

Abstract
NF-κB activation has been linked to prostate cancer (PCa) progression and is commonly observed in castrate-resistant disease. It has been suggested that NF-κB-driven resistance to androgen deprivation therapy (ADT) in PCa cells may be mediated by aberrant androgen receptor (AR) activation and AR splice variant production. Preventing resistance to ADT may therefore be achieved by utilizing NF-κB inhibitors. However, low oral bioavailability and high toxicity of NF-κB inhibitors is a major challenge for clinical translation. Dimethylaminoparthlide (DMAPT) is an oral NF-κB inhibitor in clinical development and has already shown favorable pharmacokinetic and pharmacodyanamic data in patients with heme malignancies including decrease of NF-κB in circulating leuchemic blasts. Here we report that activation of NF-κB/p65 by castration in mouse and human PCa models resulted in a significant increase in AR variant-7 (AR-V7) expression and modest upregulation of AR. In vivo castration of VCaP-CR tumors resulted in significant upregulation of phosphorylated-p65 and AR-V7, which was attenuated by combination with DMAPT and DMAPT increased the efficacy of AR inhibition. We further demonstrate that the effects of DMAPT sensitizing PCa cells to castration were dependent on the ability of DMAPT to inhibit phosphorylated-p65 function.
Introduction
Worldwide, there are approximately 300,000 deaths from metastatic castration resistant prostate cancer (mCRPC) [1]. Strategies that prevent emergence of clones that are resistant to androgen deprivation therapy (ADT) may augment the efficacy of AR targeted therapy, whether it be testosterone suppression alone or with abiraterone or new generation potent direct androgen receptor inhibitors such as enzalutamide, apalutamide or darolutamide. This in turn may decrease the number of men who relapse after adjuvant ADT for localized disease and/or prolong the survival of men with metastatic hormone sensitive prostate cancer (mHSPC). Advances identifying the aberrant biology driving resistance to therapy and poor clinical outcomes include aberrant androgen receptor (AR) activation from AR upregulation, AR splice variants (AR-Vs) such as AR-V7 and non-androgen ligands such as cytokines IL-6 [2–4]. There are currently no approved agents that can prevent androgen-mediated AR upregulation or constitutive AR activation in CRPC, and this remains a compelling strategy to prevent therapy resistance.
Activation of nuclear factor kappa B (NF-κB) is involved in a range of biological processes such as cell proliferation, inflammation, and cell invasion, and is strongly associated with oncogenesis [5, 6]. NF-κB activation is driven through either canonical or non-canonical pathways. Canonically, NF-κB activation is dependent on the IκB kinase complex (IKK), whereby, IκBα is phosphorylated by IKK in a IκKβ-dependent manner, which results in the degradation of IκBα and the release of the p65/p50 dimer to gather in the nucleus [7]. Phosphorylation of p65 at Ser536 is mediated by IKK and is required for canonical NF-κB-dependent cellular activity [8]. In the noncanonical NF-kB cascade, RelB/p100 heterodimers are processed to RelB/p52 heterodimers in an IKKα-dependent manner. Activation of the canonical NF-κB pathway is most commonly implicated in oncogenesis.
NF-κB is a prime target for inhibition to augment ADT efficacy. Over-expression of AR-V7 may also feedback and enhance NF-κB signaling resulting in bidirectional positive interactions [9–11]. Hence, combination of potent and safe NF-κB inhibitors with anti-androgen drugs is likely to be an effective strategy for the management of CRPC and to prevent or at least delay progression of hormone sensitive prostate cancer (HSPC) to mCRPC. The relevance of targeting NF-κB signaling is further increased given the development of parthenolide analogues, which are NF-κB inhibitors and have entered clinical testing. Dimethylaminoparthenolide (DMAPT) is a bioavailable parthenolide analogue, which inhibits NF-κB by binding to the cysteine residue of IκKβ and prevents one of its subunits, p65, binding to DNA, thereby inducing cell cycle arrest and promoting apoptosis of cancer cells [12]. DMAPT largely blocks the canonical NF-κB pathway by binding directly to and inhibiting IκKβ [13–15] and has been shown to have no meaningful impact on the non-canonical. Kwok et. al. [13] showed that inactivation of IκKβ abolished sensitivity of parthenolide (of which DMAPT is a derivative compound). In humans, DMAPT has demonstrated linear pharmacokinetics and no toxicity was observed with doses that were able to achieve low micromolar serum concentrations and decrease NF-κB in circulating blasts. It is also notable that DMAPT has been shown to enhance radiation anti-cancer activity in PCa [16, 17], and non-small cell lung cancer [18, 19] and chemotherapy in bladder cancer [20] while mitigating toxicity to normal tissue [21, 22].
In this manuscript, we provide novel insights into strategies to block NF-κB-mediated resistance by inhibition of NF-κB and the consequent AR upregulation and AR-V7 with an orally administered agent that is being advanced into prostate cancer clinical trials.
Materials and Methods
In vivo studies
Animal experiments were approved by and performed in accordance with the guidelines of Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee (Animal protocol #102–2015). VCaP-CR cells were implanted subcutaneously into the flank of donor male ICR-SCID mice (Taconic Biosciences) 7–8 weeks of age as a 50% Matrigel/PBS suspension and allowed to grow up to 2000 mm3 at which point the tumors were harvested and processed into tissue pieces approx. 2–3 mm in diameter. Resected tumor pieces were subcutaneously implanted into the flank of 7-week-old experimental male ICR-SCID mice. Tumors were measured with calipers thrice weekly until tumors reached 200 mm3, at which point mice were randomly sorted into four treatment groups: vehicle, castration (surgical), DMAPT (100 mg/kg in H2O, by oral gavage Q.D.) or castration plus DMAPT. Mice were treated and tumors measured until mice reached a humane tumor endpoint or the study period ended. DMAPT was obtained from Dr. Peter Crooks (University of Arkansas for Medical Sciences, Little Rock, AR) and stored at −20°C. On the day of treatment, DMAPT was made up to 10 mg/mL in sterile water.
