 cells were processed using an SL-TMT-based protocol which included an additional Fe2+-NTA phosphopeptide enrichment step following proteolytic digestion and prior to labeling (if applicable)37 (Figure S4). To enhance the breadth of the phosphopeptide data sets, the cells were treated with pervanadate (which inhibits tyrosine phosphatases) and calyculin A (which inhibits serine and threonine phosphatases). Aside from phosphopeptide enrichment with Fe2+-NTA, sample processing was identical to the whole cell proteome except for the concatenation strategy for the BPRP fractions. Here, every twelfth sample was combined, resulting in only 12 superfractions. Each superfraction was analyzed over a 90 min gradient on an Orbitrap Eclipse mass spectrometer. Again, we loaded 1 μg of peptide on-column for non-FAIMS analysis, and 2 μg of peptide for FAIMS-based analysis. 21 In total, we analyzed 72 samples, that together comprised six data sets of unlabeled, TMT11-labeled, and TMTpro16-labeled phosphopeptides, which were again analyzed with and without FAIMS.
The analysis for each data set revealed similar numbers of identified unique phosphopeptides and phosphoproteins regardless of tag or data acquisition strategy. A total of 828 228 phosphopeptides were matched to a mouse tryptic database at a peptide FDR of less than 1%. Of these phosphopeptides, 188 849 were unique in sequence and phosphorylation pattern. These phosphopeptides originated from 8318 proteins in total across the six data sets at a 1% protein FDR (Figure 3A). As in the whole proteome data sets, the use of FAIMS increased phosphopeptide and proteome depth for a given labeling strategy. However, the upset plots for the Fe2+-NTA-enriched samples showed more exclusivity of phosphopeptides to a given tag or data acquisition strategy (Figure S5A). More specifically, only 11 123 phosphopeptides were common in all six data sets, while over 25 000, over 20 000, and nearly 16 000 phosphopeptides were unique to the unlabeled + FAIMS, TMTpro16 + FAIMS, and TMT11 + FAIMS data sets, respectively. Venn diagrams showed that only 18 880 phosphopeptides (16.5%) (Figure 3B) were shared among the three data sets when not using FAIMS, while a similar number 21 805 (13.6%) (Figure 3C) were shared among the three data sets using FAIMS. These results contrasted with the ~30% and ~23% overlap in the three whole cell proteome data sets with and without FAIMS, respectively (Figure 2B and 2C). However, collapsing phosphopeptide identifications down to the phosphoprotein level (Figure 3D and 3E) showed overlap among acquisition methods consistent with that of the whole proteome analysis (Figure 2D and 2E).
Figure 3.

Overview of the Fe2+-NTA-enriched phosphopeptide data sets. (A) Table of total and unique phosphopeptides, as well as phosphoproteins quantified for all six Fe2+-NTA-enriched data sets. Proteins identified with single peptides are included in the tally. Venn diagrams illustrating the overlap of the unlabeled, TMT11, and TMTpro16 data sets at the peptide level (B) without using FAIMS and (C) with FAIMS, and at the protein level (D) without using FAIMS and (E) with FAIMS.
Distinct phosphopeptides may be identical in sequence but differ in the number of phosphate groups on the actual molecule. We limited our searches to singly, doubly, and triply phosphorylated peptides (Figure 4A). In all six data sets, we observed that ~79–83% of phosphopeptides identified were singly phosphorylated, while ~15–20% were doubly phosphorylated, and 1.4–3.7% were triply phosphorylated. We noted a higher percentage of triply phosphorylated peptides in the data sets analyzed with FAIMS than those without. Moreover, we detected an increasing trend for a higher percentage of triply phosphorylated peptides in the order: unlabeled, TMT11, and TMTpro16 irrespective of whether or not FAIMS was used. We also investigated the distribution of phosphate groups on serine, threonine, and tyrosine residues (Figure 4B). These pervanadate and calyculin-A treated cells showed a consistent ratio of ~70:20:10 with respect to pSer:pThr:pTyr peptides across all data sets. In addition, we assembled the entire data set (<0.05% peptide FDR, <1% protein FDR), tallied the phosphorylation sites of singly phosphorylated peptides, and assessed their localization (Figure 4C). In total, we identified 95 674 phosphorylation sites (including 9429 pTyr sites), of which 76 935 were localized to a single residue with >95% confidence using the AScore algorithm.31 Like the phosphopeptides (Figure 4B), the phosphorylation sites were also at a 70:20:10 ratio with respect to pSer:pThr:pTyr sites. This ratio was skewed from the more commonly accepted 86.5:11.8:1.841 or 83:15:232 ratios likely due to the effect of the pervanadate and calyculin-A treatments.
