

Abstract
Prostate cancer (PCa) genomic subtypes that stratify aggressive disease and inform treatment decisions at the primary stage are currently limited. Previously, we functionally validated an aggressive subtype present in 15% of PCa characterized by dual deletion of MAP3K7 and CHD1. Recent studies in the field have focused on deletion of CHD1 and its role in androgen receptor (AR) chromatin distribution and resistance to AR-targeted therapy, however, CHD1 is rarely lost without co-deletion of MAP3K7. Here we show that in the clinically relevant context of co-loss of MAP3K7 and CHD1 there are significant, collective changes to aspects of AR signaling. While CHD1 loss mainly impacts the expansion of the AR cistrome, loss of MAP3K7 drives increased AR target gene expression. PCa cell line models engineered to co-suppress MAP3K7 and CHD1 also demonstrated increased AR-v7 expression and resistance to the AR-targeting drug enzalutamide. Furthermore, we determined that low protein expression of both genes is significantly associated with biochemical recurrence (BCR) in a clinical cohort of radical prostatectomy specimens. Low MAP3K7 expression, however, was the strongest independent predictor for risk of BCR over all other tested clinicopathologic factors including CHD1 expression. Collectively, these findings illustrate the importance of MAP3K7 loss in a molecular subtype of PCa that poses challenges to conventional therapeutic approaches.
Introduction
The majority of men diagnosed with prostate cancer (PCa) will exhibit indolent tumors, but about 20% will eventually develop aggressive, metastatic disease (1). Although some men diagnosed with early stage PCa choose active surveillance, most patients will receive some form of definitive treatment including surgery, radiation, chemotherapy, and/or hormonal therapy, all of which have side effects that can be significant (2, 3). Therefore, identification of aggressive subtypes of PCa is of the utmost importance in order to identify men who would benefit most from aggressive treatment strategies and spare those who are less likely to succumb to their disease. Numerous studies have examined molecular features of primary prostate tumors to define PCa subtypes, but this strategy has yet to identify drivers that predict worse disease-free survival (DFS) in patients (4, 5). Translocations of ERG, for example, are the most common genomic rearrangement, occurring in about 50% of primary PCa; however, ERG rearrangements by themselves do not predict lethality (6).
We recently identified and validated a lethal subtype of PCa that harbors deletions of MAP3K7 and CHD1 (6). Co-deletion of CHD1 and MAP3K7 occurs in 15% of primary tumors and associates with poor DFS (6). In multiple cohorts of PCa patients, loss of CHD1 is significantly associated with loss of MAP3K7, and both are mutually exclusive of ERG translocations (4, 6, 7). In our tissue recombination mouse model, which recapitulates PCa development and pathology through a mix of fetal urogenital mesenchyme and mouse prostate epithelial stem cells, co-suppression of MAP3K7 and CHD1 led to histopathological evidence of invasive, aggressive disease with signs of abnormal differentiation and increased focal expression of the androgen receptor (AR) (6). Co-suppression of MAP3K7 and CHD1 in a LNCaP human xenograft model also led to increased tumor growth and decreased survival of the mice (6).
MAP3K7 encodes TGF-β activated kinase (TAK1), a serine/threonine kinase downstream of many cytokine signaling receptors including TNF and IL-1. TAK1 is known to signal to p38, JNK, NF-kβ, and others (8, 9). CHD1, chromodomain helicase DNA-binding protein 1, binds histone 3 lysine 4 trimethylation marks on chromatin as a reader to facilitate a more open chromatin structure (10). Multiple recent studies have evaluated the role of CHD1 loss in PCa including its contribution to the “normal” AR chromatin landscape, or cistrome, and its relation to resistance to the AR-targeted therapy enzalutamide (11, 12). However, CHD1 is rarely lost without co-deletion of MAP3K7, and few studies have evaluated its biological role in the context of MAP3K7 loss.
The AR pathway is a major therapeutic target in the clinic, and many therapies have been developed to disrupt its activity (13, 14). Despite initial response, most patients relapse with a castration resistant form of the disease for which there is no definitive cure (15). Current efforts are aimed at identifying molecular profiles that can indicate cases with greater potential for treatment resistance (16). In the present study, we determined that a molecular subtype of PCa with loss of MAP3K7 and CHD1 exhibits an increase in many features of AR activity including greater chromatin binding, target gene expression, and resistance to enzalutamide. Low protein expression of MAP3K7 in particular confers significant risk for disease recurrence in PCa patient samples, highlighting an aggressive PCa subtype with potential for resistance to common hormonal therapies.
Materials and Methods
Cell Culture
LNCaP cells(ATCC Cat# CRL-1740, RRID:CVCL_1379) were obtained from the ATCC, and LAPC4 (ATCC Cat# PTA-1441, RRID:CVCL_4744) and 22RV1 (ATCC Cat# CRL-2505, RRID:CVCL_1045) cell lines were acquired from the University of Colorado Cancer Center cell line repository. Cell line identity was verified by spectral karyotyping every 6 months, and cells were used below passage 50. Each batch of frozen cells is tested for mycoplasma by DAPI staining and PCR, and once thawed cells are passaged a maximum of 4 times for use in experiments. Production of lentivirus for shRNA plasmids and generation of LNCaP shControl, shMAP3K7, shCHD1, and shDouble cell lines was described previously (6). LAPC4 and 22RV1 cells were generated in the same manner. Cells were kept in shRNA antibiotic selection with 1ug/mL puromycin (Sigma #P8833) and 800 ug/mL Geneticin (InvivoGen, G-418, #ant-gn-5). ShRNA targeting constructs are listed in Table S1. Cells were maintained in RPMI-1640 (LNCaP, 22RV1) or IMDM (LAPC4) medium (Gibco #21875034 and #12440053) with 1% penicillin-streptomycin (PS) (Thermo Fisher Scientific #15140122) and 10% or 7.5% fetal bovine serum (FBS) (Gemini Bio-Products #100-106), respectively. LAPC4 cells were also supplemented with 1 nM R1881 (Perkin Elmer, #NLP005005MG). Where indicated, cells were cultured in steroid-depleted medium which consisted of phenol red-free RPMI-1640 (Gibco #11835030) or IMDM (Gibco #21056023) supplemented with 10% (LNCaP, 22RV1) or 7.5% (LAPC4) charcoal-stripped FBS (Gibco #12676029) and 1% PS. For indicated assays, cells were treated with varying doses of R1881 and enzalutamide (Selleckchem #MDV3100).
Monolayer growth assays
LNCaP monolayer growth assays were quantified by trypan blue exclusion. LAPC4 and 22RV1 growth assays and enzalutamide growth assays for all cell types were measured with IncuCyte ZOOM Live-Cell Analysis System using percent well confluence.
RT-qPCR
RNA was isolated using TRIzol Reagent (Invitrogen #15596018). Real-time quantitative PCR (RT-qPCR) was carried out using diluted cDNA, Power SYBR Green PCR Mastermix (Applied Biosystems #A25742) and the primers listed in Table S1. The primers for AR were described previously (17). Samples were run on an ABI 7500 Real-time PCR Instrument (Applied Biosystems).
siRNA constructs
LNCaP cells were transfected with Lipofectamine RNAiMAX Reagent (Invitrogen #13778150). AR-variant-targeting siRNA constructs have been described previously (18, 19). RNA was collected at 24 hrs for RT-qPCR verification, and immunoprecipitation (IP) of AR(N20) was done after 72 hrs.
Propidium Iodide (PI) and Annexin V staining
LNCaP cells were plated in 96-well plates in complete growth medium for 2 days. Medium was changed to steroid-depleted medium, and cells were transfected with AR-variant-targeting siRNA constructs. 24 hours after transfection, Annexin V fluorescent conjugate (Biotium #29005) and PI fluorescent dye (Biotium #40016) were added to culture medium and Annexin V (green), PI (red), and cell confluence were measured with the IncuCyte Zoom Live-Cell Analysis System.
Cell cycle analysis
LNCaP cells were grown in steroid-depleted medium ± 1 nM R1881 for 48 hours. Staining of cells for cell cycle analysis was performed as described previously (20).
Immunoblotting
Standard protocols were used for protein isolation, quantification and immunoblotting. Images were acquired using LI-COR Odyssey imaging systems and quantified using Image Studio software (RRID:SCR_015795). Antibodies used were: MAP3K7 (Cell Signaling Technology #5206S, RRID:AB_ 10694079), CHD1 (Bethyl # A301-218A, RRID:AB_890568), AR N-20 (Santa Cruz #sc-816, RRID:AB_1563391), AR C-19 (Santa Cruz #sc-815, RRID:AB_ 630864), AR-v7 (Precision Antibody #AG10008, RRID:AB_2631057), Lamin A/C (Cell Signaling Technology #4777, RRID:AB_10545756), α-tubulin (Cell Signaling Technology #3873, RRID:AB_1904178), β-actin (Cell Signaling Technology #3700, RRID:AB_2242334), PARP (Millipore #AB16661, RRID:AB_ 90869), cleaved-PARP (Cell Signaling Technology #5625, RRID:AB_10699459), Cleaved caspase-3 (Cell Signaling Technology #9661S, RRID:AB_2341188), goat anti-rabbit-HRP (Santa Cruz #sc-2004, RRID:AB_631746), and goat anti-mouse-HRP (Santa Cruz, #sc-2005, RRID:AB_631736).