Cell culture and reagents
VCaP-CR cells were a gift from Stephen Plymate (University of Washington). FVB-MyCCaP cells were purchased from ATCC (ATCC® CRL-3255™). VCaP-CR and LNCaP-95 cell lines were cultured in phenol-free RPMI-1640 Medium (Gibco) supplemented with 10% charcoal-stripped fetal bovine serum (Sigma-Aldrich) and 1x GlutaMAX™ (Gibco). 22Rv1 cells were cultured in RPMI-1640 Medium (ATCC® 30–2001™) supplemented with 10% fetal bovine serum (Sigma-Aldrich). FVB-MyCCaP cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1x GlutaMAX™ (Gibco). Cell lines were regularly screened for mycoplasma contamination using the LookOut® Mycoplasma PCR Detection Kit (Sigma-Aldrich).
Western Blot
Sub-confluent treated cells were washed twice with cold PBS, trypsinized and then lysed in Pierce RIPA buffer (Thermo Fisher Scientific, 89900) with PhosSTOP™ inhibitor cocktail (Sigma Aldrich, PHOSS-RO) at 4°C for 30 mins. Protein concentrations were measured by a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Proteins were separated by SDS‐PAGE (10% Mini-PROTEAN® TGX™ Precast Gel, Biorad, 4561036) and transferred to a PVDF membrane (Bio-Rad, 1620177). The membrane was blocked 3% BSA in TBST for 1 hour at room temperature and then blotted with primary antibodies overnight at 4°C. Full list of antibodies available in Supplemental Data. Blots were washed 3 × 5 minutes with TBST. The blots were then incubated with fluorescent-conjugated secondary antibody (Bio‐Rad) 1 hour at room temperature and washed 3 × 5 minutes with TBST. Proteins were visualized with a ChemiDoc MP fluorescent imager (Bio-Rad). Bio-Rad Image Lab analysis software was used to quantify protein band density. All western blots were repeated once.
qPCR gene expression analysis
Prostate tumor cells (cell lines or dissociated tumors) were collected, RNA was prepared using TRIzol reagent (Invitrogen) and quantified using a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized (iScript cDNA synthesis kit; BioRad) and used as a template for qPCR reactions using SsoFast qPCR supermix (Bio-Rad). Reactions were performed using a BioRad CFX96 Touch™ Real-Time PCR thermocycler, using an annealing temperature of 60°C and 40 cycles. List of primer sets available in Supplemental Methods Table 1. Results were analyzed by the 2−ΔΔCt method relative to β-actin as a control gene.
Immunohistochemical analysis of VCaP-CR tissues
For IHC, 4 μm thick sections were cut from paraffin-embedded blocks and dried onto positively charged microscope slides. Slides were prepared and stained using the ImmPRESS® HRP Anti-Mouse IgG (Peroxidase) Polymer Detection Kit (Vector Laboratories) as previously described [23].
For analysis, tissue sections were imaged using an EVOS M7000 automated microscope (Thermo Fisher Scientific). Images were de-identified and 50 random fields per section were run through QuPath image analysis software [24] to quantify positive DAB-stained cells (as a percentage of total cells).
Transcriptomic analysis
Prostate tumor cells (cell lines or dissociated tumors) were collected, RNA was prepared using TRIzol reagent (Invitrogen) and quantified using a NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific). RNA sequencing data from VCaP tissues were aligned with STAR to human reference genome GRCh37 (hg19) and quantified using RSEM to generate gene-level expression in raw counts and transcripts per million (TPM), and isoform-level expression. Two samples were removed due to failure of meeting sequencing and alignment metrics. Count data was first modelled to demonstrate fit with the negative binomial distribution. Samples underwent hierarchical clustering in quality control using principal component analysis, and one outlier sample was manually assessed and removed prior to downstream analysis.
Generation of stable cell lines
Stable cell lines of LNCaP-95, 22Rv1 and FVB-MyCCaP overexpressing NF-κB2/p65 were generated by transfection of pCMV4 p65 plasmids and selection of clones after application of selective pressure with antibiotics. pCMV4 p65 was a gift from Warner Greene (Addgene plasmid # 21966; http://n2t.net/addgene:21966; RRID:Addgene_21966). LNCaP-95, 22RV-1 and FVB-MyCCaP cell lines were generated by transfection of pBabe-Puro-IKBalpha-mut plasmids and selection of clones after selective pressure with antibiotics. pBabe-Puro-IKBalpha-mut (super repressor) was a gift from William Hahn (Addgene plasmid # 15291; http://n2t.net/addgene:15291; RRID:Addgene_15291). Target protein expression in generated cell lines was confirmed by western blot.
Three-dimensional in vitro tumoroid therapy assay
LNCaP-95 and 22Rv1 cells were cultured in 3D in 100% Matrigel droplets. Once tumoroids had established (2–3 days), whole tumoroids were removed from Matrigel by incubation with 2mg/mL dispase (Thermo Fisher Scientfic, 17105041) and re-plated in Matrigel in 96-well tissue culture plates (20–30 tumoroids per well). Cultures were treated for 72 hours with singlet or doublet therapy of DMAPT (5 μM) and/or Enzalutamide (10 μM), or DMSO vehicle control. After therapy period, media was removed and tumoroid cultured washed with PBS and dead vs live cells visualized using ReadyProbes™ Cell Viability Imaging Kit (Thermo Fisher Scientific, R37609). Stained cultures were imaged using an EVOS M7000 automated microscope (Thermo Fisher Scientific) and the percentage cell viability was quantified using CellProfiler image analysis software [25], 10 tumoroids per treatment group were used for analysis.
Statistical analysis
All statistical calculations were carried out in GraphPad (Prism). Statistical significance was determined by performing an unpaired two-tailed Student t-test unless otherwise noted. Comparisons between treatment arms in the Kaplan Meier analysis of the in vivo study were carried out using a Log-rank (Mantel-Cox) test to compare time-to-1cm3. A p-value of less than 0.05 was considered as significantly different from the null hypothesis across all experiments as indicated by asterisks in all figures.
Results
DMAPT blocks castration mediated NF-κB and AR-V7 upregulation in VCaP-CR cells.
