

Abstract
Severe combined immune deficiency (SCID) caused by RAG1 or RAG2 deficiency is a genetically-determined immune deficiency characterized by the virtual absence of T and B lymphocytes. Unless treated with hematopoietic stem cell transplantation (HSCT), patients with RAG deficiency succumb to severe infections early in life. However, HSCT carries the risk of graft-versus-host disease. Moreover, a high rate of graft failure and poor immune reconstitution have been reported after unconditioned HSCT. Expression of the RAG genes is tightly regulated, and preclinical attempts of gene therapy with heterologous promoters have led to controversial results. Using patient-derived induced pluripotent stem cells (iPSCs) and an in vitro artificial thymic organoid system as a model, here we demonstrate that gene editing rescues the progressive T-cell differentiation potential of RAG2 deficient cells to normal levels, with generation of a diversified T cell repertoire. These results suggest that targeted gene editing may represent a novel therapeutic option for correction of this immunodeficiency.
Keywords: RAG2, gene editing, SCID, induced pluripotent stem cells
Introduction
Severe combined immune deficiency (SCID) comprises an array of inherited genetic defects that affect the development of T lymphocytes (and, in some cases, also B and/or NK cells), thereby compromising adaptive immune responses. Patients with SCID are highly susceptible to serious infections from birth and inevitably die within the first years of life unless treated with allogeneic hematopoietic stem cell transplantation (HSCT). The Recombination Activating Genes 1 and 2 (RAG1 and RAG2) proteins initiate the process of V(D)J recombination that gives rise to a diverse repertoire of T and B cell receptors (TCRs, BCRs), thereby allowing recognition of antigens and adaptive immune responses [1]. Mutations in RAG1 or RAG2 can result in various clinical phenotypes [2]. Functionally null mutations cause a complete arrest of T and B cell development, resulting in T− B− NK+ SCID, whereas hypomorphic variants that allow residual RAG function are partially permissive to T (and in some cases, B) cell development, and often manifest with immune dysregulation as a result of faulty negative selection of self-reactive cells, in addition to infections [3].
Currently, the only definitive cure for RAG deficiency is represented by allogeneic HSCT, however this treatment comes with an array of possible complications including graft vs. host disease and transplant-related toxicities. Furthermore, challenges exist in finding matched donors for select ethnic groups, and graft failure and incomplete immune reconstitution have been frequently reported, especially after unconditioned haploidentical HSCT [4]. Previous preclinical attempts to correct RAG deficiency by gammaretrovirus- or lentivirus-mediated gene transfer in mice have led to controversial results, reflecting inadequate and/or dysregulated expression of the RAG genes using heterologous promoters [5–11].
To circumvent some of these challenges, we have taken an ex vivo editing approach to engineer patient-derived induced pluripotent stem cells (iPSCs), by knocking-in a promoterless RAG2 cDNA at the endogenous locus, thereby maintaining epigenetically-controlled expression of the RAG2 gene, while avoiding the need to tailor donor templates to correct patient-specific mutations [12–15]. To assess rescue of T-cell differentiation, patient-derived iPSCs were differentiated into human embryonic mesodermal progenitors (hEMPs) and then cultured in an artificial thymic organoid (ATO) system that includes a murine stromal cell line (MS5) engineered to express the human Notch Delta-like ligand 4 (DLL4) in serum-free medium enriched with lymphopoietic cytokines and growth factors. Progressive maturation of T-cell development was monitored by flow-cytometry and high throughput sequencing was used to analyze diversity of the T-cell repertoire. Results were benchmarked against what was observed with unedited patient-derived iPSCs and healthy donor iPSCs.
Methods
Generation and Characterization of iPSC lines
Fibroblasts were cultured from a skin biopsy specimen obtained from the RAG2 deficient patient upon informed consent according to protocol 04-09-113R approved by Boston Children’s Hospital IRB. Primary fibroblasts from patient and control foreskin fibroblasts (ATCC, Manassas, VA) were reprogrammed into iPSC using CytoTune -iSP 2.0 Sendai Reprogramming Kit (ThermoFisher) following kit instructions for feeder-dependent fibroblasts under IRB-approved protocol 16-I-N139. iPSC were analyzed by G-band karyotyping (Cell Line Genetics, Madison, WI) to ensure genomic integrity.
Gene Editing
All single guide RNAs (sgRNAs) used in this study were purchased from Trilink Biotechnologies (San Diego, CA, USA) and were HPLC purified. All sgRNAs also contained three 2’-O-Methyl phosphorothioate modifications at the 5’ and 3’ ends. The 20bp sgRNA used for this work was of the sequence 3’-TGCAGAGACATAGTTTCTGA-5’.
Recombinant High Fidelity Cas9 was used in all editing experiments from Integrated DNA Technologies. All RNPs used for editing were created using a 1:3 Cas9:sgRNA ratio and were allowed to complex at room temperature for 30 minutes prior to electroporation. 1x10^6 iPSCs were resuspended in 20uL OPTI-MEM (Thermo Fisher) and then combined with the RNP and inserted into a single well of a nucleovette strip (Lonza). Cells were electroporated using a Lonza 4D Nucleofector (program CA137) and all 1x106 cells were then recovered in one well of a 6-well plate coated with Vitronectin (Thermo Fisher) in Essential 8 Flex Medium (Thermo Fisher) supplemented with 10μM ROCK Inhibitor (Tocris Bioscience, Bristol, UK). Plated cells were immediately transduced with rAAV6 at 50,000-250,000 vector genomes/cell as tittered by qPCR.