Subcellular fractionation
Nuclear and cytoplasmic extracts were prepared as described previously (21). Aliquots of whole cell lysates were taken from each sample after hypotonic lysis and before centrifugation.
RNA-Seq
LNCaP cells in triplicate were grown in steroid-depleted medium for 3 days, then treated with either 0.1% EtOH or 1 nM R1881 for 4 hours. RNA was extracted using the PerfectPure RNA Cultured Cell kit (5Prime #2302350) including an additional DNase treatment using Turbo DNA-Free Kit (Ambion #AM1907). Samples were submitted to the University of Colorado Anschutz Medical Campus Genomics and Microarray Core. Libraries were prepared with the Illumina TruSEQ Stranded Total RNA with Ribo-Zero TM library preparation kit. Sequencing was performed on Illumina HiSEQ 2000 High-throughput mode using V3 Chemistry, PE100. Gene expression was quantified using default parameter of RSEM (v1.2.1, RRID:SCR_013027) with Bowtie2 (v2.1.0, RRID:SCR_016368) as the read aligning program and differential expression was calculated using EBSeq (v1.1.5, RRID:SCR_003526). Reads were aligned to hg19 RefSeq (RRID:SCR_003496) annotated genes. LAPC4 and 22RV1 shControl and shDouble cells were collected as described for LNCaP cells, and samples were submitted at a later date to Novogene. Libraries were prepared using Novogene’s eukaryotic RNA-Seq method, and sequencing was performed on the Illumina NovaSeq 6000 Platform, PE150. Gene expression was quantified as described for LNCaP cells. Heatmaps and further comparison of gene expression changes for all cell lines were generated with Rstudio (v1.2.5033, RRID:SCR_000432).
ChIP-seq
Details for ChIP sample collection are outlined in supplemental methods. For ChIP-seq, 1 ng of ChIP or input DNA per sample was used for library construction (NuGEN). Samples were pooled and sequenced on Illumina HiSEQ 2000 High-throughput mode using V3 Chemistry, PE100 at the University of Colorado Anschutz Medical Campus Genomics and Microarray Core. The fastq file quality was accessed using FastQC (RRID:SCR_014583) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MultiQC (RRID:SCR_014982) (https://multiqc.info/). Illumina adapters and low-quality reads were filtered out using BBDuk (RRID:SCR_016969) (http://jgi.doe.gov/data-and-tools/bb-tools). Bowtie2 (v.2.3.4.3, RRID:SCR_016368) was used to align reads to the hg38 reference human genome. Samtools (v.1.9, RRID:SCR_002105) was used to select the mapped reads. Optical and PCR duplicates were removed using Picard (RRID:SCR_006525) MarkDuplicates tool (Broad Institute, http://broadinstitute.github.io/picard/). The same number of reads for all samples was randomly extracted using samtools view. Bedtools (RRID:SCR_006646) genomecov was used to create bedgraph files, and Igv (RRID:SCR_011793) tools toTDF was used to create tdf files. Peaks were called using MACS2 (v2.1.1.20160309, RRID:SCR_013291) (22) with default parameters. Average profiles of AR ChIP peaks to their binding sites were generated using ngs.plot (RRID:SCR_011795) (23).
Clinical prostate cancer cohort
The Stanford University database of surgically removed prostates (1986–2003) was previously subjected to a comprehensive histopathologic review (24). In this current study we selected high grade cases (containing Gleason grade 4 or 5) with 5+ years of follow-up, 125 with biochemical recurrence (BCR) and 125 without BCR that were matched by age at surgery and by percent total high grade cancer. 206 of these matched cases, 112 with BCR and 94 without, had sufficient tumor tissue for all histologic analyses. The pathologist was blinded to patient clinical features including BCR status during IHC scoring of samples. The investigators did not have access to patient identifiers and the Institutional Review Board of the University of Colorado considered this as non-human subject research. Complete details regarding selection of this cohort and analysis of clinical and histopathological features are outlined in supplemental methods. Complete information for all cases is included in supplementary data.
Dataset analyses
mRNA expression data, genomic CNA data, and clinical data for MSKCC (25) and TCGA patient datasets (4) were downloaded from the cBioPortal for Cancer Genomics (RRID:SCR_014555) (26). Data were further processed using GraphPad Prism (RRID:SCR_002798) and RStudio (RRID:SCR_000432), and pathway annotations were generated with Gene Set Enrichment Analysis (GSEA, RRID:SCR_003199) (27). Metascape (RRID:SCR_016620) was used for cell line RNA-Seq and ChIP-seq pathway enrichment analysis (28). For the meta-analysis of patient genomic data, CNA data from the MSKCC/DFCI (29), Broad/Cornell (30), TCGA PanCan (31), and SU2C (32) datasets were downloaded from cBioPortal. Duplicate patient IDs were filtered to produce a final list of unique patient IDs, along with discretized calls for deep and shallow deletions for MAP3K7 and CHD1, and fusions for ERG.
Statistical analyses
Statistical analyses were performed with GraphPad Prism 7 and 8 (RRID:SCR_002798). Data were plotted as means ± SEM. Two-tailed p <0.05 was considered statistically significant. Asterisks denote levels of statistical significance as determined by one or two-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test, unpaired two-tailed t-test, one-sided Fisher’s exact test, log-rank Mantel-Cox test, Wilcoxon rank sum test, or Chi-squared test for trend. Specific tests used are indicated in the figure legends. All experiments were performed in triplicate unless noted otherwise.
Results
Loss of MAP3K7 and CHD1 increased proliferation with and without androgens
We first sought to explore the functions of MAP3K7 and CHD1 in androgen signaling using LNCaP, an AR-responsive PCa cell model. To determine if dual loss of MAP3K7 and CHD1 alters androgen-stimulated proliferation, we performed a monolayer growth assay using LNCaP cells stably expressing shRNAs against MAP3K7 and/or CHD1 (Fig. 1A) in the absence [castration condition, ethanol (EtOH)] or presence of androgen. Suppression of MAP3K7 and CHD1 (shDouble) led to a robust increase in the growth of LNCaP cells in the absence and presence of a minimally stimulatory dose (10 pM) of the synthetic androgen R1881 (Fig. 1B). This was true for seven different combinations of shRNAs against both MAP3K7 and CHD1.
Fig 1. Co-suppression of MAP3K7 and CHD1 increases androgen-independent growth and AR activity.
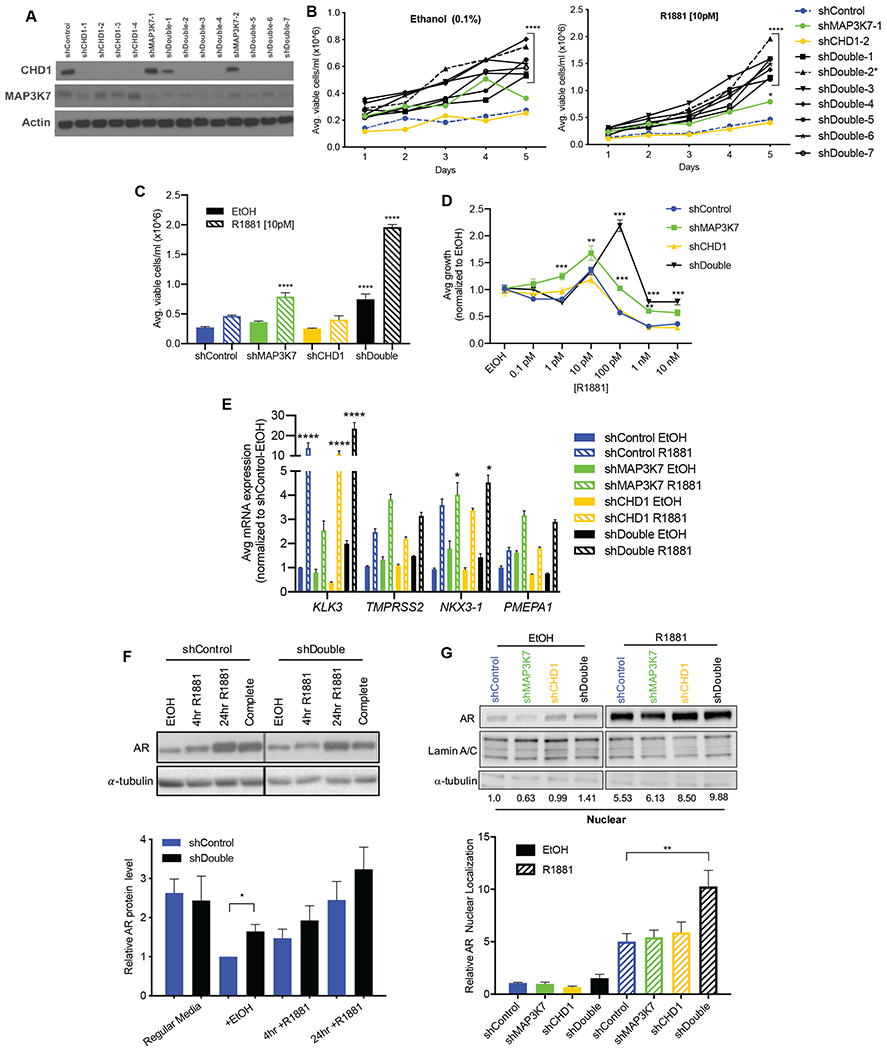
(A) Western blot of shControl, shMAP3K7, shCHD1, and shMAP3K7-shCHD1 (shDouble) LNCaP cells. Multiple shRNA constructs are shown. (B) 5-day growth assay for LNCaP shControl, shMAP3K7, shCHD1, and various shDouble combinations in steroid-depleted medium with vehicle (0.1% EtOH) or 10 pM R1881. Asterick denotes shDouble (2) combination used in subsequent experiments. (n=3; p-value indicated for all shDouble constructs versus shControl at Day 5). (C) Quantification of day 5 from growth assay in B, including the shDouble-2 combination (n=3; p-values indicate comparison to shControl EtOH). (D) Day 7 of R1881 dose response curves for LNCaP cells normalized to respective EtOH treatment (n=3; p-values indicate comparison to shControl at each dose). (E) RT-qPCR of indicated genes in LNCaP cells cultured in steroid-depleted medium ± EtOH or 1 nM R1881 for 8 hr. Ct values normalized to B2M then to shControl (n=3; p-values represent comparison to shControl EtOH). (F) Quantification (n=3) and representative western blot of AR protein in shControl and shDouble LNCaP cell lysates isolated after different treatments: complete growth medium or steroid-depleted medium ± 1 nM R1881 for 4 hr or 24 hr. (G) Quantification (n=3) and representative western blot of AR protein in nuclear fractions isolated from LNCaP cell lysates after treatment with ± 1 nM R1881 for 4 hr. AR blot of EtOH-treated samples is shown at a higher exposure than R1881-treated samples in order to visualize bands. All data represent mean ± SEM; One-way ANOVA with Tukey’s multiple comparisons test (C, G), two-way ANOVA with Dunnett’s multiple comparisons test (B, D, E), and unpaired two-tailed t-test (F); *=p<0.05; **=p<0.01; ***=p<0.001; ****=p<0.0001.