In vitro, we observed that VCaP-CR cells under pressure from prolonged enzalutamide treatment, AR inhibition showed a marked increase in expression of total and activated NF-κB (phosphorylated-p65) expression (Figure 1A). This observation matches other examples in the literature where castration-resistance is linked to NF-κB signaling [11, 26]. This increase in NF-κB activation was counteracted by NF-κB inhibitor DMAPT. Given that activation of NF-κB signaling has been observed to correlate with PCa progression and resistance to AR inhibition, we hypothesized that the combination of DMAPT and castration would provide significant tumor control in vivo. Previous work has documented that DMAPT acts by blocking p65 nuclear translocation and DNA binding [20, 27]. VCaP-CR-tumor bearing mice, a PCa cell line with some well documented castration resistance (CR) were treated with either DMAPT or vehicle control with and without surgical castration (Figure 1B, Supplementary Figure 1). DMAPT alone did not significantly slow VCaP-CR tumor growth compared to vehicle treatment (median time for tumors to reach 1cm3: 34.2±6.5 weeks and 34±6.6 weeks respectively). Castration provided a moderate level of tumor control (50.2±4.7 weeks), however the most significant reduction in tumor growth was achieved when mice were castrated in combination with DMAPT (p=0.001 compared to vehicle, p=0.0053 compared to castration alone). When treated in this combinatorial manner, remarkably only two of six tumors exceeded 1cm3 by the end of the study. Given that DMAPT alone provided minimal tumor control, this indicates a strong augmentation of anti-tumor effect of the combination therapy. IHC analysis (Figure 1C–D) of end-point tumors showed that surgical castration alone increased phosphorylated-p65 expression. We also observed significant increases in AR and AR-V7 protein expression following castration, both of which were reduced by DMAPT treatment. In the absence of castration, DMAPT still provided some reduction in AR and AR-V7 expression. The combination of castration and DMAPT treatment also resulted in a significant loss of Ki-67 in tumors compared to vehicle-treated tumors and a trend towards increased cleaved-caspase 3 (Supplementary Figure 2). Similar decreases in levels of mRNA AR-FL and AR variant expression were observed, with DMAPT treatment particularly providing the more pronounced suppression of AR-V7 expression (Figure 1E). This may indicate that changes in total AR expression are largely due to AR-V7 changes, rather than AR-FL. RNA-seq performed using end-point tumors validated our IHC results by indicating that castration and DMAPT combination resulted in significantly lower AR-V7-associated gene expression [28] (Figure 1F). Our observation of NF-κB-driven upregulation of AR-V7 aligns with previous accounts by other investigators that activation of NF-κB can drive the emergence of castration resistant disease [11, 29].
Figure 1: DMAPT blocks castration mediated NF-κB and AR-V7 upregulation in VCaP-CR cells.
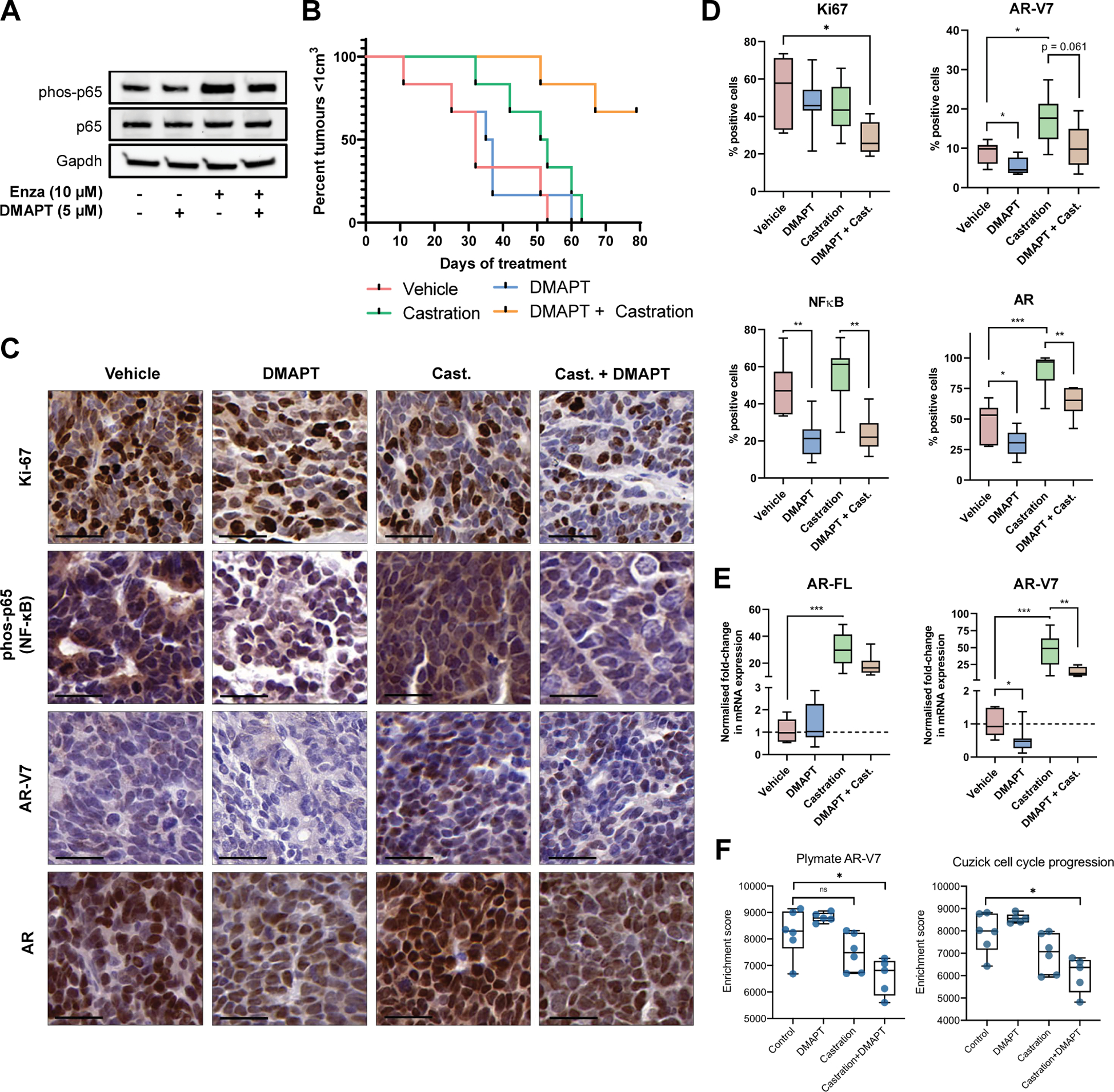
(A) Western blot targeting NF-κB VCaP-CR cells and Enzalutamide-resistant VCaP-CR cells treated +/− DMAPT (5μM) for 72 hours. (B) Kaplan-Meier curve (time until tumor sizes > 1cm3) of VCaP-CR xenograft tumors treated with DMAPT, castration, DMAPT + castration or vehicle control (n = 6–8 mice per group). (C) Ki-67, phos-p65, AR-V7 and AR IHC staining (scale bar 25 μm) and (D) quantification in VCaP-CR xenograft tumours at ethical endpoint (n = 6–8 mice per group, error bars +/−1SEM). (E) mRNA expression of AR-FL and AR-V7 in VCaP-CR xenograft tumors treated with DMAPT, castration, DMAPT + castration or vehicle control (n = 6–8 mice per group, error bars +/−1SEM). (F) AR-V7 and Cell cycle enrichment scores from RNA-seq of VCaP-CR xenograft tumors (n = 5 mice per group). * p<0.05, **p<0.01, ***p<0.001.