Quantification of homologous recombination by digital droplet PCR (ddPCR)
Analysis of the percentage of cells with successful integration of the RAG2 cassette was carried out by ddPCR using the National Institutes of Health Core Genomics Facility. Genomic DNA extracted from all target cells was purified using a DNEasy Blood and Tissue Kit (Qiagen*). 100ng of purified gDNA was then combined with WT FAM Probe: 5’ – CCCGAGGAACGTGACCATGGAGTGGC – 3’ along with forward primer: 5’ – GCACAGGAAGTTTAGCAGTG – 3’ and reverse primer: 5’ - GGGAATTCAAGACGCTCAGA – 3’ and MUT HEX Probe: 5’ – GAGCCTGCAGATGGTGACCGTGTCCA – 3’ along with forward primer: 5’ – GCACCTTCGGCTAGTCTTTA – 3’ and reverse primer: 5’ – ATCAGAGAAAAGCCTGGCTG – 3’ at a primer/probe ratio of 900nM/250nM and in a total reaction volume of 22uL including 11uL ddPCR Supermix for Probes (No dUTP) (BioRad). Droplet generation and PCR/reading was performed by the Genomics Core with cycling parameters of: 95C (10 minutes), [94C (30 seconds), 61.7C (30 seconds), 72C (2 minutes) – repeated steps in brackets 50 times], 98C (10 minutes), −4C (indefinite) on a QX200 Droplet Digital PCR System (Bio-Rad).
Analysis of INDEL frequency and identity
Freshly purified CB-derived CD34+ HSPCs, were obtained through the Binns Program for Cord Blood Research at Stanford University, under informed consent. INDEL frequencies and identities were examined using the ICE tool from Synthego. Genomic DNA from cells exposed to the target RNP was isolated 2 days post electroporation and the PCR product was then cleaned up using a Qiagen PCR Cleanup Kit* before being submitted for Sanger sequencing using the following primers (also used for PCR): forward: 5’-ATGTGGTTCTTTCAGCTGACG-3’ and reverse: 5’-CGAAAAGTAACCTTTTTGTTGT-3’ with an appropriate mock control genomic DNA sample used as the baseline for deconvolution.
Analysis of off-target CRISPR/Cas9 effects by whole genome sequencing
Whole genome sequencing of the precursor and edited cell lines was performed to a median depth of 30X using the Illumina NovaSeq 6000 system. Reads were trimmed using Trimmomatic v0.39 and mapped using BWA-MEM v07.17 to the human hg38 reference genome with the inserted construct sequence added as an additional contig [16]. PCR duplicates were marked using Samblaster v0.1.2.5 (http://broadinstitute.github.io/picard/). and GATK v4.1.9.0 was used to perform base recalibration [17]. The genomic insertion location was validated by examining split reads from the edited sample, and identifying the location of reads with one pair member mapped to the construct and the other pair member mapped to the human genome.
To identify potential off-target mutations, SNPs and INDELs were called in a paired fashion using MuTect2 from GATK v4.1.9.0 and following the GATK Best Practices (https://gatk.broadinstitute.org/hc/en-us/articles/360035531132--How-to-Call-somatic-mutations-using-GATK4-Mutect2) [18]. To reduce false-positive calls, variants were filtered with the following criteria: edited_sample_depth >15, edited_sample_alt_count > 5, edited_sample_freq >0.1, precursor_sample_depth >15, precursor_sample_alt_count = 0.
Isolation and culture of MS5-hDLL4 cells
The murine stromal cell line (MS5) edited to ectopically express human Notch ligand, delta-like 4 (hDLL4) was used as the hEMP co-culture cell type for the ATOs. MS5-hDLL4 cells were cultured in Dulbecco’s Modified Essential Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA) and 100μg/mL Primocin (InvivoGen, San Diego, CA, USA). For ATO seeding, MS5-hDLL4 cells were used at a confluency of approximately 70% and were dissociated with Tryp1E Express.
Generation and isolation of human embryonic mesodermal progenitors (hEMPs)
Mesodermal progenitors were induced from iPSCs as described [19] with certain optimizations. Briefly, iPSCs were cultured on Vitronectin coated played in Essential 8 complete medium. At the time of induction, cells are 60-70% confluent and are dissociated from the well using Tryp1E at 37C for 10 minutes. Disassociated cells are then washed and resuspended in X-Vivo 15 medium (Lonza) supplemented with rhActivin A (R&D Systems), rhBMP4 (R&D Systems), rhVEGF (R&D Systems), rhFGF (R&D Systems) at 10ng/mL and ROCK Inhibitor at 10μM (Tocris). After counting, 2.5x10^6 cells are seeded into a single Vitronectin coated well in a 6-well dish in 2mL X-Vivo 15 media supplemented with the same factors. Medium was then changed each day with the same growth factors as above without ROCK inhibitor. On day 5, cells are incubated with 1X Accutase (Stem Cell, Vancouver, Canada) for 10 minutes at 37C and washed twice with PBS before staining with EPCAM and CD56 antibodies to prepare for fluorescence activated cell sorting. Stained cells were sorted on a FACS ARIA instrument (BD Biosciences, San Jose, CA) for CD56+ EPCAM-cells.
T cell differentiation
ATOs were generated as described [19] with some minor alterations. Briefly, 10,000 EPCAM-CD56+ hEMP cells were combined with 0.5*10^6 MS5-hDLL4 washed and counted cells per ATO resuspended directly in EGM2 (Lonza) media with 10μM TGF-βRI inhibitor SB-431542 (SB Blocker) (Tocris Bioscience) and ROCK Inhibitor. Enough cells were combined in a 1.5mL Eppendorf tube to accommodate 6 ATOs. Combined cells in 1.5mL Eppendorf tubes were centrifuged at 1400RPM for 5 minutes at 23C in a swing bucket centrifuge to pool cell aggregates to the bottom of the tube. The media was aspirated out and enough complete media was added to the cell slurry such that the total volume was 36μL (6uL per ATO with 6 ATOs in each tube). Two 6μL droplets of the resuspended cell slurry were then placed with a p20 pipette onto a pre-wet Millicell Transwell Insert (EMD Millipore, Billerica, MA) Membrane sitting in 1mL of complete EGM2 media in a single well of a six well plate. The media was then changed every other day. At day 7, the media was further supplemented with hematopoietic cytokines rhTPO (Peprotech), rhFLT3L (Peprotech) both at 5ng/mL, and rhSCF (Peprotech) at 50ng/mL. At day 14 the media is changed to “RB27” which consists of RPMI 1640 (Gibco, Waltham, MA, USA), 4% B27 (ThermoFisher Scientific, Grand Island, NY), 30uM L-Ascorbic Acid (Sigma-Aldrich, St. Louis, MO) resuspended in 1X PBS, 1% Glutamax (ThermoFisher Scientific, Grand Island, NY), 100μg/mL Primocin, rhIL7 and rhFLT3L at 5ng/mL as well as SCF at 10ng/mL. Media was changed twice weekly. From weeks 3 to 5 of RB27 culture, cells were harvested from ATOs by adding 1mL MACS buffer (PBS with 5% BSA and 0.5M EDTA) to each well, physically disrupting the ATOs using two p1000 tips, and then further pipetting to dissociate the ATO fragments from the membrane. Cells were then centrifuged and resuspended and counted in FACS buffer before being stained with the antibodies listed in Table S1 on a BD LSR II Fortessa (BD Biosciences, San Jose, CA) and analyzed using FlowJo software version 10.5.3 (FlowJo, Ashland, OR, USA).