Suppression of MAP3K7 alone was also sufficient for increased growth (Fig. 1B, C) while knockdown of CHD1 was not (Fig. 1B, C), suggesting MAP3K7 suppression contributes more to this growth effect. A dose response curve of R1881 showed that MAP3K7 knockdown increased relative androgen-stimulated growth at many doses compared to shControl cells (Fig. 1D). Additionally, shDouble cells exhibited a shifted dose response curve, reaching higher doses of R1881 before a growth inhibitory effect occurred (Fig. 1D). In LAPC4 cells, another androgen sensitive PCa model, dual knockdown of MAP3K7 and CHD1 (Fig. S1A) also increased androgen-independent and -stimulated growth compared to shControl cells (Fig. S1B). shMAP3K7 and shDouble LAPC4 cells similarly displayed increased growth at various R1881 doses compared to shControl (Fig. S1C). In the castration-resistant cell line 22RV1, knockdown of MAP3K7 and CHD1 did not provide a growth advantage, likely due to the advanced stage and androgen-independence of this cell line (Fig. S1D).
Suppression of MAP3K7 and CHD1 increased AR activity
We next examined the effects loss of MAP3K7 and/or CHD1 had on gene expression of AR canonical target genes. Expression of all genes was induced by R1881 as expected, but to varying degrees with the different knockdowns (Fig. 1E). TMPRSS2, NKX3-1, and PMEPA1 were strongly induced in shMAP3K7 and shDouble cells compared to shControl cells, while KLK3 was most strongly induced in shCHD1 and shDouble cells (Fig. 1E). This suggests that knockdown of MAP3K7 and CHD1 each contribute to the increased expression of various AR target genes seen in shDouble cells. Similar gene expression trends were seen in LAPC4 and 22RV1 cells (Fig. S1E-F), demonstrating that the combined knockdown of MAP3K7 and CHD1 increases AR transcriptional activity.
AR protein levels remained largely consistent across multiple medium conditions in shDouble LNCaPs cells relative to shControl cells (Fig. 1F). Since translocation of AR from the cytoplasm to the nucleus upon ligand binding is a critical step in AR signaling, we then assessed nuclear localization of AR. A slight increase in AR nuclear localization was observed in shDouble cells in the absence of ligand, while a robust increase occurred in response to androgen stimulation in shDouble cells (Fig. 1G, Fig. S1G). The effects seen on AR nuclear localization along with increased target gene expression suggest significant alterations in AR activity induced by the combined knockdown of MAP3K7 and CHD1.
MAP3K7 and CHD1 co-suppression increased AR signature gene expression
To globally assess changes in AR-regulated gene expression, we performed RNA-Seq and AR (N-terminal) ChIP-seq on shControl, shMAP3K7, shCHD1 and shDouble LNCaP cells grown in castration conditions and supplemented with vehicle (EtOH) or 1 nM R1881 for 4 hours (Fig. 2A). We also performed RNA-Seq on 22RV1 and LAPC4 shControl and shDouble cells ± 1 nM R1881. Initially, we analyzed the mRNA expression profiles of all 3 cell lines against a set of verified AR target genes (Fig 2B) (33). Strikingly, the shDouble LNCaP, LAPC4, and 22RV1 cells all had a higher basal level of AR target gene expression than shControl cells in castration conditions, which increased even further upon R1881 administration (Fig. 2B). The increases in gene expression seen in shDouble LNCaP cells were much stronger than those induced by shMAP3K7 or shCHD1 individually, with some target genes being increased specifically by knockdown of either MAP3K7 or CHD1. Consistently, a similar pattern of expression was observed when the RNA-Seq data was analyzed against the “Hallmark Androgen Response” gene set from the Molecular Signatures Database (MSigDB) (Fig. S2A) (34).
Fig 2. Co-loss of MAP3K7 and CHD1 cooperatively increases AR signaling and AR chromatin binding.
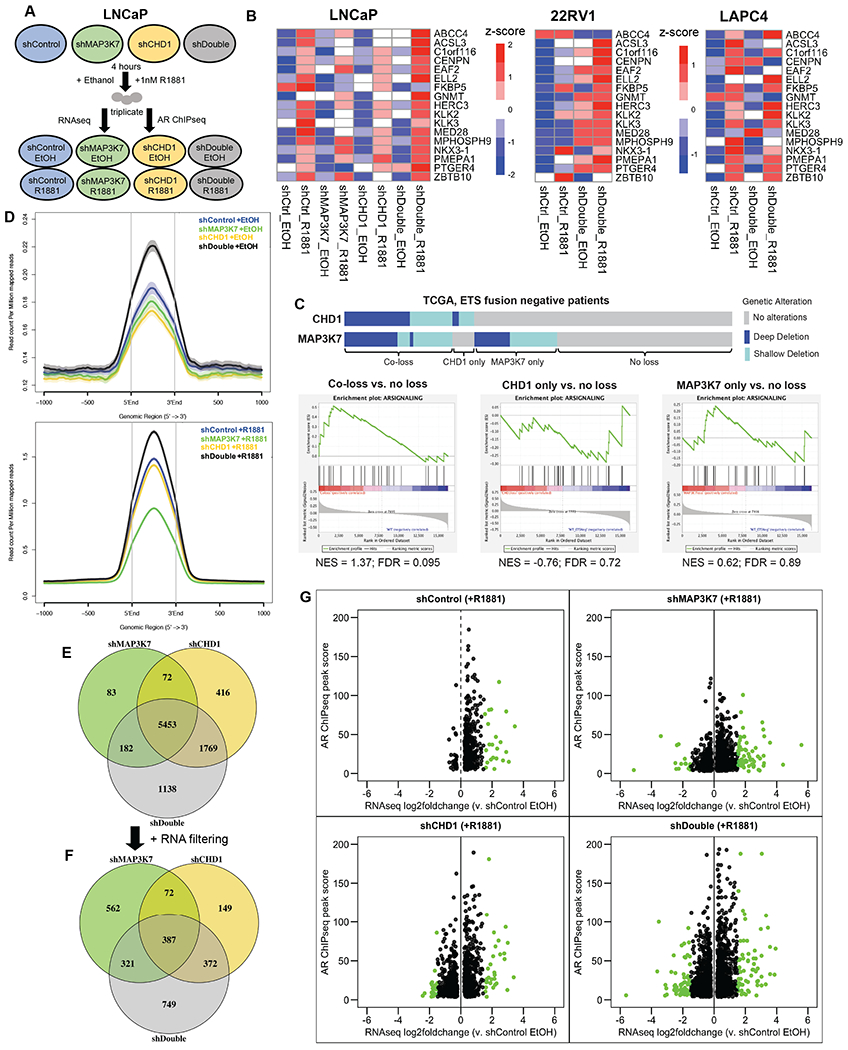
(A) Schematic of experimental setup and workflow for RNA-Seq and ChIP-seq. (B) Heatmap illustration of RNA-Seq z-score mRNA expression changes of a validated AR target gene signature in indicated cell types ± 1 nM R1881 for 4 hr (33). (C) GSEA of AR signaling gene signature (MSigDB: M15861) in indicated TCGA PCa groups with copy number loss of MAP3K7, CHD1, or both versus PCa with loss of neither. (D) Normalized signal intensity (read count per million mapped reads) of AR ChIP-seq binding events centered on the peak regions in indicated cells treated with vehicle EtOH (top) or 1 nM R1881 (bottom). (E) Venn diagram of differential, gene-annotated AR ChIP binding sites in indicated cell types treated with 1 nM R1881 compared to shControl EtOH as a background (fold-change >4, p-value <1e-04). (F) Venn diagram of the annotated AR ChIP sites from (E) that are further filtered for significant gene expression changes (FDR<0.05) over shControl EtOH. (G) Scatter plots of annotated genes from (F) including the shControl R1881 versus shControl EtOH comparison not shown in (F). Genes with log2 fold-change ± 1.5 are shown in green.