Increased NF-κB activity upregulates AR and AR-V7 expression.
To confirm that the observed co-upregulation of p65 NF-κB and AR/AR-V7 was not limited to the VCaP-CR cell line, we cultured mouse and human PCa cell lines (FVB-MyCCaP, LNCaP-95 and 22Rv1 respectively) that are known to express AR-V7 or AR-variants (Figure 2A) [30, 31] in enzalutamide. All three enzalutamide-treated lines showed significantly increased phosphorylated-p65 expression compared to the respective parental lines, along with increased AR and AR-variant expression. As was observed in prior VCaP-CR experiments, DMAPT treatment reversed AR and AR-V7 expression. Because DMAPT is also known to have biological effects that are not NF-κB-related, including induction of reactive oxygen species and STAT inhibition [32], we compared the effect of DMAPT treatment with genetic inhibition of NF-κB by generating cell lines with stable expression of an IκBα super repressor [33], which sequesters NF-κB in the cytoplasm. Both DMAPT treatment and the IκBα super repressor slowed cell proliferation relative to parental cell lines to a similar extent by 72 hours (LNCaP95 parental: p=0.0068 compared to DMAPT and p=0.0027 compared to IκBα SR; 22Rv1: p=0.0052 compared to DMAPT and p=0.0011 compared to IκBα SR; FVB-MYCCaP: p=0.0207 compared to DMAPT and p=0.0194 compared to IκBα SR, Figure 2B) and there was no significant difference in AR-variant mRNA expression between the chemical and genetic inhibitors (Figure 2C). This indicates that the anti-AR-variant effect of DMAPT is largely NF-κB-dependent. Given that the canonical NF-κB pathway, rather than non-canonical, is most commonly activated in cancer pathology, we hypothesize that NF-κB-driven upregulation of AR-variants is largely occurring through this canonical signaling cascade. As such we set out to test the reliance of this mechanism on p65.
Figure 2: Increased NF-kB activity upregulates AR and AR-V7 expression.
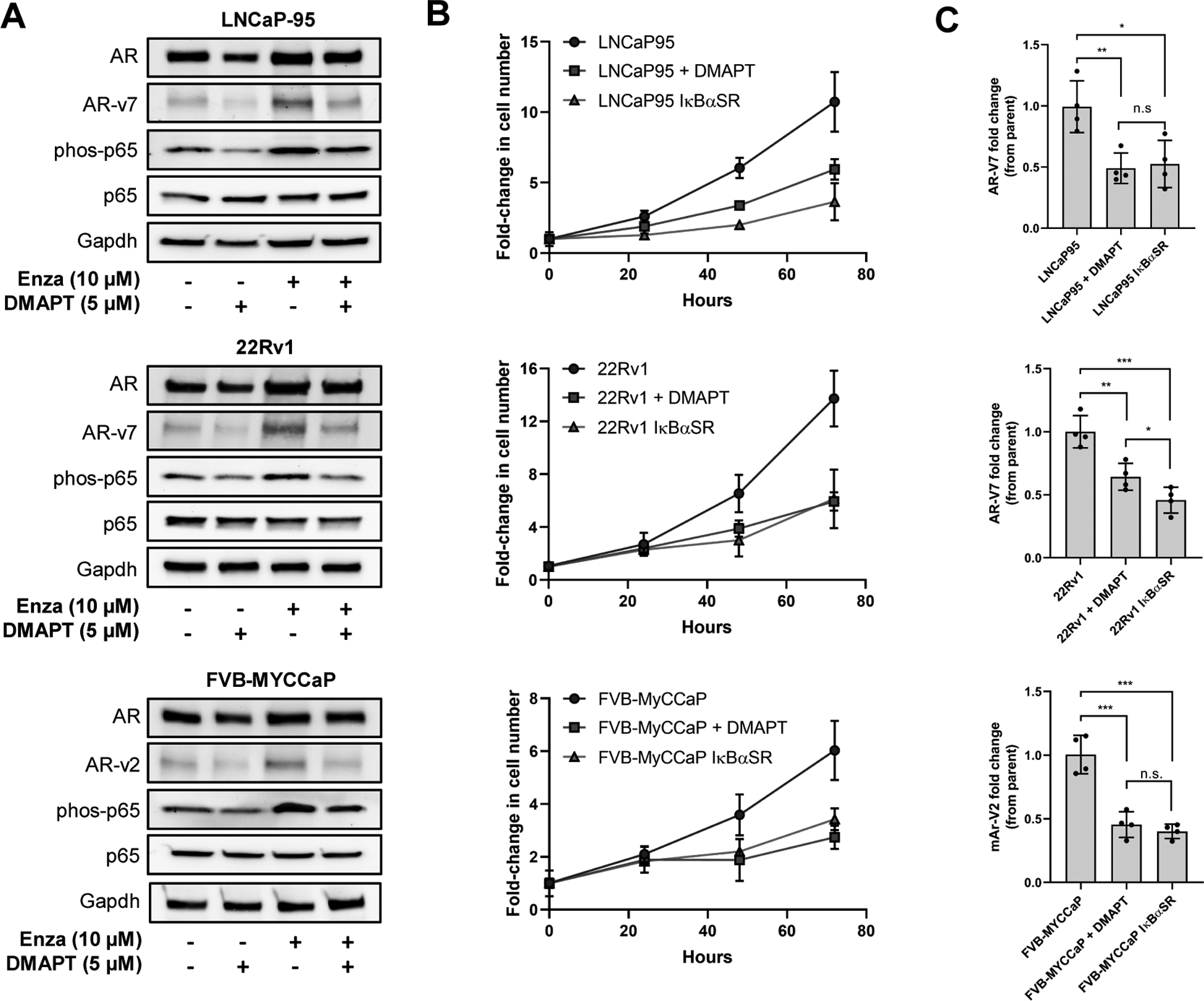
(A) Protein expression of AR, AR-V7, p65 and phos-p65 in human (LNCaP-95 and 22Rv1) and mouse (FVB-MyCCaP) PCa cell lines resistant to Enzalutamide (10 μM) and treated with DMAPT (5 μM) for 72 hours. (B) Proliferation growth curves and (C) AR-variant mRNA expression in human and mouse prostate cancer cell lines in the presence and absence of NF-κB inhibition (chemical – DMAPT, and generic – IκBa-super repressor) (n = 3 in technical triplicate, error bars +/−1SEM). * p<0.05, **p<0.01, ***p<0.001.