TCR sequencing and Data Visualization
TCR sequencing was conducted by Adaptive Biotechnology (Seattle, WA, USA) using their immunoSEQ assay service and analyzer. Approximately 1μg of genomic DNA from CD3+ sorted ATO derived T cells was submitted to their facilities for sequencing. Sample data was generated using a two-step amplification bias-controlled multiplex PCR strategy to amplify each V and J gene. Amplicons were then amplified again to adapt barcodes for Illumina next generation sequencing. Raw demultiplexed data was downloaded and analyzed using R to generate heatmaps. Briefly, recombination events between genes at the TRA/D and TRB loci were imported separately into the R programming environment. Once imported, the number of recombination events observed between each pair of genes was transformed into a percentage of total recombination events observed. Genomic loci for genes was downloaded from NCBI and genes were ordered by their chromosomal position. To aid in visualization the percentage of recombination events per gene pair was scaled by square root transformation. Software program Treemap version 2019.4.2 was used to generate treemap illustrations of CDR3 identities and frequencies within the sequenced population. The Shannon’s Entropy Index score was tabulated using software program Past4.03.
Statistical Tests
Statistical tests used in the data shown in this paper were generated using GraphPad Prism8 Software version 8.4.1. Details of each test are included in the figure description.
Results
Derivation, characterization and gene editing of patient RAG2-mutated iPSC line
Fibroblasts from a patient with T− B− NK+ SCID due to a homozygous RAG2 null mutation (c.831T>A, predicted to cause p.Y277* premature termination) were reprogrammed to iPSCs by infection with a non-integrating Sendai virus vector kit allowing transient expression of reprogramming factors OCT4, SOX2, c-MYC and KLF4. RAG2-mutated, patient-derived iPSCs were expanded and subcloned. Quantitative real-time polymerase chain reaction (qRT-PCR) demonstrated robust expression of stemness and pluripotency genes (Figure 1A), and G-banding showed an apparently normal karyotype (Figure 1B).
Figure 1. Characterization of iPSCs and gene editing.
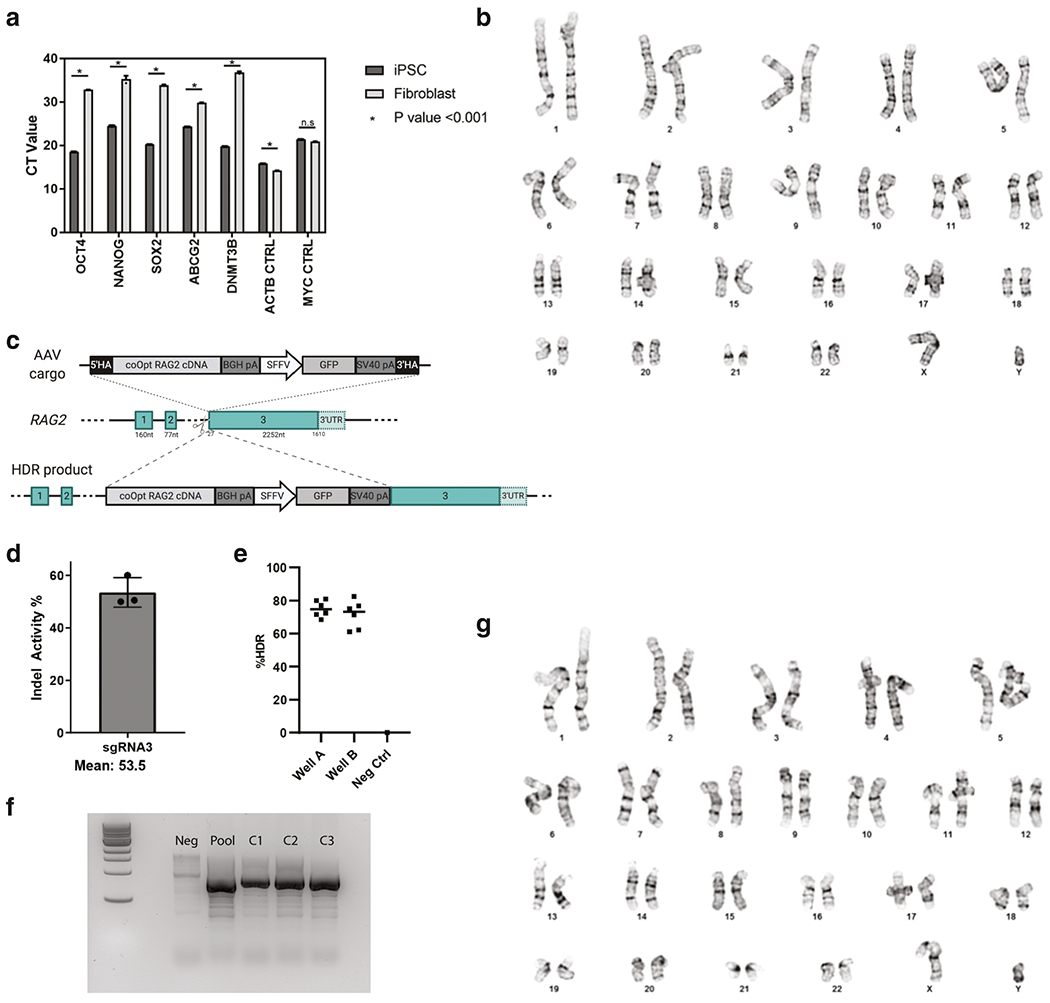
A. q-PCR based quantification of mRNA expression levels of pluripotency markers in fibroblast derived iPSCs benchmarked against fibroblast mRNA. B. Karytotype results of the RAG2 deficient patient cell line reprogrammed to pluripotency. C. Illustration of the gene editing strategy, showing the AAV cargo being integrated into the RAG2 locus. The RAG2 exons present in the germline configuration have been indicated by numbers (1, 2, 3). The donor template delivered through ad adeno-associated (AAV) vector includes a codon-optimized sequence of RAG2 cDNA (coOpt RAG2 cDNA), corresponding to the coding sequence of RAG2 exon 3. 5’HA, 3’ HA: 5’ and 3’ homology arms; BGH pA: bovine growth hormone polyadenylation signal; SFFV: spleen focus-forming virus promoter; GFP: green fluorescent protein; 3’UTR: 3’ untranslated region. D. Representative data showing on-target sgRNA activity as measured by INDEL frequencies at the RAG2 locus. E. ddPCR data showing enrichment for the coOpt RAG2 cDNA integrated cargo in a heterogeneous pool of RAG2 deficient patient iPSCs. F. DNA electrophoresis gel showing three clones with integration of the coOpt RAG2 cDNA cassette across the 3’ junction of the cargo and the chromosomal DNA. G. G-banding of the clone edited with the coOpt RAG2 cDNA cassette on both alleles showing normal karyotype.
