

SUMMARY
The assembly of nascent proteins into multi-subunit complexes is a tightly regulated process which must occur at high fidelity to maintain cellular homeostasis. The ER membrane protein complex (EMC) is an essential insertase that requires seven membrane-spanning and two soluble cytosolic subunits for function. Here we show that the kinase With no lysine 1 (WNK1), known for its role in hypertension and neuropathy, functions as an assembly factor for the human EMC. WNK1 uses a conserved amphipathic helix to stabilize the soluble subunit, EMC2, by binding to the EMC2-8 interface. Shielding this hydrophobic surface prevents promiscuous interactions of unassembled EMC2 and directly competes for binding of E3 ubiquitin ligases, permitting assembly. Depletion of WNK1 thus destabilizes both the EMC and its membrane protein clients. This work describes an unexpected role for WNK1 in protein biogenesis and defines the general requirements of an assembly factor that will apply across the proteome.
eTOC blurb:
Assembly of multisubunit complexes is tightly regulated. The ER membrane protein complex (EMC) is an essential, nine-subunit membrane protein insertase. Pleiner et al. find that the kinase, WNK1, is an assembly factor for the human EMC. WNK1 depletion perturbs EMC assembly and affects the biogenesis of its membrane protein clients.
Graphical Abstract
INTRODUCTION
Protein complex assembly is a major challenge within the crowded cellular environment, as nearly half of the eukaryotic proteome is organized into multi-subunit complexes (Havugimana et al., 2012; Marsh and Teichmann, 2015). In their unassembled state, these subunits expose hydrophobic interfaces that make them susceptible to non-productive interactions and aggregation (Juszkiewicz and Hegde, 2018). To prevent cytotoxicity, orphan subunits that are synthesized in excess or fail to assemble must be recognized and degraded by the ubiquitin-proteasome system (McShane et al., 2016; Sung et al., 2016; Xu et al., 2016; Yanagitani et al., 2017). Assembly pathways have thus evolved to balance two primary goals. First, they must provide nascent subunits a reasonable opportunity to assemble by preventing premature degradation and promiscuous interactions of normal assembly intermediates. Second, to protect the cellular proteome they must ensure that terminally unassembled subunits are efficiently degraded, which is achieved through tight coupling with quality control machinery.
In prokaryotes, the organization of genes into operons ensures the near stoichiometric expression of individual subunits and regulates the localization and timing of their assembly (Li et al., 2014; Shieh et al., 2015). In eukaryotes, the lack of the operon structure and their characteristic subcellular compartmentalization, make titrating subunit stoichiometry and coordinating assembly more challenging. In some cases, nascent subunits are assembled co-translationally (Duncan and Mata, 2011; Shiber et al., 2018; Bertolini et al., 2021). However, protein complexes that must be assembled in a particular cellular compartment, or those that cannot be assembled as they are translated, instead rely on subunit-specific chaperones for assembly (Le Tallec et al., 2007; Jäkel et al., 2002; Kihm et al., 2002). Given the enormous diversity of protein subunits, the suite of factors that regulate protein complex assembly is incompletely defined.
One type of multiprotein complex that faces additional challenges for assembly, are those that contain both soluble and membrane-spanning subunits; these complexes require the temporal and spatial coordination of protein synthesis and membrane insertion between the cytosol and endoplasmic reticulum (ER). Depending on the architecture of the complex, the presence of both soluble and membrane-spanning subunits may preclude co-translational assembly, necessitating specific assembly factors. Physiologically important examples of membrane protein complexes that contain both soluble and membrane-spanning subunits include the calcium and potassium voltage-gated ion channels, as well as the ubiquitously expressed ER membrane protein complex (EMC) (Jonikas et al., 2009; Christianson et al., 2011).
The EMC is an evolutionarily conserved ER-resident complex which plays a critical role in the biogenesis of a diverse set of membrane proteins (Chitwood and Hegde, 2019; Volkmar and Christianson, 2020; Wideman, 2015). The EMC exerts its role in membrane protein biogenesis through its three distinct functions. First, the EMC co-translationally inserts multi-pass membrane proteins that contain Nexo transmembrane domains (TMDs) (i.e. in which their N-terminus must be translocated to the exoplasmic side of the membrane). As a result, the EMC is responsible for inserting the first TMD of many G-protein coupled receptors (GPCRs) (Chitwood et al., 2018), a physiologically essential family of cell-surface receptors (Hauser et al., 2018). Second, the EMC is necessary and sufficient for the post-translational insertion of tail-anchored proteins with TMDs of moderate hydrophobicity. Tail-anchored EMC substrates include squalene synthase and sterol-O-acyltransferase 1, both essential enzymes that regulate cholesterol homeostasis (Guna et al., 2018; Volkmar et al., 2019). Finally, proteomic experiments suggest that the EMC also plays a role as an intramembrane chaperone, required for the stabilization of many cellular and viral multi-pass membrane proteins (Shurtleff et al., 2018; Tian et al., 2019; Barrows et al., 2019; Lin et al., 2019; Ngo et al., 2019).
The importance of the EMC’s clients and its central role in membrane protein biogenesis underscore the physiologic relevance of understanding how this macromolecular complex is assembled. Its ubiquitous expression and high abundance further increase the potential consequences of aberrant EMC assembly or the accumulation of orphan EMC subunits (Kulak et al., 2014). In mammals, the EMC is composed of seven integral membrane subunits (EMC1, 3-7, and 10) and two soluble cytosolic subunits (EMC2 and the closely related paralogues EMC8/9) (Figures 1A and S1A). Upon assembly, the intact EMC is characterized by a tripartite architecture that includes a large lumenal domain, an intramembrane hydrophilic vestibule critical for insertion, and a cytosolic domain that anchors and stabilizes the entire complex (Pleiner et al., 2020; Bai et al., 2020; Li et al., 2014; Miller-Vedam et al., 2020; O’Donnell et al., 2020). The cytosolic domain is organized around the soluble EMC2-8/9 heterodimer. The incorporation of the paralogues EMC8 and 9 into the EMC is mutually exclusive as they occupy an overlapping binding site on EMC2. Given that EMC8 is the predominant of the two factors in most cell types, we will refer to EMC8 throughout (Uhlén et al., 2015).
Figure 1. Unassembled EMC2 binds WNK1.
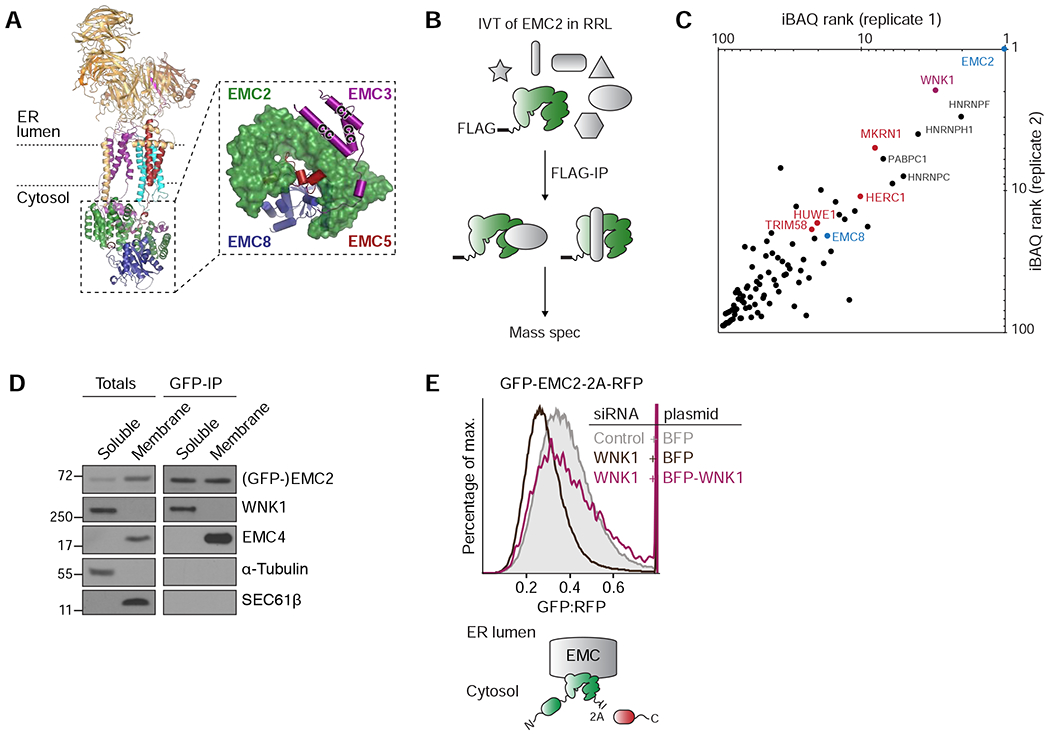
(A) Overview of the cytosolic domain of the human EMC, assembled around EMC2 (PDB 6WW7; (Pleiner et al., 2020). Inset: close-up on the cytosolic domain as viewed from the membrane. CC = coiled coil; CT = C-terminus. (B) Strategy for identification of binding partners of unassembled EMC2. 3xFLAG-tagged EMC2 was in vitro translated (IVT) in rabbit reticulocyte lysate (RRL) and isolated by native immunoprecipitation (IP) with anti-FLAG affinity resin and analyzed by mass spectrometry. (C) EMC2-specific interaction partners, identified by mass spectrometry as outlined in B, with an LFQ-ratio > 5 over controls were ranked by their iBAQ values (see Table S2). The resulting top hits from two replicates are plotted. (D) Cells stably expressing GFP-EMC2 were fractionated into cytosolic and membrane-associated fractions. GFP-tagged EMC2 was purified from both fractions under native conditions using an anti-GFP nanobody, and its interaction partners were analyzed by Western blotting with the indicated antibodies. (E) HEK293T cells stably expressing GFP-EMC2 were transiently transfected with plasmids encoding either BFP or BFP-tagged siRNA-resistant WNK1. Cells were treated with either a scrambled control or WNK1 siRNA, and BFP-positive cells were analyzed by flow cytometry. The relative levels of GFP-EMC2, normalized to an internal RFP expression control (described in methods), are plotted as a histogram and reflect changes in subunit stability.
For function of the EMC the cell thus must ensure that the nine EMC subunits are assembled at the correct stoichiometry, while preventing inappropriate interactions of isolated subunits or intermediates during this process. Previous work suggests that the assembly of the EMC is highly regulated. Depletion of individual ‘core’ EMC subunits (EMC1, 2, 3, 5, 6) leads to the post-translational degradation of the entire complex via the ubiquitin-proteasome pathway (Guna et al., 2018; Volkmar et al., 2019). The structure of the human EMC rationalizes this observed subunit interdependency and why EMC stability is particularly dependent on the soluble subunit EMC2. EMC2 is a superhelical architectural scaffold, responsible for anchoring both the cytosolic and membrane spanning subunits of the complex (Figure 1A). EMC2 forms an extensive, predominantly hydrophobic interface with EMC8 and makes interactions with the cytosolic extensions of the membrane-spanning subunits EMC3 and 5.
In its unassembled state, EMC2 thus has multiple exposed hydrophobic interfaces that ultimately interact with other EMC subunits. Such interfaces are particularly prone to non-productive interactions that could lead to aggregation if left unshielded during assembly. Due to its strong subunit interdependency, the presence of several hydrophobic subunit interfaces, and the additional challenge of coordinating assembly between the cytosol and ER membrane, we set-out to investigate the regulation of EMC2 assembly into the EMC.
RESULTS
Unassembled EMC2 binds WNK1
We hypothesized that EMC2 could require a novel assembly factor because orphan EMC2 is robustly targeted for degradation in cells (Figure S1B), leading to little to no detectable unassembled EMC2 in the cytosol. Further, when expressed in the cell-free rabbit reticulocyte lysate (RRL) system, EMC2 is soluble, displays no evidence of aggregation (Figure S1C), and does not recruit nonspecific chaperones such as HSP70 or known quality control factors such as UBE2O and BAG6 (Figure S1D) (Hessa et al., 2011; Yanagitani et al., 2017).
To identify this putative assembly factor, we leveraged the in vitro RRL system, which preserves all aspects of cellular biosynthesis and quality control, but is devoid of proteasome activity. We reasoned that when expressed in the absence of membranes, orphan EMC2 would remain stably bound to cytosolic assembly factors or quality control machinery, facilitating their identification by mass spectrometry (Figure 1B). By analyzing the factors that co-purified with EMC2 under native conditions we identified its known interaction partner EMC8, and several E3 ubiquitin ligases, consistent with its rapid turnover in cells when unassembled (Figures 1C and S1B). Unexpectedly however, the kinase With no Lysine 1 (WNK1) (Xu et al., 2000) was reproducibly the most abundant EMC2-specific binding partner, and its interaction with nascent EMC2 was validated by immunoblotting (Figure S1D). Given the relatively low expression level of EMC2 in RRL (~30 nM), we concluded that WNK1 is a selective and relatively stable interaction partner of EMC2. Using cellular fractionation, we found that WNK1 specifically interacts with cytosolic, but not membrane-bound EMC2 in human cells (Figure 1D), potentially consistent with a role in EMC assembly.
WNK1 is a ubiquitously expressed regulator of cellular ion homeostasis and cell volume (Alessi et al., 2014; Hadchouel et al., 2016; Shekarabi et al., 2017). Mutations in the WNK1 gene cause dysregulation of renal ion transport leading to hypertension (Wilson et al., 2001), as well as defects in sensory and autonomic neurons that result in peripheral neuropathy (Shekarabi et al., 2008). During osmotic stress, WNK1 initiates a kinase cascade to activate ion channels such as the Na-K-Cl cotransporter (NKCC1) to re-establish cellular ion levels. WNK1 is universally expressed in all cell types, but also has three additional paralogues that are differentially expressed across tissues and contribute to WNK kinase signalling in a potentially redundant manner (Veríssimo and Jordan, 2001; Uhlén et al., 2015). Although WNK1 has never previously been implicated in protein assembly, its pleiotropic phenotypes, particularly as related to a diversity of membrane proteins, could be consistent with a general role in membrane protein biogenesis. In fact, many previously described EMC substrates regulate ion homeostasis and cell volume (Table S1) (Jentsch, 2016; Shurtleff et al., 2018; Tian et al., 2019). Interestingly, we found that one of WNK1’s primary downstream targets, NKCC1, also relies on EMC for biogenesis (Figures S1E–F).
WNK1 is an EMC assembly factor
We reasoned that for WNK1 to be a bona fide EMC2 assembly factor it must: (i) transiently stabilize unassembled EMC2; (ii) be required for the optimal assembly and function of the entire EMC; and (iii) directly interact with unassembled, but not assembled EMC2. We thus employed several orthogonal biochemical strategies to interrogate each of these three requirements.