DMAPT mediated inhibition of AR-V7 expression and function is reversed by constitutive expression of p65.
To validate the dependence of the action of DMAPT on p65, we performed rescue experiments by generating cell lines stably expressing p65 using human LNCaP-95 and 22Rv1 PCa lines. In both cell lines, constitutive overexpression of p65 resulted in significant upregulation of AR-V7 protein expression compared to the parental and empty vector-containing cell lines (Figure 3A). In addition to upregulating AR-V7 protein expression, constitutive expression of p65 resulted in increased mRNA expression of variant and full-length transcripts (Figure 3B). The ability of DMAPT to reduce AR-V7 was significantly blunted in the cell lines constitutively overexpressing p65, indicating a potentially crucial role for p65 in NF-κB-driven castrate resistant disease. Expression of AR-V7 and AR-FL was not significantly different in the empty-vector-containing cell lines compared to the parental lines (Supplemental Figure 3). EDN2 and ETS2 have previously been shown to be specifically upregulated by AR-V7 [34]. We compared basal expression of both genes with and without p65 overexpression (Figure 3C). In both LNCaP-95 and 22Rv1 lines, constitutive p65 expression resulted in upregulation of END2 and ETS2. Interestingly, overexpression of p65 resulted in almost a 10-fold increase in expression of both genes in 22Rv1 cells, which might be due to the cell line having significantly higher basal expression of AR-V7, compared to LNCaP-95. In R1881-stimulated cells, DMAPT treatment significantly reduced expression of AR-target genes, FKBP5 and KLK3 (Figure 3D).
Figure 3: Constitutive overexpression of p65 blocks DMAPT-induced reduction of AR-V7 expression and function.
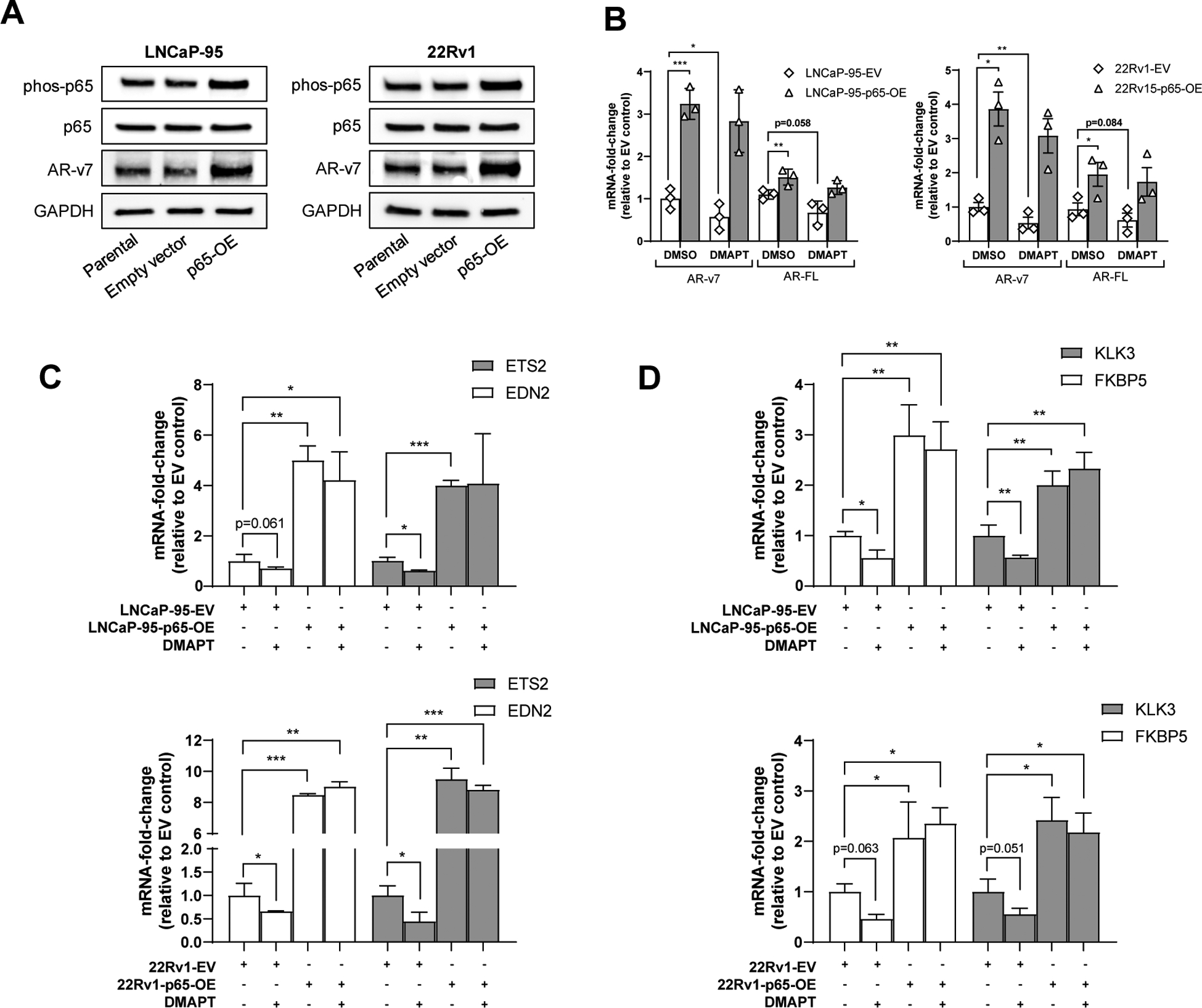
(A) Protein expression of phos-p65, p65 and AR-V7 by western blot comparing parental LNCaP-95 and 22Rv1 cell lines with lines stably expressing empty vector or p65 overexpression vector. (B) AR-V7 and AR-FL mRNA expression in LNCaP-95 and 22Rv1 cells overexpressing p65 compared to control cell lines (parental and empty vector) ± DMAPT treatment (5μM for 72 hours, n = 3 in technical triplicate, error bars +/−1SEM). (C) Basal expression of AR-V7 specific targets, ETS2 and EDN2, ± DMAPT treatment (5μM for 72 hours) ± p56 overexpression (n = 2 in technical triplicate and repeated once, error bars +/−1SEM). D) R1881-stimulated expression on AR targets, FKBP5 and KLK3 ± DMAPT treatment (5μM for 72 hours) ± p65 overexpression (n = 2 in technical triplicate and repeated once, error bars +/−1SEM). * p<0.05, **p<0.01, ***p<0.001.