Gene targeting at the RAG2 locus in patient-derived iPSCs was performed using CRISPR-Cas9 technology. All amino acids present in the RAG2 protein are encoded by exon 3. The exon 3 sequence was codon-optimized using IDT’s codon optimization tool to allow for improved translational efficiency and to diverge the cargo’s nucleotide sequence identity from that of the wild type locus to circumvent premature cross over during the homology directed repair of the Cas9-derived double stranded DNA break. This codon-optimized recoded RAG2 (r.RAG2) cDNA was cloned into adeno associated virus of serotype 6 (AAV6) vector and produced using 293T HEK cells and an iodixanol gradient for purification before tittering using ddPCR.
To target the RAG2 locus at exon 3, four single-guide RNAs were generated that recognize target DNA sequences around the translation initiation site (Figures 1C and S1). As a potential HSCT therapy, the designed sgRNAs were evaluated in umbilical cord blood (UCB)-derived hematopoietic stem cells for quantification of INDEL activity and cellular viability. SgRNA #3 was selected due to its proximity to the start codon (6 base pairs upstream of start site). In general, gRNAs with high INDEL activity have stimulated high gene targeting efficiencies [20]. Using MS modification and HPLC purification along with high fidelity spCas9 protein, previously optimized SpCas9 / sgRNA molar ratios and electroporation conditions, an average bulk allele INDEL activity of 53.5% percent was observed in iPSCs (Figure 1D). RAG2-mutated iPSCs were targeted with a high fidelity spCas9/sgRNA ribonucleoprotein (RNP) complex, with the donor repair template delivered via AAV6. Verification and quantification of the integration of the r.RAG2 cDNA into the endogenous locus was confirmed by ddPCR to be 39.87 ± 0.27% (Figure S2). This heterogeneous pool of cells was then enriched by sib-selection to contain targeted integration in approximately 80% of clones (Figure 1E). Subsequent PCR of the knock-in junction sites spanning the genomic insertion locus of subcloned populations showed targeted integration (Figure 1F). Karyotyping of an iPSC clone with homozygous integration of the r.RAG2 cDNA showed lack of cytogenetic aberrations (Figure 1G). Paired mutation calling identified no putative variants occurring in regions (within 10 kb) identified as having BLAST similarity to our sgRNA. Patient derived iPSCs demonstrated expression of pluripotency markers, efficient targeted integration of the r.RAG2 cassette, and the genomic integrity to support subsequent differentiation studies.
RAG2 gene editing rescues human T cell development in vitro
To assess whether targeted integration of the r.RAG2 cDNA at the endogenous locus rescues T cell development, we used a recently published method whereby iPSCs are differentiated into human embryonic mesodermal progenitor (hEMP) cells, and then differentiated into T cells in a 3D artificial thymic organoid (ATO) system [21]. In this platform, iPSC-derived hEMPs are co-cultured with the murine stromal cell line MS5 engineered to express the human Notch ligand DLL4 (MS5-hDLL4) in the presence of lymphopoietic cytokines and growth factors. Control-derived iPSCs, patient derived RAG2-mutated unedited, and RAG2 gene-edited iPSCs were successfully differentiated into CD56+ EPCAM− hEMPs (Figure S3). Upon co-culture with MS5-hDLL4 in the ATO system, control cells showed progressive expression of markers corresponding to distinct stages of T-cell development (pro-T: CD34+ CD7+ CD5−; pre-T: CD7+ CD5+ CD1a+; CD4+ CD8+ double positive (DP) cells; and CD3+ TCRαβ+ (or CD3+ TCRγδ+) T cells) (Figure 2A, Figure S4). Few myeloid (CD14+) and NK (CD3− CD56+) cells, and no CD19+ B cells were generated in the system from control-derived iPSCs (Figure 2A, Figure S4). RAG2-mutated, unedited cells were able to progress through CD7+ CD5+ pre-T cell stage before becoming blocked at the DP stage, with lack of maturation to CD3+ TCRαβ+ (or CD3+ TCRγδ+) T cells (Figure 2B). These are data are consistent with a severe impairment of V(D)J recombination and with the patient’s immunological phenotype, as well as with the recent demonstration by our group that RAG deficiency in humans allows differentiation to DP cells but impedes development of CD3+ TCRαβ+ cells [22]. In addition, a low cell yield per ATO was obtained during in vitro T-cell differentiation of unedited RAG2-deficient cells, suggestive of reduced viability from failure to progress past beta selection (Figure 2C). By contrast, the RAG2 gene-edited, patient-derived line showed robust progression beyond the DP stage, with generation of CD3+ TCRαβ+ in a proportion that was comparable to that obtained during differentiation of normal donor iPSCs (Figure 2B, S4). Moreover, a similar number of patient-derived gene edited cells and control-derived cells were obtained throughout the various stages of T-cell differentiation, indicating rescue of cell viability (Figure 2C, 2D). These data indicate that this cDNA gene replacement strategy restores a functional RAG complex to developing T lymphocytes.