Using GSEA we evaluated an AR signaling gene set (MSigDB: M15861) (34) in a TCGA dataset of primary PCa stratified by MAP3K7 and/or CHD1 deletions (4). When comparing cancers with deletions of MAP3K7, CHD1, or both genes to those with loss of neither, only those with the dual deletion exhibited a relative enrichment in the AR signaling signature (Fig. 2C). This was also true within a Memorial Sloan Kettering Cancer Center (MSKCC) PCa dataset (Fig. S2B) (25).
Loss of MAP3K7 and CHD1 altered AR chromatin binding
Based on the increased AR gene signature and target gene expression, we suspected an increase in AR chromatin binding in shDouble cells. Using AR ChIP-seq data from LNCaP cells, we compared AR binding events across the genome in ethanol and R1881-treated conditions. In ethanol conditions, only shDouble cells exhibited a significant increase in AR binding compared to shControl cells (Fig. 2D, top). In R1881-stimulated conditions, shDouble cells maintained this increased AR binding (Fig. 2D, bottom). Interestingly, knockdown of CHD1 did not significantly change overall AR binding intensity compared to shControl cells while knockdown of MAP3K7 decreased AR binding (Fig. 2D bottom). Examples of AR peaks at two target genes are shown in Fig. S2C. At the ELL2 locus, AR binding is slightly elevated in shCHD1 cells and significantly increased in shDouble cells (Fig. S2C, top). Conversely, both individual knockdowns decrease AR binding at the FKBP5 locus while the double knockdown greatly increases the AR peak (Fig. S2C, bottom). These findings suggest a mechanism by which the combination of MAP3K7 and CHD1 suppression cooperatively increases AR chromatin binding.
To further investigate the respective contributions of MAP3K7 and CHD1 suppression to changes in the AR cistrome, we compared all ChIP binding events in shControl, shMAP3K7, shCHD1, and shDouble R1881-stimulated cells to shControl EtOH-treated cells as a relative baseline. We compared the overlap of differential AR binding events and identified many peaks that were shared among the 3 cell types (5453). Suppression of CHD1 induced many more differential peaks overall, as well as contributed more unique peaks than shMAP3K7 to the shDouble AR cistrome (1769 versus 182) (Fig. 2E). Interestingly, shDouble cells had many unshared sites (1138) that were either binding events unique to shDouble cells, or only exhibited a significant increase over shControl EtOH cells upon dual suppression of MAP3K7 and CHD1 (Fig. 2E).
We then overlapped the differential AR ChIP binding events with RNA-Seq differential expression in order to identify functional gene expression changes potentially induced by AR. Using the ChIP peaks from Fig. 2E, we further filtered for genes that showed significant expression changes (FDR<0.05) in R1881-treated cells relative to shControl (EtOH-treated) cells (Fig. 2F). While shDouble cells still exhibited many unique ChIP sites with significant gene expression changes (749), the relative contribution of shCHD1 and shMAP3K7 to effects seen in shDouble cells shifted to a more even distribution (372 and 321, respectively) (Fig. 2F). shMAP3K7 cells displayed significantly more unique AR-bound genes with altered gene expression (562), implying that some of the shared AR-bound sites between all 3 conditions from (Fig. 2E) only exhibited significant expression changes upon loss of shMAP3K7 alone.
The individual points from Fig. 2F, including data from shControl R1881 versus shControl EtOH not shown previously, are shown in Fig. 2G. These results illustrate that while knockdown of CHD1 contributed more to chromatin binding changes, knockdown of MAP3K7 induced greater expression changes for genes bound by AR. Additionally, suppression of MAP3K7 significantly contributed to the pathway enrichments seen among genes from Fig. 2G in shDouble cells (Fig. S2D). Similarly, when comparing differential AR ChIP binding events induced by R1881 within each genetic background, a stepwise increase in AR ChIP binding intensity (fragment reads) was evident from shControl, shMAP3K7 and shCHD1 to shDouble cells (Fig. S2E).
Suppression of MAP3K7 and CHD1 increased enzalutamide resistance and AR-v7 signaling
Given the observed alterations in androgen signaling, we sought to determine whether loss of CHD1 and MAP3K7 expression altered the cellular response to enzalutamide, a common AR antagonist administered in the clinic (35). When LNCaP cells were treated with varying concentrations of enzalutamide, we observed a significant increase in resistance in shDouble cells compared to shControl cells (Fig. 3A). Knockdown of either MAP3K7 or CHD1 was sufficient for increased resistance at several doses, highlighting the impact that both genes have on AR activity. shDouble LAPC4 cells demonstrated a similar trend of increased resistance at multiple doses compared to shControl cells, while the single knockdowns were more sensitive to enzalutamide at some doses (Fig. S3A).
Fig 3. Suppression of MAP3K7 and CHD1 increases enzalutamide resistance and AR-v7 signaling.
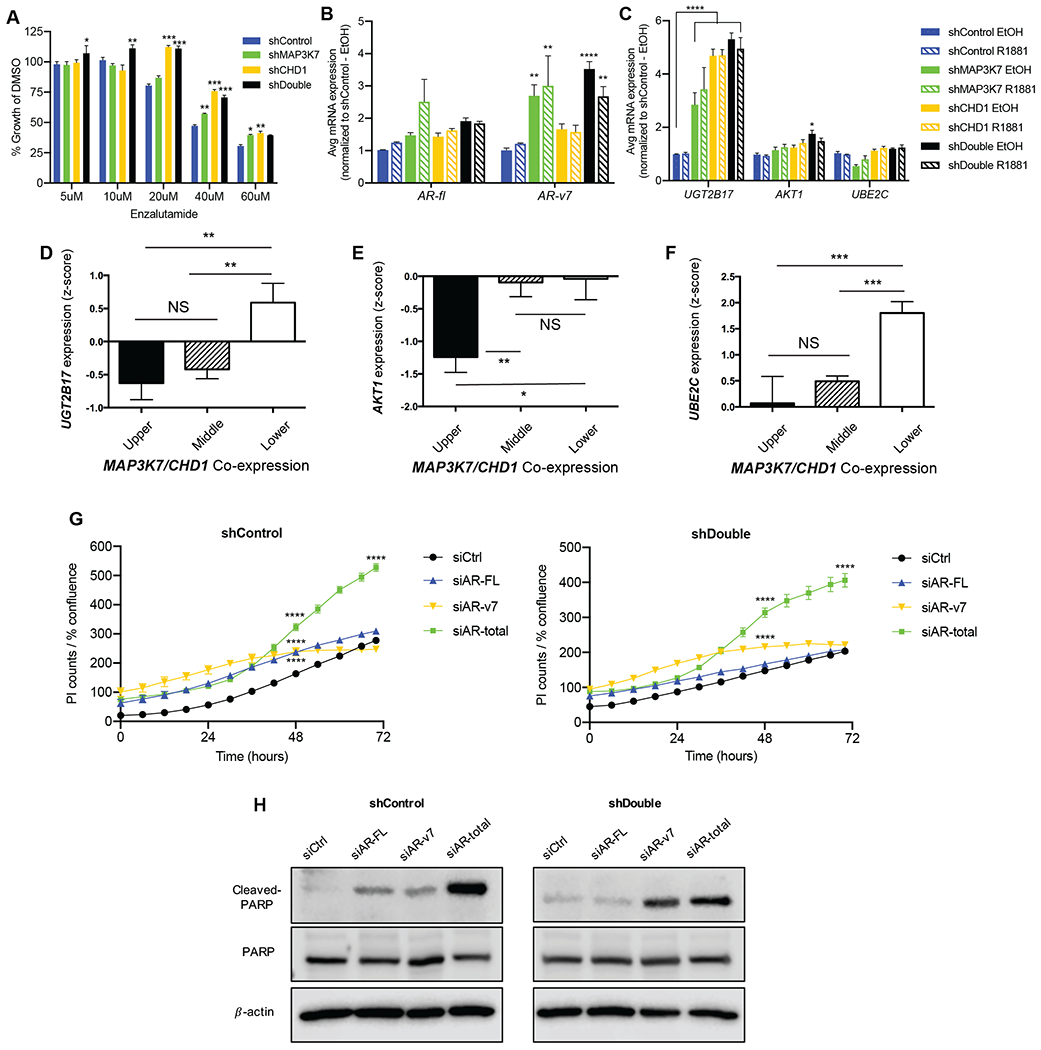
(A) Day 5 of growth assay for indicated LNCaP cells treated with 5-60 µM enzalutamide in complete medium, normalized to DMSO vehicle control (n=3; p-values indicate comparison to shControl at each concentration). (B) RT-qPCR of AR-FL and AR-v7 in indicated LNCaP cells ± 1 nM R1881 for 4 h. (C) RT-qPCR analysis of AR-v7 target genes UGT2B17, AKT1, and UBE2C in shControl and shDouble LNCaP cells ± EtOH or 1 nM R1881 for 4 h. For (B-C), Ct values normalized to B2M expression and then to shControl (n=3; p-values indicate comparison to shControl EtOH). (D) Correlation of UGT2B17, (E) AKT1, and (F) UBE2C mRNA expression with quartiles of MAP3K7 and CHD1 mRNA co-expression in the MSKCC dataset (25). (G) Propidium iodide (PI) counts normalized to cell confluence of shControl (left) and shDouble (right) LNCaP cells in castration conditions beginning 24 hours after transfection with indicated siRNAs (n=3; p-values indicate comparison to siCtrl). (H) Western blot of lysates from LNCaP cells 96 hours after transfection with indicated siRNAs showing cleaved PARP, total PARP, and the loading control β-actin. All data represent means ± SEM; unpaired two-tailed t-tests (D, E, F) and two-way ANOVA with Dunnett’s multiple comparisons test (A, B, C, G); *=p<0.05; **=p<0.01; ***=p<0.001; ****=p<0.0001.