First, we used a quantitative flow cytometry assay to measure the stability of GFP-EMC2 in cells in comparison to an internal RFP expression control (separated by a viral 2A sequence). We found that three independent small interfering RNAs (siRNAs) targeting WNK1 destabilized GFP-EMC2 (Figure S3A). This effect was rescued by exogenous expression of an siRNA-resistant version of WNK1, excluding the possibility of off-target effects (Figure 1E). Because both the GFP-tagged EMC2 and the RFP control are expressed from a single mRNA open reading frame, changes in the GFP-EMC2:RFP fluorescence ratio necessarily reflect post-translational changes in EMC2 stability. Furthermore, the observed destabilization of EMC2 upon depletion of WNK1 could be rescued by a proteasome inhibitor, consistent with degradation of unassembled EMC2 via the ubiquitin-proteasome pathway (Figure S3B).
To ensure the effects of WNK1 on EMC2 stability were not an artefact of overexpression, we generated a cell line in which a C-terminal GFP tag was introduced into the genomic locus of EMC2. Using this knock-in cell line, we used flow cytometry to verify that WNK1 depletion also destabilized EMC2 when it was expressed at endogenous levels from its native promoter (Figures 2A and S2A–B). Further, pulse-labeling of EMC2-GFP in the knock-in cell line showed that WNK1 knockdown markedly decreased the stability of nascent unassembled EMC2 in the cytosol (Figure S2C). Thus, we concluded that WNK1 specifically stabilizes both endogenous and exogenously expressed unassembled EMC2 and meets the first criterion for an assembly factor.
Figure 2. WNK1 is an EMC assembly factor.
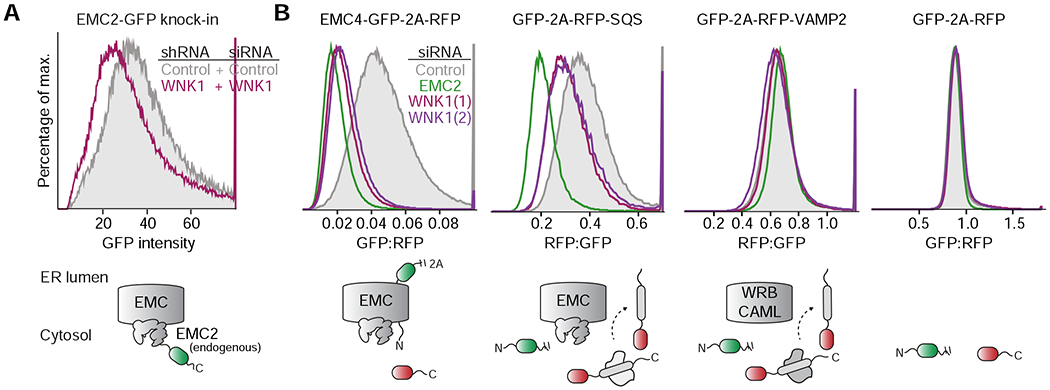
(A) HeLa EMC2-GFP CRISPR knock-in cells, in which EMC2 is expressed from its endogenous promoter, were transduced to stably express either a scrambled control or WNK1 shRNA and then analyzed for GFP fluorescence by flow cytometry. (B) HEK293T cell lines stably expressing EMC4-GFP, RFP-squalene synthase (SQS; a tail-anchored EMC substrate) and RFP-VAMP2 (a tail-anchored WRB-CAML substrate) or an unfused control (GFP-2A-RFP) were treated with the indicated siRNAs and analyzed by flow cytometry.
Second, given that EMC2 is required for the stability of the remaining eight EMC subunits (Volkmar et al., 2019), defects in EMC2 assembly would be expected to at least partially destabilize the entire EMC. Therefore, we tested whether WNK1 affects the assembly of the intact EMC, including its membrane-spanning subunits. To do this, we generated cell lines ectopically expressing EMC4- and EMC5-GFP, while EMC2 remains expressed at endogenous levels from its native promoter. After verifying incorporation of EMC4 and EMC5-GFP into the intact EMC (Figure S3D), we found that WNK1 depletion also destabilized these membrane-spanning EMC subunits, essentially phenocopying EMC2 knockdown (Figures 2B and S3C). Importantly, the observed decrease in EMC levels upon WNK1 knockdown also compromised EMC function. We demonstrated that treatment with several independent WNK1 siRNAs caused biogenesis defects for the post- and co-translational EMC substrates, SQS and OPRK1, but had no effect on the EMC independent substrates, VAMP2 and TRAM2 (Figures 2B and S3C). Critically, in these reporter cell lines, the entire EMC is expressed at endogenous levels, suggesting that the observed effects of WNK1 depletion on EMC function are unlikely to be the result of overexpression.
In line with these observations, Western blotting of WNK1 depleted cells showed specifically reduced steady-state levels of endogenous EMC subunits and substrates, but not other ER-resident or cytosolic proteins (Figure S2D). These results suggest WNK1 depletion has a specific effect on the EMC and its clients, arguing against a general dysregulation of the ER upon WNK1 knockdown. Taken together, the data suggest that WNK1 plays a role in the assembly of the intact EMC, meeting the second criterion for an assembly factor.
Finally, we biochemically interrogated the interaction between WNK1 and EMC2. Binding assays with purified recombinant components confirmed that EMC2 and WNK1 interact directly and form a stable complex in vitro (Figure 3A). Truncation analysis identified a single twenty-six amino acid region of WNK1 (amino acids 2241-2266), which was both necessary and sufficient for binding to EMC2 (Figure 3A). This previously unannotated sequence in the C-terminus of WNK1 is predicted to form a conserved amphipathic helix in metazoans (Figure S4A) and is sufficient to rescue the destabilizing effect of WNK1 knockdown on EMC2 levels in cells (Figure 3B). This amphipathic helix is conserved amongst paralogues of WNK1, and the analogous regions in WNK2 and 3 also bind to EMC2 in vitro (Figures S4A–B). Experiments comparing binding of WNK1-helix to either free EMC2 or purified EMC (normalized to EMC2 levels) confirmed that the WNK1-helix selectively interacts with unassembled EMC2 (Figure 3C), explaining why full length WNK1 only purified with cytosolic, but not membrane-bound EMC2 from cells (Figure 1D). We thus determined that WNK1 meets all the criteria of an EMC2 assembly factor.
Figure 3. A conserved amphipathic α-helix in WNK1 binds to unassembled EMC2.
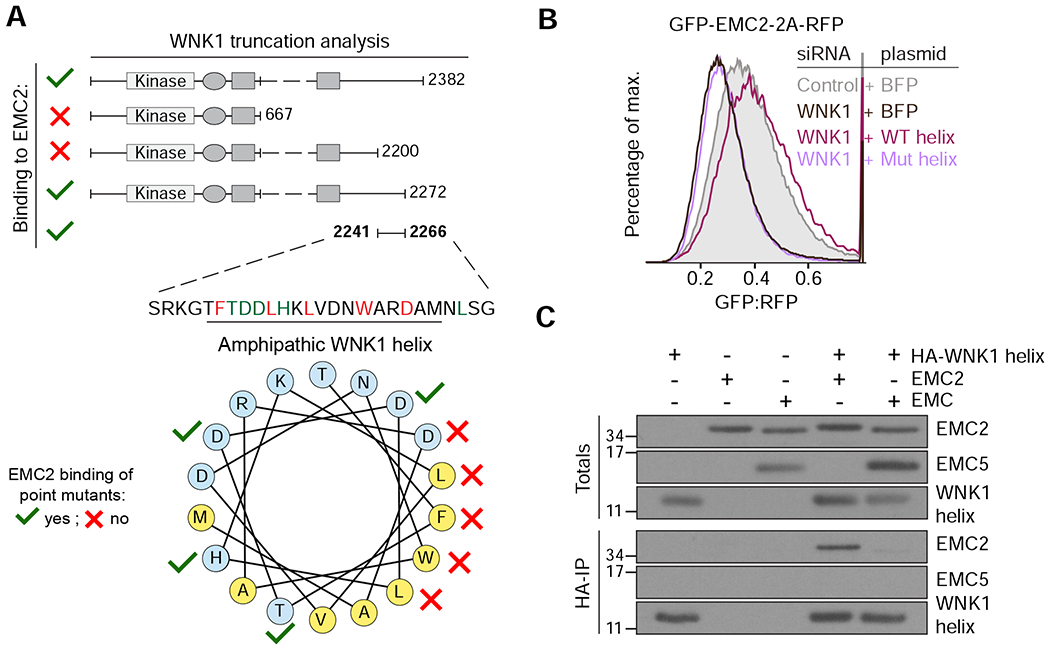
(A) FLAG-tagged WNK1 and its truncations were tested for binding to EMC2 (see methods). The EMC2 binding site was mapped to a previously uncharacterized C-terminal peptide that is predicted to form a conserved amphipathic α-helix (see Figure S4A). Polar residues are shown in blue and hydrophobic residues in yellow. A mutational analysis (Figure S4C) revealed residues required for EMC2 interaction (depicted with red crosses). (B) HEK293T cells stably expressing GFP-EMC2 were transiently transfected with plasmids encoding either BFP or BFP-tagged wild type (WT) or L2250K mutant (Mut) WNK1 helix. Cells were treated with either scrambled or WNK1 siRNA and BFP-positive cells were analyzed by flow cytometry. (C) Intact EMC, purified from stable EMC5-3xFLAG HEK293T cells, and recombinant EMC2 were incubated with 3xHA-tagged WNK1 helix and immunoprecipitated with anti-HA resin. Totals and eluates were analyzed by Western blotting with the indicated antibodies.
The amphipathic WNK1 helix binds the EMC2-8 interface
To understand the molecular function of WNK1 in EMC2 assembly, a necessary first step was to identify the surface of unassembled EMC2 that interacts with WNK1. Based on the structure of EMC2, we had hypothesized that an assembly factor would be required to shield one of its exposed subunit-interfaces to allow assembly. Mutational analysis of the WNK1-helix suggested that binding to EMC2 is mediated via the hydrophobic face of the helix, with the flanking D2260 providing a critical ionic interaction: point mutations to D2260, F2246, L2250, L2253, or W2257, but not T2247, D2248, D2249 or H2251, disrupted binding in vitro (Figures 3A and S4C).
In considering the potential location of WNK1’s binding site, we were struck by the extensive, conserved hydrophobic interface between EMC2 and EMC8 (buried surface area of 1393Å; Pleiner et al., 2020) (Figure 4A). Structure-guided mutagenesis confirmed that the WNK1 recognition site partially overlaps with that of EMC8, as mutations that disrupted WNK1 binding also decreased EMC8 binding (Figure 4B). Using purified recombinant components, we confirmed that the interaction of EMC2 with WNK1-helix is mutually exclusive to the interaction with EMC8 (Figures 4C). WNK1 thus directly shields the interface of EMC2 that is eventually occupied by EMC8.
Figure 4. WNK1 binds the hydrophobic EMC2-EMC8 interface.
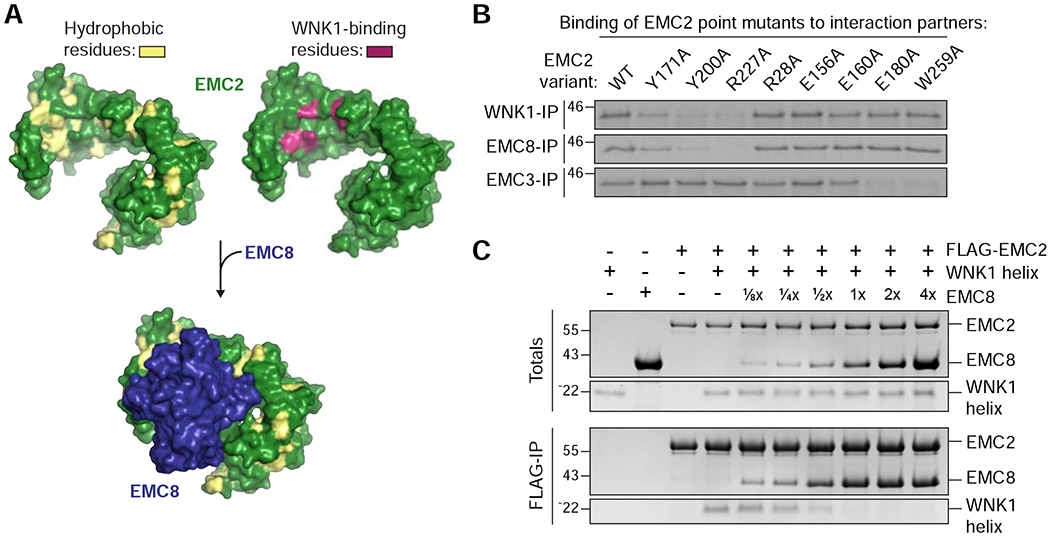
(A) Depicted is the surface representation of EMC2 (PDB 6WW7; Pleiner et al., 2020) in which hydrophobic residues are highlighted in yellow (left) and mutations that affected WNK1 binding are highlighted in purple (right, see B). The WNK1 binding site overlapped with the binding site of EMC8 (blue), which is characterized by an extended hydrophobic patch. EMC2 is viewed from the cytosol. (B) 35S-methionine-labeled wild type (WT) EMC2, or the indicated point mutants were translated in RRL and tested for binding to 3xFLAG-tagged WNK1, EMC8 or EMC3 via co-immunoprecipitation using anti-FLAG resin. EMC2 mutants that are unable to bind WNK1 also do not bind EMC8, but are capable of interacting with EMC3, indicating proper folding. (C) A stoichiometric complex of purified FLAG-tagged EMC2 and WNK1 helix was pre-formed on ice and then incubated with increasing amounts of purified EMC8. The resulting EMC2 complexes were then immunoprecipitated with FLAG resin and analyzed by SDS-PAGE and Coomassie staining.
The role of WNK1 kinase activity in EMC2 assembly
After identifying the binding site between EMC2 and WNK1, we were now in a position to directly interrogate the mechanistic basis of WNK1’s role in EMC2 assembly. We first asked whether the kinase activity of WNK1 was required for EMC assembly. As the expression of a kinase-dead mutant of WNK1 was lethal in cells, we instead used the pan-WNK kinase inhibitor WNK463 to inhibit WNK1 activity (Yamada et al., 2016). We found that under conditions where WNK1-dependent phosphorylation of its substrate kinases SPAK/OSR1 was markedly decreased, we observed no effect on EMC subunit or substrate levels using our established fluorescent reporter assays in cells (Figures S3E–F). Consistent with EMC2 assembly occurring through a kinase-independent mechanism, we found that EMC2 was not appreciably phosphorylated by WNK1 in a direct in vitro kinase assay. We next tested whether EMC2 could instead affect WNK1 activity, but found that EMC2 binding to WNK1 did not affect autophosphorylation of WNK1 or phosphorylation of OSR1 (Figure S3G). These data suggest that the role of WNK1 in EMC assembly does not depend on its kinase activity, consistent with the observation that the amphipathic helix of WNK1 alone is sufficient to compensate for WNK1 loss in cells (Figure 3B).