Figure 2: Human iPSC derived T cell differentiation in healthy donor cells and in RAG2 deficient and RAG2 edited cells.
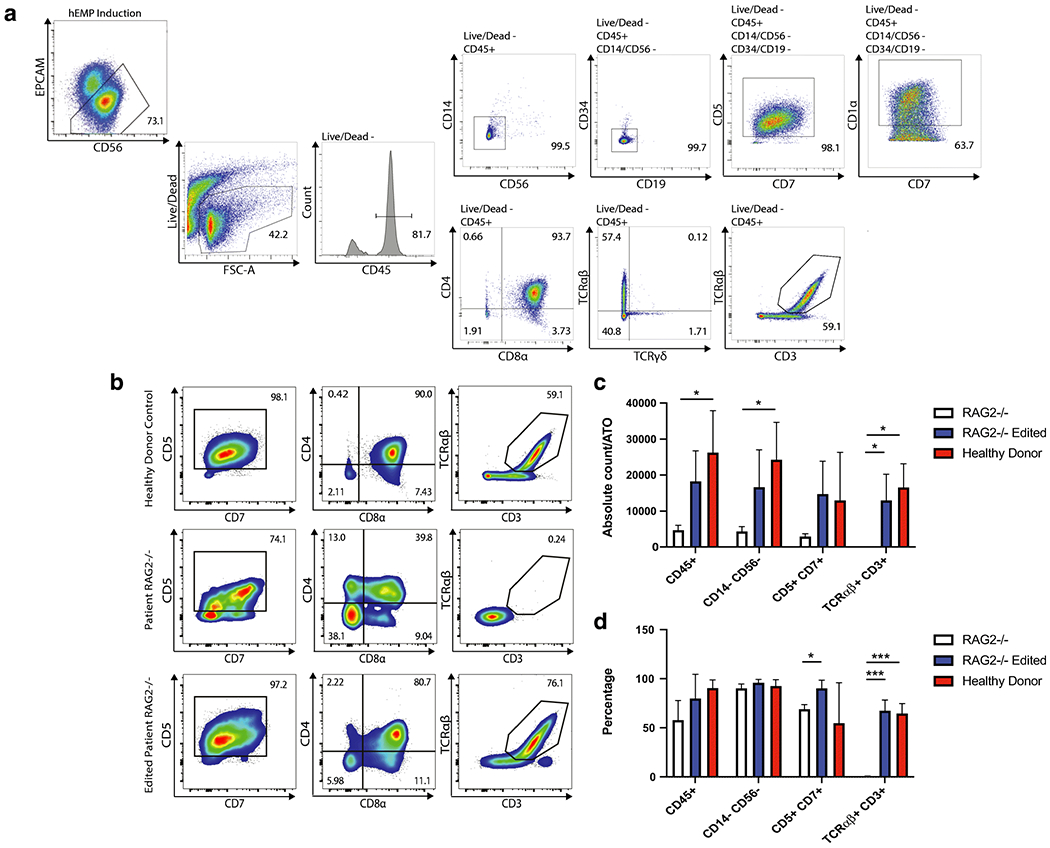
A. Representative analysis of the flow cytometry gating showing expression of early and mature T cell differentiation markers for a healthy donor sample. B. Flow cytometry results of T cell differentiation markers for the healthy donor cells, the RAG2 deficient patient cells, and the RAG2 gene-edited cells showing co-expression of CD5 and CD7, along with CD4, CD8α, and in the case of the healthy donor and edited cells, also CD3 and TCRαβ. C. Absolute count of indicated iPSC-derived T cell subsets per ATO in the healthy donor sample, the RAG2 deficient sample, and the gene-edited sample. D. Representative data showing differentiation profiles for the three examined samples over three distinct experiments. Statistical significance was computed using multiple -tests with 1% False Discovery Rate with two-stage step-up method of Benjamini, Krieger and Yekutieli. *, p<0.05; ***, p<0.001
Analysis of TCR repertoire diversity in gene-edited cells
To further investigate the robustness of T cell development rescue, we analyzed the quality and identity of TCR rearrangements at the TCRβ (TRB) and TCRα/TCRδ (TRA/TRD) loci by the high throughput immunoSEQ service offered by Adaptive Biotechnologies.
VJ recombination at the TRA locus is not entirely stochastic. Specifically, the most downstream TRAV and the most upstream TRAJ genes (located proximally to each other) are rearranged first, whereas rearrangement of the most distal genes occurs by sequential rounds of recombination in thymocytes that survive through the process [23, 24]. Prior work has demonstrated that mature T cells from patients with hypomorphic RAG mutations manifest an abnormal composition of the TRA repertoire, with reduced usage of the most distal TRAV and TRAJ genes [25]. This impairment was not observed in RAG2 deficient, gene-edited patient T cells. A polyclonal pattern of rearrangements at the TRB and TRA/TRD loci, and a similar pattern in the usage of TRAV genes, were observed in bulk CD3+ cells derived from healthy control iPSCs and from patient-derived RAG2-gene edited iPSCs that had been differentiated in the ATO system (Fig. 3A), suggesting robust rescue of VJ pairing in edited cells.
Figure 3: Analysis of VJ recombination and TCR repertoire in healthy and edited iPSC derived T cells.