Constitutive expression of p65 inhibits the anti-tumor action of DMAPT alone or in combination with castration.
Using the constitutively expressing p65 LNCaP-95 and 22Rv1 PCa cell lines cultured in 3D, we performed in vitro therapy experiments where tumoroid cultures were treated with DMAPT and/or Enzalutamide as singlet or doublet therapy (Figure 4 and Supplementary Figure 4). In empty vector-containing cell lines, DMAPT and Enzalutamide singlet treatment reduced cell survival by 47–66% and combination therapy reduced cell survival by 73–77% compared to DMSO vehicle. This result in two in vitro PCa tumoroid culture models matches the VCaP-CR in vivo data showing significant reduction in tumor growth in mice treated with DMAPT and surgical castration (Figure 1B). While some reduction of cell survival was still observed, constitutive overexpression of p65 significantly blunted anti-tumor activity of both singlet and doublet therapies. This result confirms the importance of the p65 NF-κB subunit for modulating the restoration of castration-sensitivity by DMAPT in PCa cells.
Figure 4: Constitutive overexpression of p65 inhibits the anti-tumour action of DMAPT alone or in combination with castration.
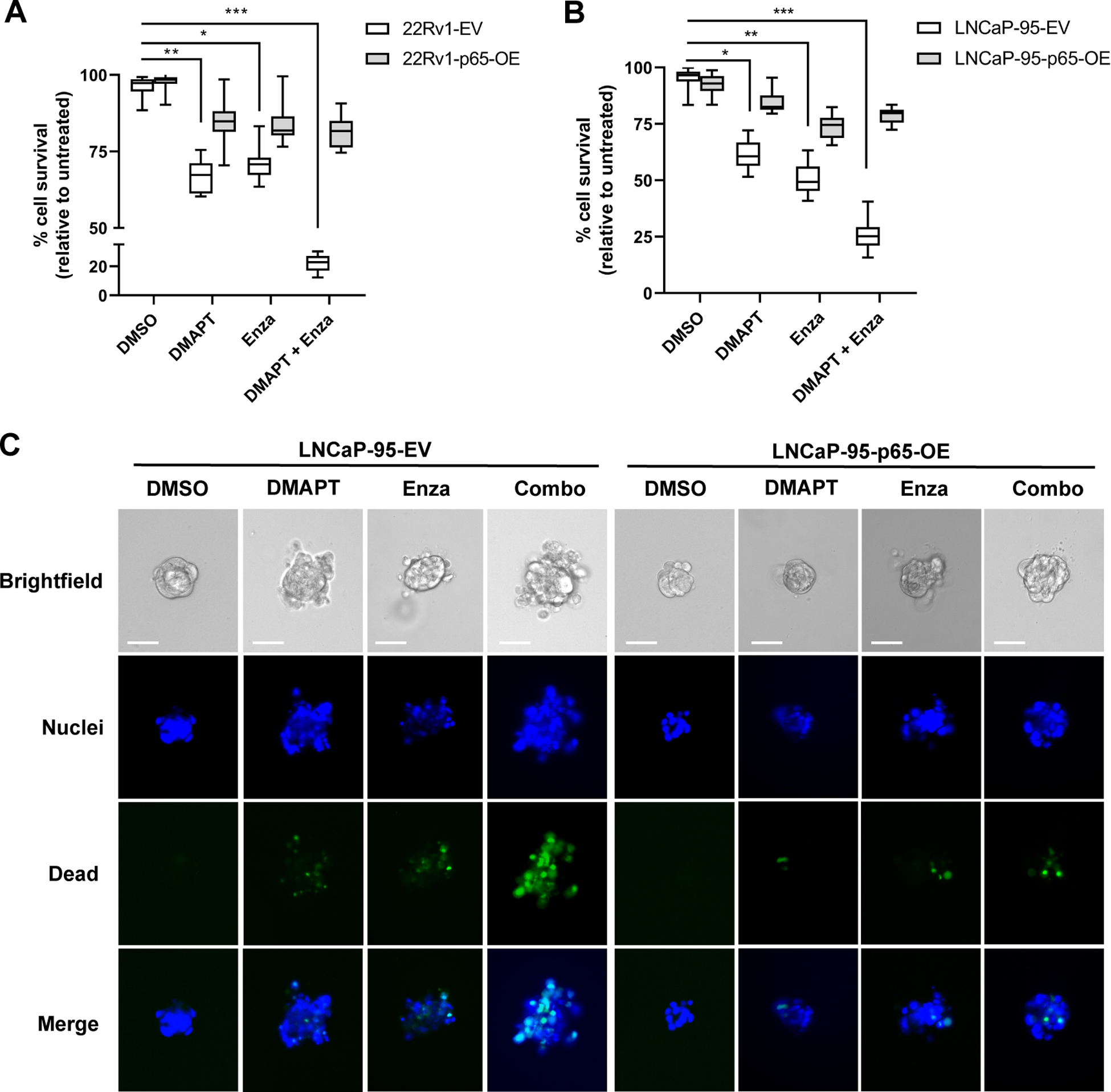
Cell survival relative to untreated parental lines of (A) LNCaP-95 cells and (B) 22Rv1 cells overexpressing p65 cultured in 3D and treated for 72 hours ± DMAPT (5 μM) ± Enzalutamide (10 μM), (n = 10 3D spheres per treatment group, +/−1SEM). (C) Representative images of cell death staining in treated LNCaP-95. Dead and total cells in tumoroid cultures were stained using ReadyProbes™ Cell Viability Imaging Kit. * p<0.05, **p<0.01, ***p<0.001.