WNK1 prevents promiscuous interactions of unassembled EMC2
In the absence of a role for its kinase activity in assembly, it was clear that WNK1 exerts its function in assembly primarily through binding of its amphipathic helix to EMC2. Based on the identified interaction surface of WNK1 with EMC2, WNK1 is shielding a hydrophobic subunit interface of EMC2, that would be exposed prior to assembly. We speculated that such increased surface hydrophobicity might make unassembled EMC2 prone to promiscuous and non-specific interactions. Using an in vitro photocrosslinking assay we could indeed capture transient interactions between free EMC2 and hydrophobic proteins containing exposed TMDs (Figures 5A–B and S5A–B). Because EMC2 does not efficiently crosslink with TMDs in the context of the intact complex (Pleiner et al., 2020), it is likely that interactions with a TMD represent aberrant and/or promiscuous interactions of unassembled EMC2.
Figure 5. WNK1 phenocopies EMC assembly to prevent promiscuous interactions of EMC2.
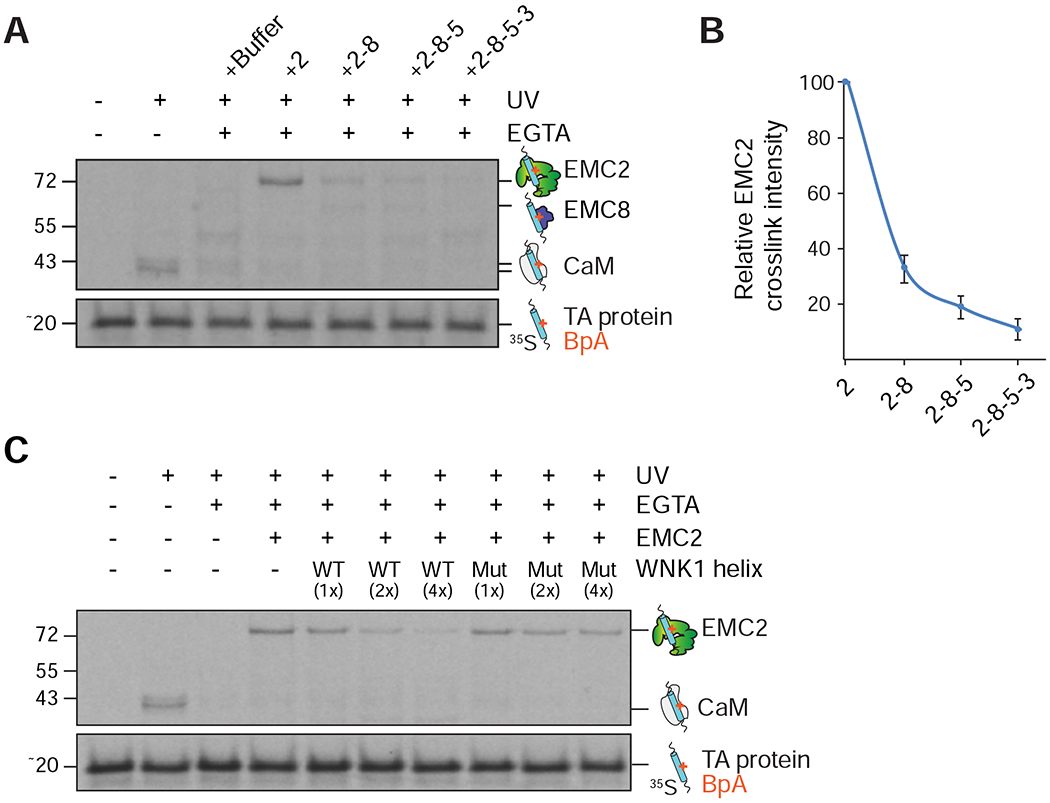
(A) A complex of calmodulin (CaM) and 35S-methionine-labeled SEC61β (a tail-anchored (TA) protein that contains an exposed transmembrane domain (TMD)), containing the photocrosslinker BpA in the TMD, was generated in the PURE in vitro translation system. The CaM-SEC61β complexes were incubated with either purified EMC2 alone or complexes of EMC2 with the indicated binding partners. After UV irradiation the samples were analyzed by SDS-PAGE and autoradiography. (B) Quantification of the EMC2-SEC61β crosslink intensity from (A) (n=3). (C) Using the same assay as in (A), the CaM-SEC61β complexes were incubated with EMC2 alone or pre-formed complexes of EMC2 with increasing concentrations of purified wild type (WT) or L2250K mutant (Mut) WNK1 helix.
We found that addition of wild type WNK1-helix, but not a matched L2250K mutant that disrupts binding to EMC2, prevented non-specific interactions of EMC2 with these hydrophobic TMDs (Figure 5C). Stepwise addition of EMC2’s cytosolic interaction partners revealed that EMC8 binding strongly reduced crosslinking of TMDs to unassembled EMC2 (Figures 5A–B and S5A–B). Addition of the EMC5 C-terminus, and to a lesser extent the cytosolic loops of EMC3, further reduced crosslinking intensity. We thus concluded that one of WNK1’s functions is to serve as a placeholder for EMC8, transiently shielding the hydrophobic EMC2-8 interface to prevent non-specific off-pathway interactions during assembly.
WNK1 competes with E3 ubiquitin ligase binding to EMC2
Another critical function of an assembly factor is to extend the time window available to nascent subunits for assembly, by preventing premature engagement and degradation by cellular quality control machinery. We therefore asked, if WNK1 binding to unassembled EMC2 affects its turnover by the ubiquitin-proteasome pathway. We found that expression of wild type WNK1-helix, but not a mutant that disrupts binding to EMC2, nearly linearly stabilized GFP-EMC2 in cells (Figure 6A). We directly demonstrated that this stabilization was due to a marked decrease in ubiquitination of EMC2 in the presence of exogenous WNK1-helix (Figure 6B). Finally, using a pulse-chase assay, we demonstrated that depletion of endogenous WNK1 decreased the half-life of nascent EMC2 in cells (Figures 6C and S2C). Together these data all suggest that WNK1 binding protects unassembled EMC2 from premature degradation by the ubiquitin-proteasome pathway.
Figure 6. WNK1 prevents EMC2 ubiquitination and directly competes for E3 ligase binding.
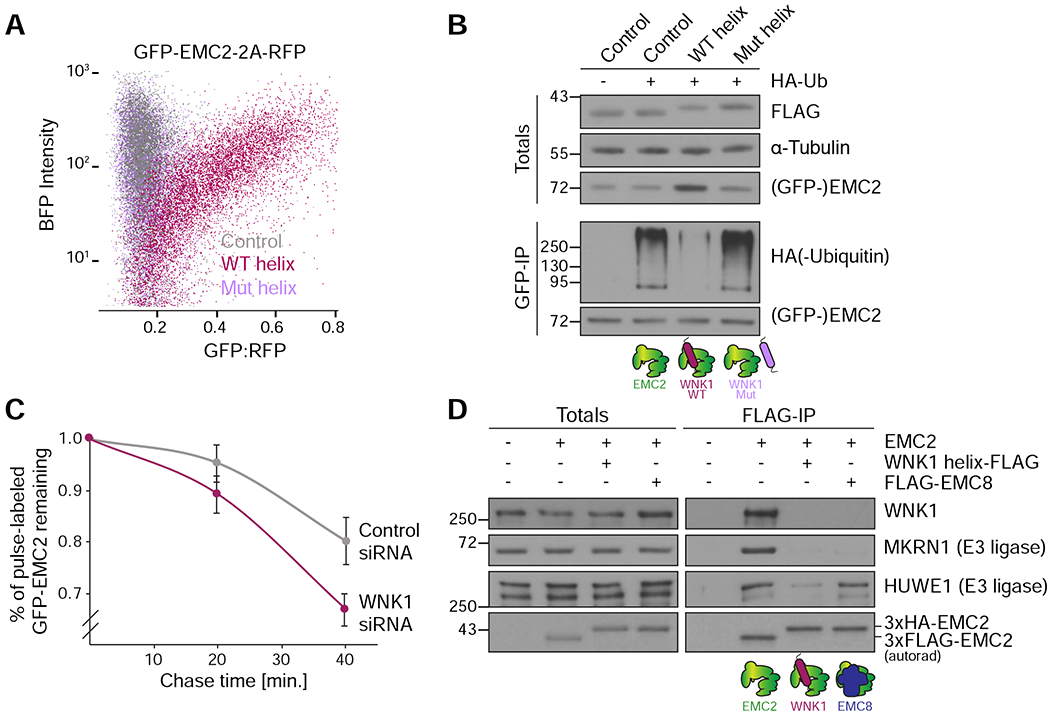
(A) HEK293T cells stably expressing GFP-EMC2 were transfected with plasmids encoding BFP (control), BFP-tagged WNK1 wild type (WT) or L2250K mutant (Mut) helix. Cells were analyzed by flow cytometry and their relative GFP intensity, normalized to an internal RFP expression control, was plotted against BFP intensity.(B) Stable HEK293T GFP-EMC2 cells were transfected with plasmids encoding BFP (control), BFP-tagged WNK1 WT or Mut helix −/+ HA-tagged ubiquitin. Cells were additionally treated with 10 nM bortezomib for 16 hours before harvest. Unassembled GFP-EMC2 was purified from cell lysate prepared in the presence of 50 μM deubiquitinase inhibitor PR-619 using an anti-GFP nanobody (as in Figure 1D), and eluates were normalized by GFP fluorescence. Samples of total lysates and eluates were analyzed by SDS-PAGE and Western blotting with the indicated antibodies. (C) Stable HEK293T GFP-EMC2 cells were treated with either scrambled or WNK1 siRNA, and newly synthesized proteins were pulse-labeled with 35S-methionine. Following a chase with excess unlabeled methionine, the amount of remaining labeled GFP-EMC2 was assessed after 0, 20 and 40 minutes by immunoprecipitation with an anti-GFP nanobody. Eluates were normalized by GFP fluorescence and then analyzed by SDS-PAGE and autoradiography (n=3). (D) 35S-methionine-labeled EMC2 was translated in RRL and purified either directly via a 3xFLAG tag or when bound to 3xFLAG-WNK1 helix or 3xFLAG-EMC8 (in those cases 3xHA-tagged EMC2 was utilized). Samples of totals and eluates were analyzed by SDS-PAGE and Western blotting with the indicated antibodies. EMC2 was detected by autoradiography (autorad). Binding of the co-purifying E3 ligases MKRN1 and HUWE1, identified by mass spectrometry (Figure 1C, Table S2), to EMC2 is incompatible with binding to WNK1.
We hypothesized that one mechanism by which WNK1 binding could prevent ubiquitination of EMC2 was by directly competing for recognition by E3 ubiquitin ligases. To test this, we translated EMC2 in RRL either alone or in the presence of wild type WNK1-helix or EMC8. Following immunoprecipitation under native conditions, we analyzed the resulting eluates for the presence of co-purifying E3 ubiquitin ligases that had been previously identified to bind unassembled EMC2 by mass spectrometry (Figure 6D, see also Figure 1C and Table S2). We found that WNK1 competed for binding of multiple E3 ubiquitin ligases to unassembled EMC2. Thus, we concluded that in addition to its role in preventing promiscuous interactions of EMC2, WNK1 also prevents premature degradation of unassembled EMC2 to allow for assembly.
Reconstitution of the assembly pathway for the EMC cytosolic domain
Finally, in order to position WNK1 in the EMC assembly pathway, we reconstituted the assembly of the tetrameric EMC cytosolic domain (EMC2, 8, 3, and 5). We first demonstrated that EMC8 can displace WNK1 from a preformed complex with EMC2 (Figures 4C and S5C), consistent with WNK1 binding preceding that of EMC8.
In vitro translation of EMC2 and 3 in the presence of canine rough microsomes established that EMC3 is sufficient to recruit EMC2 to the membrane (Figure S5D). Experiments using purified components revealed that binding to EMC2 requires both the coiled coil and C-terminus of EMC3 (Figure 7A), and is compatible with formation of a trimeric complex with either WNK1 or EMC8 (Figure S5C). In contrast, EMC2 could only bind EMC5 in the presence of EMC8, but not WNK1, suggesting that EMC2 and EMC8 form a composite interface for EMC5 (Figures 7B and S5E–F). EMC8 binding to EMC2 must thus precede interaction with EMC5. In yeast, which lack an EMC8 homologue, EMC2 binds EMC5 independently (Figures S6A–B).
Figure 7. Model for assembly of the cytosolic domain of the EMC.
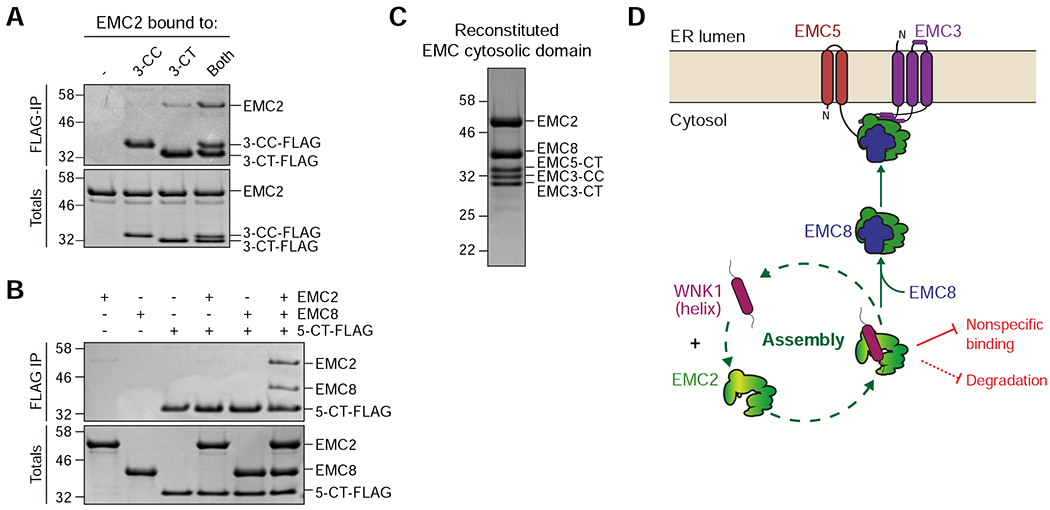
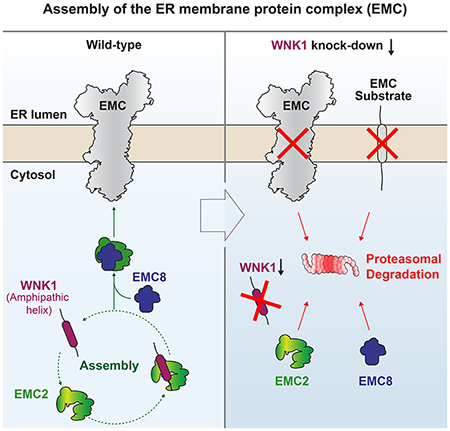
(A) Both the EMC3 coiled coil (3-CC) and C-terminus (3-CT) are needed to interact with EMC2. Purified 3xFLAG-tagged 3-CC and 3-CT were incubated with EMC2 either alone or in combination and then subjected to FLAG-IP. Eluates were analyzed by SDS-PAGE and Coomassie staining. (B) The EMC5 C-terminus (5-CT) selectively binds the EMC2-8 complex. 3xFLAG-tagged 5-CT was incubated either with buffer, EMC2, EMC8, or both and subjected to FLAG-IP. Totals and eluates were analyzed by SDS-PAGE and Coomassie staining. (C) Reconstitution of the EMC cytosolic domain from purified components: EMC2, EMC8, EMC3-coiled coil (CC), EMC3-C-terminus (CT) and EMC5-CT. The complex was analyzed by size-exclusion chromatography (SEC), followed by SDS-PAGE and Coomassie staining of the peak fraction. (D) WNK1 stabilizes unassembled EMC2 by using a conserved C-terminal amphipathic α-helix to prevent nonspecific binding and premature degradation in the cytosol. WNK1 exerts these protective functions by shielding a hydrophobic patch on EMC2 at the EMC2-8 interface and by directly competing with E3 ligase binding to EMC2, strongly reducing its ubiquitination. EMC8 then displaces WNK1 in the cytosol, allowing assembly of the EMC2-8 complex with the membrane-bound subunits EMC3 and EMC5.