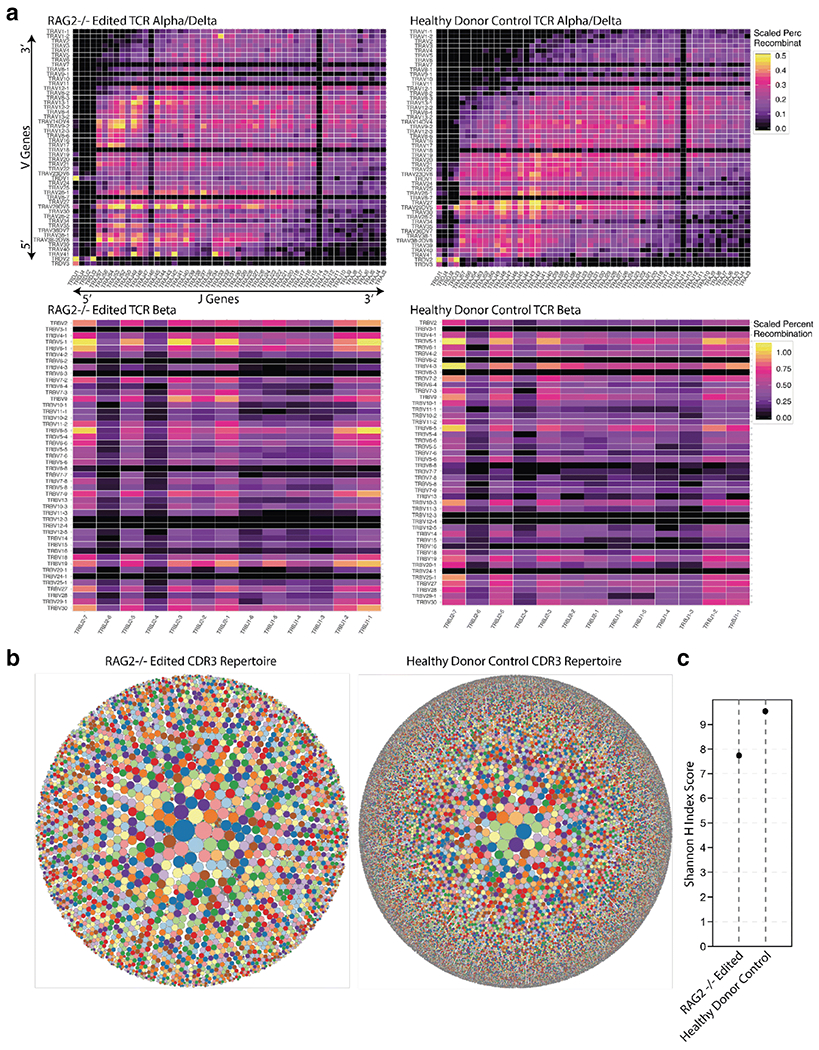
A. Heatmaps showing the pairings of V and J genes for both the TRA/TRD and TRB loci in healthy donor and RAG2 deficient gene-edited CD3+ sorted cells. V and J genes are listed in 5’ to 3’ order according to their physical location on the chromosome. B. Treemap showing diversity of TCRα CDR3 sequences in sorted CD3+ cells differentiated in vitro from healthy donor and RAG2-deficient gene-edited patient-derived iPSCs. Each dot represents a unique CDR3 sequence and the size of each dot is scaled to the frequency of that CDR3 being present in the total number of reads. C. Shannon H Index score for TCRα CDR3 sequences in sorted CD3+ cells from indicated samples, showing generation of a diverse repertoire of CD3+ cells from healthy donor and patient-derived, RAG2 gene-edited iPSCs. A Shannon index value of 8 typically denotes polyclonality. Results shown in this Figure are representative examples from three replicates.
The complementary determining region 3 (CDR3) determines the specificity of the TCR to its cognate peptide MHC complex. A tree-map profile analyzing CDR3 identities revealed a polyclonal pattern of TCR CDR3 specificities, with a Shannon’s H entropy index demonstrating a similar, albeit slightly lower, diversity of the CDR3 repertoire in sorted CD3+ cells from RAG2 gene-edited iPSCs as compared to control cells (Figure 3B–C). Furthermore, virtual spectratyping revealed similar CDR3 lengths of productive rearrangements in CD3+ cells from patient-derived gene-edited iPSCs and control iPSCs (Figure S5).
Finally, we compared the quality of in vitro T-cell differentiation of control-derived iPSCs in the ATO system and in the OP9-DLL1 monolayer system. Prior art has demonstrated similar differentiation results and kinetics using the DLL1 and DLL4 ligand [21]. As compared to the polyclonal pattern of rearrangements at both the TRB and TRA loci detected in the ATO system (Fig. 3A), healthy donor iPSCs differentiated upon co-culture with OP9-DLL1 cells demonstrated a polyclonal pattern of TRB rearrangements, but a restricted usage of TRA genes. In particular, iPSC derived T cells showed preferential usage of the most downstream TRAV most upstream TRAJ genes (Fig. S6A), suggesting reduced viability of T cell progenitors in this system as compared to cells cultured in the ATO platform. These data support superiority of the ATO method for T-cell differentiation of iPSCs.
Discussion
RAG deficiency is a prominent cause of SCID. In a recent study, it was found to account for 19.2% of all cases of SCID identified in the United States and Canada in the period 2010-2018, representing the second most common genotype after IL2RG gene defects [26]. Moreover, RAG deficiency emerged as the most common form of atypical SCID (accounting for 29.6% of these cases), a condition characterized by residual and perturbed immune function, with clinical manifestations of immune dysregulation. Patients with RAG deficiency have a poor prognosis, unless immune reconstitution is achieved. In particular, severe forms of RAG deficiency are fatal early in life; among patients with hypomorpohic mutations manifesting with combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI), treatment refractory autoimmune cytopenias are very common, and a high mortality rate has been reported in childhood and young adulthood [27]. Allogeneic HSCT represents the mainstay of treatment for RAG deficiency; however, graft-versus-host disease, graft failure and incomplete immune reconstitution remain significant challenges. In a series of patients with RAG deficiency who received haploidentical HSCT at three major centers between 1985 and 2009, a very high rate (75%) of graft failure was observed among recipients of unconditioned HSCT, so that repeat HSCT had to be frequently used, and none of the patients who engrafted in the absence of myeloablative conditioning reconstituted B- cell immunity [28]. Use of chemotherapy allowed improved immune reconstitution, but was associated with inferior survival, reflecting treatment-related toxicity [28]. Data from the Primary Immune Deficiency Treatment Consortium have indicated that improved outcome has been obtained in more recent years, however they also confirmed a high rate of graft failure and poor T- and B- cell immune reconstitution in the absence of conditioning chemotherapy [29]. It has been speculated that impaired immune reconstitution after unconditioned HSCT in patients with RAG deficiency may reflect competition between autologous, genetically-defective lymphoid progenitor cells and donor-derived cells. This competition may extend up to relatively late stages of T cell differentiation, as suggested by our observation that bone marrow-derived CD34+ cells from RAG-deficient patients can differentiate into DP T cells in vitro when cultured in the ATO system [22] and confirmed here when differentiating patient-derived iPSCs. Others have shown that in vitro differentiation of RAG2-deficient iPSCs and RAG-deficient CD34+ peripheral blood cells in the OP9-DLL1 system is associated with multiple and earlier blocks in T cell development, with modest generation of CD7+ CD5+ cells and of DP T cells [30, 31]. This apparent discrepancy may reflect dynamic changes in cell composition and survival over time. Indeed, previous work from our lab had shown that co-culture of RAG1-mutated iPSCs with OP9-DLL4 cells permits generation of CD7+ CD5+ cells, however survival and further differentiation of these cells is not sustained over time [32]. Data presented here suggest that differences in the efficiency of the ATO vs. the OP9-DLL1/DLL4 experimental platforms in supporting T cell development may also contribute to the variable presence of RAG-deficient cells reaching intermediate stages of T cell development. In any case, irrespective of the co-culture system used, decreased cellular viability of RAG2-deficient cells has been observed since the earliest stages of T cell development. In addition to failure to properly accomplish V(D)J recombination, introduction of illegitimate rearrangements due to spontaneous DNA double strand breaks may also account for reduced survival of RAG-deficient T cell progenitors [30].