A number of mechanisms of enzalutamide resistance have been proposed. These include alternative splicing of the AR gene which results in AR splice variants (AR-Vs) lacking the ligand-binding domain. This allows AR to function in a ligand-independent, constitutively active manner (19, 36). Expression of AR-v7, the most abundant AR variant, is predictive of biochemical recurrence, upregulated in castration-resistant prostate cancer (CRPC) (19, 37), and correlated with decreased survival following enzalutamide treatment (38).
We evaluated expression of AR-v7 in our models and found AR-v7 to be weakly expressed in shControl LNCaP cells in castration conditions and significantly increased in shDouble cells (Fig. 3B, black bars). The increase in AR-v7 expression is likely induced by loss of MAP3K7 as AR-v7 levels in shDouble cells are not much higher than in shMAP3K7 cells (Fig. 3B, green bars). Because LNCaP cells express AR-full length (AR-FL) at much higher levels than AR-v7, we also calculated the ratio of AR-v7 to AR-FL mRNA. We observed that multiple shDouble combinations had higher AR-v7/AR-FL ratios relative to shControl cells (Fig. S3B). Suppression of MAP3K7 and CHD1 in LAPC4 cells also resulted in increased expression of AR-v7 (Fig. S3C). 22RV1 cells already have high levels of AR-v7 expression, and shDouble cells did not exhibit a further increase. However, an increase in a similar variant AR-v567es was observed, suggesting MAP3K7 and CHD1 may regulate AR splice variant expression more broadly (Fig. S3D).
Whether or not AR-v7 transcriptional targets differ from those of AR-FL is heavily debated (39, 40), but some putative specific targets of AR-v7 include UGT2B17, UBE2C, and AKT1 (17). UGT2B17 and AKT1 were significantly upregulated in shDouble LNCaP cells relative to shControl cells, with UGT2B17 also being significantly increased in shMAP3K7 and shCHD1 cells (Fig. 3C). There was no difference in UBE2C expression. We evaluated the association of MAP3K7 and CHD1 with expression of these AR-v7 target genes in the MSKCC patient dataset (25). Expression of all three genes, UGT2B17, AKT1, and UBE2C, was significantly inversely correlated with expression of MAP3K7 and CHD1 (Fig. 3D–F).
To determine if increased AR-v7 expression contributed to the enhanced growth of shDouble cells, we utilized AR isoform-specific siRNA targeting constructs to knockdown AR-FL, AR-v7, or total AR capturing both variants (Fig. S3E-G) (18, 19). We evaluated cellular staining of propidium iodide (PI) and Annexin V as markers of apoptosis in LNCaP cells transfected with the siRNAs in castrate medium conditions. In both shControl and shDouble cells, total AR knockdown significantly increased PI staining by 48 hours (Fig. 3G). shControl cells were also somewhat sensitive to siAR-FL and siAR-v7 while shDouble cells were only sensitive to siAR-v7. Annexin V detection displayed similar results (Fig. S3H). These patterns of increased PI and Annexin V staining were recapitulated by detection of cleaved PARP. ShControl cells exhibited cleaved PARP induction with siAR-v7, siAR-FL, or most strongly with siAR-total (Fig. 3H). ShDouble cells instead exhibited PARP cleavage only with siAR-v7 or siAR-total (Fig. 3H).
MAP3K7 and CHD1 immunohistochemical (IHC) expression predicts recurrence in clinical PCa
We know from previous studies that gene expression of CHD1 and MAP3K7 correlate with each other and with poor DFS (6). We recently found that decreased MAP3K7 protein (TAK1) alone is correlated with PCa brain metastases (41), and others showed that copy number loss of MAP3K7 and CHD1 is associated with cribriform pattern PCa in the TCGA (42). Cribriform patterning is a sub-variant of Gleason pattern 4 and is an independent predictor of poor outcome (43). While these recent studies support the hypothesis that the dual loss of MAP3K7 and CHD1 drives aggressive PCa, the evaluation of CHD1 and MAP3K7 protein expression in primary PCa and its association with patient outcome has not been evaluated.
In this study we evaluated the expression of CHD1 and MAP3K7 in a cohort of well-characterized radical prostatectomy specimens with clinical follow-up to assess the impact of reduced expression of one or both proteins on biochemical recurrence (BCR). From the Stanford database we identified 206 high grade cases, 112 with BCR and 94 without BCR matched for age at surgery and percent total high grade cancer (Gleason grade 4 or 5). This cohort is outlined in detail in the supplemental methods. Table S2 shows characteristics of these cases illustrating that patients with BCR had higher Gleason scores, PSA, total cancer volume, clinical stage, and involvement of lymph nodes and seminal vesicles. Cases with BCR also more often had cribriform patterns present.
H-scores for CHD1 and MAP3K7 were dichotomized into high expressors (≥150) and low expressors (<150) for each case. Examples of samples with high and low expression of CHD1 and MAP3K7 are shown (Fig. 4A, Fig. S4A). An initial comparison of H-scores across all cases illustrated that expression of MAP3K7 and CHD1 were each significantly lower in the BCR group compared to the non-BCR group (Fig. 4B, Table S3). We further stratified patients into 4 groups based on the combination of high and low CHD1 and MAP3K7 H-scores to assess their individual and combined associations with BCR. As expected, cases with low H-scores of both proteins demonstrated the highest frequency of BCR (Fig. 4C, 84.6%). Interestingly, cases with low MAP3K7 H-scores alone also demonstrated a high percentage of BCR (Fig. 4C, 74.4%), much higher than that of cases with only low CHD1 H-scores (Fig. 4C, 46.2%). This suggests that MAP3K7 protein expression is a potential driving factor of aggressiveness in the MAP3K7/CHD1 subtype.
Fig 4. MAP3K7 and CHD1 expression are predictors of biochemical recurrence in a clinical PCa cohort.