Mutations to EMC2 at the EMC2-5 and EMC2-3 interface decreased incorporation of EMC2 into the complex both in vitro and in cells, suggesting that membrane tethering by both EMC5 and EMC3 is critical for assembly (Figures S6C–E). Using this information, we reconstituted the cytosolic domain of the EMC, including EMC2 and 8 and the loops of EMC3 and 5, with purified recombinant components and verified it forms a stable tetrameric complex, that does not bind WNK1 (Figure 7C).
DISCUSSION
We propose the following working model for the role of WNK1 in EMC assembly (Figure 7D). EMC2 serves as a cytosolic scaffold essential for stabilizing the entire EMC via interactions with four of the remaining eight EMC subunits (EMC3-5,8). As a result, EMC2 contains several exposed subunit interfaces, which are ultimately shielded by its binding partners upon assembly. In particular, the interface between EMC2 and 8 is highly hydrophobic, which may be critical for recruiting and anchoring EMC8, which forms no independent interactions with other EMC subunits. Because we found the hydrophobic EMC2-8 interface is particularly prone to promiscuous interactions, its exposure to the crowded cytosolic environment must be tightly regulated to prevent toxic aggregation or off-pathway interactions. For this reason, orphan EMC2 has a very short half-life and is rapidly degraded by the ubiquitin-proteasome pathway.
Two general strategies can be employed to prevent exposure of similar hydrophobic interfaces during assembly: (i) co-translational assembly of nascent subunits on the ribosome or (ii) reliance on specific chaperones to coordinate post-translational assembly. In the case of EMC2, the interaction between EMC2 and 8 requires the very C-terminal amino acids of EMC2. Assembly of the EMC2-8 heterodimer thus cannot occur until EMC2 is fully translated and has emerged from the ribosome, potentially precluding co-translational assembly. Instead, we propose that nascent EMC2 is first captured by WNK1.
WNK1 binds selectively to unassembled EMC2 by recognizing the exposed EMC2-8 interface. WNK1 shields this interface by using the hydrophobic face of a conserved amphipathic helix in its C-terminus. Because the opposite face of the WNK1 helix is highly polar, the resulting EMC2-WNK1 complex would display a strongly reduced surface hydrophobicity. We found that WNK1’s function in EMC assembly did not rely on its well-studied kinase activity, consistent with the fact that EMC2 binds WNK1 nearly two thousand amino acids downstream of its kinase domain, separated by what is predicted to be a largely unstructured linker.
Binding of WNK1 to nascent EMC2 has two primary molecular functions: first, it prevents nonspecific interactions of unassembled EMC2 by shielding the sticky surface on the EMC2-8 interface; and second, WNK1 stabilizes unassembled EMC2 by preventing binding of E3 ubiquitin ligases. WNK1 therefore functions to increase the efficiency of EMC assembly by providing sufficient time for EMC2 to assemble while simultaneously safeguarding the proteome by preventing potentially toxic off-pathway interactions. Lower eukaryotes, which do not express a homologue of EMC8, also do not express a homologue of any WNK family member. However, given that in yeast, the N-terminus of EMC4 occupies the analogous WNK1 binding site on EMC2 upon assembly, it is possible that the requirement for an EMC2 assembly factor is not metazoan-specific (Figure S6F).
The affinity between EMC2 and WNK1 must be precisely tuned to favor biosynthesis, while ensuring that the opportunity for assembly is time-limited such that terminally unassembled EMC2 is robustly triaged towards degradation. In comparison to the EMC2-8 interaction, the EMC2-WNK1 interaction is notably weaker and likely to bury less surface area (Pleiner et al., 2020). Binding of EMC8 can therefore efficiently displace WNK1 to assemble with EMC2.
We favor a model whereby the exposed hydrophobic EMC2-8 interface serves as a degron for recruitment of E3 ubiquitin ligases. This would be consistent with the observation that WNK1 binding reduces ubiquitination of EMC2 in cells and actively competes with E3 ligases that bind to unassembled EMC2 in vitro. We cannot formally exclude that WNK1 binding allosterically inhibits binding of E3 ligases to a distant site on EMC2. However, the dual use of an exposed inter-subunit interface for assembly and quality control is a conserved mechanism for regulating protein complex assembly. Alpha-hemoglobin-stabilizing protein (AHSP) stabilizes nascent α-hemoglobin by binding to the α-β interface (Kihm et al., 2002), while the quality control factor UBE2O targets orphan α-hemoglobin for degradation by binding to the same site (Nguyen et al., 2017; Yanagitani et al., 2017). Similarly, N-terminal acetylation (Ac/N-) degrons, that when exposed target unassembled subunits for degradation by the Ac/N-end rule pathway, are shielded upon assembly (Hwang et al., 2010; Shemorry et al., 2013).
Finally, after displacement of WNK1, the heterodimeric EMC2-8 complex can be recruited to the membrane. As EMC8 cannot independently bind to any of the membrane-spanning EMC subunits, we postulate that EMC8 is recruited to EMC2 in the cytosol. The EMC2-8 heterodimeric complex can then be targeted to the membrane via interaction with the cytosolic domains of the membrane-spanning subunits, EMC3 and EMC5. Because the EMC2-8 heterodimer interacts with the very C-terminal residues of EMC3 and 5, assembly of EMC2-8 at the membrane must occur post-translationally after EMC3 and 5 are released from the ribosome and inserted into the bilayer. Consistent with the structural importance of EMC2, depletion of WNK1 destabilized both EMC2 and the membrane-bound EMC subunits, resulting in biogenesis defects of tail-anchored and multi-pass EMC substrates. EMC-independent membrane proteins were not affected by knockdown of WNK1, excluding pleiotropic effects on protein biogenesis at the ER. To exclude potential artefacts of overexpression, we also verified that depletion of WNK1 results in specific destabilization of endogenous unassembled EMC2, the membrane-spanning EMC subunits, and its substrates. Taken together, our data thus demonstrate that WNK1 functions as an EMC assembly factor, which coordinates EMC2 assembly with EMC8, ensuring incorporation of the soluble subunits into the intact complex.
Given the identification of WNK1’s role in EMC2 binding, an important question is the potential contribution of the three additional WNK paralogues to EMC assembly. We found that both WNK2 and WNK3 contain amphipathic helices that can similarly bind to EMC2 and could thus be partially redundant to WNK1 in mediating EMC assembly. Depletion of WNK1 has been shown to up-regulate WNK3, but not WNK2 or WNK4 expression (Roy et al., 2015), which could potentially partially complement a loss of WNK1 function. However, we suspect that WNK1 is the primary assembly factor for EMC2 because (i) WNK1 is the only paralogue that, like the EMC, is ubiquitously expressed across all cell types; and (ii) an analysis of the expression levels of WNK paralogues shows that WNK1 is the most abundantly expressed member of the WNK family (Veríssimo and Jordan, 2001; Uhlén et al., 2015). Therefore, perturbation of WNK1 levels is expected to have the most significant impact on EMC assembly in human cells.
The identification of WNK1 as an EMC assembly factor has several important implications. First, many of the pleiotropic phenotypes attributed to WNK1 knockdown or knockout must be carefully evaluated. In particular, the reported role of WNK1 in surface expression of many membrane proteins could be at least partially explained by the indirect effect of WNK1 depletion on the biogenesis of any EMC-dependent substrate. Additionally, given WNK1’s central role in regulation of ion homeostasis, its role in EMC assembly begs the question whether there is a potential connection between osmotic stress and protein biogenesis in the ER. For example, the EMC is required for biogenesis of many solute carriers that are regulated by WNK1 activation. Further, mutational analysis suggested that the amphipathic helix identified to bind EMC2 is also required for recruitment of WNK1 to clathrin-coated vesicles during osmotic stress (Figure S7) (Zagórska et al., 2007). Thus, a potential relationship between EMC assembly and ion homeostasis represents an important area for future study.
Finally, it is clear that the role of WNK1 in EMC2 assembly exemplifies many of the general principles of chaperones in multi-subunit complex assembly. Other scaffold subunits like EMC2 that contain exposed hydrophobic interfaces may be uniquely vulnerable to off-pathway interactions and premature degradation and thus require engagement by assembly factors. We also envision that other protein complexes with subunits that are synthesized and assembled at distinct locations might require novel assembly factors. For example, a major class of membrane protein complexes with soluble subunits are the voltage-gated calcium and potassium ion channels (Buraei and Yang, 2010; Pongs and Schwarz, 2010). Indeed, previous data suggests that like EMC2, some of these soluble subunits are also degraded in the absence of their membrane-embedded binding partners (Menegola and Trimmer, 2006). Further, given the complexity of the membrane-spanning subunits of the EMC, and increasing evidence that membrane protein complex assembly is highly regulated (Inglis et al., 2020; Pleiner et al., 2020), it is likely that the EMC may also rely on dedicated intramembrane chaperones for assembly.
Thus, given the enormous diversity of subunits whose assembly must be tightly regulated by the cell, the known set of assembly factors is undoubtedly incomplete. The strategies reported here lay out a roadmap for their identification and validation. Characterization of these assembly factors will reveal how the cell ensures the stoichiometric assembly of intricate molecular machines in the crowded cellular milieu, and provide insight into how these processes may fail leading to disease (Harper and Bennett, 2016; Oromendia and Amon, 2014).
Limitations of the Study
Although we demonstrate that one of WNK1’s roles in assembly is to prevent the premature degradation of EMC2 by the ubiquitin proteasome pathway, one critical area for further research is the identification of the specific E3 ubiquitin ligase(s) that recognizes unassembled EMC2. Potential candidates include the E3 ligases that we found to co-purify with unassembled EMC2 from cell-free extracts including MKRN1, HUWE1, and HERC1 (Figure 1C). Identification of the responsible E3 ligase(s) will require a combination of biochemical reconstitution and genetic perturbation to address potential redundancy and the propensity of large ligases such as HUWE1 and HERC1 to non-specifically interact with affinity resins. Identification of the E3 ubiquitin ligase will provide crucial insight into how unassembled EMC2 is recognized, and more broadly reveal how protein complex assembly and quality control are coordinated.