Gene therapy represents an alternative approach to attain immune reconstitution in patients with SCID. When compared to allogeneic HSCT, edited autologous cell therapies have the advantages of overcoming difficulties in finding matched donors, as every patient provides their own donor cells, as well as mitigating the risk of GvHD and requisite immunosuppression, as autologous cells have exact HLA matches. Excellent results have been recently reported with gene therapy using self-inactivating lentiviral vectors in patients with X-linked SCID [33]. However, preclinical data in Rag-deficient animals have been less successful. In particular, variable efficiency of T and B cell reconstitution has been reported in Rag1−/− mice treated with lentivirus-based gene therapy [7], and severe immune dysregulation with lymphocytic infiltrates in multiple organs and production of autoantibodies have been reported by another group [5]. Better results have been observed with the use of lentiviral vectors to correct the immunodeficiency in Rag2−/− mice [34], though when a similar strategy was applied to a hypomorphic mouse model (Rag2R229Q) the T and B cell count of reconstituted animals remained lower than normal [6].
Of major concern for the clinical application of lentivirus-based gene therapy for RAG deficiency is the observation that RAG gene expression is tightly regulated through the cell cycle and along lymphoid development. Dysregulated expression might increase the risk of leukemic transformation and autoimmunity. Moreover, conventional gene therapy (i.e., with gene addition, as opposed to gene correction) carries the additional risk that the endogenous mutant allele might interfere with the wildtype RAG cDNA introduced with the lentiviral vector.
New approaches are needed in order to expand the clinical toolkit available for treating RAG2 deficiency. Ex vivo CRISPR/Cas9-AAV6 mediated homology directed repair gene therapy approaches have been shown to be highly efficient and precise at correcting endogenous pathogenic mutation in healthy and patient-derived HSPCs [20, 35, 36]. Ex vivo gene targeting circumvents challenges presented by in vivo Cas9 editing, such as delivery of the gene editing machinery, specificity of targeted cells, preexisting adaptive immunity to spCas9 proteins, and precision of expression times for targeted nucleases which can compound the risk of generating off-target editing [37, 38].
This study confirms and extends previous observations that homology-driven repair can correct the T cell differentiation potential of RAG2-mutated iPSCs [30], indicating that RAG2 is an exceptional target for this type of gene replacement therapy. Because the entire RAG2 amino acid sequence is encoded by a single exon, replacing the coding sequence of this exon minimizes the risk of interfering with transcriptionally required intronic regulatory elements, while still correcting all reported patient RAG2 ORF mutations [39]. Additionally, a single copy of the corrected template DNA restores RAG2 function, as indicated by the observation that the parents of RAG2-deficient patients are heterozygous for the gene defect, and yet they do not manifest clinical or immunological abnormalities. In this study, we have made use of a codon optimized donor template. Codon optimization is meant to improve the efficiency of translation.
Bioinformatic analysis was used to make sure that no cryptic splice sites were introduced in the process of codon optimization. Furthermore, the presence of silent mutations introduced by codon optimization every other 2-3 amino acids in the corrective DNA donor template will assure that no premature cross over will take place during the homologous DNA repair process, thereby increasing the frequency of the corrective DNA donor being integrated.
Taken together, this work shows that targeted on-site integration of a wild-type transgene allows by-passing of the developmental block due to RAG2 deficiency, robust RAG2 catalytic activity, and generation of a TCR repertoire with similar, albeit slightly lower, diversity as compared to that of an immunocompetent donor. The combination of these data show that this strategy effectively restores RAG function, and indicates that this is a treatment modality that warrants further investigation for future clinical application as an edited autologous HSCT therapy.
Supplementary Material
Acknowledgments
We would like to thank the lab of William James and, in particular, Sally Cowley who generously provided some preliminary stem cells to work with. Additionally, we would like to thank Julianne Cohen, who helped efficiently manage resources within the lab. Finally, we would like to thank the NIH Oxford Cambridge Scholars program for providing the tools to establish this collaborative work.
Funding
This work was supported by the Division of Intramural Research at the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant 1 ZIA AI001222-02) and by the Bench-to-Bedside grant “RAG deficiency: From pathophysiology to precise gene correction” to LDN. We would like to acknowledge the generous funding provided by the Wellcome Trust via grant WT200844/Z/16/Z to O.A. M.H.P gratefully acknowledges the support from Laurie Karass Lacob Translation Research Fund, Amon Carter Foundation and Chan-Zucherberg Biohub.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Ethics Approval
A skin biopsy specimen was obtained from the RAG2 deficient patient upon informed consent according to protocol 04-09-113R approved by Boston Children’s Hospital IRB.
Consent to participate
Not applicable
Consent for publication
Not applicable
Code availability
Not applicable
Disclosure of Conflicts of Interests: M.H.P. serves on the SAB for CRISPR Tx and Allogene Tx. Neither company had input into the design, execution, data analysis or publication of the work presented in this manuscript. The other authors declare that they have no conflict of interest.
Availability of data and material
All material will be made available upon request, and pending signature of appropriate Material and Transfer Agreement. Original data are available upon request.