Figure 4.

Characteristics of Fe2+-NTA-enriched phosphopeptide data sets. (A) Bar graphs illustrating the number of singly, doubly, and triply phosphorylated peptides across the six phosphopeptide data sets. (B) Bar graphs summarizing the number of phosphopeptides with at least one phosphorylated serine (pSer), threonine (pThr), and tyrosine (pTyr) residue across the six data sets. (C) Bar graph depicting the number of identified and localized phosphorylation sites for all six data sets. We tally the numbers of serine (pSer), threonine (pThr), and tyrosine (pTyr)-centered motifs for phosphorylation sites with a single phosphorylation site per peptide.
The low phosphopeptide overlap suggested that the identification of certain phosphopeptides may be influenced by the tag, data acquisition strategy, and/or site localization issues. Yet, like the proteins in the whole cell proteome data sets (Figure S1B), most phosphoproteins identified (n = 4985) were found in all six Fe2+-NTA-enriched data sets (Figure S5B). As such, the identification of phosphopeptides may be method-specific, but phosphoproteins can be more universally identified. At the phosphoprotein level, comparing the three labeling approaches without FAIMS revealed that 5586 phosphoproteins (71%) were common in all three differentially labeled data sets, while this value was approximately 85% considering overlap in at least two data sets (Figure 3D). Likewise, with data acquired using FAIMS, 6494 proteins (79%) were common in all differentially labeled data sets, while this value was over 90% considering overlap in at least two data sets (Figure 3E). The high overlap at the phosphoprotein level reinforced the comprehensiveness of these data sets.
We again emphasized the advantage of FAIMS for the Fe2+-NTA enriched data sets by performing pairwise comparisons of the number of phosphopeptides and phosphoproteins for each labeling approach between data acquired with and without FAIMS using a series of Venn diagrams. The peptide overlap ranged between 35% and 45% for unlabeled (Figure S6A), TMT11-labeled (Figure S6B), and TMTpro16-labeled (Figure S6C) peptides, which is approximately 10% less than that for the whole cell proteome peptide data sets (Figure S2A-C). As expected, the phosphoprotein level data again demonstrated high overlap. Specifically, the protein overlap ranged between 81% and 87% for unlabeled (Figure S6D), TMT11-labeled (Figure S6E), and TMTpro16-labeled (Figure S6F) peptides. These data supported further the advantage of FAIMS for increasing data set depth.
We noted that FAIMS separation increased the number of peptides identified in the whole cell proteome analysis, as well as in the Fe2+-NTA enriched data sets. The resolution of FAIMS CVs had been estimated to be 15–20 V,42 and we used CV = −40, −60, and −80 V. Recently, instrument software (Thermo Tune 3.4) has allowed for dynamic exclusion across CVs so as not to acquire MS2 data on the same precursor mass multiple times across different CVs. As such, we explored the overlapping peptides in all six of our FAIMS data sets across CVs (Figure S7A-G). We noted for both the whole cell proteome (Figure S7A-C) and the Fe2+-NTA enriched data sets (Figure S7D,E) that over 90% of peptides were unique to a single CV regardless of labeling strategy. These data exemplified the high level of peptide diversity achieved with FAIMS using CVs of −40, −60, and −80 V.