Finally, while knockdown of WNK1 reduced the efficiency of EMC assembly, some nascent EMC2 is still incorporated into the EMC under these conditions. This may be due to potential functional redundancy between WNK1 and its paralogues, or incomplete knockdown. Since unassembled EMC2 is a very short-lived and low-abundance species, relatively small amounts of any WNK paralogue might be sufficient to partially protect it from degradation. It may also be possible for some EMC2 to recruit and bind EMC8 even in the absence of WNK1. However, it is clear that WNK1 increases the efficiency of EMC2 assembly into the EMC. In cells and especially in living organisms, even a small decrease in EMC assembly efficiency would be potentially detrimental. Therefore, the role of WNK1 in EMC assembly exerts a physiologically important contribution to cellular homeostasis.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rebecca Voorhees (voorhees@caltech.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
The mass spectrometry data set referenced in Figure 1C is provided as Table S2. Figures 1A, 4A, S6A and S6F are based on the published structure of the human EMC (PDB 6WW7) (Pleiner et al., 2020). Figures S6A and S6F additionally make use of the published yeast EMC structure (PDB 6WB9) (Bai et al., 2020).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All adherent cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% fetal calf serum (FCS). In cases where the cells contained a doxycycline-inducible reporter, tetracycline-free FCS was used. 15 μg/ml blasticidin S and 100 μg/ml hygromycin B were included for culturing Flp-In T-Rex 293 and HeLa cell lines stably expressing reporter constructs. Suspension growth-adapted Flp-In T-Rex 293 cell lines were grown in Freestyle 293 Expression Medium. Expi293F cells were grown in Expi293 Expression Medium. All cell lines used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-WNK1 | Bethyl | Cat# A301-515A, RRID:AB_999680 |
| Rabbit polyclonal anti-EMC2 | Proteintech | Cat# 25443-1-AP, RRID:AB_2750836 |
| Mouse monoclonal anti-EMC3 | Proteintech | Cat# 67205-1-Ig, RRID:AB_2882498 |
| Rabbit polyclonal anti-EMC4 | Proteintech | Cat# 27708-1-AP, RRID:AB_2880950 |
| Rabbit polyclonal anti-EMC5 | Bethyl | Cat# A305-833A-M, RRID:AB_2890207 |
| Rabbit polyclonal anti-EMC6 | Abcam | Cat# ab84902, RRID:AB_1925516 |
| Rabbit polyclonal anti-EMC8 | Proteintech | Cat# 19889-1-AP, RRID:AB_2878618 |
| Rabbit polyclonal anti-FDFT1 | Bethyl | Cat# A305-361A, RRID:AB_2631752 |
| Rabbit polyclonal anti-VAMP2 | Cell Signaling Technology | Cat# 13508, RRID:AB_2798240 |
| Rabbit polyclonal anti-Hsp70 | Enzo Life Sciences | Cat# ADI-SPA-812, RRID:AB_10013742 |
| Rabbit polyclonal anti-Ube2O | Bethyl | Cat# A301-873-A-T, RRID:AB_2780177 |
| Mouse monoclonal anti-Bip | BD Biosciences | Cat# 610978, RRID:AB_398291 |
| Rabbit polyclonal anti-AE2 | Bethyl | Cat# A304-502A, RRID:AB_2620696 |
| Rabbit monoclonal anti-NKCC1 | Cell Signaling Technology | Cat# 8351, RRID:AB_10830068 |
| Rabbit polyclonal anti-CLINT1 | Bethyl | Cat# A301-926A, RRID:AB_1524117 |
| Rabbit polyclonal anti-γ-AP1 | Bethyl | Cat# A304-771A, RRID:AB_2620966 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| Rabbit polyclonal anti-Clathrin Heavy Chain | Bethyl | Cat# A304-743A, RRID:AB_2620938 |
| Rabbit monoclonal anti-Rps24 | Abcam | Cat# ab196652, RRID:AB_2714188 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat# T9026, RRID:AB_477593 |
| Rabbit monoclonal anti-β-Actin | Cell Signaling Technology | Cat# 8457, RRID:AB_10950489 |
| Rabbit polyclonal anti-MKRN1 | Bethyl | Cat# A300-990A RRID:AB_2142814 |
| Rabbit polyclonal anti-HUWE1 | Proteintech | Cat# 19430-1-AP, RRID:AB_2878579 |
| Sheep polyclonal anti-SPAK/OSR1 | MRC PPU Reagents and Services | Cat# S637B, RRID:AB_2890208 |
| Sheep polyclonal anti-SPAK/OSR1 pSer373/pSer325 | Sigma-Aldrich | Cat# 07-2273, RRID:AB_11205577 |
| Rabbit polyclonal anti-GFP | Gift from Ramanujan Hegde (Chakrabarti and Hegde, 2009) | N/A |
| Rabbit polyclonal anti-TRAPα | Gift from Ramanujan Hegde | N/A |
| Rabbit polyclonal anti-BAG6 | Gift from Ramanujan Hegde (Mariappan et al., 2010) | N/A |
| Rabbit polyclonal anti-Sec61β | Gift from Ramanujan Hegde | N/A |
| HRP-conjugated mouse monoclonal anti-HA | Sigma-Aldrich | Cat# H6533, RRID:AB_439705 |
| HRP-conjugated mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# A8592, RRID:AB_439702 |
| HRP-conjugated goat anti-rabbit IgG | BioRad | Cat# 170-6515, RRID:AB_11125142 |
| HRP-conjugated goat anti-mouse IgG | BioRad | Cat# 172-1011, RRID:AB_11125936 |
| Donkey polyclonal anti-mouse IgG Alexa Fluor 488 | Thermo Scientific | Cat# A-21202, RRID:AB_141607 |
| Donkey polyclonal anti-rabbit IgG Alexa Fluor 647 | Thermo Scientific | Cat# A-31573, RRID:AB_2536183 |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline | Sigma-Aldrich | Cat# D9891, CAS: 24390-14-5 |
| Digitonin | Millipore | Cat# 300410, CAS: 11024-24-1 |
| LMNG | Anatrace | Cat# NG310, CAS: 1257852-96-2 |
| Complete EDTA-free protease inhibitor cocktail | Roche | Cat# 11873580001 |
| Pierce Streptavidin Magnetic Beads | Thermo Scientific | Cat# 88817 |
| Anti-Flag M2 affinity resin | Sigma-Aldrich | Cat# A2220 |
| 3xFlag peptide | Sigma-Aldrich | Cat# F4799 |
| Anti-HA agarose | Sigma-Aldrich | Cat# A2095 |
| HA peptide | Thermo Scientific | Cat# 26184 |
| Ni-NTA agarose | QIAGEN | Cat# 30210 |
| Glutathione sepharose 4B | GE Healthcare | Cat# 17-0756-05 |
| L-Glutathione, reduced | Sigma-Aldrich | Cat# G4251 |
| Biotin | Sigma-Aldrich | Cat# B4501 |
| PMSF | Thermo Scientific | Cat# 36978 |
| EasyTag L-[35S]-Methionine | Perkin Elmer | Cat# NEG709A005MC |
| ATP, [γ-32P] | Perkin Elmer | Cat# BLU002A100UC |
| PEI MAX Mw 40,000 | Polysciences | Cat# 24765-1, CAS: 49553-93-7 |
| Poly-D-lysine | GIBCO | Cat# A3890401 |
| Amino acid kit | Sigma-Aldrich | Cat# 09416 |
| PhosSTOP Phosphatase inhibitor tablets | Roche | Cat# 4906845001 |
| Puromycin Dihydrochloride | Thermo Scientific | Cat# A1113803 |
| Hygromycin B | Millipore | Cat# 400051-100KU, CAS: 31282-04-9 |
| Blasticidin S | Thermo Scientific | Cat# A1113903, CAS: 3513-03-9 |
| MG132 Proteasomal Inhibitor | Calbiochem | Cat# 474790 |
| Bortezomib | Calbiochem | Cat# 504314, CAS 179324-69-7 |
| PR619 | Sigma-Aldrich | Cat# SML0430 |
| WNK463 | MedChemExpress | Cat# HY-100626, CAS 2012607-27-9 |
| 4-Benzoylphenylalanine (Bpa) | Bachem | Cat# 4017646.0001, CAS 104504-45-2 |
| DAPI | Sigma-Aldrich | Cat# MBD0015 |
| SlowFade Gold | Thermo Scientific | Cat# S36936 |
| Sytox Blue Dead Cell Stain | Thermo Scientific | Cat# 34857 |
| Creatine phosphate | Roche | Cat# 621714 |
| Creatine kinase | Roche | Cat# 127566 |
| His-Ubiquitin | Boston Biochem | Cat# U-530 |
| UbcH5a | Boston Biochem | Cat# E2-616 |
| GST-UBE1 (human) | Boston Biochem | Cat# E-306 |
| S7 Micrococcal Nuclease | Roche | Cat# 10107921001 |
| RNasin | Promega | Cat# N251 |
| SP6 Polymerase | New England Biolabs | Cat# M0207L |
| DNAse I | New England Biolabs | Cat# M0303L |
| 3xFLAG-Calmodulin | Shao et al., 2017 | N/A |
| Bpa-RS | Shao et al., 2017 | N/A |
| Amber suppressor tRNA | Shao et al., 2017 | N/A |
| Deposited data | ||
| CryoEM structure of the human EMC | Pleiner et al., 2020 | PDB 6WW7 |
| CryoEM structure of the yeast EMC | Bai et al., 2020 | PDB 6WB9 |
| Experimental models: cell lines | ||
| Flp-In T-REx 293 | Thermo Scientific | Cat# R78007, RRID: CVCL_U421 |
| HeLa | ATCC | Cat# CCL-2, RRID:CVCL_0030 |
| HeLa EMC2-GFP knock-in | This paper | N/A |
| HeLa Flp-In T-REx | Gift from Christian Schlieker | N/A |
| Expi 293F | Thermo Scientific | Cat# A14527, RRID:CVCL_D615 |
| Flp-In T-REx 293 dox inducible GFP-2A-mCherry | This paper | N/A |
| Flp-In T-REx 293 dox inducible GFP-EMC2-2A-mCherry | Pleiner et al., 2020 | N/A |
| Flp-In T-REx 293 dox inducible EMC4-GFP-2A-mCherry | This paper | N/A |
| Flp-In T-REx 293 dox inducible EMC5-GFP-2A-mCherry | This paper | N/A |
| Flp-In T-REx 293 dox inducible GFP-NKCC1-2A-mCherry | This paper | N/A |
| Flp-In T-REx 293 dox inducible EMC5-3xFLAG | This paper | N/A |
| Flp-In T-REx 293 dox inducible GFP-WNK1 | This paper | N/A |
| Flp-In T-REx 293 dox inducible 3xFLAG-WNK1 | This paper | N/A |
| Flp-In T-REx 293 dox inducible 3xFLAG-WNK1 (L2250K, L2253K, W2257K) | This paper | N/A |
| HeLa Flp-In dox inducible GFP-2A-mCherry-FDFT1(SQS)(378-410)-Opsin | This paper | N/A |
| HeLa Flp-In dox inducible GFP-2A-mCherry-VAMP2(91-114)-Opsin | This paper | N/A |
| HeLa Flp-In dox inducible TRAM2-GFP-2A-mCherry | This paper | N/A |
| HeLa Flp-In dox inducible OPRK1-GFP-2A-mCherry | This paper | N/A |
| E. coli Rosetta™ 2(DE3) | Novagen | Cat# 71400 |
| BL21 Competent E. coli | New England Biolabs | Cat# C2530H |
| NEBExpress® Iq Competent E. coli | New England Biolabs | Cat# C3037I |
| Oligonucleotides | ||
| On-Targetplus siRNA against WNK1: GCAGUUGUCUCAAUAUCUA | Dharmacon | Cat# J-005362-05-0002 |
| On-Targetplus siRNA against WNK1: GCAGGAGUGUCUAGUUAUA | Dharmacon | Cat# J-005362-06-0002 |
| On-Targetplus Non-targeting control siRNA #1: UGGUUUACAUGUCGACUAA | Dharmacon | Cat# D-001810-01-05 |
| Silencer Select siRNA against WNK1: CAUCAUCCCUUAGUCUACAtt | Thermo Scientific | Cat# s35233 |
| Silencer Select siRNA against WNK1: CCAGCGUAGUUUCAAGUAUtt | Thermo Scientific | Cat# s35234 |
| Silencer Select siRNA against WNK1: CAAUGAGUCAGAUAUCGAAtt | Thermo Scientific | Cat# s35235 |
| Silencer Select siRNA against EMC2: CAAUGAACAUGACUAUGCAtt | Thermo Scientific | Cat# s18670 |
| Silencer Select siRNA against EMC5: GGCCUUUGCAGUUACCUGUtt | Thermo Scientific | Cat# s41131 |
| Silencer Select negative control no. 2 siRNA | Thermo Scientific | Cat# 4390846 |
| sgRNA against EMC2 C-terminus for knock-in: TTGCAGATCACCCAGTCTTA | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3.1 HA CFP hsNKCC1 WT – source of NKCC1 coding sequence | Gift from Biff Forbush (Somasekharan et al., 2013) | Addgene ID #49077 |
| pCRISPaint-TagBFP – source of TagBFP coding sequence | Gift from Veit Hornung (Schmid-Burgk et al., 2016) | Addgene ID #67168 |
| pSpCas9(BB)-2A-Puro (pX459) | Gift from Feng Zhang (Ran et al., 2013) | Addgene ID #48139 |
| pTP602_gRNA against human EMC2 C-terminus used for knock-in in pX459 | This paper | N/A |
| pTP600_EMC2-GFP knock-in donor plasmid | This paper | N/A |
| MISSION® pLKO.1-puro Non-Mammalian shRNA Control Plasmid | Sigma-Aldrich | Cat# SHC002 |
| TRC1.5_pLKO-puro Mission shRNA targeting WNK1 | Sigma-Aldrich | Cat# TRCN0000000919 |
| pCMV6-Entry-WNK1-Myc-1xFLAG (human) ORF clone | Origene | Cat# RC218208 |
| pTP061_pCMV6-Entry-WNK1(1-2106)-Myc-1xFLAG (human) | This paper | N/A |
| pTP062_pCMV6-Entry-WNK1(1-2200)-Myc-1xFLAG (human) | This paper | N/A |
| pTP063_pCMV6-Entry-WNK1(1-2272)-Myc-1xFLAG (human) | This paper | N/A |
| pcDNA5/FRT/TO | Thermo Scientific | Cat# V652020 |
| Flp-Recombinase pOG44 | Thermo Scientific | Cat# V600520 |
| pTP045_GFP-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP021_GFP-EMC2-2A-mCherry in pcDNA5/FRT/TO | Pleiner et al., 2020 | N/A |
| pTP244_EMC4-GFP-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP022_EMC5-GFP-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP092_GFP-NKCC1-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP004_EMC5-3xFLAG in pcDNA5/FRT/TO | This paper | N/A |
| pTP428_GFP-WNK1 in pcDNA5/FRT/TO | This paper | N/A |
| pTP047_3xFLAG-WNK1 in pcDNA5/FRT/TO | This paper | N/A |
| pTP115_3xFLAG-WNK1 (L2250K, L2253K, W2257K) in pcDNA5/FRT/TO | This paper | N/A |
| pTP064_GFP-2A-mCherry-FDFT1(SQS)(378-410)-Opsin in pcDNA5/FRT/TO | This paper | N/A |
| pTP040_GFP-2A-mCherry-VAMP2(91-114)-Opsin in pcDNA5/FRT/TO | This paper | N/A |
| pTP131_TRAM2-GFP-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP498_OPRK1-GFP-2A-mCherry in pcDNA5/FRT/TO | This paper | N/A |
| pTP604_3xHA-Ubiquitin in pcDNA5/FRT/TO | This paper | N/A |
| pcDNA3.1 | Thermo Scientific | Cat# V79020 |
| pTP123_3xFLAG-TagBFP-WNK1 in pcDNA3.1 | This paper | N/A |
| pTP118_3xFLAG-TagBFP-WNK1(2241-2266) in pcDNA3.1 | This paper | N/A |
| pTP182_3xFLAG-TagBFP-WNK1(2241-2266) (L2250K, L2253K, W2257K) in pcDNA3.1 | This paper | N/A |
| pRMV409_3xFLAG-TagBFP in pcDNA3.1 | This paper | N/A |
| pHAGE2 lentiviral transfer vector | Gift from Magnus A. Hoffmann and Pamela Bjorkman | N/A |
| pΔ8.9 and pVSV-G packaging plasmids | Gift from Carlos Lois | N/A |
| pTP340_3xFLAG-TagBFP-EMC2 (human) in pHAGE2 | This paper | N/A |
| pTP377_3xFLAG-TagBFP-EMC2 (E156A) (human) in pHAGE2 | This paper | N/A |
| pTP461_3xFLAG-TagBFP-EMC2 (E180A) (human) in pHAGE2 | This paper | N/A |
| pTP355_EMC5-TagBFP-3xFLAG (human) in pHAGE2 | This paper | N/A |
| pTP357_EMC5(F90A)-TagBFP-3xFLAG (human) in pHAGE2 | This paper | N/A |