Discussion
ADT has been the main treatment strategy for HSPC but unfortunately after an initial response to AR inhibition, progression to the highly aggressive state of mCRPC is often inevitable. Several mechanisms have been identified as key driving forces in resistance to ADT and the development of mCRPC, including, AR amplification, phosphorylation of AR, and expression of AR-V7 [35, 36]. It is well documented that treating mCRPC with single-agent therapies has limited durable benefit. Interestingly, it has previously been demonstrated that activation of the AR and splice variant signaling axis by p52 (non-canonical NF-κB signaling) can induce enzalutamide resistance [37], whereas here we have demonstrated that AR inhibition with enzalutamide or castration drives canonical NF-κB activation and subsequent AR-V7 upregulation. Recent work by Liang et al [38] has shown that IκKβ blockade prevented the emergence of enzalutamide resistance in prostate cancer in vivo models. The work by Liang and colleagues identify both BCL-2 and IκKβ dependencies in clinically relevant enzalutamide-resistant prostate cancer cells in vitro and in vivo and furthermore indicate the greater relevance of IκKβ upregulation in the progression of human castrate resistant prostate cancer. This further highlights the therapeutic benefit of DMAPT, which acts largely through the canonical NF-κB pathway, in reversing resistance to castration.
Significantly, our data demonstrate that expression of AR-V7, and to a lesser degree AR-FL, was significantly reduced in CRPC cells following DMAPT treatment, suggesting that DMAPT-mediated reduction in AR-V7 expression restores CRPC responsiveness to ADT. Therefore, we would suggest that utilizing a treatment plan combining DMAPT and AR antagonism might increase therapeutic efficacy and decrease resistance to AR inhibition in both HSPC and mCRPC. It is worth noting that DMAPT, and its less soluble parental compound parthenolide, have both also previously been demonstrated to be a novel inhibitor of PCa growth [22, 39]. NF-κB has been an appealing target for prostate cancer therapy for many years and numerous studies have shown some anti-cancer efficacy of NF-κB inhibitors. However, low bioavailability and toxicity at higher concentrations of such phytochemicals and proteasome inhibitors is a major challenge for clinical translation [40–42]. In this regard, DMAPT is of particular benefit as it has been developed to have increased bioavailability and has low toxicity in mice and humans at biologically relevant doses and is thought to be one of the only promising orally dosed clinical NF-κB inhibitors. The DMAPT clinical data to date in a 28 day pilot trial patients with hematological malignancies clinical trials has shown good PK-PD data and no adverse events as serum levels above 10 μM with inhibition of NF-κB in circulating blasts was observed and this bodes well for a planned prostate cancer trial.
These data highlight, the key importance of p65 in the restoration of sensitivity to AR inhibtion in mCRPC. Cell lines with constitutively active p65 did not respond to treatment with DMAPT, either alone or in combination with enzalutamide. This loss of therapeutic benefit closely aligned with high expression of AR-V7 or AR-FL. The results of this study would recommend the use of a combination therapy approach, targeting the androgen-AR axis and NF-κB/p65 signaling pathway to treat mCRPC and mHSPC by mitigating a cause of resistance to AR targeted therapy whether it be by suppression of androgens or direct AR inhibition. Given the encouraging early clinical data for DMAPT, it is our belief that this study provides strong preclinical justification for the use of DMAPT in the treatment of mCRPC and mHSPC and suggests the need for clinical trials combining DMAPT and AR antagonists in such PCa patients.
Supplementary Material
Implications.
Our study shows that DMAPT, an oral NF-κB inhibitor in clinical development, inhibits phosphorylated-p65 upregulation of AR-V7 and delays PCa castration resistance. This provides rationale for the development of DMAPT as a novel therapeutic strategy to increase durable response in patients receiving AR targeted therapy.
Acknowledgements
We would like to thank Stephen Plymate for the gift of the VCaP-CR cell line and Michelangelo Fiorentino for support with in vivo tissue analysis, as well as the Molecular Biology Core Facility (DFCI) for assistance with Illumina RNA sequencing. This work was supported by a U.S. Department of Defense (DOD) Idea Award (W81XWH-19-1-0564) to C.J.S.