References
- 1.Alt FW, Rathbun G, Oltz E, Taccioli G, Shinkai Y. Function and control of recombination-activating gene activity. Ann N Y Acad Sci. 1992;651:277–94. doi: 10.1111/j.1749-6632.1992.tb24626.x. [DOI] [PubMed] [Google Scholar]
- 2.Wada T, Takei K, Kudo M, Shimura S, Kasahara Y, Koizumi S et al. Characterization of immune function and analysis of RAG gene mutations in Omenn syndrome and related disorders. Clin Exp Immunol. 2000;119(1):148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delmonte OM, Villa A, Notarangelo LD. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood. 2020;135(9):610–9. doi: 10.1182/blood.2019000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hassan A, Booth C, Brightwell A, Allwood Z, Veys P, Rao K et al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood. 2012;120(17):3615–24; quiz 26. doi: 10.1182/blood-2011-12-396879. [DOI] [PubMed] [Google Scholar]
- 5.van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol. 2014; 133(4):1116–23. doi: 10.1016/j.jaci.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E et al. Efficacy of lentivirus-mediated gene therapy in an Omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol. 2018;142(3):928–41 e8. doi: 10.1016/j.jaci.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25(9):1471–83. doi: 10.1038/leu.2011.106. [DOI] [PubMed] [Google Scholar]
- 8.Lagresle-Peyrou C, Benjelloun F, Hue C, Andre-Schmutz I, Bonhomme D, Forveille M et al. Restoration of human B-cell differentiation into NOD-SCID mice engrafted with gene-corrected CD34+ cells isolated from Artemis or RAG1-deficient patients. Mol Ther. 2008;16(2):396–403. doi: 10.1038/sj.mt.6300353. [DOI] [PubMed] [Google Scholar]
- 9.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci U S A. 2006;103(44):16406–11. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates F, Malassis-Seris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P et al. Gene therapy of RAG-2−/− mice: sustained correction of the immunodeficiency. Blood. 2002;100(12):3942–9. doi: 10.1182/blood-2002-03-0782. [DOI] [PubMed] [Google Scholar]
- 11.Lagresle-Peyrou C, Yates F, Malassis-Seris M, Hue C, Morillon E, Garrigue A et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. 2006;107(1):63–72. 10.1182/blood-2005-05-2032. [DOI] [PubMed] [Google Scholar]
- 12.Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384–9. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genovese P, Schiroli G, Escobar G, Tomaso TD, Firrito C, Calabria A et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–40. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rai R, Romito M, Rivers E, Turchiano G, Blattner G, Vetharoy W et al. Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott - Aldrich Syndrome. Nat Commun. 2020;11(1):4034. doi: 10.1038/s41467-020-17626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubbard N, Hagin D, Sommer K, Song Y, Khan I, Clough C et al. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127(21):2513–22. doi: 10.1182/blood-2015-11-683235. [DOI] [PubMed] [Google Scholar]
- 16.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30(17):2503–5. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell stem cell. 2019;24(3):376–89 e8. doi: 10.1016/j.stem.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun. 2019;10(1):1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nature methods. 2017;14(5):521–30. doi: 10.1038/nmeth.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 2020;4(12):2611–6. doi: 10.1182/bloodadvances.2020001730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–70. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- 24.Krangel MS. Mechanics of T cell receptor gene rearrangement. Curr Opin Immunol. 2009;21(2):133–9. doi: 10.1016/j.coi.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I et al. PROMIDISalpha: A T-cell receptor alpha signature associated with immunodeficiencies caused by V(D)J recombination defects. J Allergy Clin Immunol. 2018. doi: 10.1016/j.jaci.2018.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Dvorak CC, Haddad E, Buckley RH, Cowan MJ, Logan B, Griffith LM et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010-2018). J Allergy Clin Immunol. 2019;143(1):405–7. doi: 10.1016/j.jaci.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farmer JR, Foldvari Z, Ujhazi B, De Ravin SS, Chen K, Bleesing JJH et al. Outcomes and Treatment Strategies for Autoimmunity and Hyperinflammation in Patients with RAG Deficiency. The journal of allergy and clinical immunology In practice. 2019;7(6):1970–85 e4. doi: 10.1016/j.jaip.2019.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014;123(2):281–9. doi: 10.1182/blood-2013-01-476432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood. 2018;132(17):1737–49. doi: 10.1182/blood-2018-03-840702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Themeli M, Chhatta A, Boersma H, Prins HJ, Cordes M, de Wilt E et al. iPSC-Based Modeling of RAG2 Severe Combined Immunodeficiency Reveals Multiple T Cell Developmental Arrests. Stem Cell Reports. 2020;14(2):300–11. doi: 10.1016/j.stemcr.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bifsha P, Leiding JW, Pai SY, Colamartino ABL, Hartog N, Church JA et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv. 2020;4(12):2606–10. doi: 10.1182/bloodadvances.2020001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brauer PM, Pessach IM, Clarke E, Rowe JH, Ott de Bruin L, Lee YN et al. Modeling altered T-cell development with induced pluripotent stem cells from patients with RAG1-dependent immune deficiencies. Blood. 2016;128(6):783–93. doi: 10.1182/blood-2015-10-676304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N Engl J Med. 2019;380(16):1525–34. doi: 10.1056/NEJMoa1815408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Til NP, de Boer H, Mashamba N, Wabik A, Huston M, Visser TP et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther. 2012;20(10):1968–80. doi: 10.1038/mt.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med. 2017;9(372). doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 36.Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13(2):358–76. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat Med. 2019;25(2):249–54. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vakulskas CA, Behlke MA. Evaluation and Reduction of CRISPR Off-Target Cleavage Events. Nucleic Acid Ther. 2019;29(4):167–74. doi: 10.1089/nat.2019.0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sweeney CL, Zou J, Choi U, Merling RK, Liu A, Bodansky A et al. Targeted Repair of CYBB in X-CGD iPSCs Requires Retention of Intronic Sequences for Expression and Functional Correction. Mol Ther. 2017;25(2):321–30. doi: 10.1016/j.ymthe.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All material will be made available upon request, and pending signature of appropriate Material and Transfer Agreement. Original data are available upon request.