CONCLUSIONS
We assembled a total of 12 data sets originating from an NIH/3T3 mouse embryonic fibroblast cell line that encompassed three labeling approaches (unlabeled, TMT11-labeled, and TMTpro16-labeled) and two data acquisition strategies (ion trap MS2 with and without FAIMS-based gas-phase separation). Six data sets were of a whole cell proteome tryptic digest, while the other six were of enriched phosphopeptides. For the whole cell proteome tryptic digest data sets, we identified a total of 1 509 526 peptide-spectrum matches, of which 247 630 were unique in sequence, covering 11 482 proteins. In addition, we acquired 828 228 peptide-spectrum matches that corresponded to 188 849 nonredundant phosphopeptides from 8318 phosphoproteins. Moreover, we designed an interactive R Shiny application “NIH/3T3 Peptide and Phosphopeptide Viewer,” which can be viewed at https://wren.hms.harvard.edu/OPapp/ for querying peptides and proteins in our data sets (Figure 5). Although peptide overlap across data sets was minimal, most identified proteins were common among the data sets regardless of labeling or data acquisition strategy, which was a testament to the comprehensiveness of the data sets. We also noted some differences in the mass and length distribution of peptides that were unique to each of the six data sets. We postulate that this observation may be a consequence of the addition of the isobaric tags, which can add a mass equivalent to that of several amino acid residues. Overall, these data sets provide a rich resource of peptides and phosphopeptides which may be used as starting points for targeted assays, such as parallel reaction monitoring (PRM),43 TOMAHAQ,44 or TOMAHTO.45 Although the peptides that we analyzed here were of murine origin, 98 684 of these peptides are also found in the human proteome, and these can be used to target proteins of human origin. We anticipate future data sets to be compiled for other cell types or organisms using the technologies and strategies highlighted herein.
Figure 5.

The NIH/3T3 Mouse Cell Line Tryptic Peptide Viewer. (A) Proteins may be selected from a drop-down list by typing in key letters.. Gene symbols and UniProt accession numbers are listed in pairs. (B) Tab panels with overall information in one tab, and PSM information in the other tab. In the “Overall Info” tab, total peptide numbers are summarized. A link to the UniProt entry of the selected protein is provided. In the “PSM Percentile Rank” tab, a histogram showing the log10(PSM) distribution is presented. PSMs of the proteins with selected gene symbol and the percentile ranks of the log10(PSM) are labeled on the plot. (C) Nonphosphorylated peptides are listed in one tab, and phosphorylated peptides listed in another. The data table outputs all the peptides assigned to the selected protein and the peptide spectrum match (PSM) numbers under different analytical conditions. The search box on the right corner allows character searches, such as amino acid residues. After clicking on a row, the peptide sequence will be copied into (D), a peptide search box. Clicking the search button submits the search, and a blast hyperlink will appear below. (E) The peptide detail table displays the peptides with modifications (if any), peptide characteristics, and the phosphorylation site localization score (if applicable). The application is available at https://wren.hms.harvard.edu/OPapp/.
Supplementary Material
Figure S1: Overlap of the whole proteome tryptic digest data sets across the three labeling strategies
Figure S2: Overlap of differentially labeled peptides from the whole proteome tryptic digest data sets with respect to data acquisition with and without FAIMS-based gas phase separation
Figure S3: Peptide characteristics
Figure S4: Workflow for Fe2+-NTA-enriched data sets
Figure S5: Overlap of the Fe2+-NTA enriched data sets across the three labeling strategies
Figure S6: Overlap of differentially labeled phosphopeptides from Fe2+-NTA-enriched data sets with respect to data acquisition with and without FAIMS-based gas phase separation
Figure S7: Overlap of peptides when using different compensation voltages for analysis with FAIMS (PDF)
Phosphorylation sites identified in the Fe2+-NTA phosphopeptide enrichment data sets; Columns include: type (single or multiply phosphorylated peptides), protein ID, gene symbol, protein description, site position, motif peptide, AScore, and redundancy (XLSX)
Peptides identified in whole cell proteome tryptic digest data sets; Columns include: retention time, observed mass, observed m/z, charge state, compensation voltage (CV), RAW filename (SrchName), mass difference (PPM), Comet Expect score, XCorr, deltaCN, number of ions matched, total ions matched, protein name (reference), redundancy, peptide sequence (with flanking residues), trimmed peptide sequence, peptide length, missed cleavage, gene symbol, maximum precursor intensity, and peak width (ZIP)
Phosphopeptides identified in the Fe2+-NTA phosphopeptide enrichment data sets; Columns include those in Table S1, plus the AScore peptide, and columns for phosphorylation site position, and peptide score (ZIP)
Funding
This work was supported by NIH grant no. R01GM132129 (J.A.P.) and GM67945 (S.P.G.).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00247.
Contributor Information
Olesja Popow, Department of Cancer Biology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts 02115, United States.
Xinyue Liu, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Kevin M. Haigis, Department of Cancer Biology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts 02115, United States
Steven P. Gygi, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
Joao A. Paulo, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts 02115, United States.