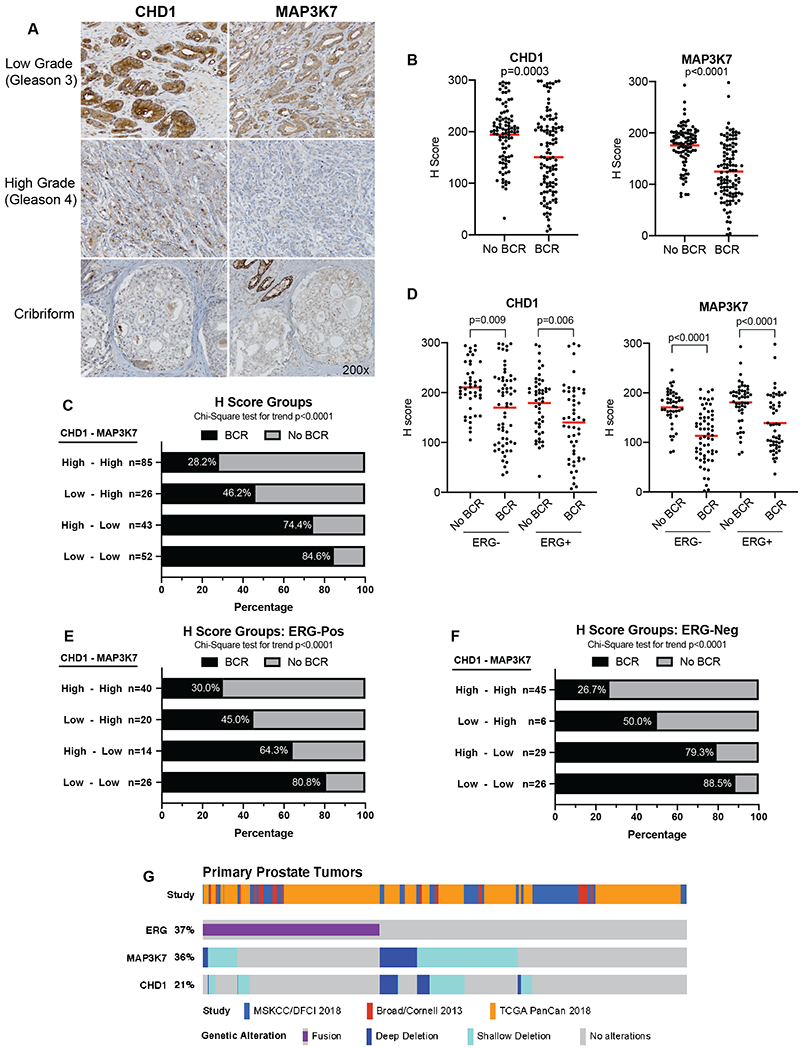
(A) IHC for CHD1 (left panels) and MAP3K7 (right panels) in representative low grade (Gleason 3), high grade (Gleason 4), and cribriform PCa. All photos taken at 200x. (B) CHD1 and MAP3K7 IHC H-scores for 207 cases stratified by biochemical recurrence (BCR) and no BCR. (C) Frequency of BCR in cases with indicated combinations of high (>150) and low (<150) H-scores for MAP3K7 and CHD1. (D) CHD1 and MAP3K7 H-scores further stratified by the presence or absence of nuclear ERG staining. (E) Frequency of BCR in ERG-positive cases with indicated combinations of H-scores for MAP3K7 and CHD1. (F) Frequency of BCR in ERG-negative cases with indicated combinations of H-scores for MAP3K7 and CHD1. Red bars (B, D) denote mean. (G) Meta-analysis of multiple PCa genomic databases displaying frequency of ERG fusions and deletions of MAP3K7 and CHD1 in 761 primary PCa cases. Wilcoxon rank sum test (B, D), Chi-Square test for trend (C, E, F).
Since our earlier analyses of patient datasets showed that coordinate loss of CHD1 and MAP3K7 was associated with ERG-negative tumors (6), we stratified BCR cases and non-BCR cases by ERG expression (Fig. 4D, Table S4). The mean H-scores for CHD1 and MAP3K7 were significantly lower in BCR cases compared to non-BCR cases in both ERG-positive and ERG-negative cases (Fig. 4D). Despite the fact that MAP3K7 and CHD1 deletions are generally mutually exclusive of ERG-fusions, 60% of ERG-positive cases had low H-scores for either MAP3K7, CHD1, or both (Fig. 4E). In both ERG-positive (Fig. 4E) and ERG-negative (Fig. 4F) subgroups, cases with only low MAP3K7 H-scores again had a higher proportion of BCR than cases with only low CHD1 H-scores, suggesting MAP3K7 expression as a key factor regardless of ERG status. It is important to note that there are very few ERG-negative cases with a low CHD1 H-score in absence of a coordinately low MAP3K7 H-score (Fig. 4F), similar to what is seen at the genomic level in patient datasets.
If cases with cribriform morphology were removed from the analysis, low MAP3K7 was significantly associated with BCR regardless of ERG status while CHD1 expression was only different between BCR and non-BCR groups in ERG-positive tumors (Fig. S4B-C). In tumors with cribriform morphology, however, H-scores for both CHD1 and MAP3K7 were only significantly lower in ERG-negative cases with BCR (Fig. S4D). Unlike in non-cribriform cases (Fig. S4C), independent low H-scores of CHD1 or MAP3K7 both resulted in high frequencies of BCR, similar to that of their combined effect (Fig. S4E). This suggests that low CHD1 expression has more impact on BCR in cases with cribriform cancer than those without.
Due to the frequency of low MAP3K7 and CHD1 H-scores we see in ERG-positive cases, we performed a meta-analysis of patient genomic data from multiple published cohorts to better evaluate the frequency of these genomic alterations. Interestingly, we found that at the primary stage, 19.4% of ERG-positive cases exhibited MAP3K7 deletions (homozygous and heterozygous together), and 11.6% had CHD1 deletions (Fig. 4G, Table S5). These frequencies are recapitulated well in our IHC cohort with 9% of the ERG-positive cases having truly low (<100) H-scores of MAP3K7 and CHD1 together. The proportion of overlap between MAP3K7 and CHD1 deletions and ERG fusions increases even more in metastatic tumors (Fig. S5, Table S5).
MAP3K7 is an independent predictor of biochemical recurrence
We performed univariate and multivariate logistic regression analyses in order to determine key variables that predict BCR. In the univariate analysis, CHD1 H-scores, MAP3K7 H-scores, total cancer volume, and clinical stage were all significantly, positively associated with BCR (Table 1). We then performed a multivariate regression analysis using an optimized combination of clinical factors with the highest predictive value as the base model, and BCR as the output. We tested all clinical variables in our study that would be available post-surgery, and the strongest combination included total cancer volume, PSA, seminal vesicle invasion, lymph node involvement, Gleason scores, age, date of surgery, and prostate weight. MAP3K7 and CHD1 H-scores were then added individually and together to determine if they added to or subtracted from the predictive value of this base model (Table 2). Interestingly, MAP3K7 H-scores added to the model while CHD1 H-scores did not. Additionally, we used an established model of post radical prostatectomy risk assessment, the CAPRA-S Score, as the base model for the multivariate analysis. The CAPRA-S score considers multiple post-operative factors including PSA, Gleason score, seminal vesicle invasion, lymph node invasion, surgical margins, and extracapsular extension (44). MAP3K7 H-scores also significantly added to the predictive value of the CAPRA-S score (Table S6). This demonstrates that MAP3K7 expression has a strong association with BCR that significantly adds to the predictive value of established post-surgical clinical factors.
Table 1.
Univariate logistic regression modeling risk for BCR. Logistic regression analysis testing the extent to which each individual variable in the study models the risk for BCR as an output (AUC value). OR=Odds ratio, CI= 95% confidence interval.
| BCR | No BCR | OR | CI (95%) | p-value | AUC | |
|---|---|---|---|---|---|---|
| Age (Years) | 112 | 94 | 1 | 0.96, 1.04 | 0.875 | 0.484 |
| Date of Surgery | 112 | 94 | 0.95 | 0.86, 1.04 | 0.284 | 0.548 |
| % High Grade (Gleason 4/5) | 112 | 94 | 1.01 | 0.99, 1.02 | 0.065 | 0.581 |
| PSA (ng/ml) | 112 | 94 | 1 | 0.99, 1.01 | 0.611 | 0.588 |
| Total Cancer Volume (cc) | 112 | 94 | 1.15 | 1.07, 1.24 | 0 | 0.701 |
| ERG presence (+/−) | 112 | 94 | 0.77 | 0.44, 1.33 | 0.346 | 0.533 |
| Cribriform presence (+/−) | 112 | 94 | 1.82 | 0.99, 3.40 | 0.056 | 0.562 |
| Gleason 3+3 and 3+4 (%) | 112 | 94 | 0.62 | 0.34, 1.10 | 0.103 | 0.554 |
| Gleason 4+3 (%) | 112 | 94 | 0.8 | 0.46, 1.39 | 0.432 | 0.528 |
| Clinical Stage T1 | 112 | 94 | 0.52 | 0.29, 0.90 | 0.022 | 0.58 |
| Clinical Stage T2 | 112 | 94 | 1.94 | 1.11, 3.43 | 0.022 | 0.58 |
| H-score MAP3K7 | 112 | 94 | 0.98 | 0.97, 0.99 | 0 | 0.772 |
| MAP3K7 H-score <150 vs 150+ | 112 | 94 | 8.22 | 4.39, 16.04 | 0 | 0.735 |
| H-score CHD1 | 112 | 94 | 0.99 | 0.98, 0.99 | 0 | 0.645 |
| CHD1 H-score <150 vs 150+ | 112 | 94 | 3.24 | 1.78, 6.05 | 0 | 0.63 |
Table 2.
Multivariate logistic regression modeling risk for BCR. The modeling starts with an optimized combination of clinical factors (total cancer volume, PSA, age, date of surgery, prostate weight (PWEI), seminal vesicle invasion, lymph node involvement, and Gleason score), and then adds MAP3K7 and CHD1 H-scores to determine if each adds to or subtracts from the overall model performance for risk of BCR (AUC value). Difference indicates value added to the base AUC.
| AUC | Difference | |
|---|---|---|
| Optimized clinical factors (base) | 0.833 | 0 |
| +MAP3K7 H-scores | 0.87 | 0.037 |
| +CHD1 H-scores | 0.842 | 0.009 |
| +MAP3K7 & CHD1 H-scores | 0.868 | 0.035 |
We also performed a multivariate regression analysis using MAP3K7 H-scores as the base model. Only the addition of seminal vesicle invasion, total cancer volume, and lymph node involvement notably added to the predictive power of this model (Table S7).