Footnotes
Conflict of interest statement:
C. J. Sweeney is a stockholder in Leuchemix. All other authors declare no financial interests
References
- 1.Welch HG, Gorski DH, and Albertsen PC, Trends in Metastatic Breast and Prostate Cancer. N Engl J Med, 2016. 374(6): p. 596. [DOI] [PubMed] [Google Scholar]
- 2.Fujita K and Nonomura N, Role of Androgen Receptor in Prostate Cancer: A Review. World J Mens Health, 2019. 37(3): p. 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paschalis A, et al. , Alternative splicing in prostate cancer. Nat Rev Clin Oncol, 2018. 15(11): p. 663–675. [DOI] [PubMed] [Google Scholar]
- 4.Smith PC, et al. , Interleukin-6 and prostate cancer progression. Cytokine Growth Factor Rev, 2001. 12(1): p. 33–40. [DOI] [PubMed] [Google Scholar]
- 5.Basseres DS and Baldwin AS, Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene, 2006. 25(51): p. 6817–30. [DOI] [PubMed] [Google Scholar]
- 6.Karin M, Nuclear factor-kappaB in cancer development and progression. Nature, 2006. 441(7092): p. 431–6. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh S and Karin M, Missing pieces in the NF-kappaB puzzle. Cell, 2002. 109 Suppl: p. S81–96. [DOI] [PubMed] [Google Scholar]
- 8.Mattioli I, et al. , Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol, 2004. 172(10): p. 6336–44. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, et al. , NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am J Pathol, 2009. 175(2): p. 489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin R, et al. , Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS One, 2013. 8(4): p. e60983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin R, et al. , Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene, 2015. 34(28): p. 3700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song JM, et al. , Dimethylaminoparthenolide, a water soluble parthenolide, suppresses lung tumorigenesis through down-regulating the STAT3 signaling pathway. Curr Cancer Drug Targets, 2014. 14(1): p. 59–69. [DOI] [PubMed] [Google Scholar]
- 13.Kwok BH, et al. , The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IkappaB kinase. Chem Biol, 2001. 8(8): p. 759–66. [DOI] [PubMed] [Google Scholar]
- 14.Pei S, et al. , Rational design of a parthenolide-based drug regimen that selectively eradicates acute myelogenous leukemia stem cells. J Biol Chem, 2016. 291(48): p. 25280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei S, et al. , Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem, 2013. 288(47): p. 33542–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morel KL, et al. , Parthenolide Selectively Sensitizes Prostate Tumor Tissue to Radiotherapy while Protecting Healthy Tissues In Vivo. Radiat Res, 2017. 187(5): p. 501–512. [DOI] [PubMed] [Google Scholar]
- 17.Mendonca MS, et al. , DMAPT inhibits NF-kappaB activity and increases sensitivity of prostate cancer cells to X-rays in vitro and in tumor xenografts in vivo. Free Radic Biol Med, 2017. 112: p. 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deraska PV, et al. , NF-kappaB inhibition by dimethylaminoparthenolide radiosensitizes non-small-cell lung carcinoma by blocking DNA double-strand break repair. Cell Death Discov, 2018. 4: p. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estabrook NC, et al. , Inhibition of NF-kappaB and DNA double-strand break repair by DMAPT sensitizes non-small-cell lung cancers to X-rays. Free Radic Biol Med, 2011. 51(12): p. 2249–58. [DOI] [PubMed] [Google Scholar]
- 20.Shanmugam R, et al. , A water soluble parthenolide analog suppresses in vivo tumor growth of two tobacco-associated cancers, lung and bladder cancer, by targeting NF-kappaB and generating reactive oxygen species. Int J Cancer, 2011. 128(10): p. 2481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel KL, et al. , DMAPT is an Effective Radioprotector from Long-Term Radiation-Induced Damage to Normal Mouse Tissues In Vivo. Radiat Res, 2019. 192(2): p. 231–239. [DOI] [PubMed] [Google Scholar]
- 22.Morel KL, et al. , Chronic low dose ethanol induces an aggressive metastatic phenotype in TRAMP mice, which is counteracted by parthenolide. Clin Exp Metastasis, 2018. 35(7): p. 649–661. [DOI] [PubMed] [Google Scholar]
- 23.Morel KL, et al. EZH2 inhibition activates a dsRNA–STING–interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer, 2021, 10.1038/s43018-021-00185-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bankhead P, et al. , QuPath: Open source software for digital pathology image analysis. Sci Rep, 2017. 7(1): p. 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carpenter AE, et al. , CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol, 2006. 7(10): p. R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCall P, et al. , NFkappaB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J Cancer, 2012. 107(9): p. 1554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holcomb BK, et al. , Dimethylamino parthenolide enhances the inhibitory effects of gemcitabine in human pancreatic cancer cells. J Gastrointest Surg, 2012. 16(7): p. 1333–40. [DOI] [PubMed] [Google Scholar]
- 28.Sharp A, et al. , Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest, 2019. 129(1): p. 192–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Austin DC, et al. , NF-kappaB and androgen receptor variant expression correlate with human BPH progression. Prostate, 2016. 76(5): p. 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu R, et al. , Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res, 2009. 69(1): p. 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson PA, et al. , Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res, 2005. 65(24): p. 11565–71. [DOI] [PubMed] [Google Scholar]
- 32.Mathema VB, et al. , Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation, 2012. 35(2): p. 560–5. [DOI] [PubMed] [Google Scholar]
- 33.Boehm JS, et al. , Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell, 2007. 129(6): p. 1065–79. [DOI] [PubMed] [Google Scholar]
- 34.Krause WC, et al. , Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol, 2014. 54: p. 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonarakis ES, et al. , AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med, 2014. 371(11): p. 1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferraldeschi R, et al. , Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene, 2015. 34(14): p. 1745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nadiminty N, et al. , NF-kappaB2/p52 enhances androgen-independent growth of human LNCaP cells via protection from apoptotic cell death and cell cycle arrest induced by androgen-deprivation. Prostate, 2008. 68(16): p. 1725–33. [DOI] [PubMed] [Google Scholar]
- 38.Liang Y, et al. , Emergence of Enzalutamide Resistance in Prostate Cancer is Associated with BCL-2 and IKKB Dependencies. Clin Cancer Res, 2021. [DOI] [PubMed] [Google Scholar]
- 39.Kawasaki BT, et al. , Effects of the sesquiterpene lactone parthenolide on prostate tumor-initiating cells: An integrated molecular profiling approach. Prostate, 2009. 69(8): p. 827–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storka A, et al. , Safety, tolerability and pharmacokinetics of liposomal curcumin in healthy humans. Int J Clin Pharmacol Ther, 2015. 53(1): p. 54–65. [DOI] [PubMed] [Google Scholar]
- 41.Soni K and Kohli K, Sulforaphane-decorated gold nanoparticle for anti-cancer activity: in vitro and in vivo studies. Pharm Dev Technol, 2019. 24(4): p. 427–438. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, et al. , Plants vs. cancer: a review on natural phytochemicals in preventing and treating cancers and their druggability. Anticancer Agents Med Chem, 2012. 12(10): p. 1281–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.