REFERENCES
- (1).Deutsch EW The PeptideAtlas Project. Methods Mol. Biol 2010, 604, 285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Todaro GJ; Green H Quantitative studies of the growth of mouse embryo cells in culture and their development into establishes lines. J. Cell Biol 1963, 17 (2), 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wells WA A cell line that is under control. J. Cell Biol 2005, 168 (7), 988–989. [Google Scholar]
- (4).Jainchill JL; Aaronson SA; Todaro GJ Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J. Virol 1969, 4 (5), 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lowy DR; Willumsen BM Function and regulation of ras. Annu. Rev. Biochem 1993, 62, 851–891. [DOI] [PubMed] [Google Scholar]
- (6).Varmus HE The molecular genetics of cellular oncogenes. Annu. Rev. Genet 1984, 18, 553–612. [DOI] [PubMed] [Google Scholar]
- (7).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; Purkayastha S; Juhasz P; Martin S; Bartlet-Jones M; He F; Jacobson A; Pappin DJ Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 2004, 3 (12), 1154–69. [DOI] [PubMed] [Google Scholar]
- (8).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Johnstone R; Mohammed AK; Hamon C Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (9).Rauniyar N; Yates JR 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res 2014, 13 (12), 5293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li J; Cai Z; Bomgarden RD; Pike I; Kuhn K; Rogers JC; Roberts TM; Gygi SP; Paulo JA TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res 2021, 20 (5), 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li J; Van Vranken JG; Pontano Vaites L; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH; Kuhn K; Pike I; Bomgarden RD; Rogers JC; Gygi SP; Paulo JA TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17 (4), 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Guevremont R High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J. Chromatogr A 2004, 1058 (1–2), 3–19. [PubMed] [Google Scholar]
- (13).Schweppe DK; Eng JK; Yu Q; Bailey D; Rad R; Navarrete-Perea J; Huttlin EL; Erickson BK; Paulo JA; Gygi SP Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res 2020, 19 (5), 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Erickson BK; Mintseris J; Schweppe DK; Navarrete-Perea J; Erickson AR; Nusinow DP; Paulo JA; Gygi SP Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res 2019, 18 (3), 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Prasad S; Belford MW; Dunyach JJ; Purves RW On an aerodynamic mechanism to enhance ion transmission and sensitivity of FAIMS for nano-electrospray ionization-mass spectrometry. J. Am. Soc. Mass Spectrom 2014, 25 (12), 2143–53. [DOI] [PubMed] [Google Scholar]
- (17).Purves RW; Prasad S; Belford M; Vandenberg A; Dunyach JJ Optimization of a New Aerodynamic Cylindrical FAIMS Device for Small Molecule Analysis. J. Am. Soc. Mass Spectrom 2017, 28 (3), 525–538. [DOI] [PubMed] [Google Scholar]
- (18).Pfammatter S; Bonneil E; McManus FP; Prasad S; Bailey DJ; Belford M; Dunyach JJ; Thibault P A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Mol. Cell Proteomics 2018, 17 (10), 2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Navarrete-Perea J; Gygi SP; Paulo JA HYpro16: A Two-Proteome Mixture to Assess Interference in Isobaric Tag-Based Sample Multiplexing Experiments. J. Am. Soc. Mass Spectrom 2021, 32 (1), 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang T; Keele GR; Churchill GA; Gygi SP; Paulo JA Strain-Specific Peptide (SSP) Interference Reference Sample: A Genetically Encoded Quality Control for Isobaric Tagging Strategies. Anal. Chem 2021, 93 (12), 5241–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schweppe DK; Rusin SF; Gygi SP; Paulo JA Optimized Workflow for Multiplexed Phosphorylation Analysis of TMT-Labeled Peptides Using High-Field Asymmetric Waveform Ion Mobility Spectrometry. J. Proteome Res 2020, 19 (1), 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach JJ; Paulo JA; Gygi SP Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem 2019, 91 (6), 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Paulo JA; Urrutia R; Banks PA; Conwell DL; Steen H Proteomic analysis of a rat pancreatic stellate cell line using liquid chromatography tandem mass spectrometry (LC-MS/MS). J. Proteomics 2011, 75 (2), 708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Paulo JA; Urrutia R; Banks PA; Conwell DL; Steen H Proteomic analysis of an immortalized mouse pancreatic stellate cell line identifies differentially-expressed proteins in activated vs non-proliferating cell states. J. Proteome Res 2011, 10 (10), 4835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hill MM; Hemmings BA Analysis of protein kinase B/Akt. Methods Enzymol. 2002, 345, 448–63. [DOI] [PubMed] [Google Scholar]