MAP3K7 loss altered cell adhesion, spliceosome, and cell cycle pathways
Since we determined that low MAP3K7 protein expression is a major driving force behind the clinical impact of the MAP3K7/CHD1 subtype, we further investigated the consequences of loss of MAP3K7 expression. We performed Metascape analysis on significant gene expression changes in LNCaP shMAP3K7 cells compared to shControl cells (both R1881-treated) in order to identify pathways altered by MAP3K7 loss (28). Pathways enriched in genes upregulated by suppression of MAP3K7 included ephrin signaling, cell adhesion, and mesenchymal cell differentiation (Fig. 5A, green bars). Genes downregulated by shMAP3K7 were heavily involved in the cell cycle, DNA repair, cytoskeletal organization and response to cytokines (Fig. 5A, blue bars).
Fig 5. MAP3K7 loss alters cell adhesion, spliceosome, and cell cycle pathways.
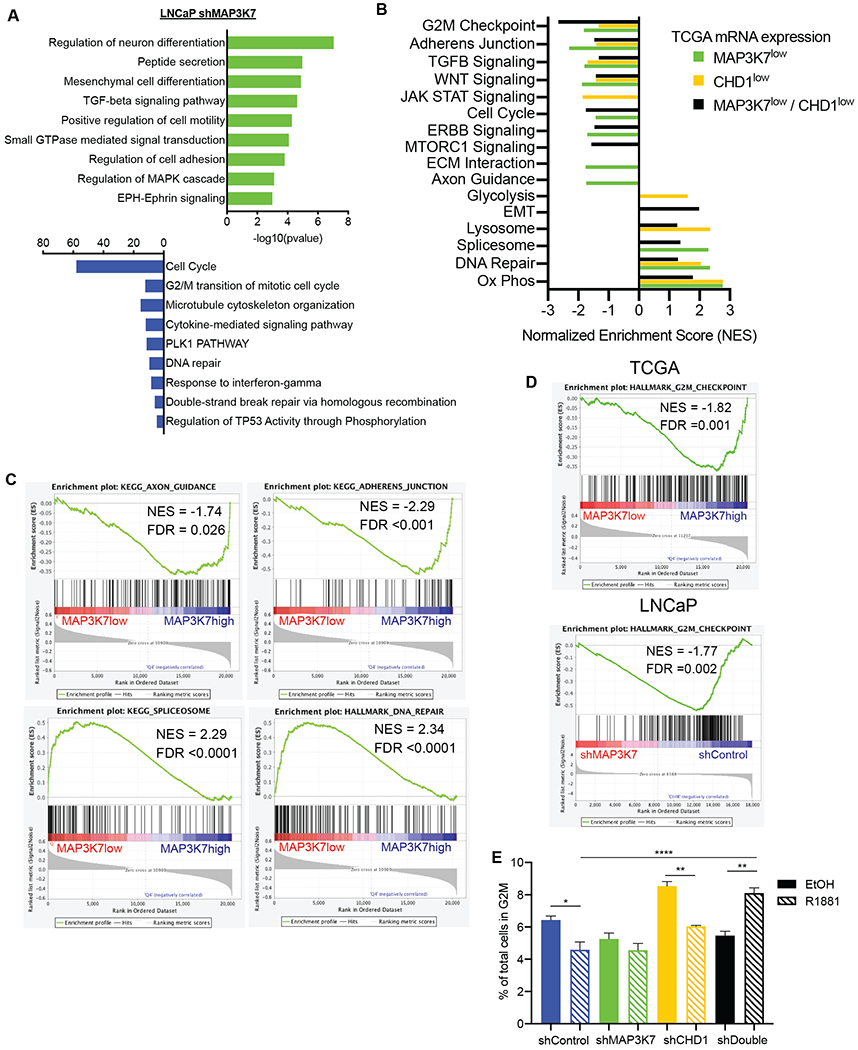
(A) Pathway enrichment analysis for LNCaP shMAP3K7 versus shControl (+ R1881) RNA-Seq changes using Metascape (28). (B) Summary of GSEA results comparing TCGA cases with lower quartile mRNA expression of MAP3K7, CHD1 or both versus cases with upper quartile expression of respective groups. Nominal p-value <0.05 for all enrichments included. (C) GSEA plots of signatures particularly enriched in TCGA cases with low versus upper quartile expression of MAP3K7. (D) GSEA plots of the G2M signature in TCGA MAP3K7 low versus high cases and LNCaP shMAP3K7 versus shControl cells (+R1881). (E) Flow cytometry cell cycle analysis of indicated LNCaP cells after 48 hours in steroid-depleted medium ± 1 nM R1881 showing percent of total cells in G2M (n=3). Data represent mean ± SEM. Log-rank Mantel-Cox test (A, B), One-way ANOVA with Tukey’s multiple comparisons test (G); *=p<0.05; **=p<0.01; ***=p<0.001; ****=p<0.0001.
We next compared enrichments seen in our cell line model to patient data. TCGA primary PCa cases were stratified by mRNA expression quartiles to perform enrichments in cancers with low (quartile 1) versus high (quartile 4) MAP3K7 expression, CHD1 expression, or combined expression (4). GSEA enrichments for these three comparisons are summarized by normalized enrichment scores showing shared and unique pathway enrichments (Fig. 5B) (27).
Many of the pathways enriched in cancers with low MAP3K7 or low MAP3K7 and CHD1 expression are similar to those seen in LNCaP cells. Some of the strongest enrichments observed in TCGA MAP3K7 low cases are highlighted (Fig. 5C). The negative enrichment scores for axon guidance and adherens junction suggest cytoskeletal and cell adhesion changes, and we have previously shown that suppression of MAP3K7 induces changes in actin cytoskeleton and increases cell motility (45). The spliceosome pathway is also enriched in cancers with low MAP3K7 expression (Fig. 5C), which could explain alterations seen in AR splice variant expression. Further studies will be needed to elucidate the specific mechanisms by which MAP3K7 may regulate splicing.
Loss of MAP3K7 induced changes in androgen-regulated cell cycle progression
Alterations in DNA repair pathways and the cell cycle (Fig. 5B–C) were of particular interest as we have recently shown that loss of MAP3K7 induces sensitivity to the combination of the cell cycle checkpoint inhibitor Dinaciclib and DNA damaging agents (46). One of the strongest negative enrichments seen in TCGA PCa data was that of the G2M transition (Fig. 5B), and GSEA analysis of LNCaP shMAP3K7 versus shControl cells showed a similar enrichment (Fig. 5D). We performed cell cycle analysis in LNCaP cells after 48 hours ± 1 nM R1881 to evaluate potential changes in cell cycle transitions. Androgen-treated shControl cells became growth-inhibited relative to vehicle-treated cells at this R1881 concentration and time point, demonstrated by a decreased G2M ratio (Fig. 5E) along with an increase in percentage of cells in G1 (Fig. S5A). ShMAP3K7 cells were not significantly affected following R1881 treatment while shCHD1 cells were growth-inhibited by this dose of R1881 (Fig. 5E, Fig. S5A). In contrast, androgen-treated shDouble LNCaP cells displayed a significantly higher G2M ratio (Fig. 5E) and lower percentage of cells in G1 (Fig. S5A) relative to untreated cells. This was also significant or trending across multiple shDouble shRNA construct combinations (Fig. S5B).
These results indicate that shDouble cells move rapidly through the cell cycle at a dose of androgen that is normally growth inhibitory to LNCaP cells. This finding suggests that suppression of MAP3K7 is significantly contributing to the shift in response to androgen. The corresponding enrichment of the G2M transition across many TCGA cases with low expression of MAP3K7 suggests a robust alteration in genes regulating the cell cycle.
Discussion
We have made a number of significant findings in the current study. We are the first, to our knowledge, to evaluate MAP3K7 and CHD1 expression by IHC in association with biochemical recurrence (BCR) in a clinical cohort of PCa patients. We showed that tumors with BCR had significantly lower expression of CHD1 and MAP3K7 than those without BCR, and that low MAP3K7 expression was the most significant independent predictor of BCR. This is a critical finding as other studies in the field have focused primarily on the loss of CHD1, which we demonstrated is a factor secondary to MAP3K7 expression for predicting BCR. However, evaluation of MAP3K7 and CHD1 expression in primary PCa and its correlation with subsequent metastasis and therapeutic response is also needed to fully understand the predictive value of this subtype. Recent studies have evaluated such clinical cohorts, but they did not include the prevalent losses of MAP3K7 and CHD1 in their analysis (47).
In our previous study we evaluated suppression of MAP3K7 and CHD1 in our mouse prostate stem cell, tissue recombination model. We found that grafts of dual knockdown cells exhibited aggressive pathology, as well as focal areas of increased AR expression and evidence of lineage plasticity (6). Using human prostate cancer cell line models in our current study, we have expanded our findings to show that combined loss of MAP3K7 and CHD1 has striking effects on AR activity. Importantly, these effects are far greater than those seen with suppression of either gene alone. While it is important to evaluate the individual effects of CHD1 and MAP3K7 for mechanistic insight, the dual loss of these genes is the condition most often seen in patients.