| pTP360_EMC5(F93A)-TagBFP-3xFLAG (human) in pHAGE2 | This paper | N/A |
| SP64 vector | Promega | Cat# P1241 |
| pRMV188_3xFLAG-hsSec61b(2-13)-ggVillin-1(792-826)-hsSec61beta(13-68) in SP64 | Shao et al., 2013 | N/A |
| RMV225_3xFLAG-hsSec61beta(2-end) in SP64 | This paper | N/A |
| pTP007_3xHA-EMC2 (human) in SP64 | This paper | N/A |
| pTP345_3xHA-EMC2 (Y171A) (human) in SP64 | This paper | N/A |
| pTP369_3xHA-EMC2 (Y200A) (human) in SP64 | This paper | N/A |
| pTP332_3xHA-EMC2 (R227A) (human) in SP64 | This paper | N/A |
| pTP370_3xHA-EMC2 (R28A) (human) in SP64 | This paper | N/A |
| pTP342_3xHA-EMC2 (E156A) (human) in SP64 | This paper | N/A |
| pTP343_3xHA-EMC2 (E160A) (human) in SP64 | This paper | N/A |
| pTP367_3xHA-EMC2 (E180A) (human) in SP64 | This paper | N/A |
| pTP368_3xHA-EMC2 (W259A) (human) in SP64 | This paper | N/A |
| pTP008_3xHA-EMC8 (human) in SP64 | This paper | N/A |
| pTP361_hsEMC5-3xFLAG (human) in SP64 | This paper | N/A |
| pTP363_hsEMC5(F90A)-3xFLAG (human) in SP64 | This paper | N/A |
| pTP366_hsEMC5(F93A)-3xFLAG (human) in SP64 | This paper | N/A |
| gb001_SP6 3xFLAG-EMC2 (human) gblock | This paper | N/A |
| gb033_SP6 EMC3-3xFLAG (human) gblock | This paper | N/A |
| gb076_SP6 3xHA-EMC2 (yeast) gblock | This paper | N/A |
| gb077_SP6 EMC5-3xFLAG (yeast) gblock | This paper | N/A |
| gb078_SP6 EMC3-3xFLAG (yeast) gblock | This paper | N/A |
| T7 PURExpress plasmid | New England Biolabs | Cat# E6800S |
| pRMV210_T7 3xHA-Sec61β(2-end)(F85Amber) human in T7 PURExpress | This paper | N/A |
| pTP112_T7 3xFLAG-Sec61β(2-59)-SQS(378-410)(Y400Amber)-Opsin in T7 PURExpress | This paper | N/A |
| All E. coli expression constructs in pQE80-derivative | Qiagen | N/A |
| pTP105_His14-bdNEDD8-EMC2 (human) | This paper | N/A |
| pTP030_His14-bdNEDD8-3xFLAG-EMC2 (human) | This paper | N/A |
| pTP024_His14-bdNEDD8-EMC8 (human) | This paper | N/A |
| pTP031_His14-bdNEDD8-3xFLAG-EMC8 (human) | This paper | N/A |
| pTP083_His14-bdSUMO-EMC3(35-117)-3xFLAG (human) | This paper | N/A |
| pTP128_His14-bdSUMO-EMC3(196-261)-3xFLAG (human) | This paper | N/A |
| pTP354_His14-bdSUMO-EMC5(66-131)-3xFLAG (human) | This paper | N/A |
| pTP189_His14-bdSUMO-OSR1(2-end)(D164A) (human) | This paper | N/A |
| pTP396_His14-Avi-Spacer-SUMOEu1-anti-GFP nanobody 3K1K | Pleiner et al., 2015; Pleiner et al., 2020 | Addgene ID #149336 |
| pTP264_His14-bdNEDD8-BirA (E. coli) | Gift from Dirk Görlich (Pleiner et al., 2015) | Addgene ID #149334 |
| pAV286_His14-Tev-SENPEuB | Gift from Dirk Görlich (Pleiner et al., 2020; Vera Rodriguez et al., 2019) | Addgene ID #149333 |
| pDG02583_His14-MBP-bdSUMO-bdNEPD1 | Gift from Dirk Görlich (Pleiner et al., 2018) | Addgene ID #104129 |
| pSF1389_His14-Tev-bdSENP1 | Gift from Dirk Görlich (Frey and Görlich, 2014) | Addgene ID #104962 |
| pTP072_His14-bdSUMO-WNK1(2241-2266)-3xFLAG (human) | This paper | N/A |
| pTP096_His14-bdSUMO-WNK1(2241-2266)-3xHA (human) | This paper | N/A |
| pTP076_His14-bdSUMO-WNK1(2241-2266) (human) | This paper | N/A |
| pTP107_His14-bdSUMO-WNK1(2241-2266) L2250K) (human) | This paper | N/A |
| pTP108_His14-bdSUMO-WNK1(2241-2266) L2253K (human) | This paper | N/A |
| pTP109_His14-bdSUMO-WNK1(2241-2266) W2257K (human) | This paper | N/A |
| pTP322_His14-bdSUMO-WNK1(2241-2266) F2246K (human) | This paper | N/A |
| pTP323_His14-bdSUMO-WNK1(2241-2266) D2248K (human) | This paper | N/A |
| pTP324_His14-bdSUMO-WNK1(2241-2266) H2251K (human) | This paper | N/A |
| pTP325_His14-bdSUMO-WNK1(2241-2266) W2257L (human) | This paper | N/A |
| pTP326_His14-bdSUMO-WNK1(2241-2266) W2257A (human) | This paper | N/A |
| pTP327_His14-bdSUMO-WNK1(2241-2266) L2264K (human) | This paper | N/A |
| pTP463_His14-bdSUMO-WNK1(2241-2266) D2249A (human) | This paper | N/A |
| pTP464_His14-bdSUMO-WNK1(2241-2266) D2249K (human) | This paper | N/A |
| pTP465_His14-bdSUMO-WNK1(2241-2266) D2260A (human) | This paper | N/A |
| pTP466_His14-bdSUMO-WNK1(2241-2266) D2260K (human) | This paper | N/A |
| pTP522_His14-bdSUMO-WNK1(2241-2266) T2247A (human) | This paper | N/A |
| pTP523_His14-bdSUMO-WNK1(2241-2266) T2247K (human) | This paper | N/A |
| pTP173_His14-bdSUMO-WNK2(2153-2178) (human) | This paper | N/A |
| pTP174_His14-bdSUMO-WNK3(1641-1666) (human) | This paper | N/A |
| Software and algorithms | ||
| FlowJo | FlowJo | https://www.flowjo.com/ |
| UCSF Chimera | UCSF | https://www.cgl.ucsf.edu/chimera/ |
| Pymol | Schrödinger | https://pymol.org/2/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/uk/creativecloud.html |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Zeiss Zen | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Other | ||
| SuperSignal West Pico Chemiluminescent substrate | Thermo Fisher Scientific | Cat# 34080 |
| Rabbit Reticulocyte Lysate Mix | Sharma et al., 2010 | N/A |
| Canine rough microsomes | Walter and Blobel, 1983 | N/A |
| PURExpress ΔRF123 Kit | New England Biolabs | Cat# E6850S |
| TransIT-293 transfection reagent | Mirus | Cat# MIR2705 |
| Lipofectamine 3000 | Thermo Scientific | Cat# L3000015 |
| RNAiMAX lipofectamine | Thermo Scientific | Cat# 13778150 |
| DMEM, high glucose, GlutaMAX Supplement, pyruvate | Thermo Scientific | Cat# 10569010 |
| DMEM, high glucose, no glutamine, no methionine, no cystine | Thermo Scientific | Cat# 21013024 |
| Expi293™ Expression Medium | Thermo Scientific | Cat# A1435102 |
| FreeStyle™ 293 Expression Medium | Thermo Scientific | Cat# 12338026 |
| Tetracycline-free Fetal Calf Serum (FCS) | BioSera | Cat# FB-1001T/500 |
METHOD DETAILS
Constructs
Constructs for expression in rabbit reticulocyte lysate (RRL) were based on the SP64 vector (Promega, USA). Constructs for translation in the PURE system were generated from the T7 PURExpress plasmid (New England Biolabs, USA). The coding sequence of full length human WNK1 was derived from a commercial expression vector (#RC218208, Origene, USA). E. coli expression constructs needed to generate and use a biotinylated anti-GFP nanobody for native purification from human cells are available from Addgene (#149336, #149334, #149333) (Pleiner et al., 2015; Pleiner et al., 2020; Vera Rodriguez et al., 2019). E. coli expression constructs for the bdNEDP1, bdSENP1 and SENPEuB proteases were kind gifts of Dirk Görlich and are available from Addgene (#104129, #104962, #149333) (Frey and Görlich, 2014; Pleiner et al., 2018; Vera Rodriguez et al., 2019). Constructs for expression in cultured mammalian cells were generated in either the pcDNA5/FRT/TO or pcDNA3.1 backbone (Thermo Scientific).
Expression and purification of proteins
To map the EMC2 binding site on WNK1, full-length human WNK1 (residues 1-2382) and its truncations (residues 1-2106, 1-2200, 1-2272) were expressed with a C-terminal Myc-DDK (1xFLAG) tag from a pCMV6-Entry backbone (#RC218208, Origene, USA) in Expi 293F suspension cells (Thermo Fisher Scientific, USA). Cells were grown in Expi 293 Expression Medium (Thermo Fisher Scientific, USA) at 37°C, 8% CO2 and 125 rpm shaking in 1 L roller bottles with vented caps (Celltreat, USA). For a small-scale prep, a 300 ml culture at 2x106 cells/ml was transfected with 330 μg DNA, which was pre-incubated with 900 μl 1 mg/ml PEI MAX 40k (Polysciences, USA) for 15 min at room temperature in 20 ml Expi 293 Expression Medium. Transfected cells were grown for 48-60 hours before harvest. The cell pellet was resuspended and incubated for 30 min in lysis buffer (50 mM HEPES/KOH pH 7.5, 125 mM KAc, 5 mM MgAc2, 0.5% (v/v) Triton-X-100, 1 mM DTT and 1x complete EDTA-free protease inhibitor cocktail [Roche, Germany]). After centrifugation (17,000 rpm, 20 min, 4°C, SS-34 rotor, Sorvall, USA), proteins were purified from the lysate with anti-FLAG M2 affinity gel (Millipore-Sigma, USA) and eluted with 0.2 mg/ml 3xFLAG peptide in elution buffer (50 mM HEPES/KoH pH 7.5, 300 mM KAc, 5 mM MgAc2, 1 mM DTT, 10% glycerol). Purified FLAG-tagged full length WNK1 or C-terminal truncation fragments were added to in vitro translation reactions of 3xHA-tagged human EMC2 in RRL. After HA immunoprecipitation and elution with 3xHA peptide, the eluates were analyzed by SDS-PAGE and Western blotting with HRP-conjugated anti-FLAG M2 antibody to detect co-purification of WNK1 fragments with EMC2.
Human EMC2, EMC8, EMC3 coiled coil (3-CC; residues 35-117), EMC3 C-terminus (3-CT; residues 196-261), EMC5 C-terminus (5-CT; residues 66-131), OSR1(D164A), and all human WNK helix variants were purified in a similar manner. Open reading frames were fused to N-terminal His14-bdSUMO or His14-bdNEDD8 tags (Frey and Görlich, 2014) and expressed in E. coli Rosetta 2 (Novagen, USA). 3-CC, 3-CT, 5-CT and all WNK1 helices were additionally fused to a C-terminal 3xFLAG-tag. Typical expressions were carried out in a 1 L scale for 6 hours at 18°C using 0.2 mM IPTG for induction. Cells were resuspended in lysis buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 20 mM imidazole, 1 mM DTT, 1 mM PMSF). Proteins were purified from lysate by Ni2+ -chelate affinity chromatography and eluted either with imidazole elution buffer (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 500 mM imidazole, 250 mM sucrose) to keep the tag or protease elution buffer (50 nM bdSENP1 or 300 nM bdNEDP1 in 50 mM Tris/HCl pH 7.5, 300 mM NaCl, 20 mM imidazole, 250 mM sucrose) to remove the N-terminal tag. bdNEDP1 and bdSENP1 proteases were expressed as described before (Frey and Görlich, 2014; Pleiner et al., 2018). For tagged proteins, the imidazole was removed using a PD-10 desalting column (GE Healthcare, USA).
The expression and purification of GST-3C protease cleavage site-3xFLAG Calmodulin (cleaved by 3C protease) and His6-Tev-Bpa-RS (cleaved by Tev protease; used for Amber suppression in the PURE system with Bpa) was carried out as previously described (Shao et al., 2017). Briefly, both proteins were expressed in E. coli BL21 in a 1 L scale for 2 hours at 37°C using 0.2 mM IPTG and cell lysates were prepared by sonication as described above. GST-3C-3xFLAG-Calmodulin was purified using Glutathione sepharose 4B (GE Healthcare, USA) and eluted using reduced glutathione. The eluted protein was then cleaved overnight at 4°C using 1:100 (w/w) 3C protease. In order to remove the cleaved GST tag, the reaction was incubated with Glutathione resin. The flow through yielded purified 3xFLAG-Calmodulin, which was then buffer exchanged into physiologic salt buffer (PSB) (50 mM HEPES/KOH pH 7.5, 130 mM KAc, 2 mM MgAc) containing 5% (v/v) glycerol using a PD-10 desalting column (GE Healthcare). His-Tev-Bpa-RS was purified by Ni2+-chelate affinity chromatography as described above and then cleaved with 1:100 (w/w) Tev protease overnight at 4°C during dialysis into Bpa-RS storage buffer (50 mM HEPES/KOH pH 7.5, 300 mM NaCl, 2 mM MgAc, 10 mM imidazole, 10% glycerol, 1 mM DTT). The cleaved protein was passed over Nickel resin again to remove the His-Tev tag.
Expression and purification of biotinylated anti-GFP nanobody
The expression of His14-Avi-SUMOEu1-anti-GFP nanobody from plasmid pTP396 (Addgene # 149336) was carried out as summarized below and described in detail before (Pleiner et al., 2020). The anti-GFP nanobody GBP1/Enhancer was described before (Kirchhofer et al., 2010) and the Avi-tag is a 15 amino acid peptide with a single lysine that can be specifically biotinylated using purified E. coli biotin ligase BirA (Beckett et al., 1999; Fairhead and Howarth, 2015). The nanobody fusion protein was expressed in E. coli NEB Express Iq cells in a 1 L scale for 18-20 hours at 18 °C using 0.2 mM IPTG. Cells were lysed by sonication and the protein purified by Ni2+-chelate affinity chromatography as described above. The expression of His14-bdNEDD8-Biotin ligase BirA from pTP264 (Addgene #149334) was carried out essentially as described above for the anti-GFP nanobody and as detailed before (Pleiner et al., 2020).
An in-solution biotinylation reaction contained 300 μl 5x biotinylation buffer (250 mM Tris/HCl, 500 mM NaCl, 62.5 mM MgCl2, 50 mM ATP, 50 mM biotin), at least 50 μM purified Avi-tagged anti-GFP nanobody (from pTP396), a 1:50 molar ratio of purified His14-bdNEDD8-BirA, and water to add up to 1.5 ml final volume. The reaction was incubated for 3 hours at 25 °C and then applied to a PD-10 desalting column (GE Healthcare, USA) equilibrated in storage buffer (50 mM Tris/HCl pH 7.5, 200 mM NaCl, 1 mM DTT, 250 mM sucrose) to remove excess biotin. Fractions with normal 260/280 ratio (comparable to starting ratio of the prep, usually ~0.6-0.7) were pooled and quantitative biotinylation was assessed by test binding to Streptavidin beads. If fully biotinylated and added below bead capacity, all of the nanobody should be in the bound fraction and almost none should remain in the unbound fraction.