- (26).Paulo JA; Navarrete-Perea J; Erickson AR; Knott J; Gygi SP An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J. Am. Soc. Mass Spectrom 2018, 29 (7), 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang Y; Yang F; Gritsenko MA; Wang Y; Clauss T; Liu T; Shen Y; Monroe ME; Lopez-Ferrer D; Reno T; Moore RJ; Klemke RL; Camp DG 2nd; Smith RD Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11 (10), 2019–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Paulo JA; O’Connell JD; Everley RA; O’Brien J; Gygi MA; Gygi SP Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteomics 2016, 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rappsilber J; Ishihama Y; Mann M Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem 2003, 75 (3), 663–70. [DOI] [PubMed] [Google Scholar]
- (30).Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak MY; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 2012, 30 (10), 918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Beausoleil SA; Villen J; Gerber SA; Rush J; Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 2006, 24 (10), 1285–92. [DOI] [PubMed] [Google Scholar]
- (32).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villen J; Haas W; Sowa ME; Gygi SP A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143 (7), 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Elias JE; Gygi SP Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol 2010, 604, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (35).Reimers M; Carey VJ Bioconductor: an open source framework for bioinformatics and computational biology. Methods Enzymol. 2006, 411, 119–34. [DOI] [PubMed] [Google Scholar]
- (36).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res 2018, 17 (6), 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kyte J; Doolittle RF A simple method for displaying the hydropathic character of a protein. J. Mol. Biol 1982, 157 (1), 105–32. [DOI] [PubMed] [Google Scholar]
- (39).Ikai A Thermostability and aliphatic index of globular proteins. J. Biochem 1980, 88 (6), 1895–1898. [PubMed] [Google Scholar]
- (40).Guruprasad K; Reddy BV; Pandit MW Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng., Des. Sel 1990, 4 (2), 155–61. [DOI] [PubMed] [Google Scholar]
- (41).Olsen JV; Blagoev B; Gnad F; Macek B; Kumar C; Mortensen P; Mann M Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127 (3), 635–48. [DOI] [PubMed] [Google Scholar]
- (42).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell Proteomics 2012, 11 (11), 1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Erickson BK; Rose CM; Braun CR; Erickson AR; Knott J; McAlister GC; Wuhr M; Paulo JA; Everley RA; Gygi SP A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65 (2), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Yu Q; Xiao H; Jedrychowski MP; Schweppe DK; Navarrete-Perea J; Knott J; Rogers J; Chouchani ET; Gygi SP Sample multiplexing for targeted pathway proteomics in aging mice. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (18), 9723–9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Overlap of the whole proteome tryptic digest data sets across the three labeling strategies
Figure S2: Overlap of differentially labeled peptides from the whole proteome tryptic digest data sets with respect to data acquisition with and without FAIMS-based gas phase separation
Figure S3: Peptide characteristics
Figure S4: Workflow for Fe2+-NTA-enriched data sets
Figure S5: Overlap of the Fe2+-NTA enriched data sets across the three labeling strategies
Figure S6: Overlap of differentially labeled phosphopeptides from Fe2+-NTA-enriched data sets with respect to data acquisition with and without FAIMS-based gas phase separation
Figure S7: Overlap of peptides when using different compensation voltages for analysis with FAIMS (PDF)
Phosphorylation sites identified in the Fe2+-NTA phosphopeptide enrichment data sets; Columns include: type (single or multiply phosphorylated peptides), protein ID, gene symbol, protein description, site position, motif peptide, AScore, and redundancy (XLSX)
Peptides identified in whole cell proteome tryptic digest data sets; Columns include: retention time, observed mass, observed m/z, charge state, compensation voltage (CV), RAW filename (SrchName), mass difference (PPM), Comet Expect score, XCorr, deltaCN, number of ions matched, total ions matched, protein name (reference), redundancy, peptide sequence (with flanking residues), trimmed peptide sequence, peptide length, missed cleavage, gene symbol, maximum precursor intensity, and peak width (ZIP)
Phosphopeptides identified in the Fe2+-NTA phosphopeptide enrichment data sets; Columns include those in Table S1, plus the AScore peptide, and columns for phosphorylation site position, and peptide score (ZIP)