Suppression of MAP3K7 resulted in decreased AR chromatin binding along with increased AR target gene expression, suggesting a potential repressive role of MAP3K7 (TAK1) on normal AR activity. We also noted that the addition of MAP3K7 suppression to that of CHD1 drives larger changes in expression of AR-targeted genes. The synergistic changes in AR activity we see upon loss of both CHD1 and MAP3K7 may be due to compound effects on the AR signaling axis. MAP3K7 loss may be increasing AR-v7 levels along with relieving repression of AR while CHD1 loss alters the chromatin, dictating where AR and its variants can bind. Further studies on this potential mechanism are warranted. Previous studies evaluating effects of CHD1 loss alone may therefore underrepresent the significance of its impact on the AR pathway in patients.
Remarkably, loss of MAP3K7 significantly impacted the cell cycle and related pathways. In PCa there is significant interplay between AR and the cell cycle with feedback that can in turn affect AR signaling (48). UBE2C, a putative AR-v7-regulated gene, is known to be involved in controlling cyclin levels, and a significant increase in UBE2C expression was observed in patients with low expression of MAP3K7 and CHD1 (49). We previously reported that suppression of MAP3K7 sensitizes cells to the combination of a cell cycle inhibitor Dinaciclib and DNA-damaging agents (46). It will be interesting to evaluate how these drugs affect the increased AR signaling we see in cells with suppression of MAP3K7 and CHD1.
The significant differences in the genetic phenotype and disease stage of our PCa cell lines did pose limitations in our ability to identify consistent mechanisms to describe the increased AR activity seen upon suppression of MAP3K7 and CHD1. The specific mechanisms may vary slightly depending on the presence of different cofactors and confounding genetic alterations of the cell lines. Additionally, the LNCaP cell line possesses an ETV1 rearrangement which is typically mutually exclusive from loss of MAP3K7 and CHD1 in genomic databases (50). However, our meta-analysis in Fig. 4G (Fig. S5) and findings in our clinical cohort in Fig. 4 demonstrate that these features are not as mutually exclusive as previously presented. We have repeated some of our studies in other cell lines to ensure the results were not specific to LNCaP cells. Correlating AR-v7 levels with genetic alterations in public datasets has also proven difficult, and extensive IHC studies are likely necessary to successfully profile the correlation of AR-v7 expression with the loss of MAP3K7 and CHD1.
The findings in this study could have significant implications in the clinical management of PCa. Measurement of low protein expression of MAP3K7 alone or in conjunction with CHD1 could be employed to stratify patients who are likely to have recurrent disease and therefore might benefit from more aggressive therapy. Additionally, the alterations seen in AR signaling along with increased AR-v7 expression and enzalutamide resistance suggest that patients with this subtype may have limited response to some AR-targeting therapies commonly used in the clinic. Based on our current and previous findings regarding changes in cell cycle progression upon loss of MAP3K7, patients with loss or lower expression of MAP3K7 and CHD1 may instead benefit from cell cycle inhibitors alone or in combination with hormonal therapies.
Supplementary Material
Implications:
These findings strongly implicate MAP3K7 loss as a biomarker for aggressive prostate cancer with significant risk for recurrence that poses challenges for conventional androgen receptor-targeted therapies.
Acknowledgments:
This project was supported by National Institutes of Health (NIH) grants R21-CA187354 and R01-CA199741 to S. Cramer, U01-CA231978 to S. Cramer and J. Costello, American Cancer Society (126572-PF-14-223-01-CCE) to L. Rider, Boettcher Foundation Webb-Waring grant to J. Costello, and University of Colorado Cancer Center Fellowship to L. Jillson. The University of Colorado Anschutz Medical Campus Genomics and Microarray Core is supported in part by the Genomics and Microarray Shared Resource of Colorado’s NIH/NCI Cancer Center Support Grant P30CA046934 and by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082. This study was also partly supported by the National Institutes of Health P30CA046934 Bioinformatics and Biostatistics Shared Resource Core. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Footnotes
Data and materials availability: Accession codes: The RNA-Seq and ChIP-seq data are available at the Gene Expression Omnibus (GEO), reference series GSE168671. The code used for our patient dataset analysis is available at GitHub: https://github.com/jillsonl/MCR_2021_publication.
Conflicts of interests: The authors disclose no competing financial interests.
References
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019. CA Cancer J Clin 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Higano CS, Side effects of androgen deprivation therapy: monitoring and minimizing toxicity. Urology 61, 32–38 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Hugosson J, Stranne J, Carlsson SV, Radical retropubic prostatectomy: a review of outcomes and side-effects. Acta Oncol 50 Suppl 1, 92–97 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Cancer N Genome Atlas Research, The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamoun A, Cancel-Tassin G, Fromont G, Elarouci N, Armenoult L, Ayadi M et al. , Comprehensive molecular classification of localized prostate adenocarcinoma reveals a tumour subtype predictive of non-aggressive disease. Ann Oncol 29, 1814–1821 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour-Fard A, Loda M et al. , Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res 75, 1021–1034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu W, Lindberg J, Sui G, Luo J, Egevad L, Li T et al. , Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 31, 3939–3948 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N et al. , The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature 399, 798–802 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N et al. , Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 270, 2008–2011 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Zhou J, Li J, Serafim RB, Ketchum S, Ferreira CG, Liu JC et al. , Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acids Res 46, 3891–3905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S et al. , CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 35, 817–819 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E et al. , Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 37, 584–598 e511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP et al. , Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med 377, 338–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K et al. , Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367, 1187–1197 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Silberstein JL, Taylor MN, Antonarakis ES, Novel Insights into Molecular Indicators of Response and Resistance to Modern Androgen-Axis Therapies in Prostate Cancer. Curr Urol Rep 17, 29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.C. A. Reichard, A. J. Stephenson, E. A. Klein, Applying precision medicine to the active surveillance of prostate cancer. Cancer 121, 3403–3411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD et al. , Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One 6, e27970 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Compagno D, Merle C, Morin A, Gilbert C, Mathieu JR, Bozec A et al. , SIRNA-directed in vivo silencing of androgen receptor inhibits the growth of castration-resistant prostate carcinomas. PLoS One 2, e1006 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E et al. , Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69, 16–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishan A, Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol 66, 188–193 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tao J, Oladimeji P, Rider L, Diakonova M, PAK1-Nck regulates cyclin D1 promoter activity in response to prolactin. Mol Endocrinol 25, 1565–1578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen L, Shao N, Liu X, Nestler E, ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamey T, McNeal J, Freiha F, Redwine E, Morphometric and clinical studies of 68 consecutive radical prostatectomies. J Urol 139, 1235–1241 (1988). [DOI] [PubMed] [Google Scholar]
- 25.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS et al. , Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O et al. , Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E et al. , The long tail of oncogenic drivers in prostate cancer. Nat Genet 50, 645–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y et al. , Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E et al. , Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I et al. , Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A et al. , Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litwin MS, Tan HJ, The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 317, 2532–2542 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W et al. , Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer Res 75, 3663–3671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H et al. , A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 69, 2305–2313 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC et al. , AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371, 1028–1038 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q et al. , Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc Natl Acad Sci U S A 115, 6810–6815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cato L, de Tribolet-Hardy J, Lee I, Rottenberg JT, Coleman I, Melchers D et al. , ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 35, 401–413 e406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ormond DR, Kleinschmidt-DeMasters BK, Cavalcante D, Smith EE, Cramer SD, Lucia MS, Prostatic adenocarcinoma CNS parenchymal and dural metastases: alterations in ERG, CHD1 and MAP3K7 expression. J Neurooncol 142, 319–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elfandy H, Armenia J, Pederzoli F, Pullman E, Pertega-Gomes N, Schultz N et al. , Genetic and Epigenetic Determinants of Aggressiveness in Cribriform Carcinoma of the Prostate. Mol Cancer Res 17, 446–456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Humphrey PA, Cribriform adenocarcinoma of the prostate. J Urol 193, 1655–1656 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Cooperberg MR, Hilton JF, Carroll PR, The CAPRA-S score: A straightforward tool for improved prediction of outcomes after radical prostatectomy. Cancer 117, 5039–5046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu M, Shi L, Cimic A, Romero L, Sui G, Lees CJ et al. , Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res 72, 2833–2843 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Washino S, Rider LC, Romero L, Jillson LK, Affandi T, Ohm AM et al. , Loss of MAP3K7 Sensitizes Prostate Cancer Cells to CDK1/2 Inhibition and DNA Damage by Disrupting Homologous Recombination. Mol Cancer Res 17, 1985–1998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mateo J, Seed G, Bertan C, Rescigno P, Dolling D, Figueiredo I et al. , Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest 130, 1743–1751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schiewer MJ, Augello MA, Knudsen KE, The AR dependent cell cycle: mechanisms and cancer relevance. Mol Cell Endocrinol 352, 34–45 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J et al. , Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245–256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS et al. , Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448, 595–599 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.