The engineered SUMOEu1 module that is fused to the anti-GFP nanobody in pTP396 is resistant to cleavage by endogenous eukaryotic deSUMOylases and thus allows stable isolation of nanobody-target complexes from eukaryotic cell lysates, followed by proteolytic release on ice in physiological buffer using the cognate SENPEuB protease (Vera Rodriguez et al., 2019). The expression of His14-Tev-SENPEuB protease (Addgene #149333) was carried out as described before (Pleiner et al., 2020; Vera Rodriguez et al., 2019). Briefly, the protein was expressed in E. coli NEB express Iq in a 1 L scale for 6 hours at 18°C using 0.2 mM IPTG. Cells were lysed by sonication and the lysate subjected to Ni2+-chelate affinity chromatography using imidazole elution. The buffer of the eluate was then exchanged to storage buffer (50 mM Tris/HCl pH 7.5, 200 mM NaCl, 1 mM DTT, 250 mM sucrose) using a PD-10 desalting column (GE Healthcare, USA).
Mammalian in vitro translation
In vitro translations were carried out in RRL as previously described (Sharma et al., 2010). Briefly, templates for in vitro transcription were generated by PCR using primers that anneal 5’ of the SP6 promotor and 3’ of the stop codon of an open-reading frame encoded on either a gblock (Integrated DNA Technologies, USA) or SP64-derived plasmid. Transcription reactions were carried out using SP6 Polymerase (New England Biolabs, USA) for 1.5 hours at 37 °C and then added directly to nucleased RRL in the presence of radioactive 35S-methionine for 20 min at 32°C. Nascent proteins were subsequently analyzed by SDS-PAGE and autoradiography. For native immunoprecipitation of FLAG-tagged proteins from RRL for analysis by mass spectrometry or Western blot, translations were carried out for 20 min at 32°C. The reactions were then spun for 20 min at 120,000 rpm at 4°C (TLA120.1 rotor, Beckman Coulter, USA) to remove aggregates and ribosomes. Anti-FLAG M2 affinity gel was added to the supernatant and incubated for 1.5 hours at 4°C. After washing with physiologic salt buffer (PSB) (50 mM HEPES/KOH pH 7.5, 130 mM KAc, 2 mM MgAc), bound proteins were eluted with 0.2 mg/ml 3xFLAG peptide in PSB. (Yanagitani et al., 2017)
In order to test assembly of EMC2 with membrane-bound EMC subunits, canine rough ER microsomes (cRMs) were prepared as previously described (Walter and Blobel, 1983) and added to translations in RRL. After incubation for 45 min at 32°C, membranes were pelleted through a 20% (w/v) sucrose cushion, prepared in PSB, for 20 min at 55,000 rpm (TLA-55 rotor, Beckman Coulter, USA). Membrane pellets were carefully resuspended in PSB, mixed with an equal volume of solubilization buffer (2% [w/v] digitonin in PSB) and incubated for 10 min on ice. After a 10 min spin at 55,000 rpm to remove non-solubilized material, the detergent lysate was subjected to FLAG-IP as described above using 0.1% (w/v) digitonin in wash and elution buffers.
E3 ubiquitin ligase co-purification assay
Detection of co-purifying E3 ubiquitin ligases with EMC2 (Figure S1C) was carried out as follows. A 200 μl translation of 35S-methionine-labeled 3xFLAG-tagged EMC2 in RRL as described above was layered on top of a 2 ml 10-50% (w/v) sucrose gradient in PSB, and centrifuged for 2 hours at 55,000 rpm (TLS-55 rotor, Beckman Coulter, USA). 11x 200 μl fractions were harvested and subjected to FLAG-IP. The beads were washed with PSB and then incubated for 45 min at 32°C with a purified ubiquitination mix that lacked an E3 ubiquitin ligase (PSB containing 10 μM His-tagged Ubiquitin [Boston Biochem, USA], 1 mM ATP, 40 μg/ml Creatine kinase, 10 mM Creatine phosphate, 100 nM GST-tagged Ubiquitin-activating enzyme E1, and 250 nM Ubiquitin-conjugating enzyme E2 UbcH5 [both from Boston Biochem, USA]). The beads were transferred to 95°C for 5 min in SDS-Buffer (1% [w/v] SDS, 0.1 M Tris/HCl pH 8.4) to terminate ubiquitination and to elute FLAG resin-bound proteins. The samples were then diluted 10-fold with IP-Buffer (50 mM HEPES/KOH pH 7.5, 300 mM NaCl, 0.5% [v/v] Triton-X-100) and subjected to Ni2+-chelate affinity chromatography to purify His-ubiquitin-fused proteins. Nickel-bound proteins were eluted with SDS-PAGE sample buffer containing 50 mM EDTA.
In vitro translation in the PURE System and photocrosslinking
Site-specific UV crosslinking was carried out as previously described (Shao et al., 2017). A T7 promotor-based construct, encoding human SEC61β with an amber stop codon at position 85 in its TMD, was translated in the presence of radioactive 35S-methionine, 100 nM Ca2+, and 10 μM purified Calmodulin (CaM) in the coupled transcription/translation PURExpress system (New England Biolabs, USA), which contains a defined set of purified translation factors and E. coli ribosomes, but lacks all release factors ΔRF123 kit). The release factors RF2 and RF3, but not RF1 (which recognizes the UAG [amber] stop codon) were added back to the reaction. The unnatural amino acid 4-Benzoylphenylalanine (Bpa, used at 100 μM) was incorporated at the amber stop codon using recombinant Bpa synthetase (100 μg/ml) and suppressor tRNA purified as described before (Shao et al., 2017). After translation for 2 hours at 32°C and addition of 1 mM puromycin, the reaction was layered on top of a 20% (w/v) sucrose cushion prepared in 1xPSB with 100 nM CaCl2 and spun for 1 hour at 55,000 rpm (TLS-55 rotor, Beckman Coulter, USA) to remove aggregates. SEC61β-CaM complexes were removed from the cushion and incubated in the presence of 1 mM EGTA with either buffer, 3 μM purified EMC2 alone or pre-formed complexes of 3 μM EMC2 and 3 μM of any binding partner (Figures 5A and S5A). For Figure 5C, 3 μM EMC2 was pre-incubated with either 3, 6, or 12 μM wild type or L2250K His14-bdSUMO-tagged WNK1 helix (residues 2241-2266). Except for the -UV control sample, all reactions were irradiated at a distance of ~7-10 cm with a UVP B-100 series lamp (Analytik Jena, Germany) for 15 min on ice before quenching with SDS-PAGE sample buffer. Samples were analyzed by SDS-PAGE and autoradiography.
For the experiments in Figure S5A–B, a T7-promoter-based construct was generated in which the squalene synthase (SQS/FDFT1) transmembrane domain including flanking residues (378-410) with an amber stop codon at position 400, was inserted into the inert cytosolic linker region of SEC61β (Guna et al., 2018).
Stable cell line generation with the Flp-In system
Flp-In 293 T-Rex cells were purchased from Thermo Fisher Scientific (USA). HeLa Flp-In cells were a gift from Christian Schlieker. Both cell lines were grown in DMEM supplemented with 2 mM glutamine, 10% FCS, 15 μg/ml Blasticidine S and 100 μg/ml Zeocine. The open-reading frame (ORF) to be integrated into the genomic FRT site was cloned into the pcDNA5/FRT/TO vector backbone and transfected together with pOG44 Flp-In recombinase in a 9:1 ratio using either Trans-IT 293 transfection reagent (Mirus, USA) or Lipofectamine 3000 (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. 48 hours following transfection, the medium was exchanged to contain 100 μg/ml Hygromycin B to select for cells with a successfully integrated ORF. The resulting cell lines expressed a protein-of-interest from a single genomic integration site under control of a doxycycline-inducible CMV promoter.
Generation of a EMC2-GFP knock-in cell line
A single guide (sg) RNA targeting the endogenous EMC2 C-terminus (TTGCAGATCACCCAGTCTTA) was designed using the GPP Web portal (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) and cloned into pX459. In parallel a donor plasmid was created that encodes GFP and a puromycin resistance cassette flanked by 764 and 574 bp homology arms. Silent mutations were introduced into the sgRNA binding site of the left homology arm to prevent Cas9 cleavage after donor integration. 48 hours after transfection of both plasmids in a 1:1 ratio into HeLa wild type cells using Lipofectamine 3000 (Thermo Fisher Scientific, USA), successfully transfected cells were selected by addition of 1 μg/ml puromycin for three consecutive days. Cells were then recovered in medium without puromycin for two weeks. Another puromycin selection as described above was then applied to select for cells with a successfully integrated puromycin resistance cassette that express GFP-tagged EMC2 from its genomic locus.
Flow cytometry analysis of reporter cell lines
In order to measure protein stability in cells, stable cell lines were generated as described above containing a fusion of a protein-of-interest with either GFP or RFP in a GFP-2A-RFP backbone (Itakura et al., 2016). Note, the mCherry sequence is used throughout the manuscript, but is referred to as RFP for simplicity. The viral 2A sequence induces the skipping of a peptide bond by the ribosome (de Felipe et al., 2006), resulting in two separate polypeptide chains being produced from a single transcript in a 1:1 ratio. The GFP and RFP fluorescence intensity can be measured by flow cytometry to derive a GFP:RFP or RFP:GFP ratio. Changes in these ratios after perturbation, e.g. siRNA transfection, reflect differences in post-translation stability of the fusion protein. For the analysis of the effect of full length WNK1 (Figure 1E) or WNK1 helix (Figures 3B and 6A) on the stability of EMC2, the respective reporter cell line was transiently transfected with pCDNA3.1-derived plasmids encoding TagBFP-fusions of these proteins. As a control, a pcDNA3.1 plasmid encoding only TagBFP was used. The GFP:RFP ratio of BFP-positive cells was then compared.
All cells for flow cytometry were trypsinized, washed in 1xPBS, resuspended in Hank’s balanced salt solution containing 50 μg/ml DNAse I and analyzed using a MACSQuant VYB flow cytometer (Miltenyi Biotec, Germany). Sytox Blue dead cell stain (ThermoFisher Scientific, USA) was used at 2 μM, where possible, to gate out dead cells. Unstained wild type Flp-In cells and Flp-In cells transiently transfected with either GFP, RFP (or BFP if needed) were analyzed separately along every run as single-color controls for multicolor compensation using the FlowJo software package.
Lenti-viral transduction
Lenti-viral transfer plasmids encoding a scrambled control or WNK1 shRNA were purchased from Millipore-Sigma (USA) and sequenced to verify the insert. A 2nd generation packaging system (plasmids Δ8.9 and pVSV-G) was used to generate lenti-viral particles. 48 hours after lenti-viral particle addition, successfully transduced cells with genomically integrated shRNAs were selected by addition of 1 μg/ml puromycin for three consecutive days. Cells were recovered from selection for 48 hours and either analyzed directly (Figures 2A and S2C) or additionally treated with scrambled or WNK1 siRNA for 72 hours (Figure S2D).
To test incorporation of mutant EMC subunits into intact EMCs in cells (Figures S6D–E), lenti-viral transfer vectors in a pHAGE2 backbone were generated that stably integrate and express either wild type or mutant subunit from a CMV promotor-driven open-reading frame. EMC2 variants were fused to an N-terminal 3xFLAG-TagBFP tag, while EMC5 variants carried a C- terminal TagBFP-3xFLAG tag. Lenti-viral particles were generated as above and cells were transduced with these particles such that around 20% of all cells were BFP-positive, likely indicating integration of a single copy of the transgene. After 48 hours, cells were harvested, solubilized as described above and subjected to native FLAG-IP. Bound proteins were eluted with 3xFLAG peptide and the eluates analyzed by SDS-PAGE and Western blotting.
Affinity purifications from stable cell lines
To validate incorporation of GFP-tagged subunits into the EMC, we isolated the EMC under native conditions from detergent-solubilized cells using a biotinylated, protease-cleavable anti-GFP nanobody, expressed and purified as described above (Pleiner et al., 2020). Cells were grown in 15 cm dishes and GFP-EMC2/4/5 expression was induced for at least 48 hours with 1 μg/ml DOX. Cells were then harvested by centrifugation, washed with 1x PBS, weighed and resuspended with 6.8 μl solubilization buffer per 1 mg cell pellet (50 mM HEPES/KOH pH 7.5, 200 mM NaCl, 2 mM MgAc2, 1% [w/v] LMNG, 1 mM DTT, 1x complete EDTA-free protease inhibitor cocktail [Roche, Germany]). After 30 min of head-over-tail incubation with solubilization buffer, the lysate was cleared by centrifugation for 20 min at 4°C and 18,000 x g in a tabletop centrifuge. The supernatant was then added to pre-equilibrated magnetic Streptavidin beads (ThermoFisher Scientific, USA) onto which biotinylated anti-GFP nanobody had been pre-immobilized before blocking all free binding sites with biotin. After 1 hour of binding head-over-tail at 4°C, the beads were washed three times with wash buffer (solubilization buffer with 0.01% [w/v] LMNG). Finally, the anti-GFP nanobody along with all bound proteins was released under native conditions and in minimal volume by cleavage with 250 nM SENPEuB protease (Vera Rodriguez et al., 2019) in wash buffer for 20 min at 4°C. The eluate was then analyzed by SDS-PAGE and Sypro Ruby staining (Bio-Rad Laboratories, USA).
Alternatively, the EMC was purified from HEK293T cells stably expressing human EMC5-3xFLAG as previously described (Guna et al., 2018).
In order to purify unassembled cytosolic EMC2 (Figures 1D and 6B), HEK293T cells stably expressing GFP-EMC2 were resuspended in Digitonin lysis buffer (50 mM HEPES/KOH pH7.5, 10 mM KAc, 2 mM MgAc, 1 mM DTT, 0.01% [w/v] digitonin and 1x complete EDTA-free protease inhibitor cocktail). Resuspended cells were then passed 12x times through a 27Gx1-¼ needle (BD Biosciences, #305136) at 4°C. Lysed cells were then spun for 15 min at 4°C in a tabletop centrifuge to remove cellular debris. The cytosolic supernatant was then adjusted to contain 150 mM KAc for IP of GFP-tagged EMC2 as described above. The same approach was used for the analysis of unassembled nascent EMC2-GFP in the EMC2-GFP knock-in cell line (Figure S2C).
For the analysis of unassembled EMC2 ubiquitination (Figure 6B), ubiquitinated species were accumulated by treatment of the stable HEK293T cell line with 10 nM of the proteasome inhibitor bortezomib for 16 hours before harvest. Unassembled GFP-EMC2 was then purified from a cytosolic cell lysate prepared as above in the presence of 50 μM deubiquitinase inhibitor PR-619.
Radioactive pulse-chase assay
For the experiment in Figure 6C, HEK293T GFP-EMC2 Flp-In cells were treated with either scrambled or WNK1 siRNA in triplicate for a total of 72 hours. The fluorescent reporter was induced 24 hours after siRNA transfection by the exchange to fresh medium containing 1 μg/ml doxycycline. For the pulse-chase assay, 5x106 cells of each condition were harvested, washed to remove traces of full growth medium, and starved in-solution by resuspension in medium lacking methionine (Thermo Scientific, USA, # 21013024 supplemented with 200 μM cysteine). Following 30 min of starvation, newly synthesized proteins were labeled for 30 min by addition of ~60 nM fresh 35S-methionine (10.2 Ci/L). The cells were then chased by addition of 5 mM unlabeled methionine. Aliquots were taken 0, 20 or 40 minutes after the chase and snap-frozen immediately after addition of ice-cold lysis mix (4 mM HEPES/KOH pH 7.5, 1% (v/v) Triton-X-100, 1 mM DTT, 1x complete EDTA-free protease inhibitor cocktail [Roche, Germany]). All aliquots were then thawed together and incubated for 30 min at 4°C for cell lysis and solubilization. Debris was pelleted by centrifugation and the resulting cleared cell lysate was subjected to purification via anti-GFP nanobody as described above (wash buffer with 0.5 M NaCl and 0.05 % [v/v] Triton-X-100 instead of LMNG). Eluates were normalized by GFP fluorescence on a plate reader and analzyed by SDS-PAGE and autoradiography. Western blotting for total immunoprecipitated EMC2 was used to validate successful normalization.
Kinase assays
Full-length human WNK1, EMC2 and OSR1(D164A) were incubated in kinase assay buffer (50 mM HEPES/KOH pH 7.5, 150 mM KAc, 10 mM MgAc, 1 mM DTT, 1x PhosSTOP [Roche, Germany], 100 μM ATP and 200 μCi/ml 32P-ATP) for 30 min at 30°C. Reactions were stopped by addition of SDS-PAGE sample buffer and were analyzed by autoradiography.
Immunofluorescence and confocal microscopy
Lab-Tek II 8-well chamber slides (ThermoFisher Scientific, USA) were coated with 300 μl 100 μg/ml poly-D-lysine per well for 1.5 hours at 37°C and then washed three times with ddH2O. Stable HEK293T cells expressing 3xFLAG-tagged wild type or L2250K, L2253K, W2257K mutant WNK1 were seeded onto coated slides 24 hours before analysis in medium containing 1 μg/ml DOX. For the hyper-osmotic shock treatment, the medium was aspirated and exchanged to new medium additionally containing 200 mM sorbitol and incubated at 37°C for 5 min. All medium was aspirated and cells fixed with 4% (w/v) paraformaldehyde in 1x PBS for 15 min at room temperature. After two washes with 1x PBS, a 5 min quenching step with 50 mM NH4Cl in 1x PBS and two further washes with 1x PBS, cells were permeabilized with 0.2% (v/v) Triton-X-100 in 1x PBS for 3 min. Following three washes with 1x PBS, cells were blocked for 30 min with 1% (w/v) bovine serum albumin fraction V (BSA) in 1x PBS. Then cells were incubated with mouse anti-FLAG M2 antibody (1:500) and rabbit anti-Clathrin heavy chain antibody (1:100) diluted in 1x PBS containing 1% (w/v) BSA. After three 10 min washes with 1x PBS, cells were stained with the respective fluorophore-coupled secondary antibodies and 2.5 μg/ml DAPI diluted in 1x PBS containing 1% (w/v) BSA. Following another three 10 min washes with 1x PBS, cells were mounted for analysis by confocal microscopy using SlowFade Gold (Thermo Fisher Scientific, USA). Imaging was performed using a LSM880 confocal microscope (Zeiss, Germany).
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantification of the autoradiography signal, radioactive gels were exposed to X-ray film and developed films were scanned. Densitometry was performed using Fiji/ImageJ software and graphs were plotted using Excel. No statistical analyses were performed in this study. As indicated in the figure legends, error bars represent the mean and standard deviation from three independent replicates (n=3). All flow cytometry experiments and binding assays were performed at least two times with the same outcome.
Supplementary Material
Table S2. EMC2 affinity purification mass spectrometry analysis, Related to Figure 1C.
Highlights:
WNK1 functions as an EMC assembly factor in a kinase-independent manner
WNK1 binds the soluble subunit EMC2 using a conserved amphipathic α-helix
WNK1 shields the hydrophobic EMC2-8 interface to prevent promiscuous interactions
WNK1 stabilizes unassembled EMC2 by competing for binding of E3 ubiquitin ligases
ACKNOWLEDGMENTS
We thank Christian Schlieker for reagents and John O’Neill, Ramanujan Hegde, Henry Lester, and the Voorhees lab for thoughtful discussions. Confocal imaging was performed in the Caltech Biological Imaging Facility with the support of the Caltech Beckman Institute and the Arnold and Mabel Beckman Foundation. Flow cytometry was performed at the Caltech Flow Cytometry Facility. This work was supported by the Heritage Medical Research Institute, the Kinship Foundation, the Pew-Stewart Foundation, and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number DP2GM137412. T.P. is supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft and received prior funding from a Caltech Ross fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
REFERENCES
- Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y and Kahle KT (2014) The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal, 7, re3. [DOI] [PubMed] [Google Scholar]
- Bai L, You Q, Feng X, Kovach A and Li H (2020) Structure of the ER membrane complex, a transmembrane-domain insertase. Nature, 584, 475–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrows NJ et al. (2019) Dual roles for the ER membrane protein complex in flavivirus infection: viral entry and protein biogenesis. Sci Rep, 9, 9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E and Schatz PJ (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci, 8, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini M, Fenzl K, Kats I, Wruck F, Tippmann F, Schmitt J, Auburger JJ, Tans S, Bukau B and Kramer G (2021) Interactions between nascent proteins translated by adjacent ribosomes drive homomer assembly. Science, 371, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraei Z and Yang J (2010) The ß subunit of voltage-gated Ca2+ channels. Physiol Rev, 90, 1461–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti O and Hegde RS (2009) Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell, 137, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood PJ and Hegde RS (2019) The Role of EMC during Membrane Protein Biogenesis. Trends Cell Biol, 29, 371–384. [DOI] [PubMed] [Google Scholar]
- Chitwood PJ, Juszkiewicz S, Guna A, Shao S and Hegde RS (2018) EMC Is Required to Initiate Accurate Membrane Protein Topogenesis. Cell, 175, 1507–1519.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW and Kopito RR (2011) Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol, 14, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe P, Luke GA, Hughes LE, Gani D, Halpin C and Ryan MD (2006) E unum pluribus: multiple proteins from a self-processing polyprotein. Trends Biotechnol, 24, 68–75. [DOI] [PubMed] [Google Scholar]
- Duncan CD and Mata J (2011) Widespread cotranslational formation of protein complexes. PLoS Genet, 7, e1002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhead M and Howarth M (2015) Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol, 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S and Görlich D (2014) A new set of highly efficient, tag-cleaving proteases for purifying recombinant proteins. J Chromatogr A, 1337, 95–105. [DOI] [PubMed] [Google Scholar]
- Guna A, Volkmar N, Christianson JC and Hegde RS (2018) The ER membrane protein complex is a transmembrane domain insertase. Science, 359, 470–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadchouel J, Ellison DH and Gamba G (2016) Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu Rev Physiol, 78, 367–389. [DOI] [PubMed] [Google Scholar]
- Harper JW and Bennett EJ (2016) Proteome complexity and the forces that drive proteome imbalance. Nature, 537, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE and Babu MM (2018) Pharmacogenomics of GPCR Drug Targets. Cell, 172, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC et al. (2012) A census of human soluble protein complexes. Cell, 150, 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E and Hegde RS (2011) Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature, 475, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A and Varshavsky A (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science, 327, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis AJ, Page KR, Guna A and Voorhees RM (2020) Differential Modes of Orphan Subunit Recognition for the WRB/CAML Complex. Cell Rep, 30, 3691–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ and Hegde RS (2016) Ubiquilins Chaperone and Triage Mitochondrial Membrane Proteins for Degradation. Mol Cell, 63, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäkel S, Mingot JM, Schwarzmaier P, Hartmann E and Görlich D (2002) Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J, 21, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ (2016) VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol, 17, 293–307. [DOI] [PubMed] [Google Scholar]
- Jonikas MC et al. (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science, 323, 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S and Hegde RS (2018) Quality Control of Orphaned Proteins. Mol Cell, 71, 443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihm AJ, Kong Y, Hong W, Russell JE, Rouda S, Adachi K, Simon MC, Blobel GA and Weiss MJ (2002) An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature, 417, 758–763. [DOI] [PubMed] [Google Scholar]
- Kirchhofer A et al. (2010) Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol, 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N and Mann M (2014) Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods, 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Le Tallec B, Barrault MB, Courbeyrette R, Guérois R, Marsolier-Kergoat MC and Peyroche A (2007) 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol Cell, 27, 660–674. [DOI] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C and Weissman JS (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell, 157, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DL, Inoue T, Chen YJ, Chang A, Tsai B and Tai AW (2019) The ER Membrane Protein Complex Promotes Biogenesis of Dengue and Zika Virus Non-structural Multi-pass Transmembrane Proteins to Support Infection. Cell Rep, 27, 1666–1674.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, Keenan RJ and Hegde RS (2010) A ribosome-associating factor chaperones tail-anchored membrane proteins. Nature, 466, 1120–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA and Teichmann SA (2015) Structure, dynamics, assembly, and evolution of protein complexes. Annu Rev Biochem, 84, 551–575. [DOI] [PubMed] [Google Scholar]
- McShane E et al. (2016) Kinetic Analysis of Protein Stability Reveals Age-Dependent Degradation. Cell, 167, 803–815. [DOI] [PubMed] [Google Scholar]
- Menegola M and Trimmer JS (2006) Unanticipated region- and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knockout mouse brain. J Neurosci, 26, 12137–12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Vedam LE et al. (2020) Structural and mechanistic basis of the EMC-dependent biogenesis of distinct transmembrane clients. Elife, 9, e62611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo AM, Shurtleff MJ, Popova KD, Kulsuptrakul J, Weissman JS and Puschnik AS (2019) The ER membrane protein complex is required to ensure correct topology and stable expression of flavivirus polyproteins. Elife, 8, e48469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT et al. (2017) UBE2O remodels the proteome during terminal erythroid differentiation. Science, 357, [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JP, Phillips BP, Yagita Y, Juszkiewicz S, Wagner A, Malinverni D, Keenan RJ, Miller EA and Hegde RS (2020) The architecture of EMC reveals a path for membrane protein insertion. Elife, 9, e57887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB and Amon A (2014) Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech, 7, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiner T, Bates M and Görlich D (2018) A toolbox of anti-mouse and anti-rabbit IgG secondary nanobodies. J Cell Biol, 217, 1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiner T, Bates M, Trakhanov S, Lee CT, Schliep JE, Chug H, Böhning M, Stark H, Urlaub H and Görlich D (2015) Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. Elife, 4, e11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiner T, Pinton Tomaleri G, Januszyk K, Inglis AJ, Hazu M and Voorhees RM (2020) Structural basis for membrane insertion by the human ER membrane protein complex. Science, 369, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongs O and Schwarz JR (2010) Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev, 90, 755–796. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Goodman JH, Begum G, Donnelly BF, Pittman G, Weinman EJ, Sun D and Subramanya AR (2015) Generation of WNK1 knockout cell lines by CRISPR/Cas-mediated genome editing. Am J Physiol Renal Physiol, 308, F366–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Ohba A, Liu Z, Inagaki T and Satoh AK (2015) dPob/EMC is essential for biosynthesis of rhodopsin and other multi-pass membrane proteins in Drosophila photoreceptors. Elife, 4, e06306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Höning K, Ebert TS and Hornung V (2016) CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat Commun, 7, 12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Rodrigo-Brenni MC, Kivlen MH and Hegde RS (2017) Mechanistic basis for a molecular triage reaction. Science, 355, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, von der Malsburg K and Hegde RS (2013) Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell, 50, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Mariappan M, Appathurai S and Hegde RS (2010) In vitro dissection of protein translocation into the mammalian endoplasmic reticulum. Methods Mol Biol, 619, 339–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekarabi M et al. (2008) Mutations in the nervous system--specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest, 118, 2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E and Kahle KT (2017) WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab, 25, 285–299. [DOI] [PubMed] [Google Scholar]
- Shemorry A, Hwang CS and Varshavsky A (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell, 50, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A, Döring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G and Bukau B (2018) Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature, 561, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh YW, Minguez P, Bork P, Auburger JJ, Guilbride DL, Kramer G and Bukau B (2015) Operon structure and cotranslational subunit association direct protein assembly in bacteria. Science, 350, 678–680. [DOI] [PubMed] [Google Scholar]
- Shurtleff MJ et al. (2018) The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife, 7, e37018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasekharan S, Monette MY and Forbush B (2013) Functional expression of human NKCC1 from a synthetic cassette-based cDNA: introduction of extracellular epitope tags and removal of cysteines. PLoS One, 8, e82060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MK, Porras-Yakushi TR, Reitsma JM, Huber FM, Sweredoski MJ, Hoelz A, Hess S and Deshaies RJ (2016) A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife, 5, e19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Wu Q, Zhou B, Choi MY, Ding B, Yang W and Dong M (2019) Proteomic Analysis Identifies Membrane Proteins Dependent on the ER Membrane Protein Complex. Cell Rep, 28, 2517–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M et al. (2015) Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vera Rodriguez A, Frey S and Görlich D (2019) Engineered SUMO/protease system identifies Pdr6 as a bidirectional nuclear transport receptor. J Cell Biol, 218, 2006–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veríssimo F and Jordan P (2001) WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene, 20, 5562–5569. [DOI] [PubMed] [Google Scholar]
- Volkmar N and Christianson JC (2020) Squaring the EMC - how promoting membrane protein biogenesis impacts cellular functions and organismal homeostasis. J Cell Sci, 133, jcs243519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmar N, Thezenas ML, Louie SM, Juszkiewicz S, Nomura DK, Hegde RS, Kessler BM and Christianson JC (2019) The ER membrane protein complex promotes biogenesis of sterol-related enzymes maintaining cholesterol homeostasis. J Cell Sci, 132, jcs223453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P and Blobel G (1983) Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol, 96, 84–93. [DOI] [PubMed] [Google Scholar]
- Wideman JG (2015) The ubiquitous and ancient ER membrane protein complex (EMC): tether or not. F1000Res, 4, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson FH et al. (2001) Human hypertension caused by mutations in WNK kinases. Science, 293, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ and Cobb MH (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem, 275, 16795–16801. [DOI] [PubMed] [Google Scholar]
- Xu Y, Anderson DE and Ye Y (2016) The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov, 2, 16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K et al. (2016) Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol, 12, 896–898. [DOI] [PubMed] [Google Scholar]
- Yanagitani K, Juszkiewicz S and Hegde RS (2017) UBE2O is a quality control factor for orphans of multiprotein complexes. Science, 357, 472–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagórska A et al. (2007) Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol, 176, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. EMC2 affinity purification mass spectrometry analysis, Related to Figure 1C.
Data Availability Statement
The mass spectrometry data set referenced in Figure 1C is provided as Table S2. Figures 1A, 4A, S6A and S6F are based on the published structure of the human EMC (PDB 6WW7) (Pleiner et al., 2020). Figures S6A and S6F additionally make use of the published yeast EMC structure (PDB 6WB9) (Bai et al., 2020).